

Abstract
We recently described a new neurodevelopmental syndrome (TAF1/MRXS33 intellectual disability [ID] syndrome) (MIM# 300966) caused by pathogenic variants involving the X-linked gene TATA-box binding protein associated factor 1 (TAF1), which participates in RNA polymerase II transcription. The initial study reported 11 families, and the syndrome was defined as presenting early in life with hypotonia, facial dysmorphia, and developmental delay that evolved into ID and/or autism spectrum disorder. We have now identified an additional 27 families through a genotype-first approach. Familial segregation analysis, clinical phenotyping, and bioinformatics were capitalized on to assess potential variant pathogenicity, and molecular modeling was performed for those variants falling within structurally characterized domains of TAF1. A novel phenotypic clustering approach was also applied, in which the phenotypes of affected individuals were classified using 51 standardized Human Phenotype Ontology terms. Phenotypes associated with TAF1 variants show considerable pleiotropy and clinical variability, but prominent among previously unreported effects were brain morphological abnormalities, seizures, hearing loss, and heart malformations. Our allelic series broadens the phenotypic spectrum of the TAF1/MRXS33 ID syndrome and the range of TAF1 molecular defects in humans. It also illustrates the challenges for determining the pathogenicity of inherited missense variants, particularly for a gene mapping to chromosome X.
Keywords: Cornelia de Lange, exome sequencing, MRXS33 intellectual disability syndrome, TAF1, transcriptomopathy
1 |. INTRODUCTION
We recently identified individuals from nine families in which missense or splice-site variants in the X-linked gene TAF1 were associated with hypotonia, developmental delay (DD), and facial dysmorphia, followed by later diagnoses of intellectual disability (ID), autism spectrum disorder (ASD), or both (Hu et al., 2016; O’Rawe et al., 2015). Given the limited number of subjects, the large size of the gene (1,893 amino acids), and the fact that TATA-box binding protein-associated factor 1 (TAF1) is centrally involved in global RNA polymerase II (pol II) transcription, we hypothesized that there could be a wider range of phenotypes associated with mutations in different regions of the TAF1 protein and that missense variant alleles mapping outside functional domains might have less detrimental phenotypic consequences. The very size and complexity of the protein, however, make it challenging to dissect the effects of missense mutations. This syndrome has also been designated as X-linked syndromic mental retardation-33 (MRXS33; MIM: 300966), caused by mutation in TAF1 mapping to chromosome Xq13.
TAF1 encodes the largest subunit of the basal transcription factor II D (TFIID), which directs the assembly of the pol II preinitiation complex (Papai, Weil, & Schultz, 2011) and is likely required for all pol II gene promoters (Warfield et al., 2017). Human TFIID is composed of TATA-binding protein (TBP) in association with 13 TBP-associated factors (TAFs). The TAF1’s N-terminal TAND region interacts with TBP to inhibit its DNA-binding activity; a central region interacts with the TAF7 protein; a winged-helix and a zinc knuckle domain can interact with DNA; and the C-terminus contains tandem bromodomains (BrDs) that bind to acetylated histone tails (Jacobson, Ladurner, King, & Tjian, 2000; Vermeulen et al., 2007), which are present at active promoters. TAF1 is extremely conserved in evolution. To date, only missense variants have been found; the lack of hemi- and homozygous loss-of-function variants in the protein coding part of the canonical TAF1 isoform in human population databases suggests that the complete loss of TAF1 may be embryonic-lethal. This is supported by a recent study in which complete loss of TAF1 causes embryonic lethality in zebrafish (Gudmundsson et al., 2019). Some recent data have suggested that TAF1 might be involved in altering the morphology and function of the cerebellum and cerebral cortex (Janakiraman et al., 2019).
To acquire a more expansive allelic series, we used a genotype-first approach to identify more individuals with TAF1 variants from 27 unrelated families. This is the largest cohort of TAF1/MRXS33 ID syndrome cases amassed to date; we then applied computational algorithms and modeling approaches to interpret variant pathogenicity, including phenotypic clustering analysis. Despite this panoply of tools implemented, we were able to ascribe pathogenicity with confidence to only eleven of the identified de novo missense rare variant alleles, with nine in males and two in females. The following results exemplify the strengths and limitations of current approaches to determining pathogenicity for missense variants.
2 |. MATERIALS AND METHODS
2.1 |. Clinical characterization
Twenty-seven families were identified through a collaboration between 15 institutions in seven countries (Table 1). Most individuals were initially referred to clinics for the investigation of idiopathic-DD and/or ID. Individuals 3, 9, 12, and 22 were identified and sequenced through the Deciphering Developmental Disorders (DDD) study (Wright, FitzPatrick, & Firth, 2018). Individual 21 was identified in a neonatal/pediatric intensive care unit through rapid turnaround time genomic testing for patients thought likely to have an underlying genetic condition. Preliminary phenotypic information was obtained from clinical records, which ranged in level of detail from a list of key clinical features to detailed history and examination findings. Further clinical information was obtained via email communications with the probands’ parents, physicians, or both. Clinical features for each subject are summarized in Supporting Information File S1. The study was performed in accordance with protocols approved by the institutional review boards of the participating institutions. Some cases were sequenced on clinical grounds, with retrospective chart review by the participating clinician. Written informed consent was obtained for publication of photographs in all cases. Patient clinical data have been obtained in a manner conforming with IRB and/or granting agency ethical guidelines.
TABLE 1.
TAF1 variants in 24 Individuals from 22 families
| Individual ID | Gender | Inheritance | Nucleotide change (NM_004606.3) | Predicted amino acid change | Genomic coordinates ChrX(GRCh37) | CADD score | MPC score | Classification |
|---|---|---|---|---|---|---|---|---|
| 1 | Male | Maternal | c.613A>G | p.(Ser205Gly) | g.70596880A>G | 23.7 | 1.73 | Uncertain |
| 2 | Male | Maternal | c.862C>T | p.(Arg288Cys) | g.70597540C>T | 27.4 | 1.93 | Likely benign |
| 3 | Male | Maternal | c.952G>A | p.(Ala318Thr) | g.70597630G>A | 26.8 | 1.78 | Uncertain |
| 4 | Male | Maternal | c.1297G>A | p.(Asp433Asn) | g.70598758G>A | 23.6 | 0.92 | Uncertain |
| 5 | Male | De novo | c.1580A>G | p.(Asp527Gly) | g.70601752A>G | 29.3 | 2.08 | Likely pathogenic |
| 6 | Female | De novo (skewed X inactivation) | c.2039G>A | p.(Gly680Asp) | g.70603843G>A | 28.5 | 2.08 | Likely pathogenic |
| 7 | Male | Maternal | c.2180G>C | p.(Arg727Pro) | g.70603984G>C | 35 | 2.11 | Uncertain |
| 8 | Male | Maternal (skewed X inactivation in mother) | c.2617T>G | p.(Phe873Val) | g.70608216T>G | 26.2 | 3.25 | Uncertain |
| 9 | Male | De novo | c.2668C>T | p.(Arg890Cys) | g.70608626C>T | 34 | 3.33 | Likely pathogenic |
| 10 | Male | Maternal | c.2833G>A | p.(Asp945Asn) | g.70609507G>A | 29.2 | 2.50 | Uncertain |
| 11 | Male | De novo | c.2954C>T | p.(Ser985Phe) | g.70612531C>T | 28.6 | 2.91 | Likely pathogenic |
| 12 | Female | De novo (skewed X inactivation) | c.3035C>T | p.(Thr1012Ile) | g.70612768C>T | 27.2 | 2.81 | Likely pathogenic |
| 13 | Male | De novo | c.3568C>T | p.(Arg1190Cys) | g.70617204C>T | 33 | 3.14 | Likely pathogenic |
| 14 (1) | Male | Maternal | c.3760C>T | p.(Arg1254Trp) | g.70618501C>T | 26.2 | 2.80 | Likely pathogenic |
| 14 (2) | Male | De novo | c.3760C>T | p.(Arg1254Trp) | g.70618501C>T | 26.2 | 2.80 | Likely pathogenic |
| 15 | Male | De novo | c.4033G>A | p.(Val1345Ile) | g.70621564G>A | 26.2 | 1.55 | Likely pathogenic |
| 16 | Male | Maternal | c.4052T>A | p.(Ile1351Asn) | g.70621583T>A | 28.5 | 3.96 | Uncertain |
| 17 (1) | Male | Maternal (skewed X inactivation in mother) | c.4190G>A | p.(Arg1397Gln) | g.70627446G>A | 24.1 | 2.09 | Uncertain |
| 17 (2) | Male | Maternal (skewed X inactivation in mother) | c.4190G>A | p.(Arg1397Gln) | g.70627446G>A | 24.1 | 2.09 | Uncertain |
| 18 | Male | De novo | c.4442A>T | p.(Asn1481Ile) | g.70627999A>T | 31 | 3.15 | Likely pathogenic |
| 19 | Male | Maternal (skewed X inactivation in mother) | c.4442A>G | p.(Asn1481Ser) | g.70627999A>G | 25.3 | 2.51 | Uncertain |
| 20 | Male | De novo | c.4454A>G | p.(His1485Arg) | g.70641168A>G | 23.5 | 2.69 | Likely pathogenic |
| 21 | Male | De novo | c.4580C>T | p.(Ala1527Val) | g.70643034C>T | 27.7 | 2.23 | Likely pathogenic |
| 22 | Male | Maternal | c.4726A>G | p.(Lys1576Glu) | g.70643914A>G | 24 | 2.80 | Uncertain |
| BENIGN 1 | Male | Maternal, present in unaffected brother | c.1251_1253del | p.(Leu418del) | g.70598712_70598714del | 22.6 | 0.63 | Likely benign |
| BENIGN 2 | Male | Maternal, absent in brother with quadriplegia | c.1825A>G | p.(Ile609Val) | g.70602710A>G | 22.6 | 0.63 | Likely benign |
| BENIGN 3 | Male | Questionable: found in unaffected father | c.2365A>G | p.(Arg789Gly) | g.70607189A>G | 23.7 | 1.15 | Likely benign |
| BENIGN 4 | Male | Maternal, not found in a separate affected sibling | c.5364G>C | p.(Glu1788Asp) | g.70680558G>C | 14.95 | 0.94 | Likely benign |
| BENIGN 5 | Male | Maternal, seen in gnomAD in 3 males (0.0021%) | c.5659A>T | p.(Ser1887Cys) | g.70683873A>T | 27.8 | 0.86 | Likely benign |
Abbreviations: CADD, combined annotation dependent depletion.
Each individual’s phenotypic features were correlated to a Human Phenotype Ontology (HPO) ID number with the aid of the Human Ontology Browser and PhenoTips. Both positive and negative features were noted, with phenotypic features classified as pertinent negatives if this was explicitly mentioned in the records; otherwise, they were classified as “unknown.” The relative prevalence of each phenotype was calculated by dividing the number of individuals positive for the phenotype by the sample size. For the quantitative endophenotypic traits of head circumference, weight and height, percentiles were calculated using CDC growth charts.
2.2 |. Bioinformatics methodology
Variants were identified using exome sequencing (ES) or genome sequencing (GS) primarily through clinical diagnostic testing. The sequencing kits and technology varied according to the laboratories/companies involved (see Supporting Information File S1), and the variants of interest were highlighted in the molecular diagnostic test reports. For those individuals sequenced at GeneDx, the method analyzed trios using genomic DNA from the proband and both parents, and the exonic regions and flanking splice junctions of the genome were captured using the SureSelect Human All Exon V4 (50 Mb) (Individual 18), the Clinical Research Exome Kit (Agilent Technologies, Santa Clara, CA) (Individuals 4, 11, 15, and 20) or the IDT xGen Exome Research Panel v1.0 (Individual 1). Massively parallel (NextGen) sequencing was done on an Illumina system with 100 bp or greater paired-end reads. Reads were aligned to the human genome build GRCh37/UCSC hg19 and analyzed for sequence variants using a custom-developed analysis tool. Additional details of the sequencing technology and variant interpretation protocol have been previously described (Retterer et al., 2016). The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/).
Variants were confirmed by Sanger sequencing and parental testing was carried out where possible. Polyphen-2 and SIFT scores were retrieved from the dbNSFP33a database (Liu, Wu, Li, & Boerwinkle, 2016). For the combined annotation dependent depletion (CADD) scores, we used the CADD model GRCh37-v1.4. Missense severity, PolyPhen-2, and Constraint (MPC) scores–retrieved for each variant using the official MPC values provided by the authors (Samocha et al., 2017)–were interpreted and classified according to American College of Medical Genetics (ACMG) 2015 Guidelines (Richards et al., 2015). Variants were submitted to the ClinVar database (SUB6338091).
2.3 |. X-chromosome inactivation (XCI) assay
XCI was assessed for degree of skewing by a standard clinical assay at different clinical sites, as previously described (Allen, Zoghbi, Moseley, Rosenblatt, & Belmont, 1992) utilizing the HUMARA locus at Xq12, with TAF1 in close linkage at Xq13.1.
2.4 |. HPO analyzes
Table S3 summarizes the observed phenotypes for 24 affected individuals using 51 standardized phenotype terms in HPO (Köhler et al., 2017). To explore the relationships between affected individuals, these data were analyzed using scipy and scikit-learn, the software tools in Python to perform various machine-learning tasks. First, these data were transformed into a matrix with 24 rows and 51 columns. Each number in the matrix is either 1 (for an individual having the corresponding clinical feature) or 0 (for an individual not having the corresponding clinical feature), multiplied by the information content (Sánchez, Batet, & Isern, 2011) of the corresponding HPO terms. Then, the matrix is transformed into 24 rows by 24 columns using principal component analysis and subjected to hierarchical clustering. Euclidean distance was used as the metric among the 24 individuals, and 3.7 was the threshold Euclidean distance to form a cluster. Ward’s Method was used as the linkage criterion in hierarchical clustering.
2.5 |. Molecular modeling
TAF1 variants located in the TAF1-TAF7 interaction domain were mapped using the human TAF1 DUF3591-TAF7 crystal structure (resolution: 2.3 Å; PDB entry: 4RGW; Wang, Curran, Hinds, Wang, & Zheng, 2014), based on the TAF1 reference sequence NM_004606.4. Variants located in the TAF1 BrDs were mapped using the human TAF1 crystal structure (resolution: 2.1 Å; PDB entry: 1EQF; Jacobson et al., 2000), based on the reference sequence NM_138923.3. The double BrD sequence NM_138923.3 was aligned with the reference NM_004606.4 and amino acids were labeled accordingly.
We used YASARA View (Krieger, Koraimann, & Vriend, 2002) to map the mutated residues on the corresponding structures, which are shown using a ribbon representation overlaid with the molecular surface. Hydrogen atoms have been added to the structures to delineate hydrogen bonds. Atoms are shown only for the mapped residues and their interacting amino acids.
Free energy difference between the folded and unfolded protein (DG) was analyzed using FoldX (Van Durme et al., 2011). After energy minimization, protein stability was expressed as the free energy difference between the wild-type and the mutant protein (DDG). A variant is considered to destabilize the overall structure when the DDG is >0. The error margin of the FoldX stability calculation is 0.5 kcal/mol, so differences in this range are not significant.
3 |. RESULTS
3.1 |. TAF1 variants
Since the publication of the original cohort (O’Rawe et al., 2015), we gathered 27 unrelated families with missense variants in TAF1 who were referred for evaluation. Five TAF1 variants were initially assessed as likely benign (LB) based on familial segregation analysis and in silico prediction tool results (Tables 1 and S1 and Supporting Information File S1). The remaining 22 families were then numbered based on the order of the amino acid affected from the N- to C-terminus of TAF1 (Figure 1 and Table 1). In silico prediction analysis results regarding these 22 variants is provided as Table S1, with two families having two affected individuals, for a total of 24 individuals.
FIGURE 1.
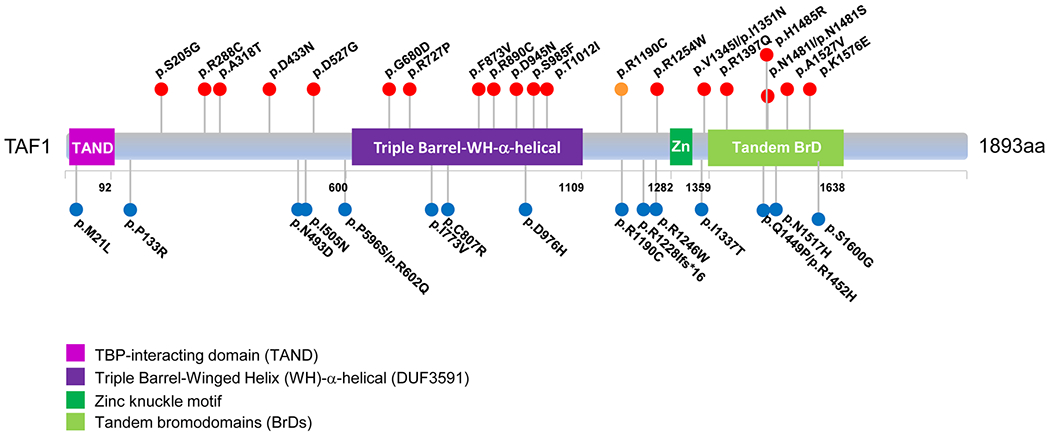
TAF1 functional motifs and the variants identified in the study cohort. Solid red circles in the top indicate new variants identified in this study, and the solid orange circle indicates a previously reported variant that is also present in the current study cohort. Solid blue circles listed below indicate previously reported variants. Boxed parts indicate structural TAF1 domains of known functions (RefSeq: NM_004606.4). TAF1, TATA-box binding protein associated factor 1
These new variants are shown in Figure 1, along with the published variants (Deciphering Developmental Disorders Study, 2017; He et al., 2015; Hu et al., 2016; Kahrizi et al., 2019; Kosmicki et al., 2017; Niranjan et al., 2015; O’Rawe et al., 2015; Okamoto, Arai, Onishi, Miyake, & Matsumoto, 2019), including those currently annotated in the Human Genome Mutation Database v.2019.1 (Stenson et al., 2014). Twelve variants were maternally inherited; ten variants were de novo, including two identified in female patients (Family 6 and 12), with significantly skewed XCI (>90:10) in the one female (Individual 6) who could be tested. XCI studies were carried out for the mother of the two affected brothers in Family 17 and for the mothers in Families 8 and 19, showing skewing (97:3, 100:0, and 92:8, respectively). The mother in Family 3 had random X-inactivation. When performing the assay on Individual 8’s mother, the same assay was conducted with Individual 8’s DNA, which revealed that the allele transmitted to her son was the one preferentially inactivated in the mother, indicating that the direction of skewing favored the normal allele in this mother. The other XCI assays were performed on a clinical basis only on the maternal DNA, so the direction of skewing was not ascertained for the mothers in Families 17 or 19.
None of these 22 variants were reported in males in the gnomAD population variant database. Only the variants in Individuals 2 and 4 and Family 17 were present in heterozygous females, and in only 1–3 females in gnomAD for each variant (Table S1). Although the variant p.Ala318Thr in Individual 3 was not present in gnomAD, it was noted that the variant p.Ala318Gly was present with minor allele frequency (MAF = 0.416%), in 11 homozygous females and in 212 hemizygous males (Table S1). This was by far the most common same-site alteration in gnomAD among these 22 TAF1 variants, and the p.Ala318Thr variant changes a hydrophobic amino acid (Ala) to a polar uncharged amino acid (Thr), which is predicted by Polyphen-2 to be possibly damaging, in comparison to a p.Ala318Gly change (Table S1). All variants resulted in substitutions affecting highly conserved nucleotide and amino acid sequences (Table S1); CADD-scores were all above 23 (Kircher et al., 2014; Rentzsch, Witten, Cooper, Shendure, & Kircher, 2019), and MPC (Samocha et al., 2017) scores were ≥2 in most of them, except those in Individuals 1, 2, 3, 4, and 15. (Table S1). SIFT, Polyphen, gDNA phastCons, and phyloP provided supportive evidence for pathogenicity for these variants (Table S1). We classified variants according to the ACMG and Genomics and the Association for Molecular Pathology (ACMG/AMP) framework (Richards et al., 2015), using Intervar (Li & Wang, 2017), which rendered a “Likely Pathogenic” diagnosis for 11 of them, including 10 of these that were de novo and one of them that was maternally inherited (Family 14) (Tables 1 and S1). The remaining 11 maternally inherited variants were initially classified as variants of uncertain significance (VUS), as there was insufficient evidence to confirm pathogenicity. In this regard, while this manuscript was under peer review, additional segregation studies were performed on Family 2, which showed that the maternal uncle of the proband has the variant and is a healthy adult, thus indicating that this variant is likely benign.
Prior genetic analysis of these research participants had included karyotyping, Fragile X testing, and/or chromosome microarray testing, with these results being normal in the vast majority of individuals in which this information was available (exceptions are listed in Table S2, see also Supporting Information File S1). This includes a paternally inherited 15q11.2 microdeletion (father is unaffected) in Individual 14-1 but a normal microarray in the affected uncle 14-2, and a de novo terminal deletion of at least 3.6 Mb extending from cytogenetic band 1p36.33 to 1p36.32, associated with the 1p36 microdeletion syndrome, in Individual 15. Other variants of interest that were found during the course of ES or GS are listed in Table S2, with the most notable among these being: (a) a RAC1 de novo variant, c.116A>G p.(Asn39Ser) in Individual 3, with in silico modeling, mouse fibroblasts spreading assays, and in vivo overexpression assays in zebrafish having demonstrated that this variant functions as a dominant-negative allele, resulting in microcephaly, reduced neuronal proliferation, and cerebellar abnormalities in vivo (Reijnders et al., 2017); (b) a HNRNPU de novo c.837_839delAGA variant in Individual 11, leading to deletion of glutamic acid at position 279, with LOF mutations associated with ID (Bramswig et al., 2017; Hamdan et al., 2014; Leduc et al., 2017; Yates et al., 2017); and (c) a genetically confirmed diagnosis of mucopolysac-charidosis type IIIA–Sanfilippo syndrome due to compound heterozygous mutations in SGSH in Individual 14-1. It is well known that multiple molecular diagnoses can be found during the course of ES or GS, and it is likely that the clinical presentation is a blended phenotype in any given individual (Karaca et al., 2018; Posey et al., 2017). To be conservative in our assignment of “pathogenicity” using the ACMG/AMP framework (Richards et al., 2015), we have included for these cases the criterion of “BP5-Variant found in a case with an alternate molecular basis for disease” in Table S1. For Family 3 in particular, the presence of the RAC1 de novo variant, in conjunction with a random XCI pattern, casts doubt on possible pathogenicity for this TAF1 variant.
We also assessed the overall frequency of variants in TAF1 that might be involved in neurodevelopmental delay by querying the clinical databases of two large providers of clinical ES, namely GeneDx and Baylor Genetics (BG). In GeneDx, 18,256 probands with a referral indication of neurodevelopmental delay were sequenced by 26th March 2019, of which 71 cases were reported as having a VUS (n = 58), a likely pathogenic variant (n = 12), or a likely benign variant (n = 1) in TAF1. Zero cases were reported as “pathogenic.” BG had sequenced ~8,100 probands with neurodevelopmental deficits as of January 2019 and identified 15 TAF1 variants, classified as one likely pathogenic and 14 VUS.
3.2 |. Clinical features
Clinical features are organized by system and summarized in Table 2 (see also Table S2 and Supporting Information File S1). The prevalence estimates are calculated with a denominator of all 24 cases from the 22 families in which there had initially been some suspicion that these variants might be pathogenic (thus likely an underestimate given that some of the variants might turn out to be benign, such as what happened with Family 2).
TABLE 2.
Phenotypic findings observed in 24 individuals with TAF1 variants from 22 families
| HPO ID | Phenotype | Positive count | Prevalence (% positive/(negative + unknown) |
|---|---|---|---|
| Developmental | |||
| HP:0001263 | Global developmental delay | 23 | 95.8 |
| HP:0000750 | Delayed speech and language development | 22 | 91.7 |
| HP:0002194 | Delayed gross motor development | 12 | 66.7 |
| HP:0001288 | Gait disturbance | 7 | 29.2 |
| HP:0007010 | Poor fine motor coordination | 2 | 8.3 |
| Neurological | |||
| HP:0001249 | Intellectual disability | 14 | 63.7 |
| HP:0001250 | Seizures | 6 | 25.0 |
| HP:0002079 | Hypoplasia of the corpus callosum | 5 | 22.7 |
| HP:0002119 | Ventriculomegaly | 5 | 22.7 |
| Behavioral | |||
| HP:0000729 | Autistic behavior | 4 | 16.7 |
| HP:0100716 | Self-injurious behavior | 3 | 12.5 |
| HP:0100023 | Recurrent hand flapping | 2 | 8.3 |
| Feeding | |||
| HP:0011968 | Feeding difficulties | 15 | 62.5 |
| HP:0002020 | Gastroesophageal reflux | 5 | 20.8 |
| Growth | |||
| HP:0001511 | Intrauterine growth retardation | 7 | 29.2 |
| HP:0008897 | Postnatal growth retardation | 7 | 29.2 |
| Craniofacial | |||
| HP:0000252 | Microcephaly | 10 | 41.7 |
| HP:0000219 | Thin upper lip vermilion | 6 | 25 |
| HP:0000316 | Hypertelorism | 5 | 20.8 |
| HP:0000278 | Retrognathia | 5 | 20.8 |
| HP:0000490 | Deep-set eye | 5 | 20.8 |
| HP:0000414 | Bulbous nose | 5 | 20.8 |
| HP:0000486 | Strabismus | 5 | 20.8 |
| HP:0000431 | Wide nasal bridge | 4 | 16.7 |
| HP:0005469 | Flat occiput | 4 | 16.7 |
| HP:0000218 | High palate | 3 | 16.7 |
| HP:0000343 | Long philtrum | 3 | 12.5 |
| HP:0002307 | Drooling | 3 | 12.5 |
| HP:0000463 | Anteverted nares | 2 | 8.3 |
| Hearing | |||
| HP:0000407 | Sensorineural hearing impairment | 5 | 20.8 |
| HP:0000410 | Mixed hearing impairment | 2 | 8.3 |
| Musculoskeletal | |||
| HP:0001290 | Generalized hypotonia | 17 | 70.8 |
| HP:0004322 | Short stature | 10 | 41.7 |
| HP:0000960 | Sacral dimple | 8 | 33.3 |
| HP:0000921 | Missing ribs | 4 | 16.7 |
| HP:0002650 | Scoliosis | 3 | 12.5 |
| HP:0001763 | Pes planus | 3 | 12.5 |
| HP:0001382 | Joint hypermobility | 2 | 8.3 |
| HP:0000768 | Pectus carinatum | 2 | 8.3 |
| HP:0000767 | Pectus excavatum | 2 | 8.3 |
| HP:0002808 | Kyphosis | 2 | 8.3 |
| HP:0001385 | Hip dysplasia | 2 | 8.3 |
| HP:0001770 | Toe syndactyly | 1 | 4.2 |
| HP:0010442 | Polydactyly | 1 | 4.2 |
| HP:0001156 | Brachydactyly | 1 | 4.2 |
| Cardiac | |||
| HP:0001629 | Ventricular septal defect | 6 | 25.0 |
| HP:0001631 | Atrial septal defect | 2 | 4.2 |
| HP:0001680 | Coarctation of the Aorta | 1 | 4.2 |
| HP:0001636 | Tetralogy of Fallot | 1 | 4.2 |
| Genitourinary | |||
| HP:0000028 | Cryptorchidism | 7 | 29.2 |
| HP:0000047 | Hypospadias | 4 | 16.7 |
Abbreviations: HPO, Human Phenotype Ontology; TAF1, TATA-box binding protein associated factor 1.
In keeping with the previously reported patients with TAF1/MRXS33 ID syndrome (O’Rawe et al., 2015), the majority of this cohort presented with hypotonia and DD during infancy, followed by later diagnoses of ID and/or ASD. Delayed speech and language development were common. Dysmorphic facial features, which are particularly prone to observer bias, were more variable, but many patients were noted to have prominent supraorbital ridges, low-set and protruding ears, and a prominent (sometimes anteverted) nasal tip (Figure 2). Roughly a third of the subjects have a sacral dimple. Some form of gait disturbance was present in around a third as well. Other less common features included seizures, hearing loss, strabismus, and cardiac and genitourinary anomalies. Digital anomalies (including brachydactyly and preaxial polydactyly) were observed in three cases (Table 2).
FIGURE 2.

Photographs of individuals with TAF1 variants. Facial features are quite variable, but many individuals had prominent supraorbital ridges, low-set and protruding ears, and usually a prominent (and sometimes anteverted) nasal tip. TAF1, TATA-box binding protein associated factor 1
3.3 |. Clustering of affected individuals by HPO terms
The phenotypes of 24 affected individuals were classified using 51 standardized terms in HPO (Köhler et al., 2017; Table S2). Hierarchical clustering was implemented (see Methods), and as a result, the 24 individuals were divided into four main clusters (Figure S1). Cluster 1 had six individuals (most among four clusters) with a TAF1 variant interpreted as “pathogenic” by ACMG/AMP criteria. The individuals from Families 3 and 22 were in Cluster 1 with TAF1 variants with “uncertain significance” according to ACMG/AMP criteria. Based on the phenotype-clustering analysis, the TAF1 variants in Families 3 and 22 might be considered likely pathogenic, although we still rate them as “uncertain” (Table S1), as this kind of analysis is exploratory and not currently a part of the ACMG/AMP guidelines (Richards et al., 2015). It is also noted above that Family 3 has a possible alternative molecular explanation, namely a RAC1 de novo variant, although oligogenic explanations (with two or more mutations in any one individual) are also possible.
We also summarized the HPO phenotypes for each cluster (Tables 2, S3, and S4). An HPO phenotype is dominant in a cluster, if more than half of individuals in the cluster are positive. Some phenotypic features, such as “Global developmental delay,” “Delayed speech and language development,” “Generalized hypotonia,” “Intellectual disability,” “Feeding difficulties,” “Thin upper lip vermilion,” “Strabismus,” and “Sacral dimple” are more frequent in Cluster 1. The phenotypes in Cluster 1 are most similar to the phenotype previously reported in the TAF1/MRXS33 ID syndrome (O’Rawe et al., 2015). It is possible that cluster 1 represents a more “classic” TAF syndrome, while other clusters consist of individuals with fewer features present, likely due to variable expressivity of the alleles. It is worth noting that Family 2 was in Cluster 4 (the furthest away from the originally reported core TAF1/MRXS33 ID syndrome phenotype), and additional segregation data recently showed this maternally inherited variant to be in a healthy adult uncle, thus rendering this variant classification as Likely Benign.
3.4 |. Variants located in the TAF1-TAF7 interaction domain
This first set of variants (from Families 6 to 12) includes seven residues located in the TAF1-TAF7 interaction domain. On the basis of the available crystal structure (Wang et al., 2014), it was possible to map the residues p.Gly680, p.Arg727, p.Phe873, p.Arg890, and p.Ser985. The first four residues are located on the surface of the structure (Figure 3a; residues p.Asp945 and p.Thr1012 are not visible in the structure). p.Gly680 and p.Arg727 are located in linker loops following an α-helix (a4) and a β-sheet (b3), respectively. The residues p.Phe873 and p.Arg890 are located in the winged helix of TAF1, a structure with DNA binding function (Wang et al., 2014). The residue p.Phe873 is located within β-sheet b11, while p.Arg890 is located in the linker loop between β-sheet b12 and α-helix a9. When calculating the Gibbs free energy difference (ΔΔG) of the ID-associated substitutions, the variants p.Gly680Asp, p.Phe873Val, and p.Arg890Cys do not seem to differ from the wild-type molecule (Table 3). The substitution of proline to arginine in position 727 is instead predicted to increase the steric hindrance in the variant area, resulting in a ΔΔG of 1.56 (Table 3), suggesting that this p.(Arg727Pro) variant could destabilize the overall protein structure.
FIGURE 3.
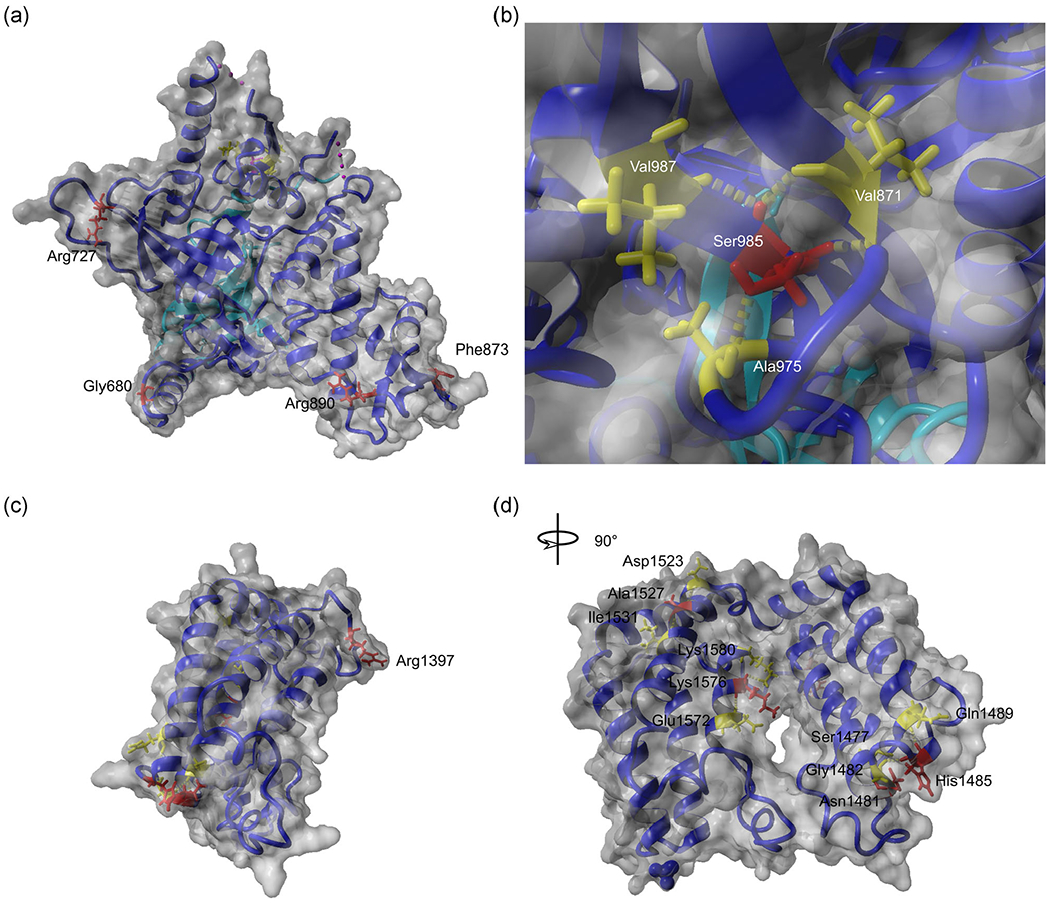
Molecular modeling of TAF1 variants. TAF1 mutations located in the TAF1-TAF7 interaction domain are mapped in the crystal structure deposited by Wang et al. (2014) (PDB: 4RGW) (a,b). Mutations located in the double BrDs are mapped using the structure deposited by Jacobson et al. (2000) (PDB: 1EQF) (c,d). The TAF1 molecule is depicted in blue, the TAF7 molecule in cyan (a,b). TAF1 residues altered in patients with intellectual disability are indicated in red and their interacting amino acids are in yellow. BrD, bromodomain; TAF1, TATA-box binding protein associated factor 1
TABLE 3.
Predicted stability values for wild-type and mutated molecules
| Molecule and substitution | ΔG | ΔΔG |
|---|---|---|
| TAF1-TAF7 | −58.66 | |
| TAF1 p.Gly680Asp-TAF7 | −55.49 | −3.17 |
| TAF1 p.Arg727Pro-TAF7 | −60.22 | 1.56 |
| TAF1 p.Phe873Val-TAF7 | −56.97 | −1.69 |
| TAF1 p.Arg890Cys-TAF7 | −58.44 | −0.22 |
| TAF1 p.Ser985Phe-TAF7 | −48.65 | −10.01 |
| TAF1 double bromodomain | −28.51 | |
| TAF1 double bromodomain p.Arg1397Gln | −27.67 | −0.84 |
| TAF1 double bromodomain p.Asn1481Ile | −24.38 | −4.13 |
| TAF1 double bromodomain p.Asn1481Ser | −27.32 | −1.19 |
| TAF1 double bromodomain p.His1485Arg | −28.23 | −0.28 |
| TAF1 double bromodomain p.Ala1527Val | −28.21 | −0.30 |
| TAF1 double bromodomain p.Lys1576Glu | −27.47 | −0.04 |
Abbreviations: TAF1, TATA-box binding protein associated factor 1.
The residue p.Ser985 is buried within the crystal structure and contacts the residue p.Val987 within the same β-sheet (Figure 3b). The residue is the last amino acid of the Gly-rich motif (from p.Gly973 to Ser985), located at the C terminus of the Triple Barrel-Winged Helix (WH)-α-helical domain (Figure 1). The Gly-rich motif forms a protein-protein interaction surface, which directly contacts the TAF7 Arg-rich motif (Wang et al., 2014). In addition, residue p.Ser985 may be involved in maintaining structural stability in this region, as it contacts the residues p.Val971 and p.Ala975, located in the neighboring β-sheets and in the related linker loop, respectively (Figure 3b). The replacement of this residue by Phe possibly impairs the H-bond distributions in the area, losing contact with the residue p.Ala975. On the basis of the structural prediction, it is likely that the Ser-Phe substitution at residue 985 affects the interaction between TAF1 and TAF7.
3.5 |. Variants located in TAF1 double bromodomain
The second set of variants (from Families 17 to 22) disrupt the double BrD of TAF1, which mediates binding to acetylated lysines of histone tails (Jacobson et al., 2000). All the disease-associated residues in this domain could be mapped in the available crystal structure. The residue p.Arg1397 is located in an accessible loop region on the surface of the structure (Figure 3c). This substitution seems not to alter the free energy of the molecule (Table 3).
Residues p.Asn1481 and p.His1485 are located within BrD1, in the loop connecting α-helices B and C (B-C loop), a structure that has been proposed to be involved in the direct binding of acetylated lysines (Jacobson et al., 2000). Residue p.Asn1481 is particularly important as it anchors the acetyl group. An asparagine at this position is conserved in many BrDs and its mutation into alanine or tyrosine abrogates acetyl-lysine binding (Filippakopoulos & Knapp, 2012; Filippakopoulos et al., 2012; Flynn et al., 2015). Therefore, mutating p.Asn1481 into isoleucine or serine should also affect acetyl-lysine binding (Figure 3d). In contrast, residue p.His1485 has a structural function, contacting the residues p.Gln1489 and p.Gly1482. The substitution of this residue by arginine could impair this function, and based on its localization in the B-C loop and the proximity to the p.Asn1481, it is possible that the anchoring function of p.Asn1481 and the binding of the B-C loop to acetylated lysines are impaired.
Residues p.Ala1527 and p.Lys1576 are located in BrD2 (Figure 3d). The residue p.Ala1527 is located in α-helix Z, and is proposed to participate in interactions with the residues p.Asp1523 and p.Ile1531 (Jacobson et al., 2000). These variants do not seem to affect overall structural stability (Table 3). The residue p.Lys1576 is located in the α-helix A (Figure 3d), at the interface between the two BrDs, and it has been proposed to participate in electrostatic interactions with glutamate residues in BrD1 (1464, 1465, and 1468). Substitution of p.Lys1576 into glutamic acid could abolish the BrD1-BrD2 interaction, as it introduces a repulsive negative charge at this interface.
4 |. DISCUSSION
One of the most difficult challenges currently facing human genetics is determining whether or not a given missense variant in an individual with a disease is pathogenic and assigning a potential etiological molecular diagnosis. Extensive allelic heterogeneity and genotype/phenotype correlations are more readily interpretable for loss-of-function truncating variants, now that some population datasets of normal control individuals are large enough to enable case-control calculations for this class of variants (DeBoever et al., 2018; Karczewski, Francioli, Tiao, & Cummings, 2019; Lek et al., 2016; Van Hout et al., 2019). A population-scale study ranked TAF1 53rd among the top 1,003 constrained human genes (Samocha et al., 2014), indicating a critical role for this protein in normal cellular functioning, and TAF1 is highly conserved, with a calculated probability of loss-of-function intolerance (pLI) of 1.0 (genes with pLI ≥ .9 are considered extremely loss of function intolerant) (Lek et al., 2016). Although missense variants are present in lower numbers in normal control individuals–TAF1 has a GnomAD (v2.1.1) calculated z score of 5.49, with 748 missense variants expected, but only 326 observed–it is nonetheless the case that many apparently healthy individuals may still carry missense variants in TAF1 (perhaps with favorably skewed X inactivation), and the current control datasets are not large enough to enable accurate domain-specific and/or amino acid-level calculations of case-control associations for particular missense variants. As such, it becomes essential to amass other pieces of evidence to classify any specific missense variant as “pathogenic” or “likely pathogenic” according to the ACMG/AMP framework (Richards et al., 2015).
It is worth highlighting that two clinical ES providers (GeneDx and Baylor Genetics) reported the vast majority of variants in TAF1 as being of uncertain significance, either because of insufficient information or because of a mismatch between the reported phenotype(s) and those reported in OMIM. Just as with disease gene discovery, the construction of an allelic series at each disease gene locus will likely expand the utility of clinical genomic sequencing by enabling clinical interpretation of more variant alleles at a locus. It is also interesting to note that mutations in several other TFIID subunits have been implicated in both neurodevelopmental and neurodegenerative conditions (El-Saafin et al., 2018; Hsu et al., 2014; Roon-Mom, van, Reid, Faull, & Snell, 2005; Zuhlke & Burk, 2007). Pathogenic variants within TAF2, TAF6, and TAF13 are all associated with autosomal recessive ID with microcephaly (Hellman-Aharony et al., 2013; Najmabadi et al., 2011; Rooms et al., 2006; Yuan et al., 2015). TBP is involved in spinocerebellar ataxia type 17 (SCA17), which is a movement disorder with typically later onset (Zuhlke & Burk, 2007).
Only one of the variants investigated here has previously been reported, including in two brothers who are hemizygous for the TAF1 variant, c.3568C>T p.(Arg1190Cys) (Okamoto et al., 2019). Both brothers had global DD, ID, dysmorphic features, and lower limb spasticity. The facial features in Individual 13 and these two boys are similar, with large protruding ears and upturned nasal tip. In addition, this same variant was reported in six individuals with ID and dysmorphic facial features in one large family, as part of a much larger ES study (Hu et al., 2016), and more clinical details were recently published (Gudmundsson et al., 2019). This one variant has, therefore, now been identified in three separate families. Individual 13 had been given a clinical diagnosis of Cornelia de Lange syndrome (CdLS), which is based on the presence of craniofacial features, growth failure, ID, and limb malformations/anomalies, which include the classical preaxial hand oligodactyly/radial ray/upper limb anomaly. According to the recently published international consensus statement on CdLS diagnosis and management (Kline et al., 2018), he does meet the criteria for nonclassic CdLS, as he has an overall score of 10 points: synophrys, mild (2 points), short nose and upturned nasal tip (2 points), long, smooth philtrum (2 points), possibly downturned corners of mouth (2 points), global DDs (1 point), and microcephaly (1 point). In the prior cohort paper, Individual 4A had also been given a clinical diagnosis of CdLS (O’Rawe et al., 2015), and Individual 4 in this current cohort has brachydactyly (Table 1). Of note, given the CdLS clinical diagnosis in Individuals 13 and 4A, a recent study reported homozygous variants within TAF6 in two families with CdLS-like features including ID, microcephaly, growth failure, and facial dysmorphism with arched eyebrows, synophrys, a thin upper lip, and long philtrum (Yuan et al., 2015). This shared clinical gestalt between mutations in TAF1 and homozygous mutations in TAF6 in an autosomal-recessive disorder with CdLS-like features has led some to refer to CdLS as a transcriptomopathy (Yuan et al., 2015).
Skewed X-inactivation was seen in the mother of the two boys (Family 1) from the original paper describing the TAF1/MRXS33 ID syndrome, which was an initial clue to search for variants on the X-chromosome (O’Rawe et al., 2015). A prior study with RNA-sequencing data (Hurst et al., 2018) also revealed a 96:4 skewed XCI in the mother of a child with a p.Ser1600Gly missense variant, and phased allele expression analysis confirmed extreme skewing in the mother toward the wild-type allele, covering the entire X-chromosome. It is noteworthy that X-chromosome skewing was observed in almost every affected female that could be tested, including Individual 6 and the mothers in Family 8, 17, and 19. It is likely that the variable expression of any phenotype and penetrance of any clinical features in maternal carriers depends upon tissue-specific levels of XCI. The fact that the mother of Individual 3 had random X-inactivation is one argument against pathogenicity for that variant (p.Ala318Thr). The X-chromosome skewing data, even from blood DNA, for Family 2 is unavailable and thus uninformative, but the genetic prediction would potentially be a random pattern, given that the variant is present in a healthy adult uncle.
We mapped the variants onto available crystal structures of domains within TAF1, but many of the novel ones fall in regions outside of known crystal structures. Of seven TAF1 missense variants previously reported (O’Rawe et al., 2015), four fell within domains predicted to be important for TAF7 binding. Three of these were located in the triple barrel-WH-α-helical domain. Five of the variants described here also fall within this evolutionarily highly conserved region. A recent study described a second DNA-binding module in the C-terminal half of TAF1, encoded by a newly characterized conserved zinc knuckle domain (Curran, Wang, Hinds, Zheng, & Wang, 2018). Mutation within this zinc knuckle reduces TFIID binding to promoters, which, in turn, leads to a decrease in transcription and cell viability. The TAF1 zinc knuckle therefore appears to play a role in transcription initiation. None of the TAF1 variants described in this cohort, previously published patients, or reported in ClinVar fall within this domain, which spans residues 1,261–1,300. It is, therefore, not known whether mutations within this region are compatible with survival or, if they are, whether the clinical phenotype is similar to that seen in patients described to date. Identification of disease variants falling outside of the currently characterized functional domains may lead to the discovery of novel functions for TAF1.
Although there is substantial overlap in the phenotype between the patients in our cohort, the range of dysmorphic features, neurological problems, and congenital anomalies is quite broad; moreover, the facial gestalt is currently nonspecific for a recognizable pattern. Whether this is due to allelic heterogeneity, locus heterogeneity for a second site suppressor, other epistatic interactions, genetic background, or environmental influences remains unknown. Phenotypic variability among TAF1/MRXS33 ID syndrome probands is expected, as it is common for syndromes whose pathogenesis is linked to mutations in large genes, or in genes with multiple interacting partners spanning many functional domains, to vary widely in their phenotypic effect. Indeed, in a recent study of a Chinese patient cohort with West syndrome, two individuals were found to have maternally inherited TAF1 missense variants (Peng et al., 2018). However, whether TAF1 is a candidate gene for this classic form of early infantile epileptic encephalopathy warrants further investigation, as one of the TAF1 variants c.4771G>A was present in a hemizygote in gnomAD.
Interestingly, reduced TAF1 expression has also been associated with X-linked dystonia Parkinsonism (XDP), a disorder found exclusively in patients with Filipino ancestry, and characterized by progressive dystonia and Parkinson-like symptoms such as tremor and rigidity (Aneichyk et al., 2018; Evidente et al., 2002; Lee et al., 2011; Pasco et al., 2011). XDP is a neurodegenerative disease associated with an antisense insertion of a SINE-VNTR-Alu (SVA)-type retrotransposon within an intron of TAF1 (Domingo et al., 2015; Makino et al., 2007; Nolte, Niemann, & Müller, 2003). It was recently shown that there is polymorphic variation in the length of a hexanucleotide repeat domain, (CCCTCT)n, in the SVA, and that the number of repeats in these cases ranges from 35 to 52, showing a highly significant inverse correlation with age at disease onset (Bragg et al., 2017) and disease expressivity (Westenberger et al., 2019). In addition, TAF1 mRNA is subjected to alternative splicing, including neuron-specific splicing of ‘microexon 34’ to produce the NTAF1 isoform (Makino et al., 2007). It is conceivable that reduced or increased expression of TAF1 might have different neurologic effects than that seen with missense variants.
In summary, we have constructed a detailed allelic series to characterize potential contributions to clinical phenotypic outcome and assist with interpretation of TAF1 variants in other families. We are far from a “saturation mutagenesis” view of the TAF1 gene, however, and the clinical consequences of individual variants remain difficult to predict. The continued accumulation of clinical and molecular data is critical to understanding the contribution of specific variants to TAF1-related phenotypes. Future research using in vitro and in vivo functional studies will be needed to elucidate the consequences of these variants.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by funds from the Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory (G. J. L.) and the New York State Office for People With Developmental Disabilities at the Institute for Basic Research in Developmental Disabilities (IBR) in Staten Island, NY (G. J. L.). The DDD study presents independent research commissioned by the UK Health Innovation Challenge Fund (Grant #HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (Grant #WT098051). L. E. thanks the Genome. One clinical team for their assistance with sequencing. S. C. was supported through a research grant from the CC-XDP consortium to H. T. M. T. H. T. M. T. acknowledges financial support by the Deutsche Forschungsgemeinschaft (SFB850 and SFB992). G. B. and the Undiagnosed Diseases Program WA are supported by the Angela Wright Bennett Foundation and the McCusker Charitable Foundation. This study was jointly funded by the United States National Human Genome Research Institute (NHGRI), a National Heart, Lung, and Blood Institute (NHLBI) grant to the Baylor-Hopkins Center for Mendelian Genomics (J. R. L., UM1 HG006542), and the National Institute of Neurological Disorders and Stroke (J. R. L., R35 NS105078). J. E. P. is supported by NHGRI K08 HG008986. K. W., M. Z., and C. W. are supported by NIH grant LM012895. Individuals from family 17 were tested as part of the Australia EPIC-ID study, and Individual 21 was tested as part of the Australian Genomics Acute Care Flagship (National Health and Medical Research Council GNT1113531). The authors extend their appreciation to the families and clinicians who contributed to this paper. G. J. L. thanks Melissa Nashat and Vicky Brandt for providing critical comments on the manuscript. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by the Wellcome Trust.
Funding information
United States National Human Genome Research Institute, Grant/Award Numbers: HG006542, HG008986; NIH, Grant/Award Number: LM012895
Footnotes
CONFLICT OF INTERESTS
G. J. L. served on advisory boards for GenePeeks, Inc., Seven Bridges Genomics, Inc., and Fabric Genomics, Inc. P. B. A. serves on an advisory board for Illumina, Inc. J. R. L. has stock ownership in 23andMe, is a paid consultant for Regeneron and Novatis, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The other authors declare no conflict of interests. N. A., A. B., G. D., and M. G. S. are employees of GeneDx, Inc.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, & Belmont JW (1992). Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. American Journal of Human Genetics, 51, 1229–1239. [PMC free article] [PubMed] [Google Scholar]
- Aneichyk T, Hendriks WT, Yadav R, Shin D, Gao D, Vaine CA, … Penney EB (2018). Dissecting the causal mechanism of X-linked dystonia-Parkinsonism by integrating genome and transcriptome assembly. Cell, 172, 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragg DC, Mangkalaphiban K, Vaine CA, Kulkarni NJ, Shin D, Yadav R, … Acuna P (2017). Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proceedings of the National Academy of Sciences of the United States of America, 114, E11020–E11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Ludecke HJ, Hamdan FF, Altmuller J, Beleggia F, Elcioglu NH,… Li Y (2017). Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Human Genetics, 136, 821–834. [DOI] [PubMed] [Google Scholar]
- Curran EC, Wang H, Hinds TR, Zheng N, & Wang EH (2018). Zinc knuckle of TAF1 is a DNA binding module critical for TFIID promoter occupancy. Scientific Reports, 8, 4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoever C, Tanigawa Y, Lindholm ME, McInnes G, Lavertu A, Ingelsson E, . Rivas MA (2018). Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nature Communications, 9(1), 1612 10.1038/s41467-018-03910-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature, 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo A, Westenberger A, Lee LV, Braenne I, Liu T, Vater I, … Schmidt TG (2015). New insights into the genetics of X-linked dystonia-parkinsonism (XDP, DYT3). European Journal of Human Genetics, 23(1), 1334–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Saafin F, Curry C, Ye T, Garnier JM, Kolb-Cheynel I, Stierle M,… Voss AK (2018). Homozygous TAF8 mutation in a patient with intellectual disability results in undetectable TAF8 protein, but preserved RNA Polymerase II transcription. Human Molecular Genetics, 27(12), 2171–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evidente VG, Advincula J, Esteban R, Pasco P, Alfon JA, Natividad FF, … Singleton A (2002). Phenomenology of “Lubag” or X-linked dystonia-parkinsonism. Movement Disorders, 17, 1271–1277. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, & Knapp S (2012). The bromodomain interaction module. FEBS Letters, 586, 2692–2704. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, . Arrowsmith CH (2012). Histone recognition and large-scale structural analysis of the human bromodomain family. Cell, 149, 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn EM, Huang OW, Poy F, Oppikofer M, Bellon SF, Tang Y, & Cochran AG (2015). A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure, 23, 1801–1814. [DOI] [PubMed] [Google Scholar]
- Gudmundsson S, Wilbe M, Filipek-Górniok B, Molin A-M, Ekvall S, Johansson J, … Bondeson M-L (2019). TAF1, associated with intellectual disability in humans, is essential for embryogenesis and regulates neurodevelopmental processes in zebrafish. Scientific Reports, 9, 10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, … Dionne-Laporte A (2014). De novo mutations in moderate or severe intellectual disability. PLOS Genetics, 10, e1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Person TN, Hebbring SJ, Heinzen E, Ye Z, Schrodi SJ, … Robison RJ (2015). SeqHBase: A big data toolset for family based sequencing data analysis. Journal of Medical Genetics, 52, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman-Aharony S, Smirin-Yosef P, Halevy A, Pasmanik-Chor M, Yeheskel A, Har-Zahav A, … Basel-Vanagaite L (2013). Microcephaly thin corpus callosum intellectual disability syndrome caused by mutated TAF2. Pediatric Neurology, 49, 411–416. [DOI] [PubMed] [Google Scholar]
- Hsu TC, Wang CK, Yang CY, Lee LC, Hsieh-Li HM, Ro LS,… Su MT (2014). Deactivation of TBP contributes to SCA17 pathogenesis. Human Molecular Genetics, 23, 6878–6893. [DOI] [PubMed] [Google Scholar]
- Hu H, Haas SA, Chelly J, Van Esch H, Raynaud M, Brouwer AP,… Kalscheuer VM (2016). X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Molecular Psychiatry, 21(1), 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst SE, Liktor-Busa E, Moutal A, Parker S, Rice S, Szelinger S,… Perez-Miller S (2018). A novel variant in TAF1 affects gene expression and is associated with X-linked TAF1 intellectual disability syndrome. Neuronal Signaling, 2(3), NS20180141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson RH, Ladurner AG, King DS, & Tjian R (2000). Structure and function of a human TAFII250 double bromodomain module. Science, 288, 1422–1425. [DOI] [PubMed] [Google Scholar]
- Janakiraman U, Yu J, Moutal A, Chinnasamy D, Boinon L, Batchelor SN,… Nelson MA (2019). TAF1-gene editing alters the morphology and function of the cerebellum and cerebral cortex. Neurobiology of Disease, 132, 104539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahrizi K, Hu H, Hosseini M, Kalscheuer VM, Fattahi Z, Beheshtian M, … Akhtarkhavari T (2019). Effect of inbreeding on intellectual disability revisited by trio sequencing. Clinical Genetics, 95, 151–159. [DOI] [PubMed] [Google Scholar]
- Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, . Lupski JR (2018). Phenotypic expansion illuminates multilocus pathogenic variation. Genetics in Medicine, 20(12), 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, … The Genome Aggregation Database Consortium. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv, 10.1101/531210 [DOI] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, … Wierzba J (2018). Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nature Reviews, Genetics, 19(10), 649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Aymé S , … Buske OJ (2017). The human phenotype ontology in 2017. Nucleic Acids Research, 45, D865–D876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmicki JA, Samocha KE, Howrigan DP, Sanders SJ, Slowikowski K, Lek M,… Neale BM (2017). Refining the role of de novo protein-truncating variants in neurodevelopmental disorders by using population reference samples. Nature Genetics, 49, 504–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E, Koraimann G, & Vriend G (2002). Increasing the precision of comparative models with YASARA NOVA–a self-parameterizing force field. Proteins, 47, 393–402. [DOI] [PubMed] [Google Scholar]
- Leduc MS, Chao HT, Qu C, Walkiewicz M, Xiao R, Magoulas P,… Scaglia F (2017). Clinical and molecular characterization of de novo loss of function variants in HNRNPU. American Journal of Medical Genetics, Part A, 173, 2680–2689. [DOI] [PubMed] [Google Scholar]
- Lee LV, Rivera C, Teleg RA, Dantes MB, Pasco PM, Jamora RD, … Peralta O (2011). The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”). International Journal of Neuroscience, 121(Suppl 1), 3–11. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T , … Birnbaum DP (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, & Wang K (2017). InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP Guidelines. American Journal of Human Genetics, 100, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, & Boerwinkle E (2016). dbNSFP v3.0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Human Mutation, 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, … Ogasawara K (2007). Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. American Journal of Human Genetics, 80, 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, … Mohseni M (2011). Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478, 57–63. [DOI] [PubMed] [Google Scholar]
- Niranjan TS, Skinner C, May M, Turner T, Rose R, Stevenson R, … Wang T (2015). Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLOS One, 10, e0116454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte D, Niemann S, & Müller U (2003). Specific sequence changes in multiple transcript system DYT3 are associated with X-linked dystonia parkinsonism. Proceedings of the National Academy of Sciences of the United States of America, 100, 10347–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto N, Arai H, Onishi T, Miyake N, & Matsumoto N (2019). Intellectual disability and dysmorphic features in male siblings arising from a novel TAF1 mutation. Congenital Anomalies. Advance online publication; 10.1111/cga.12330 [DOI] [PubMed] [Google Scholar]
- O’Rawe JA, Wu Y, Dorfel MJ, Rope AF, Au PY, Parboosingh JS, … Schuette JL (2015). TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. American Journal of Human Genetics, 97, 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papai G, Weil PA, & Schultz P (2011). New insights into the function of transcription factor TFIID from recent structural studies. Current Opinion in Genetics and Development, 21, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasco PM, Ison CV, Munoz EL, Magpusao NS, Cheng AE, Tan KT, … Demaisip C (2011). Understanding XDP through imaging, pathology, and genetics. International Journal of Neuroscience, 121(Suppl 1), 12–17. [DOI] [PubMed] [Google Scholar]
- Peng J, Wang Y, He F, Chen C, Wu L-W, Yang L-F,… Guo H (2018). Novel West syndrome candidate genes in a Chinese cohort. CNS Neuroscience & Therapeutics, 24, 1196–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, … Beaudet AL (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. New England Journal of Medicine, 376, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijnders MRF, Ansor NM, Kousi M, Yue WW, Tan PL, Clarkson K,… Marcelis C (2017). RAC1 missense mutations in developmental disorders with diverse phenotypes. American Journal of Human Genetics, 101, 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, & Kircher M (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, … Bai R (2016). Clinical application of whole-exome sequencing across clinical indications. Genetics in Medicine, 18, 696–704. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J,… Rehm HL (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooms L, Reyniers E, Scheers S, Luijk R, van Wauters J, Van Aerschot L, . Kooy RF (2006). TBP as a candidate gene for mental retardation in patients with subtelomeric 6q deletions. European Journal of Human Genetics, 14, 1090–1096. [DOI] [PubMed] [Google Scholar]
- Roon-Mom WM, van, Reid SJ, Faull RL, & Snell RG (2005). TATA-binding protein in neurodegenerative disease. Neuroscience, 133, 863–872. [DOI] [PubMed] [Google Scholar]
- Samocha KE, Kosmicki JA, Karczewski KJ, O’Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, … Daly MJ (2017). Regional missense constraint improves variant deleteriousness prediction. bioRxiv, 10.1101/148353 [DOI] [Google Scholar]
- Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, . MacArthur DG (2014). A framework for the interpretation of de novo mutation in human disease. Nature Genetics, 46, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez D, Batet M, & Isern D (2011). Ontology-based information content computation. Knowledge-Based Systems, 24, 297–303. [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, & Cooper DN (2014). The human gene mutation database: Building a comprehend-sive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics, 133, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Durme J, Delgado J, Stricher F, Serrano L, Schymkowitz J, & Rousseau F (2011). A graphical interface for the FoldX forcefield. Bioinformatics, 27, 1711–1712. [DOI] [PubMed] [Google Scholar]
- Van Hout CV, Tachmazidou I, Backman JD, Hoffman JX, Ye B, Pandey AK, … Colm O (2019). Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. BioRxiv, 10.1101/572347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WWMP, Schaik FMAV, Varier RA, … Timmers HTM (2007). Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell, 131, 58–69. [DOI] [PubMed] [Google Scholar]
- Wang H, Curran EC, Hinds TR, Wang EH, & Zheng N (2014). Crystal structure of a TAF1-TAF7 complex in human transcription factor IID reveals a promoter binding module. Cell Research, 24, 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield L, Ramachandran S, Baptista T, Devys D, Tora L, & Hahn S (2017). Transcription of nearly all yeast RNA polymerase II-transcribed genes is dependent on transcription factor TFIID. Molecular Cell, 68, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenberger A, Reyes CJ, Saranza G, Dobricic V, Hanssen H, Domingo A, . Begemann K (2019). A hexanucleotide repeat modifies expressivity of X-linked dystonia parkinsonism. Annals of Neurology, 85, 812–822. [DOI] [PubMed] [Google Scholar]
- Wright CF, FitzPatrick DR, & Firth HV (2018). Paediatric genomics: Diagnosing rare disease in children. Nature Reviews Genetics, 19, 253–268. [DOI] [PubMed] [Google Scholar]
- Yates TM, Vasudevan PC, Chandler KE, Donnelly DE, Stark Z, Sadedin S, … Broad Center for Mendelian, Genomics, D. D. D. Study (2017). De novo mutations in HNRNPU result in a neurodevelopmental syndrome. American Journal of Medical Genetics, Part A, 173, 3003–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng W-L, Gambin T, … Koparir A (2015). Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. Journal of Clinical Investigation, 125, 636–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke C, & Burk K (2007). Spinocerebellar ataxia type 17 is caused by mutations in the TATA-box binding protein. Cerebellum, 6, 300–307. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.