

Upregulated expression of efflux pumps, lpxC target mutations, LpxC protein overexpression, and mutations in fabG were previously shown to mediate single-step resistance to the LpxC inhibitor CHIR-090 in P. aeruginosa. Single-step selection experiments using three recently described LpxC inhibitors (compounds 2, 3, and 4) and mutant characterization showed that these mechanisms affect susceptibility to additional novel LpxC inhibitors.
KEYWORDS: LpxC inhibitor, Pseudomonas aeruginosa, cumulative resistance, decreased susceptibility, efflux, target mutation
ABSTRACT
Upregulated expression of efflux pumps, lpxC target mutations, LpxC protein overexpression, and mutations in fabG were previously shown to mediate single-step resistance to the LpxC inhibitor CHIR-090 in P. aeruginosa. Single-step selection experiments using three recently described LpxC inhibitors (compounds 2, 3, and 4) and mutant characterization showed that these mechanisms affect susceptibility to additional novel LpxC inhibitors. Serial passaging of P. aeruginosa wild-type and efflux pump-defective strains using the LpxC inhibitor CHIR-090 or compound 1 generated substantial shifts in susceptibility and underscored the interplay of efflux and nonefflux mechanisms. Whole-genome sequencing of CHIR-090 passage mutants identified efflux pump overexpression, fabG mutations, and novel mutations in fabF1 and in PA4465 as determinants of reduced susceptibility. Two new lpxC mutations, encoding A214V and G208S, that reduce susceptibility to certain LpxC inhibitors were identified in these studies, and we show that these and other target mutations differentially affect different LpxC inhibitor scaffolds. Lastly, the combination of target alteration (LpxCA214V) and upregulated expression of LpxC was shown to be tolerated in P. aeruginosa and could mediate significant decreases in susceptibility.
INTRODUCTION
Success in the identification and development of novel antibacterial agents for treating serious Gram-negative infections has remained elusive. This is in part due to the inability of potent small molecule inhibitors of specific intracellular targets to accumulate adequately in the cell to fully engage their target. Gram-negative pathogens can exclude toxic molecules through the combination of a highly impermeable outer membrane (OM) and active efflux (1, 2).
The genome of P. aeruginosa encodes at least 12 members of the resistance nodulation cell division (RND) family efflux pumps, along with many pumps of other families (3). Of the RND family, the MexAB-OprM, MexXY-OprM, MexCD-OprJ, and MexEF-OprN pumps have been clearly associated with resistance to antibacterial compounds (4). RND family pumps are tripartite in structure with an inner membrane pump (e.g., MexB), an outer membrane channel (e.g., OprM), and a membrane fusion protein that links these (e.g., MexA) (5). The RND outer membrane channel component traverses the Gram-negative outer membrane, and therefore RND family pumps can expel toxic molecules that are entering cells back out across the OM to outside the cells. The OM is an asymmetrical bilayer with a phospholipid inner leaflet and an outer leaflet made up primarily of lipopolysaccharide (LPS), also referred to as endotoxin. LPS is an amphipathic molecule composed of a relatively conserved lipid A region that forms the outer leaflet of the outer membrane bilayer to which is attached a core oligosaccharide and highly variable O-antigen polysaccharide repeating units that extend outward from the cell (6). The OM permeability barrier reduces the influx of many toxic molecules, which enhances the effectiveness of efflux in preventing intracellular accumulation of antibacterial compounds (1).
Along with being a key factor in establishing the permeability barrier of the OM, LPS is also essential for growth and virulence in many Gram-negative pathogens such as Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa. Targets in the lipid A biosynthetic pathway, in particular LpxC (7–12), or in the machinery that transports LPS and assembles it in the OM (13) have been the focus of intense drug discovery efforts. Early efforts to identify LpxC inhibitors yielded compounds that had activity against E. coli but not P. aeruginosa (14), raising the possibility that these inhibitors did not accumulate sufficiently in P. aeruginosa (as is often the case) or that LpxC was not essential in this pathogen. Further studies showed that the differential activity was related to differences in the LpxC amino acid sequences between these pathogens (15, 16). Efforts taking the P. aeruginosa LpxC protein into account initially yielded the LpxC inhibitor CHIR-090 that had potent activity against this pathogen (17) (Fig. 1), and since then many others have been reported (7, 18, 19). Recent examples from our group include compound 1 (9 ) and compound 2 (oxazolidinone 13f), compound 3 (isoxazoline 25d), and compound 4 (isoxazoline 25e) described previously (12) (Fig. 1) that have potent in vitro activity against P. aeruginosa. These compounds are somewhat atypical in terms of novel antibacterial discovery for their ability to potently kill P. aeruginosa wild-type cells, which are notoriously recalcitrant to most novel target inhibitors. Their antibacterial activity may relate in part to a currently not well understood ability to accumulate in Gram-negative cells.
FIG 1.
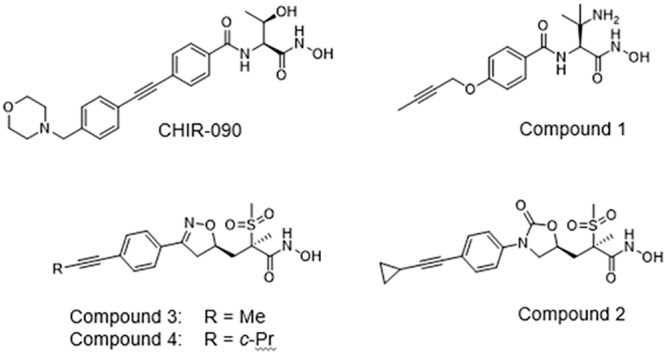
Chemical structures of LpxC inhibitors in this study.
Several mechanisms are known to reduce the in vitro susceptibility of P. aeruginosa to CHIR-090, including upregulated efflux by MexAB-OprM (via mutations in the repressor gene mexR), MexCD-OprJ (via mutations in the repressor gene nfxB), or MexEF-OprN (via mutations in mexS), mutations in the fatty acid pathway gene fabG and lpxC structural gene mutations (encoding L18V), and upstream mutations causing LpxC overexpression (20). We also noted that in vitro passaging of cells in the presence of CHIR-090 can lead to cumulative and substantial reductions in susceptibility. Here, we used single-step mutant selections and passaging approaches, along with targeted and genome sequencing, to examine the role of efflux and nonefflux mechanisms in mediating decreased susceptibility to CHIR-090 and the more recently described LpxC inhibitors compound 1 (9), compound 2 (oxazolidinone 13f), compound 3 (isoxazoline 25d), and compound 4 (isoxazoline 25e) (12) (Fig. 1) that have potent in vitro activity against P. aeruginosa.
RESULTS
Selection of single-step P. aeruginosa mutants using novel LpxC inhibitors.
Single-step mutants of two different P. aeruginosa PAO1 lineages (K767 and PAO1V) or the clinical isolate K2153 (Table 1 ) were selected using novel LpxC inhibitors shown in Fig. 1. Different PAO1 lineages were used at different times over the course of several years to isolate mutants to survey mechanisms reducing susceptibility. The mutant frequencies obtained for these selections are shown in Table 2 and are consistent with what would be expected given the multiplicity of mechanisms likely to affect these compounds. Of the two PAO1 lineages used, PAO1V appeared to have a higher frequency of mutant selection for compounds 2 and 3 at lower multiples of the MIC than did K767. Different lineages of PAO1 are known to diverge over time (21), so this is perhaps not surprising, but we did not investigate this further for these two strains. The likely resistance loci lpxC, fabG, mexR (repressor of MexAB-OprM efflux pump expression), and nfxB (repressor of MexCD-OprJ efflux pump expression) genes were then PCR amplified and sequenced from a set of selected mutants. The identified mutations are summarized in Table S1 in the supplemental material. Mutations in mexR and nfxB were identified among the isolates, consistent with the involvement of MexAB-OprM and MexCD-OprJ in reducing P. aeruginosa susceptibility to other LpxC inhibitors (20, 22). Some P. aeruginosa PAO1 lineages have inactive variants of the mexT gene that encodes the positive activator of mexEF-oprN efflux pump gene expression (23), and selection of MexEF-OprN pump upregulated mutants can be infrequent in such strains. These include PAO1 strain K767 (Table 1), which has an inactive mexT gene due to the presence of one or both of two point mutations (24), and PAO1V, which has an inactive mexT presumably due to an 8-bp insertion in mexT (3). The clinical isolate K2153 (Table 1) has an active mexT allele, and MexEF-OprN pump upregulated mutants can be selected via loss-of-function mutations in the mexS gene whose product suppresses MexT (20, 25). The LpxC inhibitors CHIR-090 and LpxC-4 selected for upregulated expression of MexEF-OprN in an appropriate strain background (20, 22), so we tested this for compound 4 using strain K2153. Mutants were selected at a frequency of approximately 1 × 10−7 at 4× MIC (Table 2) and of 9 mutants examined, 5 harbored mutations in mexS (Table S1). Along with these efflux related mutations, several mutations in fabG were found, as well as the upstream lpxC mutation causing overexpression of LpxC (20, 22) (PAO1V-CDJ0028) and a novel lpxC structural gene mutation (encoding LpxC A214V) selected by compounds 3 and 4 (K767-CDJ0037 and PAO1V-CDJ0042).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmida | Relevant characteristicsb | Source or reference |
|---|---|---|
| Strains | ||
| P. aeruginosa | ||
| K767 | PAO1 prototroph (described in reference 48) | K. Poole |
| K1454 | K767 mexR (nalB), upregulated MexAB-OprM | 49 |
| K767-CDA0033 | K767 nfxB, upregulated MexCD-OprJ | This study |
| K767-CDR0026 | K767 overexpressing LpxC | 20 |
| K767-LpxCG208S | K767 engineered to encode LpxCG208S | This study |
| K767-CDJ0011 | K767 engineered to encode LpxCL18V | 20 |
| K767-CDJ0037 | K767 encoding LpxCA214V, selected on compound 3 | This study |
| K767-CDR0061 | K767 fabG (C494T), selected on CHIR-090 | 20 |
| K2732 | PAO1 prototroph | K. Poole |
| K2732-P2 | K2732 FabF1T306A, passage 2, selected on CHIR-090 | This study |
| K2732-P6 | K3732 nfxB stop TGA-Cys; FabGD190G; PA4465N193T, passage 6, selected on CHIR-090 | This study |
| K2732-P13 | K2732 MexRT130P; nfxB stop TGA-Cys; FabGD190G; PA4465N193T, passage 13, selected on CHIR-090 | This study |
| K2733 | K2733 (K2732 ΔmexB ΔmexX ΔmexCD-oprJ ΔmexEF-oprN) | K. Poole |
| K2733-P6a (NC) | K2733 PA4465N193T, passage 6, selected on CHIR-090 | This study |
| K2733-P6b (SC) | K2733 FabGA159V, passage 6, selected on CHIR-090 | This study |
| K2733-P13a (NC) | K2733 FabGA167V; PA4465N193T, passage 13, selected on CHIR-090 | This study |
| K2733-P13b (SC) | K2733 FabGA167V; PA4465N193T, passage 13, selected on CHIR-090 | This study |
| K2733-PA4465N193T | K2732 engineered to encode PA4465N193T | This study |
| PAO1V | PAO1 prototroph | 50 |
| PAO1V-CDJ0042 | PAO1V encoding LpxCA214V, selected on compound 2 | This study |
| K2153 | Clinical isolate, MexXY upregulated, active mexT | 25 |
| K2376 | K2153 mexS in-frame deletion, upregulated MexEF-OprN expression | 51 |
| K2153-CDR0063 | K2153 mexS, upregulated MexEF-OprN expression | 20 |
| E. coli | ||
| Stellar | High transformation efficiency HST08 strain | Clontech |
| Plasmids | ||
| pRK2013 | Helper plasmid | 52 |
| pAK1900 | E. coli-P. aeruginosa shuttle vector; Apr | A. Kropinski |
| pAK-lpxC | pAK1900 with lpxC and 100 bp upstream sequence from K767 | 20 |
| pAK-lpxC WT | pAK1900 with WT lpxC and 100 bp upstream sequence from K767 | 20 |
| pAK-lpxC WT UP | pAK-lpxC WT with C→A substitution 11 bp upstream of lpxC; LpxC upregulated) | 20 |
| pAK-LpxCA214V | pAKlpxC WT altered so that lpxC encodes A214V | This study |
| pAK- LpxCA214V UP | pAK-LpxCA214V with C→A substitution 11 bp upstream of lpxC; LpxCA214V upregulated) | This study |
| pMMB-10 | Gateway adapted pMMB low-copy-number expression vector; Apr Cbr; IPTG inducible | 53 |
| pMM-10-fabF1 | IPTG inducible fabF1 expression vector | This study |
| pMM-10-PA4465 | IPTG inducible PA4465 expression vector | This study |
| pEX18ApGW | Gateway adapted gene replacement vector, Apr Cbr, sacB | 54 |
| pEX18ApGW-lpxC-G622A | Gene replacement vector with lpxC G622A allele | This study |
| pEX18ApGW-PA4465-A578C | Gene replacement vector with PA4465 A578C allele | This study |
Additional mutants are summarized in Table S1 in the supplemental material. NC, normal-size colony; SC, small colony.
Cbr, carbenicillin resistance; Apr, ampicillin resistance.
TABLE 2.
Frequencies of selecting P. aeruginosa single-step mutants with decreased susceptibility to LpxC inhibitors
| Strain | Compound | Frequenciesa
at: |
|||
|---|---|---|---|---|---|
| 2× MIC | 4× MIC | 8× MIC | 16× MIC | ||
| K767 | 2 | 2.16 × 10–8 | 3.6 × 10–10 | <1.79 × 10–10 | <1.79 × 10–10 |
| K767 | 3 | 7.27 × 10–8 | 3.16 × 10–9 | 3.51 × 10–10 | <7 × 10–10 |
| PAO1V | 2 | 1.98 × 10–7 | 1 × 10–8 | 4.77 × 10–10 | <9.55 × 10–10 |
| PAO1V | 3 | 3.77 × 10–7 | 2.28 × 10–8 | <1.14 × 10–9 | <1.14 × 10–9 |
| PAO1V | 4 | TNTC | 3.97 × 10–10 | 7.93 × 10–11 | ND |
| K2153 | 4 | TNTC | 1.13 × 10–7 | 3.31 × 10–9 | ND |
TNTC, too numerous to count; ND, not determined.
Table 3 compares the shift in susceptibility to CHIR-090 and compounds 2 to 4 in a representative fabG mutant and in mutants upregulated for MexAB-OprM, MexCD-OprJ, or MexEF-OprN efflux pump expression (mutations related to lpxC are discussed in later sections). The fabG mutant K767-CDA0033 was 4- to 8-fold less susceptible to all four compounds. Upregulation of MexAB-OprM expression (engineered strain K1454) also mediated a 4- to 8-fold decrease in susceptibility to these compounds. Upregulated expression of MexCD-OprJ (K767-CDA0033) reduced susceptibility to CHIR-090 and compound 3 by 16-fold but had little impact on susceptibility to compounds 2 or 4. This suggested that these are weak substrates for MexCD-OprJ, however, several nfxB mutants were selected using these compounds (Table S1), suggesting that different nfxB mutations may lead to a range of susceptibility shifts. Two previously characterized mutants upregulated for MexEF-OprN expression (K2376 and K2153-CDR0063) were >4-fold less susceptible to all compounds.
TABLE 3.
Impact of fabG mutation and efflux pumps on susceptibility to LpxC inhibitor compounds in P. aeruginosa
| Strain | Relevant factor | MIC (μg/ml)a
|
|||
|---|---|---|---|---|---|
| CHIR-090 | Compound 4 | Compound 3 | Compound 2 | ||
| K767 | Wild type | 1 | 0.25 | 0.25 | 0.25 |
| K767-CDR0061 | fabG mutation | 8 | 2 | 2 | 1 |
| K1454 | MexAB-OprM up | 8 | 2 | 2 | 2 |
| K767-CDA0033 | MexCD-OprJ up | 16 | 0.25 | 4 | 0.5 |
| K2153 | Wild type | 2 | 0.5 | 1 | 0.5 |
| K2376 | MexEF-OprN up | 16 | 8 | 4 | 4 |
| K2153-CDR0063 | MexEF-OprN up | >16 | 16 | 16 | >4 |
Susceptibility shifts ≥ 4-fold are indicated in boldface.
Cumulative effect of mechanisms reducing susceptibility to LpxC inhibitors that emerge during serial passage.
Consistent with the range of mechanisms that individually reduce susceptibility to CHIR-090 and other LpxC inhibitors, in vitro serial passaging experiments using CHIR-090 led to substantial cumulative decreases in susceptibility in P. aeruginosa (MIC shifts to 128 μg/ml) (20). Here, we undertook serial passage studies using a wild-type P. aeruginosa PAO1 strain K2732 and its efflux-deficient mutant (lacking MexAB-OprM, MexXY-OprM, MexCD-OprJ, and MexEF-OprN function), K2733 to ascertain the impact of efflux on the magnitude of decreased susceptibility and to isolate and characterize additional mutants. Like K767, this parent strain K2732 has an inactive mexT gene, and as such the emergence of MexEF-OprNM upregulation was not anticipated during passaging. Susceptibility to CHIR-090 decreased steadily over 13 passages for K2732, with the MIC of CHIR-090 starting at 1 μg/ml and reaching 128 μg/ml (Fig. 2A). The susceptibility of the efflux defective strain K2733 also decreased in a stepwise fashion, but the lack of efflux appeared to limit the ultimate level of insusceptibility reached over a similar time frame. CHIR-090 was much more potent against the efflux defective mutant culture at the beginning of the experiment (MIC 0.0156 μg/ml) and ultimately the CHIR-090 MIC stabilized at 1 to 2 μg/ml from passages 6 to 13 (Fig. 2A). Therefore, there was a similar circa 64-fold shift in susceptibility whether efflux was active or not, but with efflux the ultimate level of susceptibility was about 32- to 64-fold lower. A similar pattern was seen when experiments were conducted using compound 1 (9) except that there was only a 16-fold decrease in susceptibility over 13 passages in the pump defective strain (Fig. 2B).
FIG 2.

Cumulative decrease in the susceptibility of P. aeruginosa wild type (K3732, blue lines) and an efflux-deficient mutant (K2733, orange lines) to CHIR-090 (A) or compound 1 (B) during serial passaging. The y axes show the susceptibility (MIC, μg/ml) of mixed populations, as described previously (20).
Interplay of mechanisms leads to substantially reduced susceptibility to LpxC inhibitors.
Several mutants were isolated from different stages of the CHIR-090 serial passaging experiment and genome sequenced (Table 4). Qualitatively there appeared to be some differences in colony sizes on some of the selection plates, and this is noted in Table 1. As expected, mutations in the regulators of efflux appeared in isolates derived from the wild-type strain (nfxB in K2732-P6 and mexR/nfxB in K2732-P13; Table 4). Intriguingly, the combination of nfxB and mexR mutations in K2732-P13 suggests that MexAB-OprM and MexCD-OprJ can work together to increase efflux of CHIR-090. As anticipated, no mutations in the regulatory genes controlling expression of the four pumps that are inactivated in strain K2733 emerged. In isolates selected from both the parent and efflux deficient mutant, mutations in fabG were identified (K2732-P6, K2732-P13, K2733-P6b, and K2733-P13ab, Table 5), which are known to reduce susceptibility to LpxC inhibition (20). We did not find mutations in lpxC or the region upstream of lpxC in the mutants studied here, but two new mutations were identified in genes not previously associated with decreased susceptibility to LpxC inhibitors. One of these was in fabF1 (encoding FabF1T306A) and occurred in one isolate derived from the wild-type strain at passage 2 (K2732-P2, Table 4). This conferred a 4-fold decrease in susceptibility (CHIR-090 MIC shift from 1 to 4 μg/ml, Table 4). The wild-type fabF1 gene was then cloned into an inducible plasmid and expressed in trans in the passage-selected fabF1 mutant K2732-P2. As expected, the mutant containing empty vector or the uninduced fabF1 plasmid grew fairly well in the presence of 1 μg/ml of CHIR-090, whereas growth of the parent K2732 containing empty vector was much less (Fig. S1). Induction of fabF1 expression (complementation) reduced growth of K2732-P2 to levels comparable to the parent strain, consistent with a role for FabF1T306A. The other new mutation was found in the hypothetical gene PA4465 (3) (encoding N193T), and this occurred together with mutations in nfxB, mexR, and fabG in (K2732-P6 or K2732-P13, Table 4) or singly and with fabG mutations in mutants from K2733 (K2733-P6ab and K2733-P13ab, Table 4). The single mutation in K2733-P6a mediated an ∼8-fold decrease in susceptibility. This mutation was engineered singly back onto the genome of the pump plus parent K2732 (designated K2732-PA4465N193T). These cells were complemented in trans with an inducible plasmid-borne copy of wild-type PA4456 (Fig. S2). K2732-PA4465N193T harboring either the empty control vector or the uninduced PA4465 vector grew in 0.5 μg/ml of CHIR-090, whereas K2732 containing empty vector did not, consistent with this engineered mutation reducing susceptibility. Induction of PA4465 expression strongly reduced growth of K2732-PA4465N193T consistent with a role for PA4465N193T (Fig. S2).
TABLE 4.
Mutations identified in mutants isolated during serial passage in CHIR-090
| Straina | Passage step (CHIR-090 MIC in μg/ml) | Mutation or amino acid substitution(s) |
|---|---|---|
| K2732 (WT efflux) | Parent (1) | |
| K2732-P2 | 2 (4) | FabF1T306A |
| K2732-P6 | 6 (32) | nfxB stop TGA-Cys; FabGD190G; PA4465N193T |
| K2732-P13 | 13 (64) | MexRT130P; nfxB stop TGA-Cys; FabGD190G; PA4465N193T |
| K2733 (efflux deficient) | Parent (0.0313) | |
| K2733-P6a (nc) | 6 (0.25) | PA4465N193T |
| K2733-P6b (sc) | 6 (0.5) | FabGA159V |
| K2733-P13a (nc) | 13 (0.5) | FabGA167V; PA4465N193T |
| K2733-P13b (sc) | 13 (1) | FabGA167V; PA4465N193T |
nc, normal colony size; sc, small colony size.
TABLE 5.
Impact of LpxCG208S on susceptibility of P. aeruginosa PAO1 to CHIR-090 and on LpxC enzymatic activity and binding to CHIR-090
| Strain | CHIR-090 MIC (μg/ml) | Protein | kcat (s−1) | Km (μM) | kcat/Km (s−1 M−1) | Mean Ki app (nM) ± SD (CHIR-090) |
|---|---|---|---|---|---|---|
| K767 | 0.5 | LpxC | 0.89 | 5.89 | 1.51 x 105 | 0.13 ± 0.01 |
| K767-LpxCG208S | 2 | LpxCG208S | 0.53 | 32.4 | 1.64 x 104 | 9.64 ± 2.08 |
An lpxC mutation encoding LpxCG208S reduces susceptibility to CHIR-090.
A serial passage experiment in CHIR-090 using a P. aeruginosa mutS hypermutator strain (CDR0017) generated two mutants with upregulated expression of LpxC (20). Here, we examined a third mutant from this experiment that did not have upregulated LpxC expression and lacked the associated lpxC upstream mutation, which revealed a mutation within the structural lpxC gene encoding LpxCG208S. For further study, we engineered it into the P. aeruginosa nonhypermutator parent PAO1 strain K767 (K767-LpxCG208S, Table 1), where it shifted susceptibility to CHIR-090 4-fold (MIC from 0.5 to 2 μg/ml, Table 5). Purified LpxCG208S was ∼9-fold less active than the wild-type LpxC (kcat/Km of 1.64 × 104 compared to 1.51 × 105 s−1 M–1, Table 5) but also bound CHIR-090 ∼74-fold less than did the wild-type protein (Ki app of 9.64 ± 2.08 compared to 0.13 ± 0.01 nM, Table 5). Therefore, the G208S substitution decreased binding to CHIR-090. Although the LpxCG208S protein was less catalytically active, no difference in growth curves of cells expressing LpxCG208S (Mueller-Hinton broth) was seen compared to the parent by standard growth curve (data not shown).
Different LpxC protein variants have differential impacts on susceptibility to LpxC inhibitor scaffolds.
To date, we have selected three different LpxC target alterations across this and previous studies (L18V, G208S, and A214V) using different compounds. We examined the specificity of these changes for different LpxC inhibitor scaffolds (Table 6). As a control, strain K767-CDR0026, which demonstrates constitutive upregulation of wild-type LpxC, was included (20). Since the impact of LpxC upregulation on susceptibility to LpxC inhibitors is presumably mediated by a compound titration effect, it should impact all LpxC inhibitors regardless of scaffold. As expected, susceptibility to CHIR-090, compound 3 and compound 2 are all reduced by 16- to 32-fold against K767-CDR0026 compared to its parent (Table 6). In contrast, the specific LpxC variants tested here had differential impacts that correlated generally with which compound was used to select the mutation. The engineered strain (K767-CDJ0011) expressing LpxCL18V (20) was 8-fold less susceptible to CHIR-090 (Table 6). This is consistent with the LpxCL18V variant having been originally identified through a selection experiment with CHIR-090. This strain was not less susceptible to compound 3, but there was a 4-fold shift in susceptibility to compound 2. The LpxC variant of K767-LpxCG208S was also originally identified from mutants selected using CHIR-090. This engineered mutant was 4-fold less susceptible to CHIR-090 but was not less susceptible, and was possibly slightly more susceptible, to both compound 3 and compound 2 (Table 6). Two separate mutants selected on compound 3 (K767-CDJ0037 and PAO1V-CDJ0042, Table 6) that express the LpxCA214V variant were substantially less susceptible (≥16-fold) to compound 3 and to compound 2 (8- to 16-fold) but were not less susceptible to CHIR-090. Although not tested directly here, mutants expressing LpxCA214V were also selected using compound 4 at 4× or 8× the MIC (Table S1), and therefore LpxCA214V also impacts susceptibility to compound 4. We utilized existing crystal structures of LpxC bound to CHIR-090 (9) and compound 2 (12), and an unpublished crystal structure of compound 3 in an effort to explain the impact of A214V, L18V, and G208S amino acid substitutions on the susceptibility to these compounds. Residue A214 is located opposite the α-helix of the domain II insert that encapsulates the acyl chain of the natural substrate. This region of the protein is flexible and for CHIR-090, compound 2, and compound 3 envelops the “tail” portions of these ligands. We manually modified residue 214 from Ala to Val using PyMOL (The PyMOL molecular graphics system (v2.0; Schrödinger, LLC) in the CHIR-090 LpxCPa crystal structure and rendered the surface of the binding pocket. We then overlaid the crystal structures of ligands compound 2 and compound 3 bound to LpxCPa to visually inspect whether they can fit into the altered binding pocket. As illustrated in Fig. 3, the phenyl group of compounds 2 and 3 would clash with A214V, and a rearrangement of the flexible region of the protein would need to occur to accommodate this change. We hypothesize this causes a reduction in the activity of compounds 2 and 3 against LpxCA214V. Interestingly, CHIR-090 is agnostic to this change because the acetylene substituent can tolerate this modification. In fact, this modification is a better complement to the ligand and could be the reason for the slightly improved MIC against mutants expressing this variant. These observations are consistent with previous reports that a different tunnel residue modification differentially affected susceptibility to LpxC inhibitors, depending on the position of acetylene groups (26, 27). The L18V substitution reduces the activity of CHIR-090 and to a potentially lesser degree for compound 2 but had no impact on compound 3. The L18V substitution increases the binding cavity of ligands (Fig. 4). We hypothesize that Leu18 for CHIR-090 acts as a scaffolding residue to keep the ligand in place, whereas for compounds 2 and 3, the ligand is interacting with His19 (via CH—O interaction or NH—O interaction at 3.4 Å) and is less reliant on Leu18 to maintain the binding mode (Fig. 4). Therefore, the more rigid compounds 2 and 3 are not as impacted by this transformation as is CHIR-090. The crystal structure information does not provide a clear notion as to the impact or selectivity of G208S since it involves a side chain that points away from the binding site of the ligand.
TABLE 6.
Impact of different LpxC target mutations on susceptibility to LpxC inhibitor scaffolds
| Strain | LpxC variant | MIC (μg/ml)a
|
||||
|---|---|---|---|---|---|---|
| CHIR-090 | Compound 3 | Compound 2 | MER | TOB | ||
| K767 | LpxCWT | 1 | 0.50 | 0.25 | 1 | 0.125 |
| K767-CDR0026 | LpxCWT upregulated | 16 | 16 | 8 | 1 | 0.25 |
| K767-CDJ0011 | LpxCL18V | 8 | 0.5 | 1 | 1 | 0.25 |
| K767-LpxCG208S | LpxCG208S | 4 | 0.125 | 0.125 | 0.5 | 0.125 |
| K767-CDJ0037 | LpxCA214V | 0.5 | >16 | 4 | 0.5 | 0.125 |
| PAO1V | LpxCWT | 1 | 0.25 | 0.25 | 1 | 0.125 |
| PAO1V-CDJ0042 | LpxCA214V | 0.5 | 16 | 2 | 1 | 0.125 |
Susceptibility shifts ≥ 4-fold are indicated in boldface. MER, meropenem; TOB, tobramycin.
FIG 3.

Overlay of the CHIR-090 structure (green, PDB 5U39) with compound 2 (orange, PDB 6MAE) and compound 3 (blue, unpublished). The A214V location is modeled on the CHIR-090 structure and highlighted by gray sticks; the protein surface with this modification is illustrated in gray.
FIG 4.

Overlay of the CHIR-090 structure (green, PDB 5U39) with compound 2 (orange, PDB 6MAE) and compound 3 (blue, unpublished). The L18V location is modeled on the CHIR-090 structure and highlighted in gray sticks. The distance between compound 2 and His19 (3.4 Å) is indicated by a yellow dashed line.
Upregulation of LpxC expression combined with LpxCA214V can substantially decrease susceptibility to compound 1.
To explore the potential impact of upregulated expression of a resistant variant of LpxC, we compared the impact of LpxCA214V expressed in trans to that of wild-type LpxC on susceptibility to compound 1 in P. aeruginosa K767 (summarized in Table 7). The MIC of compound 1 against PAO1 harboring the empty plasmid vector pAK1900 was 1 μg/ml and rose to 4 μg/ml against cells containing pAK1900 with the wild-type lpxC gene with native upstream region (pAK-LpxC WT). This shift is likely due to the copy number of pAK1900, which is approximately 4 to 5 copies per cell. Adding the mutation encoding LpxCA214V (pAK-LpxCA214V) further reduced susceptibility 4- to 8-fold, confirming the direct role for A214V in reducing susceptibility to compound 1. As expected, the MIC of compound 1 against cells containing pAK1900 with the wild-type lpxC gene but with the upstream mutation (pAK-LpxC WT UP) was ∼8-fold higher than against cells with the same vector lacking the upstream mutation (an MIC of 32 μg/ml compared to an MIC of 4 μg/ml), presumably reflecting further increased expression of LpxC. Adding the A214V-encoding lpxC variant (pAK-LpxCA214V UP) decreased susceptibility at least an additional 8-fold, providing further confirmation of the role of A214V but also showing that LpxCA214V and protein overexpression together is tolerated and generates substantial decreases in susceptibility to compound 1. This also extends the specificity of this mutation to include compound 1 and, consistent with the data in Table 6, the A214V mutation did not affect CHIR-090, even upon overexpression of LpxCA214V.
TABLE 7.
Impact of LpxC overexpression and LpxCA214V amino acid substitution on susceptibility to compound 1 and CHIR-090 in PAO1 strain K767
| Strain | MIC (μg/ml) |
|
|---|---|---|
| Compound 1 | CHIR-090 | |
| K767 | 1 | 1 |
| K767 (pAK1900) | 1 | 1 |
| K767 (pAK-LpxC WT) | 4 | 4 |
| K767 (pAK-LpxCA214V) | 16–32 | 2 |
| K767 (pAK-LpxC WT UP) | 32 | 64 |
| K767 (pAK-LpxCA214V UP) | >128 | 32 |
DISCUSSION
In this study, we show that previously identified mechanisms decreasing the in vitro susceptibility to the LpxC inhibitor CHIR-090 also impact the susceptibility to several newer LpxC inhibitor scaffolds. Upregulation of efflux pumps MexAB-OprM, MexCD-OprJ, or MexEF-OprN reduced the susceptibility to CHIR-090 (20), and we show that these pumps affect the newer LpxC inhibitors under study here. Single-step resistance selections with another inhibitor, LpxC-4, yielded mutants upregulated for MexEF-OprN expression (nfxC mutants) and MexAB-OprM expression (nalB mutants) but did not select mutants upregulated for MexCD-OprJ (nfxB mutants) (22). Very recently reported single-step and passage selection experiments with another series of newer LpxC inhibitors, typified by ACHN-975, yielded mutants upregulated for MexCD-OprJ and MexEF-OprN but not MexAB-OprM (19). Although not definitive, this suggests that there may be substantial differences in pump recognition profiles across different LpxC structures. Consistent with this, we also saw a differential impact of MexCD-OprJ upregulation on the different compounds studied here. A thorough evaluation of the impact of these pumps on activity across a broad range of LpxC inhibitors might therefore yield insights into pump evasion. As is the case for many antibacterial compounds, efflux can work together with nonefflux mechanisms to generate significant levels of resistance, and this was shown here for CHIR-090 and compound 1.
Among the nonefflux mechanisms identified here are novel alterations in the LpxC protein target, and we show that these can be scaffold specific, as might be expected. The remaining mechanisms of upregulated LpxC protein expression, as well as mutations in fabG and the newly identified mutations in fabF1 and PA4465, could differ from specific target mutations in that they may be more scaffold independent. We and and others (19) have found that upregulation of LpxC mediated a generally consistent shift in susceptibility to a range of LpxC inhibitors, as expected. The fatty acid biosynthetic cycle, within which FabG resides, is intricately related to lipid A biosynthesis, and selection of fabG mutants is reminiscent of the selection of fabZ mutants by LpxC inhibitors in Escherichia coli (28) and Klebsiella pneumoniae (22, 29). Alterations in fabZ may mediate resistance in E. coli by altering intermediate flux between pathways (28) and/or via intricate regulatory pathways controlling phospholipid and lipid A homeostasis (30); however, the interrelationships of these pathways and the mechanism(s) by which fabG mutations mediate resistance in P. aeruginosa are not currently well understood. There appears to be a large number of fabG mutations that are tolerated and, in keeping with this, they were consistently selected in studies with CHIR-090 (20) and other compounds in this study, as well as by the ACHN series of LpxC inhibitors (19). Intriguingly, although LpxC-4 did select fabZ mutants in K. pneumoniae, no fabG mutations were reported for P. aeruginosa (22), which could suggest that some scaffolds are less impacted by mechanisms mediated by fatty acid pathway mutations, but this remains to be more fully explored.
Two new mutations in fabF1 (encoding beta-ketoacyl-acyl carrier protein synthase II [3]) and the hypothetical gene PA4465 (3) were identified in this study. Cells lacking FabF1 are defective in the production of cis-vaccenic acid (31) and become nonmotile (32), but an understanding of the specific function of FabF1 in P. aeruginosa awaits more study (33). Although the effect of the alteration in FabF1 on susceptibility to CHIR-090 appears to be subtle, the identification of a mutation in a gene related to fatty acid biosynthesis is consistent with the role of fabG mutations in P. aeruginosa and fabZ mutations in E. coli (28, 30, 34) and K. pneumoniae (22, 29) in decreasing susceptibility to LpxC inhibitors. Mutations in fabZ in E. coli may increase metabolic flux into the lipid A pathway and/or rebalance phospholipid and lipid A homeostasis to confer resistance, underscoring the intricate relationship between these pathways (28, 30, 35). This notion has been further supported by the finding that the majority of proteins in the E. coli fatty acid elongation cycle were found as part of a broad LpxC interactome (36). The gene PA4465 encodes a hypothetical protein that appears to be an analog (50% identity, 65% similarity) to E. coli RapZ (37). RapZ is important for controlling the cellular levels of GlmS (glucosamin-6-phosphate synthase) and therefore the cellular levels of glucoasamine-6-phosphate (GlcN6P), an important precursor for the synthesis of both the peptidoglycan cell wall and LPS. GlmS is controlled directly by the small RNA GlmZ, which binds and activates glmS mRNA. GlmZ levels are controlled by RapZ, an RNase adaptor protein that targets GlmZ to RNase E for degradation. Another small RNA, GlmY, can act as an antiadaptor and sequester RapZ under conditions of GlcN6P depletion, thereby stabilizing the GlmS mRNA (3, 37–42). Therefore, this regulatory circuit is important in controlling GlcN6P homeostasis through GlmS. The involvement of the putative RapZ homolog of P. aeruginosa, which may link these cell envelope biosynthetic pathways through a common pathway intermediate, is reminiscent of the interrelationship between the lipid A and fatty acid biosynthesis pathways mentioned above. Indeed, several proteins related to cell wall biosynthesis, including RapZ, GlmS, GlmU (bifunctional glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridylyltransferase), and GlmM (phosphoglucosamine mutase) were identified in an E. coli LpxC interactome study (36). It was also recently shown that E. coli cells remodel their peptidoglycan in response to defects in LPS transport to the OM, which provides protection against cell lysis (43). It is interesting to note that alterations in FabF1 and PA4465 have so far only emerged from serial passage studies. FabF1 is not essential for growth of P. aeruginosa (31), RapZ is not essential in E. coli (44), and transposon insertions have been reported in PA4465 (Pseudomonas Genome DB [www.pseudomonas.com]), suggesting that these functions could presumably be lost at high frequency. Their lack of appearance during single-step experiments could possibly reflect the subtle effect such mutations have on susceptibility, making them less likely to survive single-step selections at multiples of the MIC. Nonetheless, the identification of multiple-pathway interrelated mutations indicates that adjustments of these interrelationships contributes to survival upon LpxC inhibition. This notion is further expanded by the recent finding that mutations in acpP, which encodes acyl carrier protein required for many steps of the lipid biosynthetic pathway and which interacts with FabG, reduced the susceptibility to ACHN-95 and related molecules (19). The exact mechanism(s) by which these mutations mediate decreased susceptibility warrants further study.
Relating to the intricate pathway interrelationships described above, overexpression of LpxC in E. coli can be toxic, and therefore LpxC levels are tightly controlled via proteolytic degradation by FtsH and other mechanisms (36). This suggested that target upregulation might not be readily selected by LpxC inhibitors, and this has not to our knowledge been reported in E. coli. P. aeruginosa LpxC lacks this proteolytic control by FtsH (45) and, perhaps reflecting this, we found that overexpression of LpxC is well tolerated in P. aeruginosa and that upregulated expression mediates a substantial decrease in susceptibility to LpxC inhibitors (20). This ability to tolerate upregulated LpxC could therefore combine with alterations of the LpxC protein to mediate more substantial decreases in susceptibility, and indeed we show this to be the case using LpxCA214V, although the contribution of target mutations can be scaffold specific. Although we did not find an instance of upregulated expression of LpxCA214V in our passaging studies, we did identify the LpxCG208S variant from passaging and upon reviewing historical data from studies with compound 1, we had a P. aeruginosa mutant that emerged during a hollow-fiber model experiment that harbored the lpxC upstream mutation, overexpressed LpxC, and had the mutation encoding LpxCA214V, all suggesting that this combination does emerge under selection pressure.
In summary, we have assembled a range of some available information on mechanisms and interactions that may decrease susceptibility to LpxC inhibitors from studies over several years with different LpxC inhibitor compounds. An understanding of which in vitro selected mechanisms would ultimately prove to emerge during use of an LpxC inhibitor in the clinic of course awaits further progress in this area. These studies can, however, inform our understanding of antibacterial targets and pathways and provide a framework for establishing dosing requirements intended offset the development of resistance.
MATERIALS AND METHODS
Strains, growth conditions, and susceptibility testing.
The strains and plasmids used in this study are listed in Table 1. Media for routine genetic manipulations was lysogeny broth (LB) or agar unless specified otherwise and media for mutant selections and susceptibility testing was cation-adjusted Mueller-Hinton broth or agar. Single-step mutant selections, serial passage studies and MIC determinations were carried out as previously described (20). Compound CHIR-090 was previously described (20). Compound 1 (9) and compounds 2 to 4 (12) were synthesized at Novartis. Other antibiotics and IPTG (isopropyl-β-d-thiogalactopyranoside) were purchased commercially. Susceptibility testing was carried out as previously described (20).
Whole-genome sequencing.
Genomic DNA isolation, fragment library preparation, Illumina sequencing, and variant calling were performed as described previously (20).
DNA methods.
The lpxC G622A mutation encoding LpxCG208S and the PA4465 A578C mutation encoding PA4465N193T were introduced onto the genome of P. aeruginosa using the sac-based gene exchange vector pEX18ApGW as follows. The lpxC gene harboring the A578C mutation was PCR amplified from the genome of serial passage mutant CDR0017 (colony 3 described in reference 20) using the Gateway adapted primers lpxC-5′ (TACAAAAAAGCAGGCTCAGGAGTAGAGATGTGATTGGTG) and lpxC-3′ (TACAAGAAAGCTGGGTGAGATCGTCGGCAATCCACGCCTG). Gateway adapters were added with a second round of PCR using the attB1 (GGGGACAAGTTTGTACAAAAAAGCAGGCT)/attB2 (GGGGACCACTTTGTACAAGAAAGCTGGGT) primer pair, and then this fragment was cloned into pDON221 using Gateway cloning (Invitrogen). A sequence-confirmed pDON221-lpxC A578C construct was then used to create pEX18ApGW-lpxC-G622A using Gateway cloning following the supplied instructions. The PA4465 gene harboring the A578C mutation was PCR amplified from the genome of K732-6 using Gateway-adapted primers PA4465-5′ (GGCCAGTGCCAAGCTTgaccgaagaacatctcgaac) and PA4465-3′ (ACGAATTCGAGCTCGGTACCgtctgtccttcttgttatggg). Gateway adapters were added in a second round of PCR using the attB1/attB2 primers, and this fragment was cloned into pDON221 using Gateway technology. A sequence-confirmed pDON221-PA4465-A578C construct was then used to create pEX18ApGW-PA4465-A578C using Gateway cloning. Plasmids pEX18ApGW-lpxC-G622A and pEX18ApGW-PA4465-A578C were then mated into P. aeruginosa PAO1 strains K767 and K2732, respectively, using the E. coli Stellar strain (Clontech) that harbored the helper plasmid pRK2013 (46). P. aeruginosa merodiploids were selected on L-agar containing 25 μg/ml Irgasan and 150 μg/ml carbenicillin, and double crossovers were then selected on L-agar with 5% sucrose. Sucrose-resistant, carbenicillin-sensitive resolvants were then screened for growth on L-agar containing 2 μg/ml CHIR-090, and mutations were confirmed on the genome by PCR amplification and sequencing. IPTG-inducible complementation plasmids were made as follows. The fabF1 gene was PCR amplified from P. aeruginosa wild-type PAO1 using the Gateway-adapted primers 5′ fabF1 GW (TCGAGGAGGATATTCgtgtcgcgtagacgcgtcgtcattactg) and 3′ fabF1 GW (CAAGAAAGCTGGGTTtcagtcggcgaacctgcggaacac), and the PA4465 gene was PCR amplified from PAO1 using the Gateway-adapted primers 5′ PA4465 GW ( TCGAGGAGGATATTCatgcgcctgatcatcgtcag) and 3′ PA4465 GW (CAAGAAAGCTGGGTTctagctgctgagatcgcggtg). Gateway adapters were then added in a second round of PCR using the primers attB1-01 (GGGGACAAGTTTGTACAAAAAAGCAGGCTCGAGGAGGATATTC) and attB2-02 (GGGGACCACTTTGTACAAGAAAGCTGGGTTTCA), and the resulting fragments were cloned into pDON221 using Gateway cloning. Sequence-confirmed pDON221-fabF1 and pDON221-PA4465 were then used to create pMM-10-fabF1 and pMM-10-PA4465, respectively, using Gateway cloning. Plasmids were electroporated into P. aeruginosa strains as previously described (47). The lpxC genes on plasmids pAK-lpxC and pAK-lpxCRBS (20) were altered to encode the LpxCA214V protein using a Stratagene QuikChange site-directed mutagenesis kit with the primer pair RC271 (CGTCGACCACGATCACGTTCTCCACGCTAC) and RC272 (GTAGCGTGGAGAACGTGATCGTGGTCGACG) according to the manufacturer’s protocol.
Purification of P. aeruginosa LpxC and LpxCG208S and biochemical characterization.
P. aeruginosa wild-type LpxC and LpxCG208S were expressed in E. coli and purified as described previously (20). In vitro LpxC reactions and determination of inhibitor activity was conducted as described previously (20) except that determinations of the CHIR-090 apparent inhibitor constant (Ki app) were made using 2 nM LpxCG208S due to the reduced enzymatic activity of this variant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Keith Poole and Mike Vasil for strains; Herbert Schweizer, Steve Lory, and Andrew Kropinski for genetic tools; and Xia Yang, Daniel Pevear, and Laura McDowell for helpful discussions.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Li XZ, Plesiat P, Nikaido H. 2015. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev 28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poole K. 2002. Outer membranes and efflux: the path to multidrug resistance in Gram-negative bacteria. Curr Pharm Biotechnol 3:77–98. doi: 10.2174/1389201023378454. [DOI] [PubMed] [Google Scholar]
- 3.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar A, Schweizer HP. 2005. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv Drug Deliv Rev 57:1486–1513. doi: 10.1016/j.addr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kalinin DV, Holl R. 2017. LpxC inhibitors: a patent review (2010-2016). Expert Opin Ther Pat 27:1227–1250. doi: 10.1080/13543776.2017.1360282. [DOI] [PubMed] [Google Scholar]
- 8.Erwin AL. 2016. Antibacterial drug discovery targeting the lipopolysaccharide biosynthetic enzyme LpxC. Cold Spring Harb Perspect Med 6:a025304. doi: 10.1101/cshperspect.a025304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piizzi G, Parker DT, Peng Y, Dobler M, Patnaik A, Wattanasin S, Liu E, Lenoir F, Nunez J, Kerrigan J, McKenney D, Osborne C, Yu D, Lanieri L, Bojkovic J, Dzink-Fox J, Lilly MD, Sprague ER, Lu Y, Wang H, Ranjitkar S, Xie L, Wang B, Glick M, Hamann LG, Tommasi R, Yang X, Dean CR. 2017. Design, synthesis, and properties of a potent inhibitor of Pseudomonas aeruginosa deacetylase LpxC. J Med Chem 60:5002–5014. doi: 10.1021/acs.jmedchem.7b00377. [DOI] [PubMed] [Google Scholar]
- 10.Kawai T, Kazuhiko I, Takaya N, Yamaguchi Y, Kishii R, Kohno Y, Kurasaki H. 2017. Sulfonamide-based non-alkyne LpxC inhibitors as Gram-negative antibacterial agents. Bioorg Med Chem Lett 27:1045–1049. doi: 10.1016/j.bmcl.2016.12.059. [DOI] [PubMed] [Google Scholar]
- 11.Kurasaki H, Tsuda K, Shinoyama M, Takaya N, Yamaguchi Y, Kishii R, Iwase K, Ando N, Nomura M, Kohno Y. 2016. LpxC Inhibitors: design, synthesis, and biological evaluation of oxazolidinones as Gram-negative antibacterial agents. ACS Med Chem Lett 7:623–628. doi: 10.1021/acsmedchemlett.6b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee PS, Lapointe G, Madera AM, Simmons RL, Xu W, Yifru A, Tjandra M, Karur S, Rico A, Thompson K, Bojkovic J, Xie L, Uehara K, Liu A, Shu W, Bellamacina C, McKenney D, Morris L, Tonn GR, Osborne C, Benton BM, McDowell L, Fu J, Sweeney ZK. 2018. Application of virtual screening to the identification of new LpxC inhibitor chemotypes, oxazolidinone and isoxazoline. J Med Chem 61:9360–9370. doi: 10.1021/acs.jmedchem.8b01287. [DOI] [PubMed] [Google Scholar]
- 13.Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Kach A, Eberl L, Riedel K, DeMarco SJ, Robinson JA. 2010. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327:1010–1013. doi: 10.1126/science.1182749. [DOI] [PubMed] [Google Scholar]
- 14.Onishi HR, Pelak BA, Gerckens LS, Silver LL, Kahan FM, Chen MH, Patchett AA, Galloway SM, Hyland SA, Anderson MS, Raetz CR. 1996. Antibacterial agents that inhibit lipid A biosynthesis. Science 274:980–982. doi: 10.1126/science.274.5289.980. [DOI] [PubMed] [Google Scholar]
- 15.Mdluli KE, Witte PR, Kline T, Barb AW, Erwin AL, Mansfield BE, McClerren AL, Pirrung MC, Tumey LN, Warrener P, Raetz CR, Stover CK. 2006. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:2178–2184. doi: 10.1128/AAC.00140-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kline T, Andersen NH, Harwood EA, Bowman J, Malanda A, Endsley S, Erwin AL, Doyle M, Fong S, Harris AL, Mendelsohn B, Mdluli K, Raetz CR, Stover CK, Witte PR, Yabannavar A, Zhu S. 2002. Potent, novel in vitro inhibitors of the Pseudomonas aeruginosa deacetylase LpxC. J Med Chem 45:3112–3129. doi: 10.1021/jm010579r. [DOI] [PubMed] [Google Scholar]
- 17.McClerren AL, Endsley S, Bowman JL, Andersen NH, Guan Z, Rudolph J, Raetz CR. 2005. A slow, tight-binding inhibitor of the zinc-dependent deacetylase LpxC of lipid A biosynthesis with antibiotic activity comparable to ciprofloxacin. Biochemistry 44:16574–16583. doi: 10.1021/bi0518186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen F, Aggen JB, Andrews LD, Assar Z, Boggs J, Choi T, Dozzo P, Easterday AN, Haglund CM, Hildebrandt DJ, Holt MC, Joly K, Jubb A, Kamal Z, Kane TR, Konradi AW, Krause KM, Linsell MS, Machajewski TD, Miroshnikova O, Moser HE, Nieto V, Phan T, Plato C, Serio AW, Seroogy J, Shakhmin A, Stein AJ, Sun AD, Sviridov S, Wang Z, Wlasichuk K, Yang W, Zhou X, Zhu H, Cirz RT. 2019. Optimization of LpxC inhibitors for antibacterial activity and cardiovascular safety. ChemMedChem 14:1560–1572. doi: 10.1002/cmdc.201900287. [DOI] [PubMed] [Google Scholar]
- 19.Krause KM, Haglund CM, Hebner C, Serio AW, Lee G, Nieto V, Cohen F, Kane TR, Machajewski TD, Hildebrandt D, Pillar C, Thwaites M, Hall D, Miesel L, Hackel M, Burek A, Andrews LD, Armstrong E, Swem L, Jubb A, Cirz RT. 2019. Potent LpxC inhibitors with in vitro activity against multi-drug resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 63:e00977-19. doi: 10.1128/AAC.00977-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caughlan RE, Jones AK, Delucia AM, Woods AL, Xie L, Ma B, Barnes SW, Walker JR, Sprague ER, Yang X, Dean CR. 2012. Mechanisms decreasing in vitro susceptibility to the LpxC inhibitor CHIR-090 in the gram-negative pathogen Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:17–27. doi: 10.1128/AAC.05417-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F, Larbig KD, Heeb S, Schock U, Pohl TM, Wiehlmann L, Tummler B. 2010. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol 192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomaras AP, McPherson CJ, Kuhn M, Carifa A, Mullins L, George D, Desbonnet C, Eidem TM, Montgomery JI, Brown MF, Reilly U, Miller AA, O’Donnell JP. 2014. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid A biosynthesis in Gram-negative pathogens. mBio 5:e01551. doi: 10.1128/mBio.01551-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maseda H, Saito K, Nakajima A, Nakae T. 2000. Variation of the mexT gene, a regulator of the MexEF-oprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol Lett 192:107–112. doi: 10.1111/j.1574-6968.2000.tb09367.x. [DOI] [PubMed] [Google Scholar]
- 24.Li XZ, Barre N, Poole K. 2000. Influence of the MexA-MexB-oprM multidrug efflux system on expression of the MexC-MexD-oprJ and MexE-MexF-oprN multidrug efflux systems in Pseudomonas aeruginosa. J Antimicrob Chemother 46:885–893. doi: 10.1093/jac/46.6.885. [DOI] [PubMed] [Google Scholar]
- 25.Sobel ML, Neshat S, Poole K. 2005. Mutations in PA2491 (mexS) promote MexT-dependent mexEF-oprN expression and multidrug resistance in a clinical strain of Pseudomonas aeruginosa. J Bacteriol 187:1246–1253. doi: 10.1128/JB.187.4.1246-1253.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang X, Lee CJ, Chen X, Chung HS, Zeng D, Raetz CR, Li Y, Zhou P, Toone EJ. 2011. Syntheses, structures, and antibiotic activities of LpxC inhibitors based on the diacetylene scaffold. Bioorg Med Chem 19:852–860. doi: 10.1016/j.bmc.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee CJ, Liang X, Chen X, Zeng D, Joo SH, Chung HS, Barb AW, Swanson SM, Nicholas RA, Li Y, Toone EJ, Raetz CR, Zhou P. 2011. Species-specific and inhibitor-dependent conformations of LpxC: implications for antibiotic design. Chem Biol 18:38–47. doi: 10.1016/j.chembiol.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clements JM, Coignard F, Johnson I, Chandler S, Palan S, Waller A, Wijkmans J, Hunter MG. 2002. Antibacterial activities and characterization of novel inhibitors of LpxC. Antimicrob Agents Chemother 46:1793–1799. doi: 10.1128/aac.46.6.1793-1799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mostafavi M, Wang L, Xie L, Takeoka KT, Richie DL, Casey F, Ruzin A, Sawyer WS, Rath CM, Wei JR, Dean CR. 2018. Interplay of Klebsiella pneumoniae fabZ and lpxC mutations leads to LpxC inhibitor-dependent growth resulting from loss of membrane homeostasis. mSphere 3:e00508-18. doi: 10.1128/mSphere.00508-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng D, Zhao J, Chung HS, Guan Z, Raetz CR, Zhou P. 2013. Mutants resistant to LpxC inhibitors by rebalancing cellular homeostasis. J Biol Chem 288:5475–5486. doi: 10.1074/jbc.M112.447607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kutchma AJ, Hoang TT, Schweizer HP. 1999. Characterization of a Pseudomonas aeruginosa fatty acid biosynthetic gene cluster: purification of acyl carrier protein (ACP) and malonyl-coenzyme A:ACP transacylase (FabD). J Bacteriol 181:5498–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Overhage J, Lewenza S, Marr AK, Hancock RE. 2007. Identification of genes involved in swarming motility using a Pseudomonas aeruginosa PAO1 mini-Tn5-lux mutant library. J Bacteriol 189:2164–2169. doi: 10.1128/JB.01623-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang YM, Rock CO. 2012. Will the initiator of fatty acid synthesis in Pseudomonas aeruginosa please stand up? J Bacteriol 194:5159–5161. doi: 10.1128/JB.01198-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao Z, Davis RM, Kishony R, Kahne D, Ruiz N. 2012. Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proc Natl Acad Sci U S A 109:E2561–E2568. doi: 10.1073/pnas.1209742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Emiola A, Andrews SS, Heller C, George J. 2016. Crosstalk between the lipopolysaccharide and phospholipid pathways during outer membrane biogenesis in Escherichia coli. Proc Natl Acad Sci U S A 113:3108–3113. doi: 10.1073/pnas.1521168113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomanek N, Arends J, Lindemann C, Barkovits K, Meyer HE, Marcus K, Narberhaus F. 2018. Intricate crosstalk between lipopolysaccharide, phospholipid and fatty acid metabolism in Escherichia coli modulates proteolysis of LpxC. Front Microbiol 9:3285. doi: 10.3389/fmicb.2018.03285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalamorz F, Reichenbach B, Marz W, Rak B, Gorke B. 2007. Feedback control of glucosamine-6-phosphate synthase GlmS expression depends on the small RNA GlmZ and involves the novel protein YhbJ in Escherichia coli. Mol Microbiol 65:1518–1533. doi: 10.1111/j.1365-2958.2007.05888.x. [DOI] [PubMed] [Google Scholar]
- 38.Urban JH, Vogel J. 2008. Two seemingly homologous noncoding RNAs act hierarchically to activate glmS mRNA translation. PLoS Biol 6:e64. doi: 10.1371/journal.pbio.0060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gopel Y, Khan MA, Gorke B. 2014. Menage a trois: posttranscriptional control of the key enzyme for cell envelope synthesis by a base-pairing small RNA, an RNase adaptor protein, and a small RNA mimic. RNA Biol 11:433–442. doi: 10.4161/rna.28301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gopel Y, Papenfort K, Reichenbach B, Vogel J, Gorke B. 2013. Targeted decay of a regulatory small RNA by an adaptor protein for RNase E and counteraction by an anti-adaptor RNA. Genes Dev 27:552–564. doi: 10.1101/gad.210112.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reichenbach B, Gopel Y, Gorke B. 2009. Dual control by perfectly overlapping sigma 54- and sigma 70- promoters adjusts small RNA GlmY expression to different environmental signals. Mol Microbiol 74:1054–1070. doi: 10.1111/j.1365-2958.2009.06918.x. [DOI] [PubMed] [Google Scholar]
- 42.Reichenbach B, Maes A, Kalamorz F, Hajnsdorf E, Gorke B. 2008. The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli. Nucleic Acids Res 36:2570–2580. doi: 10.1093/nar/gkn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.More N, Martorana AM, Biboy J, Otten C, Winkle M, Serrano CKG, Monton Silva A, Atkinson L, Yau H, Breukink E, den Blaauwen T, Vollmer W, Polissi A. 2019. Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. mBio 10:e02729-18. doi: 10.1128/mBio.02729-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langklotz S, Schakermann M, Narberhaus F. 2011. Control of lipopolysaccharide biosynthesis by FtsH-mediated proteolysis of LpxC is conserved in enterobacteria but not in all gram-negative bacteria. J Bacteriol 193:1090–1097. doi: 10.1128/JB.01043-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan Y, Sachdeva M, Leeds JA, Meredith TC. 2012. Fatty acid biosynthesis in Pseudomonas aeruginosa is initiated by the FabY class of beta-ketoacyl acyl carrier protein synthases. J Bacteriol 194:5171–5184. doi: 10.1128/JB.00792-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enderle PJ, Farwell MA. 1998. Electroporation of freshly plated Escherichia coli and Pseudomonas aeruginosa cells. Biotechniques 25:954–958. doi: 10.2144/98256bm05. [DOI] [PubMed] [Google Scholar]
- 48.Masuda N, Ohya S. 1992. Cross-resistance to meropenem, cephems, and quinolones in Pseudomonas aeruginosa. Antimicrob Agents Chemother 36:1847–1851. doi: 10.1128/aac.36.9.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Srikumar R, Paul CJ, Poole K. 2000. Influence of mutations in the mexR repressor gene on expression of the MexA-MexB-oprM multidrug efflux system of Pseudomonas aeruginosa. J Bacteriol 182:1410–1414. doi: 10.1128/jb.182.5.1410-1414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Preston MJ, Fleiszig SM, Zaidi TS, Goldberg JB, Shortridge VD, Vasil ML, Pier GB. 1995. Rapid and sensitive method for evaluating Pseudomonas aeruginosa virulence factors during corneal infections in mice. Infect Immun 63:3497–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fetar H, Gilmour C, Klinoski R, Daigle DM, Dean CR, Poole K. 2011. mexEF-oprN multidrug efflux operon of Pseudomonas aeruginosa: regulation by the MexT activator in response to nitrosative stress and chloramphenicol. Antimicrob Agents Chemother 55:508–514. doi: 10.1128/AAC.00830-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Figurski DH, Helinski DR. 1979. Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci U S A 76:1648–1652. doi: 10.1073/pnas.76.4.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fuchs EL, Brutinel ED, Klem ER, Fehr AR, Yahr TL, Wolfgang MC. 2010. In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J Bacteriol 192:2779–2790. doi: 10.1128/JB.00168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choi KH, Schweizer HP. 2005. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.