

Abstract
Serlyticin-A is a secondary metabolite first isolated from a culture of Serratia ureilytica grown using squid pen as the sole carbon/nitrogen source. A previous study by Kuo et al. demonstrated that it has antioxidative and antiproliferative properties. However, the proposed chemical structure of serlyticin-A is likely incorrect based on the thermodynamic instability of its three contiguous heteroatom-heteroatom bonds. Here, we use quantum chemical calculations to predict 1H and 13C chemical shifts for serlyticin-A, and demonstrate a discrepancy between the calculated and experimental chemical shifts. We then propose several reasonable alternative structures for serlyticin-A. Considering the known antioxidant and antiproliferative activity of hydroxamic acids as well as their stability and prevalence in natural products of bacterial origin, we believe that serlyticin-A is most likely 3-indolylacetohydroxamic acid (4). We provide our rationale for this assignment as well as experimental data for pure 3-indolylacetohydroxamic acid obtained via de novo synthesis. This study highlights the power of computational NMR shift prediction to revise chemical structures for natural products like serlyticin-A.
Graphical Abstract
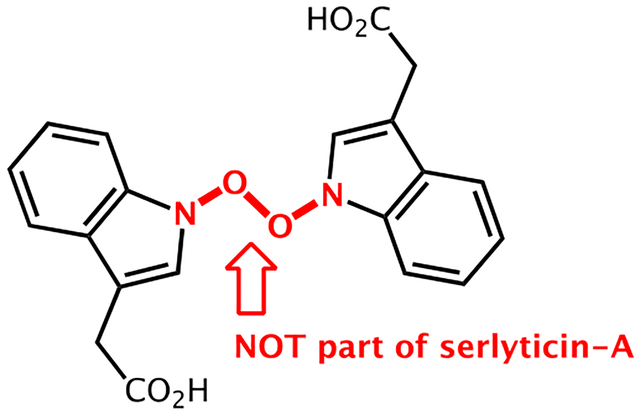
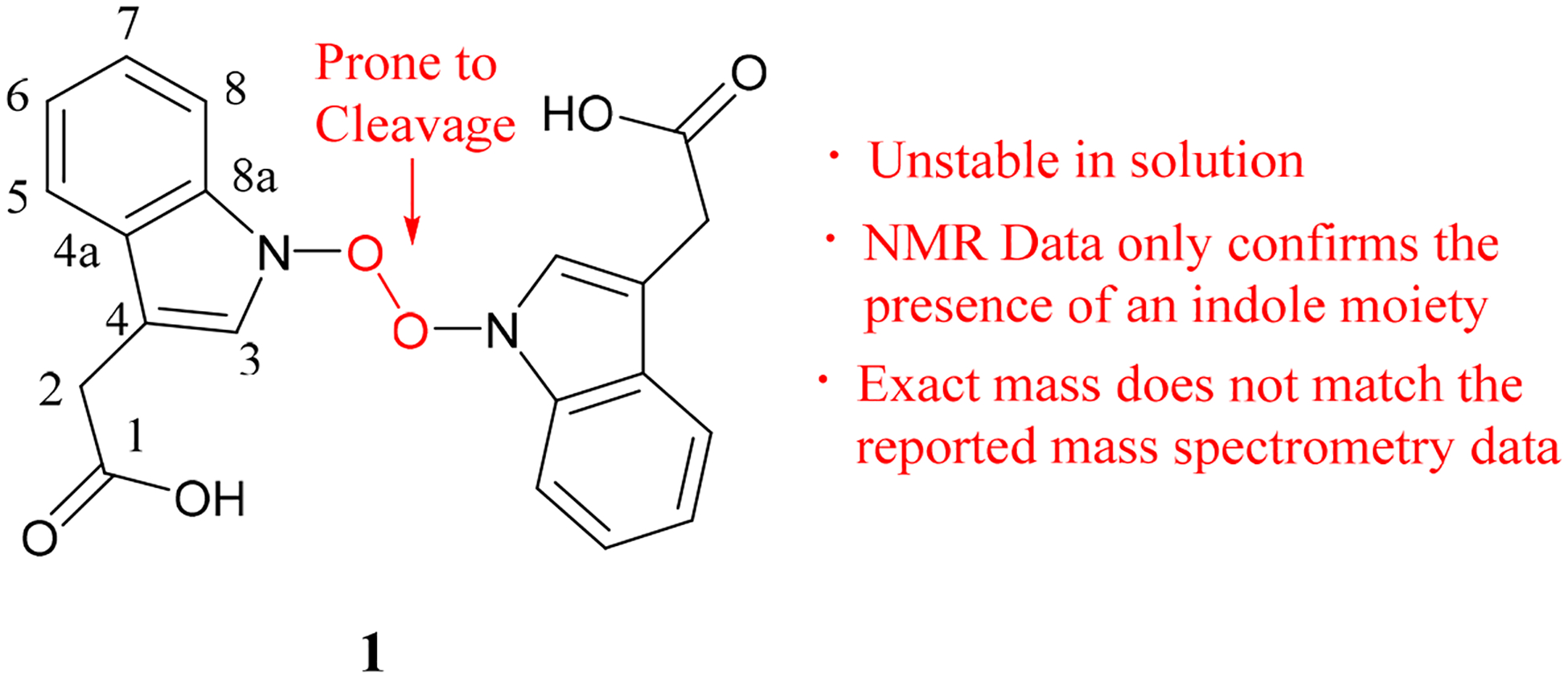
The identification of a novel natural product with antioxidant properties is highly beneficial to both the food and pharmaceutical industries. Previous efforts in this area have utilized a variety of organisms with plants being a major source of antioxidant compounds.1–4 Despite the fact that shellfish waste is a rich source of phenolic antioxidants, there have been relatively few attempts to isolate novel antioxidant natural products from this source.5,6 In 2012, serlyticin-A (1, Figure 1) was identified by Kuo and co-workers as one of the compounds in the s. ureilytica culture using squid pen waste as carbon/nitrogen source, and they demonstrated that it has antioxidative and antiproliferative properties.5 While serlyticin-A certainly has the potential for medicinal and food science applications, we suspect that the reported structure is incorrect for several reasons. First, the structure contains an O–O bond, which, based on the results of our calculations, would be prone to cleavage in solution. Second, the characterization data reported for serlyticin-A, including 1D and 2D NMR, mass spectrometry, UV-vis, and IR, are consistent with multiple potential structures. For example, 1D and 2D NMR data confirm the presence of an indole moiety but do not necessarily support the assignment of the proposed N–O–O–N substructure. Moreover, the reported mass spectrometry data do not match the exact mass of 1. While select tabulated numerical data were provided in the original report, the actual spectra were not.7 These factors coupled with the lack of X-ray crystallography data contribute to the ambiguity in the assignment of structure 1 to serlyticin-A. While it is an accepted practice to elucidate chemical structures relying on information obtained from NMR, IR, and UV-Vis, it is not uncommon for structures to be misassigned, leading to the otherwise unnecessary expenditure of additional resources in correcting them.8–10 Here, we report the use of quantum chemical calculations of NMR chemical shifts11–13 to demonstrate that the original structure of serlyticin-A may have been misassigned, and we provided several plausible alternative structures.
Figure 1.
The previously proposed structure of serlyticin-A.
RESULT AND DISCUSSION
Our investigations began with the structural optimization of 1. We observed a long O–O bond (~2.10 Å) in all relevant conformers of 1 optimized in the gas phase and solution (Figure 2). Computed Wiberg bond orders for the O–O bond were less than 0.32 for all conformers within 3 kcal/mol of the lowest energy conformer, suggesting that 1 is prone to fragment in both gas phase and solution. Optimization of these conformers while constraining the O–O bond to 1.48 Å (a reasonable distance for a covalent O–O bond) led to structures that are at least 15 kcal/mol higher in free energy than their unconstrained versions. Additionally, the calculated 13C and 1H NMR shifts for 1 deviate greatly from the reported NMR shifts (Table 1, Figure 2, and Table S1). For the constrained structure of 1, although the 13C shifts are close to the experimental shifts, several 1H shifts deviate greatly from the experimental shifts (Figure 2 and Table S1). Given the accuracy of such calculations,11–13 serlyticin-A is likely misassigned.
Figure 2.
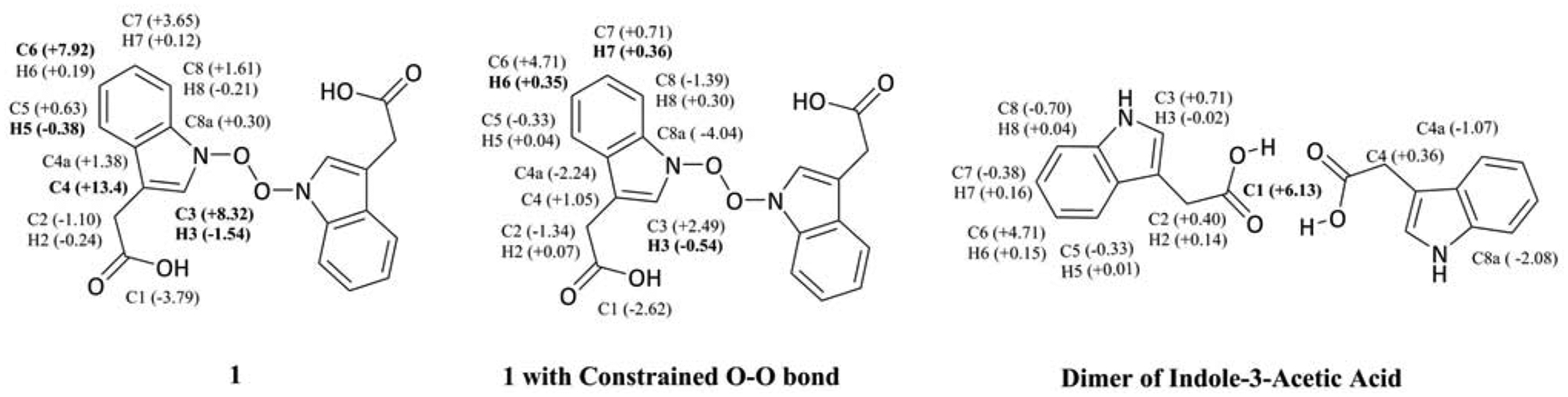
Deviations between calculated 13C and 1H chemical shifts and the reported experimental shifts for 1, constrained 1, and a dimer of indole-3-acetic acid. Deviations of less than 6 ppm for 13C and less than 0.3 ppm for 1H shifts are considered acceptable.13–15 Deviations exceeding these limits are bolded.
Table 1.
Summary of Computed NMR Results for 1–4
| Atom Label | Exp. δ (p.p.m) | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| C1 | 177.5 | 173.71 | 183.54 | 175.33 | 174.74 |
| C2 | 32.8 | 31.70 | 32.50 | 33.94 | 32.58 |
| C3 | 124.5 | 132.82 | 130.96 | 126.86 | 125.76 |
| C4 | 109.6 | 123.00 | 107.00 | 104.31 | 108.17 |
| C4a | 128.8 | 130.18 | 125.56 | 123.76 | 127.78 |
| C5 | 119.5 | 120.13 | 119.4 | 119.68 | 119.75 |
| C6 | 119.7 | 127.62 | 118.30 | 118.62 | 118.87 |
| C7 | 122.3 | 125.95 | 122.11 | 122.36 | 122.18 |
| C8 | 112.1 | 113.71 | 109.64 | 108.70 | 111.45 |
| C8a | 138.0 | 138.30 | 137.32 | 134.07 | 135.98 |
| MAD | 4.21 | 2.34 | 2.46 | 1.07 | |
| H2 | 3.68 | 3.44 | 3.44 | 3.54 | 3.78 |
| H3 | 7.14 | 5.60 | 7.10 | 7.14 | 7.18 |
| H5 | 7.54 | 7.15 | 7.42 | 7.52 | 7.61 |
| H6 | 6.99 | 7.18 | 7.11 | 7.16 | 7.15 |
| H7 | 7.07 | 7.19 | 7.28 | 7.27 | 7.24 |
| H8 | 7.33 | 7.12 | 7.56 | 7.41 | 7.40 |
| MAD | 0.45 | 0.16 | 0.10 | 0.10 |
Abbreviation: MAD, mean absolute deviation. Deviations of less than 6 ppm for 13C and less than 0.3 ppm for H shifts are considered acceptable.9−11 Chemical shifts exceeding the accepted deviations are bolded.
Kuo et al. reported 1H-1H COSY (correlation spectroscopy) and HMQC (heteronuclear multiple bond coherence) correlations, that are consistent with serlyticin-A containing an indole-3-acetic acid moiety. This led us to consider structural alternative structures possessing this group. Our predicted 13C chemical shifts for the indole-3-acetic acid dimer (Figure 2 and Table S1) matched the experimental shifts very well with only one calculated 13C chemical shift (C1) falling outside the acceptable range. Therefore, derivatives of this structure were examined further. Such structures included hydrazine, 2, hydroxylamine 3, and hydroxamic acid 4 (Figure 3). All these structures have computed chemical shifts that are consistent with the reported shifts for serlyticin-A, though 4 is the closest match (Table 1, Figures 3–4 and Table S3). Additional structures considered that were not good matches are described in the SI (Figure S1, Tables S5 and S6). Serlyticin-A has demonstrated antioxidant properties in a DPPH radical scavenging assay.5 Compounds 2–4 all have the potential to serve as effective radical scavengers due to the presence of N–O or N–N bonds. However, we deemed 4 to be the most likely structure of serlyticin-A for several reasons. First, to the best of our knowledge, N-amino indole natural products have not previously been reported. Second, while N-hydroxy indole natural products are known, these are rare and often difficult to isolate.14 Examples of notable N-hydroxyindole-containing natural products include stephacidin B, versicoamide F, nocathiacin-I, thiazomycin, coproverdine, notoamide G, and N-hydroxy-β-carbolines.15–23 The overwhelming majority of N-hydroxyindole-containing natural products possess substitution at the 2-position of the indole, likely a consequence of the tendency of indoles to be oxidized at C2. Finally, 4 contains a hydroxamic acid group—a structural motif that is found in numerous natural products of bacterial origin.24–27 Hydroxamic acids are excellent siderophores and bacteria often produce them to sequester iron from their environment. In fact, 4 itself has been shown to coordinate metals.28 Furthermore, many hydroxamic acids possess anti-proliferative properties, like that observed for serlyticin-A, due to their ability to potently inhibit histone deacetylases (HDACs).29,30 Next, we synthesized 4 from the methyl ester of indole acetic acid by converting the ester into the hydroxamic acid.31 We have characterized the product and the data agree with that available from previous studies (see SI for details).32,33 The 1H and 13C NMR chemical shifts for synthesized 4 agree with the calculated shifts (Table 1 and Figure 4; see SI for additional detail). While Kuo et al. described serlyticin-A as a yellow powder with IR absorption bands of 3366 cm−1 and 1713 cm−1 corresponding to a hydroxy group and carbonyl moiety, respectively, we found compound 4 to be a tan powder with IR absorption bands at 3422, 3218, 1636, and 740 cm−1. Unfortunately, given the lack of raw characterization data in the original report,5 we cannot be sure if these differences are meaningful. Kuo and co-workers reported UV absorption bands (solvent not indicated) for serlyticin-A at 244, 261, and 299 nm, implying that the molecule could contain an indole moiety. We analyzed 4 by UV-Vis absorption in MeOH and found absorption bands at 218, 230, and 280 nm, a reasonable match to the reported values assuming different solvents were used. Structure 1, with the molecular formula C20H16N2O6, has a calculated exact mass of 380.1008. The reported ESI-MS [m + H]+ m/z was 381.2629, a difference of > 0.15 from the calculated m/z of 1 [m + H]+. We suspect that the mass reported by Kuo and co-workers reflected a noncovalent dimer of the natural product. This type of dimerization is commonly detected in ESI-MS,34,35 and would be anticipated for a hydroxamic acid such as 4. The dimer of 4 has a calculated m/z [2m + H]+ of 381.1563, which is closer to the reported mass for serlyticin-A than the calculated m/z for 1. While our liquid chromatography mass spectrometry (LC-MS) analysis of 4 did not identify a dimer, this is not unexpected, as different instruments are known to yield different ionization patterns for the same molecule.36
Figure 3.
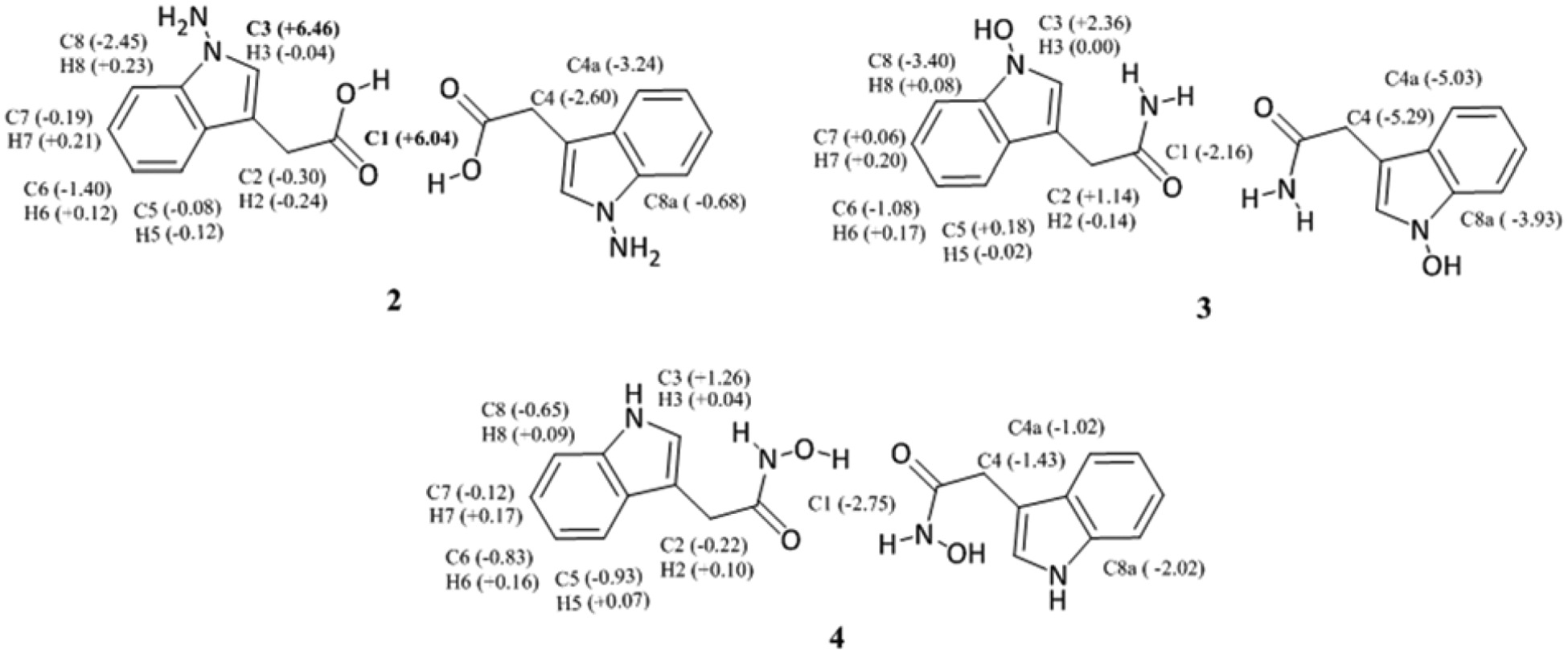
Alternative structures examined. Deviations of less than 6 ppm and less than 0.3 ppm between experimental and computed 13C and 1H shifts, respectively, are considered acceptable.13–15 Deviations exceeding these limits are bolded. For unsymmetrical dimers, shifts for the two monomers were averaged.
Figure 4.
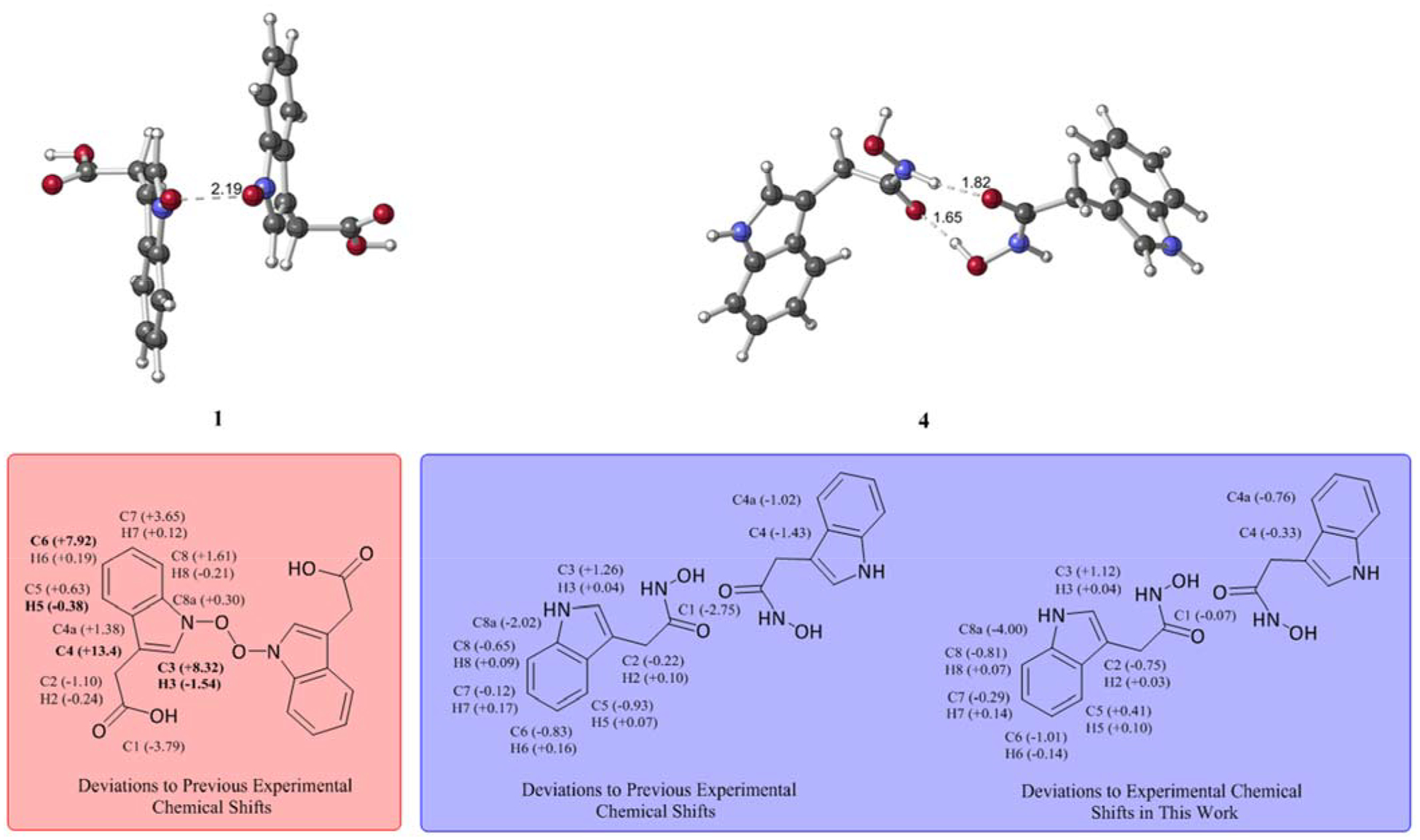
Summary of NMR results. Deviations between calculated chemical shifts of 1 and its reported experimental shifts (left). Deviations between calculated chemical shifts of 4 and the previously reported experimental shifts for serlyticin-A (center) and newly determined experimental shifts for 4 (right). Deviations of less than 6 ppm for 13C and less than 0.3 ppm for 1H shifts are considered acceptable.13–15 Deviations exceeding these limits are bolded. For unsymmetrical dimers, shifts for the two monomers were averaged.
Overall, the chemical and biological data reported for serlyticin-A5 are consistent with a compound such as 4, but comparison with an authentic sample will be necessary to confirm this hypothesis.7 Regardless, the variety of factors described above make it highly unlikely that 1 is the correct structure of serlyticin-A. In the field of organic chemistry, structural misassignments are obviously problematic.8 Many such misassignments can be avoided through judicious application of NMR chemical shifts calculations.9–10,11–13 Here, we have demonstrated that the originally proposed structure for serlyticin-A is likely incorrect, a conclusion based on (1) computations that point to the likelihood that the central O–O bond in the proposed structure is weak, (2) deviations between the reported and calculated chemical shifts for 1, and (3) the unprecedented nature of such an N-oxidized indole natural product lacking substitution at C2. Furthermore, the high prevalence and known antioxidant/antiproliferative properties of naturally occurring hydroxamic acids is consistent with our proposal that serlyticin-A is likely 3-indolylacetohydroxamic acid (4) or a closely related compound.
EXPERIMENTAL SECTION
Computational Methods.
Quantum chemical calculations were performed using Gaussian09.37 Structural optimizations and frequency calculations were performed with both B3LYP/6–31+G(d,p)38 in the gas phase and PCM(MeOH)-B3LYP/6–31+G(d,p).39,40 Both sets of optimized structures were subjected to NMR calculations using the gauge-including atomic orbital (GIAO) method.12 Results from both approaches are consistent with each other. Shown in the text are the results from NMR calculations on optimizations performed with PCM(MeOH); for gas phase results, please see Supporting Information (SI). Since the NMR experiments reported by Kuo and co-workers were performed on a sample dissolved in deuterated methanol, our 13C and 1H shifts were calculated with PCM(MeOH)-mPW1PW91/6–311+G(2d,p).41 Chemical shifts were linearly scaled using scaling factors obtained from cheshirenmr.info (slope = −1.0754 for 1H and −1.0399 for 13C; intercept = 31.8463 for 1H and 186.5993 for 13C).11,42 In order to sufficiently sample conformational space, multiple systematic conformational search runs were performed on each compound using Spartan10.43 These conformational search runs used molecular mechanics, explicitly the Merck Molecular Force Field (MMFF). However, all resulting conformers were subjected to single point calculations at the B3LYP/6–31+G(d,p) level. Then, geometric optimizations were performed on conformers within 5 kcal/mol of the lowest energy conformer, with and without implicit solvent (i.e., PCM(MeOH)). NMR calculations were only performed on the optimized conformers within 3 kcal/mol of the lowest energy conformer. The geometries of all conformers for each structure were confirmed to be minima (no imaginary frequencies). The chemical shifts of these contributing conformers were averaged using Boltzmann-weighting. This procedure is well precedented for computational NMR studies.11–13 In addition, an optimization constraining the fragmentable O–O bond of the originally proposed structure for serlyticin-A, 1 (Figure 1), was performed to assess its thermodynamic stability. Natural Bond Orbital (NBO)44 analysis calculations were performed on all contributing conformers of 1 to obtain Wiberg bond orders.45
General Experimental Procedures.
All reagents were obtained commercially unless otherwise noted. Reactions were performed using glassware that was oven dried (120oC) unless otherwise stated. Air- and moisture-sensitive liquids and solutions were transferred via syringe. Organic solutions were concentrated under reduced pressure (∼5 Torr) by rotary evaporation. Solvents were purified by passage under 12 psi N2 through activated alumina columns. Chromatography was performed using Fisher Chemical™ Silica Gel Sorbent (230–400 Mesh, Grade 60). Compounds purified by chromatography were typically applied to the adsorbent bed using the indicated solvent conditions with a minimum amount of added dichloromethane as needed for solubility. Thin layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates (250 μm). Visualization of the developed chromatogram was accomplished by fluorescence quenching or by staining with butanolic ninhydrin, or aqueous ceric ammonium molybdate (CAM).
Nuclear magnetic resonance (NMR) spectra were acquired on either a Bruker 400 operating at 400 and 100 MHz for 1H and 13C, respectively, and are referenced internally according to residual solvent signals. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s, singlet; br s, broad singlet; d, doublet; t, triplet; q, quartet; quint, quintet; sext, sextet; m, multiplet), integration, and coupling constant (Hz). Data for 13C NMR are reported in terms of chemical shift (δ, ppm). Infrared spectra were recorded using a Thermo Scientific Nicolet iS10 spectrometer with Smart iTX Accessory (diamond ATR) and are reported in frequency of absorption. Low-resolution mass spectra were obtained using a Waters Acuity Arc LC-MS.
Synthesis of Methyl 2-(1H-indol-3-yl)acetate.
To a solution of indole acetic acid (0.400 g, 2.28 mmol, 1.0 equiv) in MeOH (22.8 mL, 0.1 M) at room temperature was added concentrated hydrochloric acid (0.76 mL, 9.14 mmol, 4.0 equiv). The mixture was stirred overnight at 65 °C, cooled to room temperature, and then concentrated under reduced pressure. 100 mL of saturated NaHCO3 was added and then extracted with Et2O (3 × 100 mL). The combined organic extracts were washed with brine (1 × 150 mL), dried over Na2SO4, and concentrated under reduced pressure to yield a brown oil (0.410 g, 95%). 1H NMR (CD3OD, 400 MHz) δ 7.51 (d, 1H, J = 8.0 Hz), 7.33 (d, 1H, J = 8.0 Hz), 7.14 (s, 1H), 7.10 (t, 1H, J = 7.5 Hz), 7.01 (t, 1H, J = 7.5 Hz), 3.75 (s, 2H), 3.66 (s, 3H) ppm; 13C NMR (CD3OD, 100 MHz) δ 174.81, 139.98, 128.54, 124.64, 122.47, 119.88, 119.34, 112.25, 108.50, 52.32, 31.83 ppm; IR (Smart iTX Diamond) 3404, 2952, 2683, 1719 cm-1; LC-MS (ES+) calcd for C11H11NO2 [M + H] 190.08 found 190.23.
Synthesis of N-hydroxy-2-(1H-indol-3-yl)acetamide.
To a solution of methyl 2-(1H-indol-3-yl)acetate (0.062 g, 0.33 mmol, 1.0 equiv) and NaOH (0.066 g, 1.64 mmol, 5.0 equiv) in (1:1) MeOH/THF (1.3 mL, 0.25 M) at room temperature was added 50% aqueous hydroxylamine (0.65 mL, 10.49 mmol, 32.0 equiv). The mixture was stirred for 4.5 h at room temperature, and then concentrated under reduced pressure. 10 mL of 2 M HCl(aq) was added and the solution was extracted with (3 × 20 mL) EtOAc. The combined organic extracts were washed with brine (1 × 20 mL), dried over Na2SO4, and concentrated under reduced pressure to yield a tan solid. (0.061 g, 98%). 1H NMR (CD3OD, 400 MHz) δ 7.57 (d, 1H, J = 8.0 Hz), 7.33 (d, 1H, J = 8.0 Hz), 7.17 (s, 1H), 7.09 (t, 1H, J = 7.4 Hz), 7.01 (t, 1H, J = 7.4 Hz), 3.57 (s, 2H) ppm; 13C NMR (CD3OD, 100 MHz) δ 171.66, 138.07, 128.49, 124.77, 122.50, 119.87, 119.35, 112.26, 108.88, 30.92 ppm; IR (Smart iTX Diamond) 3422, 3218, 3057, 1636, 1544, 1081, 740 cm-1; LC-MS (ES+) calcd for C10H10N2O2 [M + H] 191.07 found 191.29.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge computational support from NSF XSEDE program. R.J.T. is supported by NIH Training Grant T32GM113770.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Details of quantum chemical calculations and characterization data for 4 including 1H and 13C NMR spectra (PDF).
The author declare no conflicts of interest.
REFERENCES AND NOTES
- (1).(a) Chang SS; Ostric-Matijasevic B; Hsieh OL; Huang CL J. Food Sci 1977, 42, 1102–1106. [Google Scholar]; (b) Inatani R; Nakatani N; Fuwa H; Seto H Agric. Biol. Chem 1982, 46, 1661–1666. [Google Scholar]; (c) Cuvelier ME; Berset C; Richard H J. Agric. Food Chem 1994, 42, 665–669. [Google Scholar]
- (2).Farombi EO; Britton G; Emerole GO Food Res. Int 2000, 33, 493–499. [Google Scholar]
- (3).Canadanovic-Brunet JM; Djilas SM; Cetkovic GS; Tumbas VT; Mandic AI; Canadanovic VM Int. J. Food Sci.Technol 2006, 41, 667–673 [Google Scholar]
- (4).Diouf PN; Stevanovic T; Cloutier A Food Chem 2009, 113, 897–902. [Google Scholar]
- (5).Kuo Y-H; Hsu H-C; Chen Y-C; Liang T-W; Wang S-L J. Agric. Food Chem 2012, 60, 9043–9047. [DOI] [PubMed] [Google Scholar]
- (6).Seymour TA; Li SJ; Morrissey MT J. Agric. Food Chem 1996, 44, 682–685. [Google Scholar]
- (7).Attempts to contact the authors of ref. 5 requesting an authentic sample and/or original spectra went unanswered.
- (8).Nicolaou KC; Snyder SA Angew. Chem. Int. Ed 2005, 44, 1012–1044. [DOI] [PubMed] [Google Scholar]
- (9).Elyashberg M; Blinov K; Molodtsov S; Smurnyy Y; Williams AJ; Churanova T J. Cheminformatics. 2009, 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Elyashberg M; Williams AJ; Blinov K Nat. Prod. Rep 2010, 27, 1296–1328 [DOI] [PubMed] [Google Scholar]
- (11).Lodewyk MW; Siebert MR; Tantillo DJ Chem. Rev 2012, 112, 1839–1862. [DOI] [PubMed] [Google Scholar]
- (12).Grimblat N; Sarotti AM Chem. Eur. J 2016, 22, 12246–12261. [DOI] [PubMed] [Google Scholar]
- (13).Willoughby PH; Jansma MJ; Hoye TR Nat. Protoc 2014, 9, 643–660. [DOI] [PubMed] [Google Scholar]
- (14).Somei M Adv. Heterocycl. Chem 2002. 82, 101–155. [Google Scholar]
- (15).Qian-Cutrone J; Huang S; Shu YZ.; Vyas D; Fairchild C; Menendez A; Krampitz K; Dalterio R; Klohr SE; Gao Q J. Am. Chem. Soc 2002, 124, 14556–14557. [DOI] [PubMed] [Google Scholar]
- (16).Liu L; Wang L; Bao L; Ren J; Bahadur Basnet B; Liu R; He L; Han J; Yin WB; Liu H Versicoamides F-H Org Lett 2017, 19, 942–945. [DOI] [PubMed] [Google Scholar]
- (17).Singh SB; Herath K; Yu NX; Walker AA; Connors N Tetrahedron. Let 2008, 49, 6265–6268. [Google Scholar]
- (18).Zhang C; Herath K; Jayasuriya H; Ondeyka JG; Zink DL; Occi J; Birdsall G; Venugopal J; Ushio M; Burgess B; Masurekar P; Barrett JF; Singh SB J. Nat. Prod 2009, 72, 841–847. [DOI] [PubMed] [Google Scholar]
- (19).Urban S; Blunt JW; Munro MH J. Nat. Prod 2002, 65, 1371–1373. [DOI] [PubMed] [Google Scholar]
- (20).Tsukamoto S; Kato H; Samizo M; Nojiri Y; Onuki H; Hirota H; Ohta T J. Nat. Prod 2008, 71, 2064–2067. [DOI] [PubMed] [Google Scholar]
- (21).Susanna TS; Chan A; Pearce N; Page MJ; Kaiser M; Copp BR J. Nat. Prod 2011, 74, 1972–1979. [DOI] [PubMed] [Google Scholar]
- (22).Santos AK; Machado LL; Bizerra AM; Monte FJ; Santiago GM; Braz-Filho R; Lemos TL Nat. Prod. Commun 2012, 7, 729–730. [PubMed] [Google Scholar]
- (23).Costa EV; Pinheiro ML; Xavier CM; Silva JR; Amaral AC; Souza AD; Barison A; Campos FR; Ferreira AG; Machado GM; Leon LL J. Nat. Prod 2006, 69, 292–294. [DOI] [PubMed] [Google Scholar]
- (24).Neilands JB Science. 1967, 156, 1443–1447. [DOI] [PubMed] [Google Scholar]
- (25).Challis GL Microbiology. 2008, 154, 1555–1569. [DOI] [PubMed] [Google Scholar]
- (26).Miller MJ Chem. Rev 1989, 89, 1563–1579. [Google Scholar]
- (27).Tsuji N; Kobayashi M; Nagashima K; Wakisaka Y; Koizumi K J. Antibiot 1974, 49, 14691. [DOI] [PubMed] [Google Scholar]
- (28).Arrabal MJ; González PV; Gámez CC; Misiego AS; de la Peña AM Analyst, 1994, 119, 1537–1540. [Google Scholar]
- (29).Marks PA; Richon VM; Rifkind RA J. Natl. Cancer. Inst 2000, 92, 1210–1216. [DOI] [PubMed] [Google Scholar]
- (30).Marks PA Expert Opin. Investig. Drugs 2010, 19, 1049–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Holson E and Olson D U.S. Patent 9745613B2, 2017.
- (32).Cohen W and Erlanger BF J. Am. Chem. Soc 1960, 82, 3928–3934. [Google Scholar]
- (33).Boularot A; Giglione C; Petit S; Duroc C; Alves de Sousa R; Larue C; Cresteil T; Daredel F; Artaud I; Meinnel T. J. Med. Chem 2007, 50, 10–20. [DOI] [PubMed] [Google Scholar]; In this report, NMR were taken in DMSO rather than chloroform, only a carbonyl peak was reported in IR (1638 cm−1), no UV data was reported, and elemental analysis was reported instead of MS data.
- (34).Pan H Rapid Commun. Mass Spectrom 2008, 22, 3555–3560. [DOI] [PubMed] [Google Scholar]
- (35).Ding J; Anderegg RJ J. Am. Soc. Mass Spectrom 1995, 6, 159. [DOI] [PubMed] [Google Scholar]
- (36).Jiang H; Somogyi A; Timmermann BN; Gang David. R. Rapid Commun. Mass Spectrom 2006, 20, 3089–3100. [DOI] [PubMed] [Google Scholar]
- (37).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; et al. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- (38).(a) Becke AD J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]; (b) Lee C; Yang W; Parr RG Phys. Rev. B: Condens. Matter Mater. Phys 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]; (c) Miehlich B; Savin A; Stoll H; Preuss H Chem. Phys. Lett 1989, 157, 200–206. [Google Scholar]
- (39).(a) Cammi R; Mennucci B; Tomasi J Chem. Rev 2005, 105, 2999–3093 [DOI] [PubMed] [Google Scholar]; (b) Leszczynski J Ed.; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2003; Vol. 8.
- (40).Cheeseman JR; Trucks GW; Keith TA; Frisch MJ J. Chem. Phys 1996, 104, 5497–5509. [Google Scholar]
- (41).Adamo C; Barone V J. Chem. Phys 1998, 108, 664–675. [Google Scholar]
- (42).Scaling factors were obtained from chesirenmr.info.
- (43).Spartan, Version10 (Wavefunction, Ind.: Irvine, CA, USA, 2010). [Google Scholar]
- (44).NBO 7.0. Glendening ED, Badenhoop J,K, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Karafiloglou P, Landis CR, and Weinhold F, Theoretical Chemistry Institute, University of Wisconsin, Madison: (2018). [Google Scholar]
- (45).(a) Reed AE; Weinstock RB; Weinhold F J. Chem. Phys 1985, 83, 735–746. [Google Scholar]; (b) Glendening DE; Weinhold F, F. J. Comp. Chem 1998, 19, 610–627. [Google Scholar]; (c) Weinhold F J. Comp. Chem 2012. 33, 2363–2379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.