

Abstract
Wnt/β-catenin signaling pathway is implicated in the etiology and progression of metabolic disorders. While lines of genetic evidence suggest that blockage of this pathway yields favorable outcomes in treating such ailments, few inhibitors have been used to validate the promising genetic findings. Here, we synthesized and characterized a novel class of triazole-based Wnt/β-catenin signaling inhibitors, and assessed their effects on energy metabolism. One of the top inhibitors, compound 3a, promoted Axin stabilization, which led to the proteasome degradation of β-catenin and subsequent inhibition of the Wnt/β-catenin signaling in cells. Treatment of hepatocytes and high fat diet-fed mice with compound 3a resulted in significantly decreased hepatic lipid accumulation. Moreover, compound 3a improved glucose tolerance of high fat diet-fed mice without noticeable toxicity, while downregulating the genes involved in the glucose and fatty acid anabolism. The new inhibitors are expected to be further developed for the treatment of metabolic disorders.
Graphical Abstract
INTRODUCTION
The Wnt/β-catenin pathway plays a pivotal role in cell proliferation, differentiation and growth.1 It regulates the expression of target genes through the transcriptional factor β-catenin that forms a cytoplasmic “destruction complex” with other proteins including Axin and adenomatous polyposis coli (APC). This complex facilitates the phosphorylation of β-catenin by casein kinase 1α (CK1α) and glycogen synthase kinase 3β (GSK3β), leading to proteasome degradation of β-catenin during the “off state” of the pathway. The “on-state”, on the other hand, involves enhanced stability and accumulation of β-catenin in the cytoplasm, resulting in its increased translocation to the nucleus where it binds to LEF/TCF transcriptional factors and activates the expression of target genes. Wnt/β-catenin pathway crosstalks with many other pathways via key proteins such as β-catenin, Axin, and GSK3β.1,2 Given the crucial function of Wnt/β-catenin pathway, it is not surprising that a strong link between this pathway and metabolic disorders has been recognized in recent years.3–5
The association of the Wnt/β-catenin pathway to metabolic disorders was first established by the identification of a human polymorphism (rs7903146) in the TCF7L2 gene, which encodes a major nuclear partner protein of β-catenin, as a strong risk factor for type-2 diabetes.6 This polymorphism has been known to enhance TCF7L2 transcription.7–9 Later, Savic et al showed that partial knockdown of Tcf7l2 results in metabolic phenotypes of smaller body weights, decreased fasting glucose, and improved glucose tolerance in mice.10 We have published similar observations in mice with the genetic haploinsufficiency of Tcf7l2.11 However, the role of TCF7L2 in maintaining metabolic homeostasis in specific tissues remains controversial. TCF7L2 knockdown was reported to increase glucose production and gluconeogenic gene expression in cultured hepatocytes,12 and the transgenic mice overexpressing a dominant negative Tcf7l2 mutant in the proglucagon gene-expressing cells exhibited defective glucose homeostasis.13 However, Boj et al reported that liver-specific Tcf7l2 knockout led to reduced hepatic glucose production during fasting and improved glucose homeostasis in adult mice on a high-fat diet.14 Recently, Thompson et al reported that liver-specific knockout of β-catenin led to a striking protection from fibrosis and liver injury in mice,15 while Popov et al showed that the hepatic downregulation of β-catenin by anti-sense oligonucleotides could improve insulin sensitivity and glucose tolerance in mice.16 Therefore, although the exact role of the Wnt/β-catenin pathway in metabolism is yet to be illustrated, overall evidence indicates that downregulation of Wnt/β-catenin signaling may provide a new therapeutic strategy for the treatment of metabolic disorders such as fatty liver diseases and diabetes.
Therapeutics targeting the Wnt/β-catenin signaling pathway is still in a state of infancy. One reason is that this pathway is bewilderingly complex, with crosstalks to numerous others.17–19 In addition, because of the severe phenotypes observed in genetic knockout animal models,20 safety concern is historically present. Approximately one dozen of Wnt/β-catenin pathway inhibitors with distinct mechanisms have been discovered.21–33 For example, porcupine inhibitors (e.g., LGK-974, Figure 1) decrease the secretion of the Wnt ligands.34 At the plasma membrane, the inhibition of LRP5/6 binding to Wnt proteins by inhibitors such as niclosamide and salinomycin can curb the amount of active β-catenin.22,35 In the cytoplasm, stabilization of the destruction complex (e.g., tankyrase inhibitor XAV93925 and CK1α activator pyrvinium36) can also attenuate β-catenin levels. Of note, the safety concern about Wnt inhibitors has not been borne out either preclinically or clinically, and recently the β-catenin/CBP inhibitor PRI-724 has entered clinical trials as a potential new treatment of various cancers.17 Several FDA-approved drugs, such as glucocorticoids, retinoids, and celecoxib, are also found to be Wnt/β-catenin pathway inhibitors.17,19 Interestingly, Wnt/β-catenin pathway inhibitors have emerged to replicate the metabolic outcomes of the above-mentioned genetic manipulation in mice. The CK2 inhibitor CX-4945 has been shown to cause mice to be resistant to high fat diet-induced obesity and metabolic disorders.37,38 Treatment of mice with a selective tankyrase inhibitor G007-LK has resulted in profound improvement of glucose tolerance and insulin sensitivity.39 However, further studies are needed to fully establish the applicability of these small molecule inhibitors in the treatment of metabolic disorders.
Figure 1.
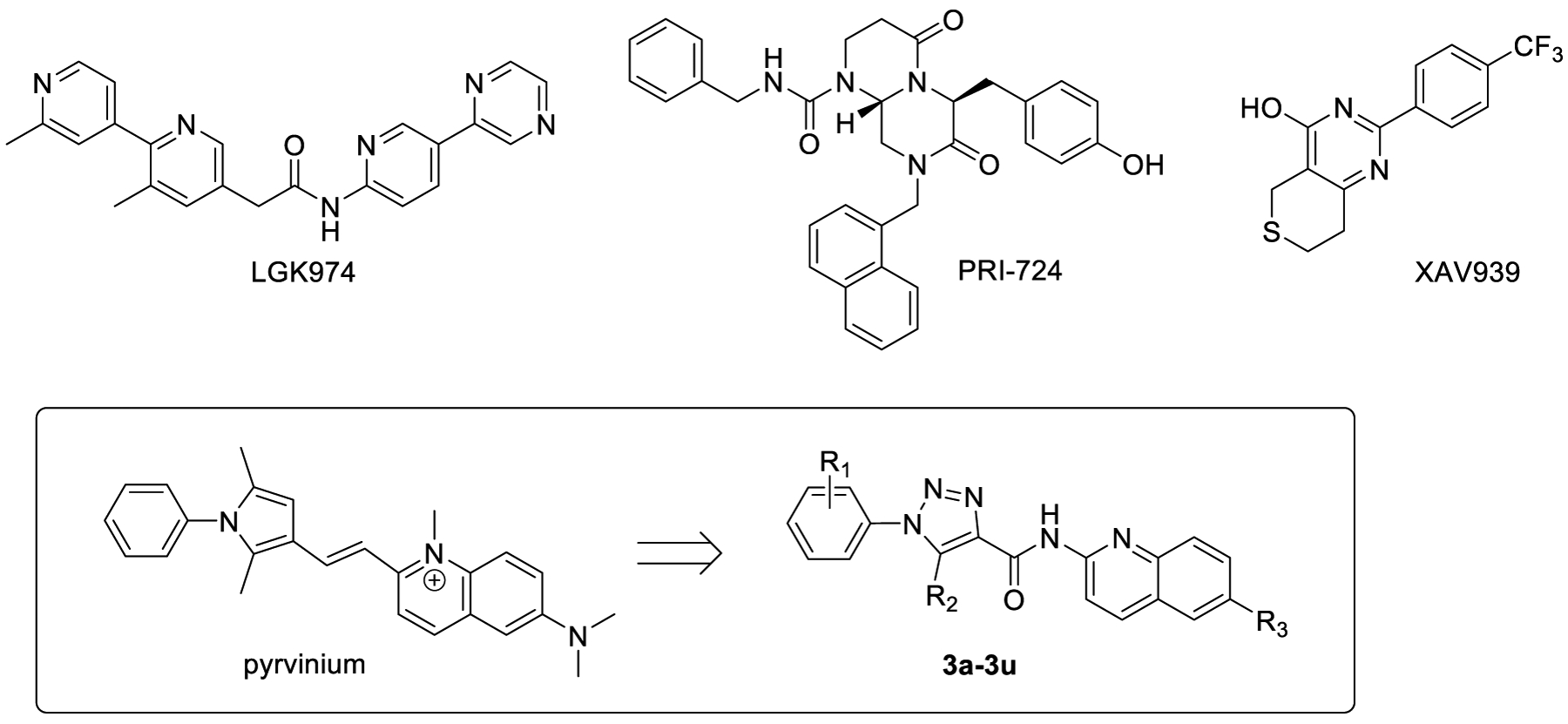
Structures of known Wnt/β-catenin pathway inhibitors LGK974, PRI724, XAV939, pyrvinium, and the design of new triazole-based inhibitors 3a-3u.
Herein we report a series of novel Wnt/β-catenin pathway inhibitors based upon a triazole scaffold (Figure 1). New compounds were designed by modifying the chemical structure of pyrvinium, an FDA approved anthelmintic effective for pinworm infection. The drug has been reported to inhibit Wnt signaling with a high potency36. However, further development of pyrvinium as a therapeutic agent for metabolic disorders is prohibited by several reasons related to its chemical structure. Pyrvinium possesses a permanently charged N-methylquinolone group that causes extremely low bioavailability of the compound.40 The double bond connecting the two ring systems leads to poor solubility. Moreover, the 2,5-dimethyl-1-phenyl-1H-pyrrole is prone to oxidation41 and known as one of the pan assay interference compounds (PAINs).42 Herein we describe the synthesis of new inhibitors 3a-3u employing a neutral aromatic amino group, an amide linker, and a substituted triazole core. Several of the new compounds showed excellent inhibitory potency against Wnt signaling. One of the new compounds, 3a, was further characterized by various in vitro and in vivo biological assays. Compound 3a showed improved bioavailability, promising efficacy against diet-induced metabolic disorders in mice. In addition, compound 3a was well tolerated in mice without any notable toxicity.
RESULTS AND DISCUSSION
Synthesis.
The synthesis of compounds 3a-3u is detailed in Scheme 1. Diazotization of the amino group of anilines 1a-1n using sodium nitrite (NaNO2) and aqueous HCl, followed by the treatment of the intermediate with sodium azide (NaN3) gave azides, which underwent cyclization with β-ketoester in EtONa/EtOH yielded trizaole-3-carboxylic acids 2a-2p. Next, PyCIU-mediated coupling of compounds 2a-2p with various aromatic amines in dichloroethane (DCE) provided target compounds 3a-3r in moderate to good yields. Finally, Pd-catalyzed cross-coupling of compound 3r with various potassium trifluoroborate derivatives yielded products 3s-3u in good yields.
Scheme 1.
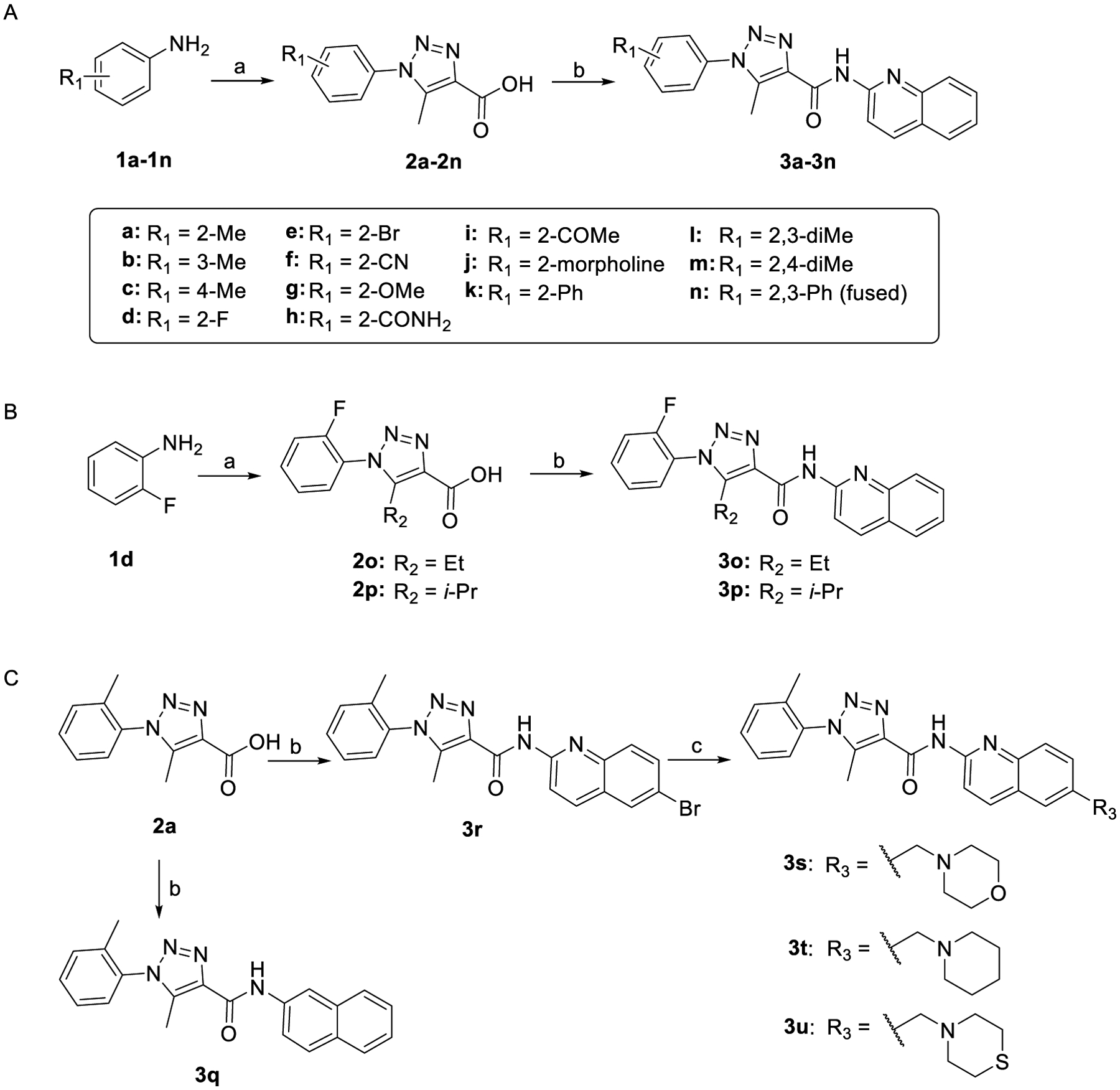
Synthesis of compounds 3a-3ua aReagents and conditions: (a) (i) NaNO2, HCl, NaN3, H2O, 0 °C; (ii) ethyl 3-oxobutanoate for 2a-2n, ethyl 3-oxopentanoate for 2o, ethyl 4-methyl-3-oxopentanoate for 2p, EtONa, EtOH, 80 °C; (b) 2-aminoquinoline for 3a-3p, naphthalen-2-amine for 3q, 6-bromoquinolin-2-amine for 3r, PyCIU, DIPEA, DCE, 80 °C; (c) potassium trifluoroborate derivatives, Pd(OAc)2, XPhos, Cs2CO3, THF/H2O, 80 °C, 24–48 h.
Structure-Activity Relationship.
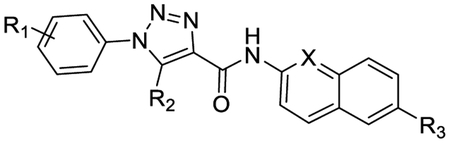
Compound 3a indicated an excellent IC50 value of 4.1 nM in the luciferase gene reporter assay for Wnt signaling activity, which is over 1,000-fold higher than that of 3b (3-methyl group) and 3c (4-methyl group), and 180-fold higher than the parent compound pyrvinium (Table 1). Additional methyl group at the 3-(3l) or 4-position (3m) of the phenyl ring led to new inhibitors with 34- and 66-fold decreased potencies, respectively. The naphthyl analog 3n was a weak inhibitor with an IC50 value of 5.5 μM. These results indicated that single ortho-substitution is preferred on the phenyl ring. Substitution of the 2-methyl group with various functionalities generated new inhibitors 3d-3k. Among them, the F-analog (3d) demonstrated excellent potency with an IC50 value in the sub-nanomolar range. The Br- (3e), NC- (3f), and MeO- (3g) analogs also showed low nM potencies. However, large substituents such as amide (3h), ketone (3i), morpholine (3j) and Ph (3k) turned out to be detrimental to the inhibitory activity.
Table 1.
Inhibition of Wnt/β-Catenin Signaling Pathway by Compounds 3a-3u
| |||||
|---|---|---|---|---|---|
| Cmpds | R1 | R2 | R3 | X | IC50 (nM)α |
| 3a | 2-Me | Me | H | N | 4.1 ± 0.4 |
| 3b | 3-Me | Me | H | N | >10,000 |
| 3c | 4-Me | Me | H | N | >10,000 |
| 3d | 2-F | Me | H | N | 1.2 ± 0.2 |
| 3e | 2-Br | Me | H | N | 7.6 ± 0.3 |
| 3f | 2-CN | Me | H | N | 34 ± 4.3 |
| 3g | 2-OMe | Me | H | N | 18 ± 3.3 |
| 3h | 2-CONH2 | Me | H | N | >10,000 |
| 3i | 2-COMe | Me | H | N | 8,800 ± 1322 |
| 3j | 2-morpholine | Me | H | N | 1,900 ± 5.4 |
| 3k | 2-Ph | Me | H | N | 390 ± 68.1 |
| 3l | 2,3-diMe | Me | H | N | 140 ± 5.8 |
| 3m | 2,4-diMe | Me | H | N | 270 ± 23.8 |
| 3n | 2,3-Ph (fused) | Me | H | N | 5,500 ± 1186 |
| 3o | 2-F | Et | H | N | 4.7 ± 0.7 |
| 3p | 2-F | i-Pr | H | N | 25 ± 0.5 |
| 3q | 2-Me | Me | H | CH | 4,900 ± 743 |
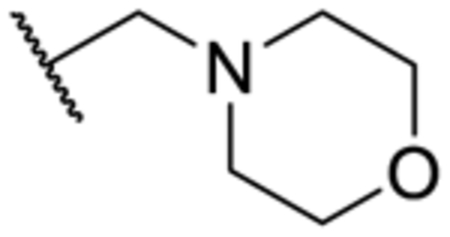
| 3r | 2-Me | Me | Br | N | 830 ± 98.2 |
| 3s | 2-Me | Me |
|
N | 1,300 ± 169 |
| 3t | 2-Me | Me |
|
N | 1,200 ± 168 |
| 3u | 2-Me | Me |
|
N | 230 ± 28 |
| pyrvinium | 750 ± 137 | ||||
The values of IC50 for each compound to inhibit the Wnt signaling activity, as determined from the luciferase reporter gene assay, were calculated and data are expressed as mean IC50 (nM) ± SE of each compound from three independent experiments. Note that compounds 3a-3u did not present any apparent cytotoxicity during the short treatment duration used for the luciferase gene reporter assay (Table S2).
Next, substitution effects of the triazole core (R2) were studied using compounds 3o and 3p. Compared to inhibitor 3d, the ethyl analog 3o was slightly less potent. However with the branched isopropyl group, compound 3p turned out to be over 20-fold less potent than compound 3d. These results indicated that small group was preferred at the R2 position.
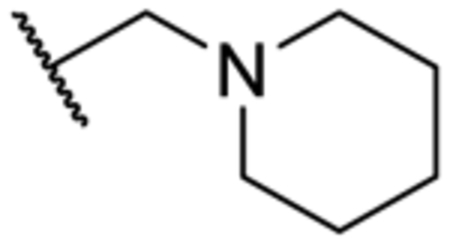
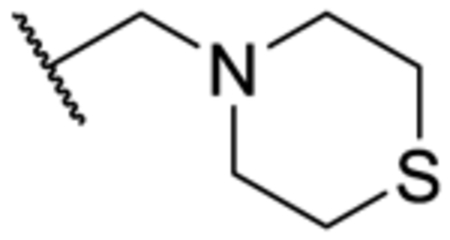
In addition, removal of the quinoline nitrogen yielded compound 3q that totally lost inhibitory activity. This result highlighted the importance of the quinoline nitrogen in maintaining high potency. Finally, we sought to explore the possibility of expanding the quinoline ring system. The Br-substituted compound 3r indicated similar potency as that of the parent compound pyrvinium. Similarly, potent inhibitors were obtained when morpholine (3s), piperidine (3t), or thiomorpholine (3u) were included at the same R3 position. These results indicated that further expansion of the quinoline ring could impair the activity of the inhibitor.
Compound 3a, a Potent Inhibitor of the Wnt/β-Catenin Signaling Pathway.
As model compounds, we next chose compounds 3a and 3n and further evaluated them in various biological assays. We chose compound 3a over the more potent compound 3d (Table 1) because in the assessment of their potential cytotoxicity in unstimulated normal HEK293 cells with a longer incubation time of 72 h relative to that for the above reporter assay, we found that 3d was more cytotoxic than 3a (Figure S1). While compound 3d was more potent than 3a, their potencies remained in the same magnitude. Compound 3a showed superior potency in HEK293 cells in the presence of Wnt signaling activators LiCl and Wnt3a (Figure 2A, B). Compound 3a inhibited the Wnt/β-catenin signaling pathway by stabilization of Axin and subsequent β-catenin degradation (Figure 2C–E and Figure S2A–B). Stabilization of Axin was confirmed by the increased cytoplasmic punctas as has been demonstrated previously by other Axin stabilizers.43 The expression of the pathway target genes including CycD1 and Axin2 was repressed at the messenger RNA (mRNA) levels. In the presence of LiCl, which activates Wnt/β-catenin pathway via the inhibition of GSK3β, compound 3a decreased the cellular level of β-catenin while the inactive analog 3n had little effect (Figure 2F).
Figure 2.
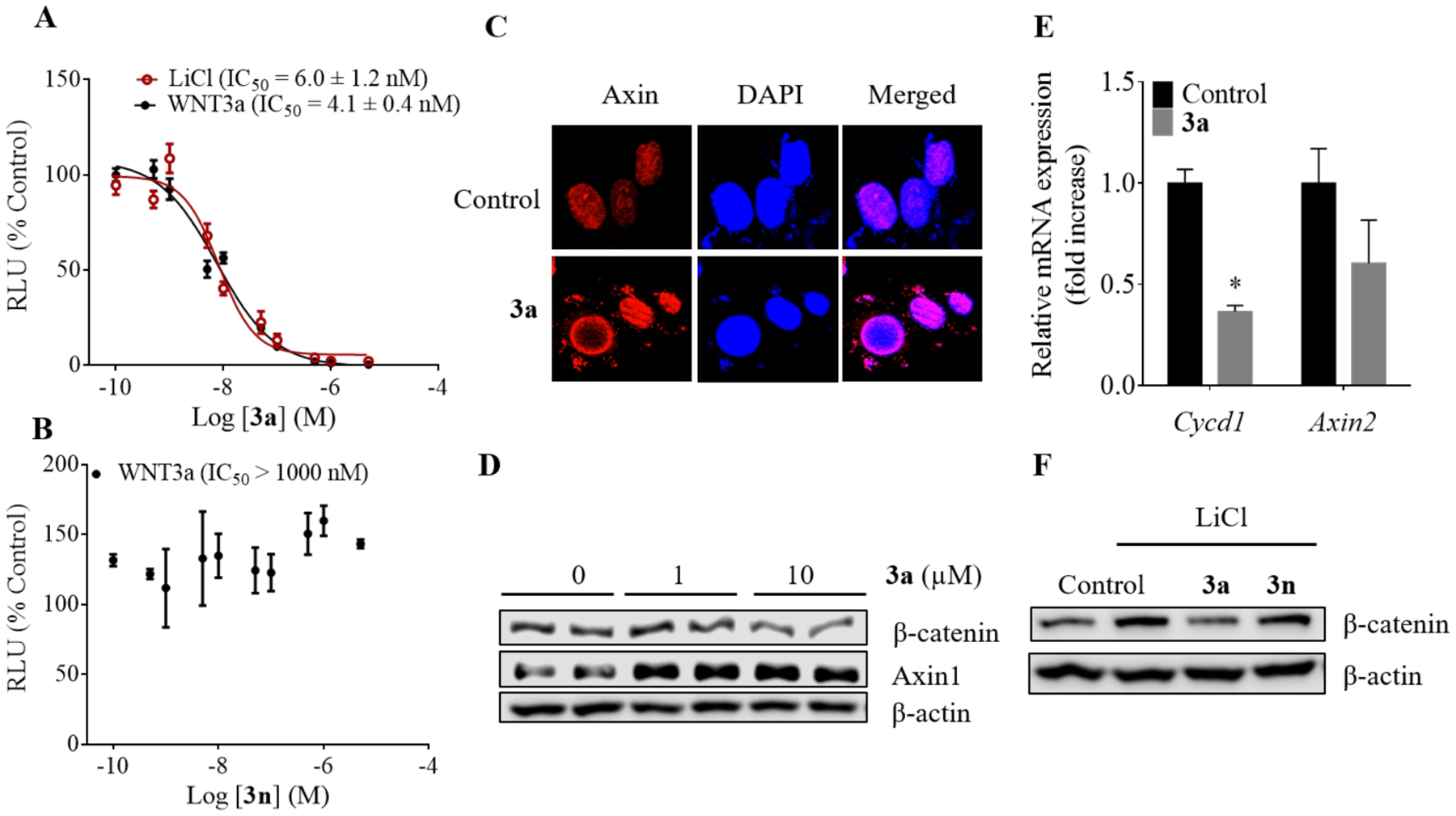
Characterization of compound 3a as an inhibitor of Wnt/β-catenin signaling pathway. (A) TCF/LEF responsive luciferase reporter assay in HEK293 cells with varying concentration of compound 3a and (B) compound 3n in Wnt3a- or LiCl-conditioned medium. The data were fitted to determine the IC50 of inhibition of LiCl- and Wnt3a-induced activation of the Wnt/β-catenin signaling pathway (mean ± S.D, n = 4). (C) Confocal microscope images of 3a-treated HEK293 cells for detection of Axin (Axin1). The cells were incubated with 1 μM of compound 3a for 14 h. The nucleus was stained by DAPI. (D) Western blot analysis showing the effect of compound 3a treatment on the levels of β-catenin and Axin1 proteins in HEK293 cells in the presence of WNT3a. The cells were treated with the compound for 2 h. (E) The mRNA expression of the target genes of Wnt/β-catenin signaling pathway. The cells were treated for 48 h and harvested with TriZol reagent. *p < 0.05. (F) Western blot analysis of HEK293 cells treated with compounds 3a and 3n in the presence of lithium chloride for 24 h. RLU-Relative light units.
The downregulation of β-catenin levels and the consistent efficacy of compound 3a in both Wnt3a- and LiCl-conditioned medium suggested a mechanism different from that of the parent compound pyrvinium, whose effect is diminished in the presence of LiCl.36 We performed additional studies with varying concentrations of LiCl, and observed no effect by LiCl in changing the inhibitory potency of compound 3a towards Wnt/β-catenin signaling pathway (Figure S2).
Mechanism of Wnt/β-catenin Pathway Inhibition by Compound 3a.
Sequential phosphorylation of β-catenin on residues Ser33, Ser37 and Thr41 by GSK3β is a critical step in β-catenin degradation. By causing increase in the phosphorylation at these sites, many small molecule Wnt inhibitors facilitate the degradation of β-catenin. To further ascertain if our new inhibitors function through a similar mechanism, we examined the inhibitory potency of compound 3a in HEK293 cells overexpressed with wild-type and mutant (S33Y) β-catenin (Figure S3). Overexpression of both exogenous β-catenin plasmids led to significant increases in Wnt signaling activities as reflected by the luciferase reporter gene assay, with the S33Y mutant giving a larger increase than the wild type. Nonetheless, compound 3a inhibited Wnt signaling with a similar potency irrespective of overexpression of either β-catenin, suggesting that the inhibitory effects of compound 3a may be independent of GSK3β phosphorylation of at least Ser33 on β-catenin. Note that aside from Ser33 phosphorylation, GSK3β also phosphorylates Ser37 and Thr41 of β-catenin. Thus the mutated β-catenin construct used in our assay may not be sufficient to impair the function of GSK3β, as immunoprecipitation of β-catenin showed that compound 3a strengthened the binding of GSK3β and Axin to β-catenin (Figure 3A). Thus, the results suggest that compound 3a treatment may lead to fortification of the destruction complex to propagate the degradation of β-catenin.
Figure 3.
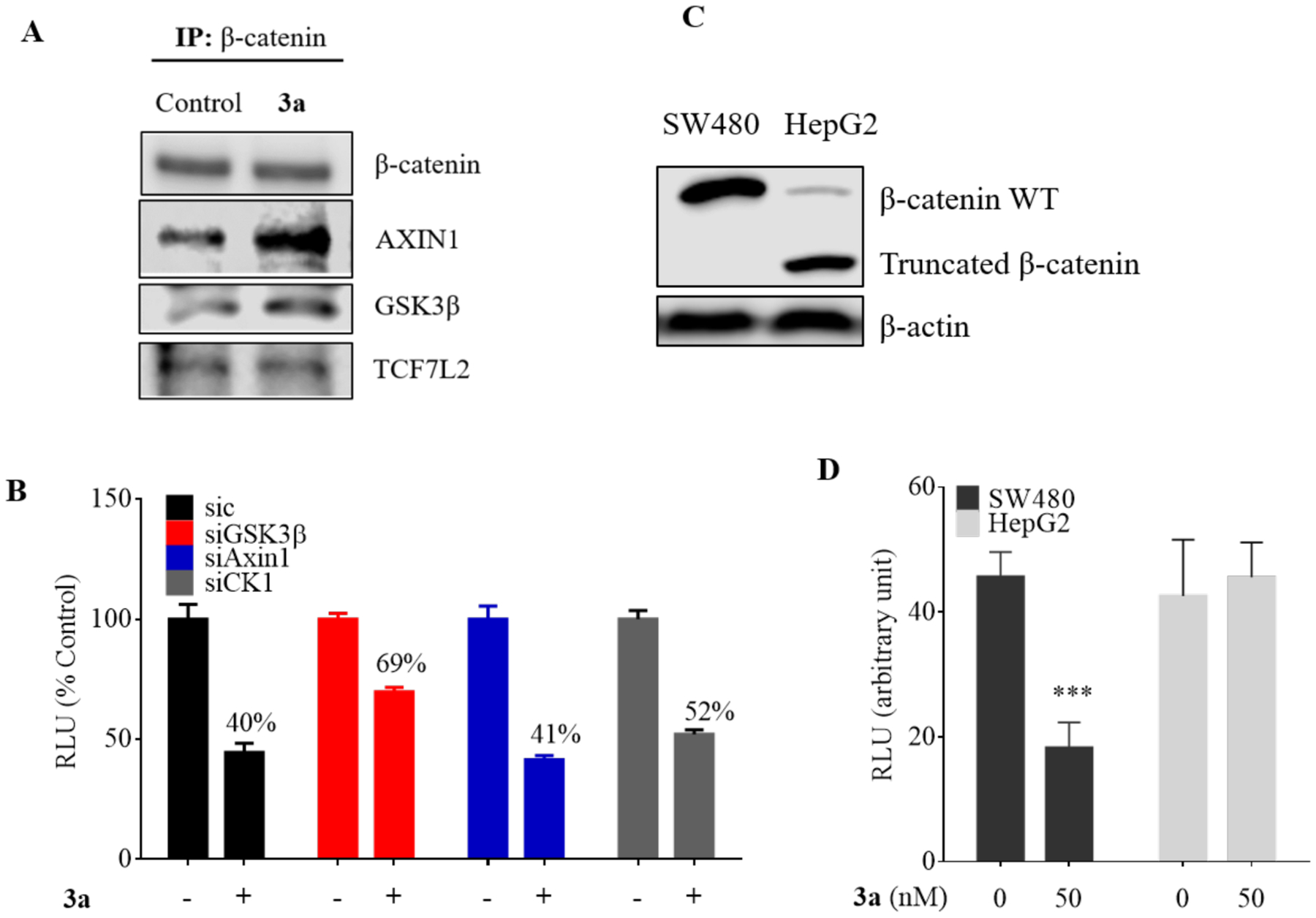
Role of Wnt/β-catenin pathway effector proteins in the inhibition by compound 3a. (A) Immunoprecipitation of β-catenin in HEK293 to determine the effect of compound 3a treatment (12 h) on the interaction of β-catenin with other pathway effector proteins. (B) TCF/LEF gene reporter assay of compound 3a in the presence of knockdown of GSK3β, Axin and CK1α. The HEK293 cells were treated with or without compound 3a (20 nM) for 24 h. (C) Western blot analysis of protein expression of β-catenin in SW480 and HepG2 cells. (D) TCF/LEF gene reporter assay of SW480 and HepG2 cells treated with compound 3a. Data are represented as mean ± S.D, n = 3. RLU-relative light units. *** p < 0.001.
We next silenced major components of the β-catenin destruction complex, CK1α, GSK3β and Axin1, to assess their contribution to the effect of inhibitor 3a. Only the knockdown of CK1 and GSK3β partially abolished the Wnt inhibitory effect while Axin1 silencing had no effect on the gene reporter assay of inhibitor 3a (Figure 3B). Next, we performed surface plasmon resonance (SPR) analysis using recombinant CK1α, GSK3β, and Axin to determine the binding affinity of compound 3a to these proteins. We found that compound 3a could not bind to CK1α and Axin (data not shown), while exhibiting a weak binding (KD = 4.3 μM) for recombinant GST-tagged GSK3β (Figure S4A–C). Further, we knocked down GSK3β and performed the reporter assay for compound 3a. The results show that exclusion of GSK3β only decreased the effect of compound 3a by 1.7 fold (IC50 of si-control = 20 nM vs. si-GSK3β = 34 nM) (Figure S4D). The non-correlation of the IC50 value from the reporter assay to the KD of weak binding to GSK3β suggests that GSK3β is probably not the main target of compound 3a, although it may play a direct role in the inhibitory effect of compound 3a on Wnt/β-catenin signaling pathway.
In order to determine the inhibitory effect of compound 3a in other cells bearing mutations that lead to an activated Wnt/β-catenin pathway, we overexpressed the luciferase reporter constructs in SW480 and HepG2 cell lines. The SW480 cells have inactivating mutations in APC, which lead to ineffective tethering of β-catenin to the destruction complex and subsequent increase in cytoplasmic and nuclear β-catenin levels.44 In contrast, HepG2 cells contain a deletion of the amino acids 25–140 of the CTNNB1 (encoding β-catenin) gene, which includes the binding sites of GSK3β and CK1α (Figure 3C).45 Inhibition of the reporter gene activity by compound 3a was well observed in SW480 cells, but insignificant in HepG2 cells (Figure 3D). These results suggest the necessity of the full length of β-catenin with intact GSK3β and CK1α sites for the inhibitory activity of compound 3a.
Compound 3a Decreased Lipid Accumulation and Altered the Expression of Lipogenic and Gluconeogenic Genes in Hepatocytes.
It has been reported that the Wnt/β-catenin pathway may play a role in cellular metabolism. Specifically, Axin has been implicated in lipid metabolism. In mice, knockdown of Axin results in significantly increased hepatic lipid accumulation.46 Because compound 3a stabilized the cellular level of Axin, we performed a Nile red staining assay to examine the effect of compound 3a on lipid accumulation in human hepatic Huh7 cells (Figure 4A). Treatment of the cells with compound 3a dramatically decreased lipid accumulation. Similar effects were observed by a known Axin stabilizer XAV939. Next, we performed a quantitative PCR to determine the effect of compound 3a on the gene expression of lipogenesis and gluconeogenesis in mouse hepatocytes. Our results showed that compound 3a downregulated the mRNA levels of the gluconeogenic (PEPCK and G6PASE) and lipogenic genes (FASN, ACAA1A, ACOT4, and SCD1) (Figure 4B). These results suggested that compound 3a might decrease the accumulation of lipids in hepatocytes by downregulating gluconeogenic and lipogenic pathways as a consequence of Axin stabilization.
Figure 4.
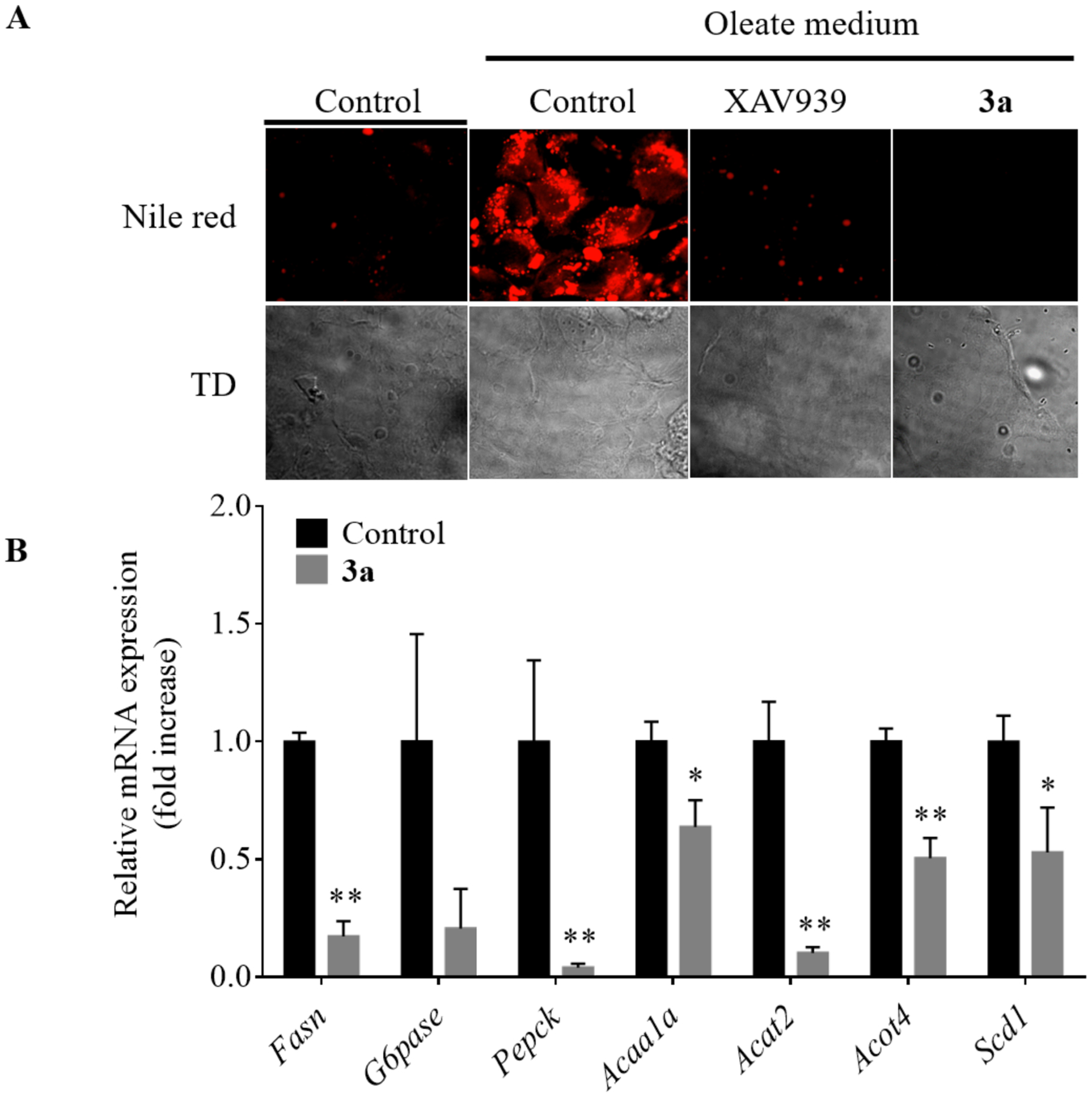
Compound 3a decreased lipid accumulation and the expression of lipogenic and gluconeogenic genes in hepatocytes. (A) Nile red staining assay of the Huh7 cells treated with 5 μM of compounds XAV939 or 3a for 36 h and together with 200 μM oleate for 16h. TD, transmitted light differential interference contrast image. (B) The mRNA expression of various lipogenic and gluconeogenic genes in normal mouse hepatocytes treated with 5 μM of compound 3a. Data represents mean ± S.D. of triplicates.
In Vivo Efficacy of Compound 3a against Diet-Induced Metabolic Disorders in Mice.
With the promising in vitro effects of compound 3a on metabolism in hepatocytes, we further studied its efficacy in vivo against metabolic disorders using a mouse model. Initially, we briefly assessed the pharmacokinetic and physicochemical properties of compound 3a. Compared to the reported parameters for pyrvinium, our new analogue showed improved physicochemical and pharmacokinetic properties with an oral bioavailability of 21% (pyrvinium has less than 1%). The plasma half-life of compound 3a is 2.8 – 3.3 h, with an oral maximal concentration of 2.2 μg/mL in the plasma after a single dose of 10 mg/kg (Table S3). We then proceeded to efficacy studies in the high fat diet-fed mouse model. We performed a pilot dose escalation study to determine the optimal dose for compound 3a and to ensure little or no toxicity to the mice (data not shown). While we did not observe any noticeable toxicity at up to 200 mg/kg of compound 3a, we found promising efficacy and hepatic improvement at 40 mg/kg so we chose this dose to conduct a more comprehensive study in mice. Wild type C57BL/6J mice were fed with a high fat diet or normal chow diet for 6 weeks before treatment. The mice were then divided into four groups: two were fed normal chow and the other two were fed with the high fat diet. Compound 3a was administered intraperitoneally every 2 days at 40 mg/kg for 11 weeks, a dose selected based on the pilot dose escalation studies. After 11 weeks of treatment, compound 3a significantly improved glucose tolerance in the high fat diet group (Figure 5A–B). The inhibition of Wnt signaling and the improvement of glucose tolerance by compound 3a were confirmed by the decreased hepatic mRNA levels of Wnt target genes and those of gluconeogenesis and lipogenesis in the mice fed with the high fat diet, respectively (Figure 6A). Of note, the effects of compound 3a on gene expression were insignificant in the mice fed with the normal chow diet, suggesting a selectivity of compound 3a towards metabolic disorders (Figure 6B).
Figure 5.
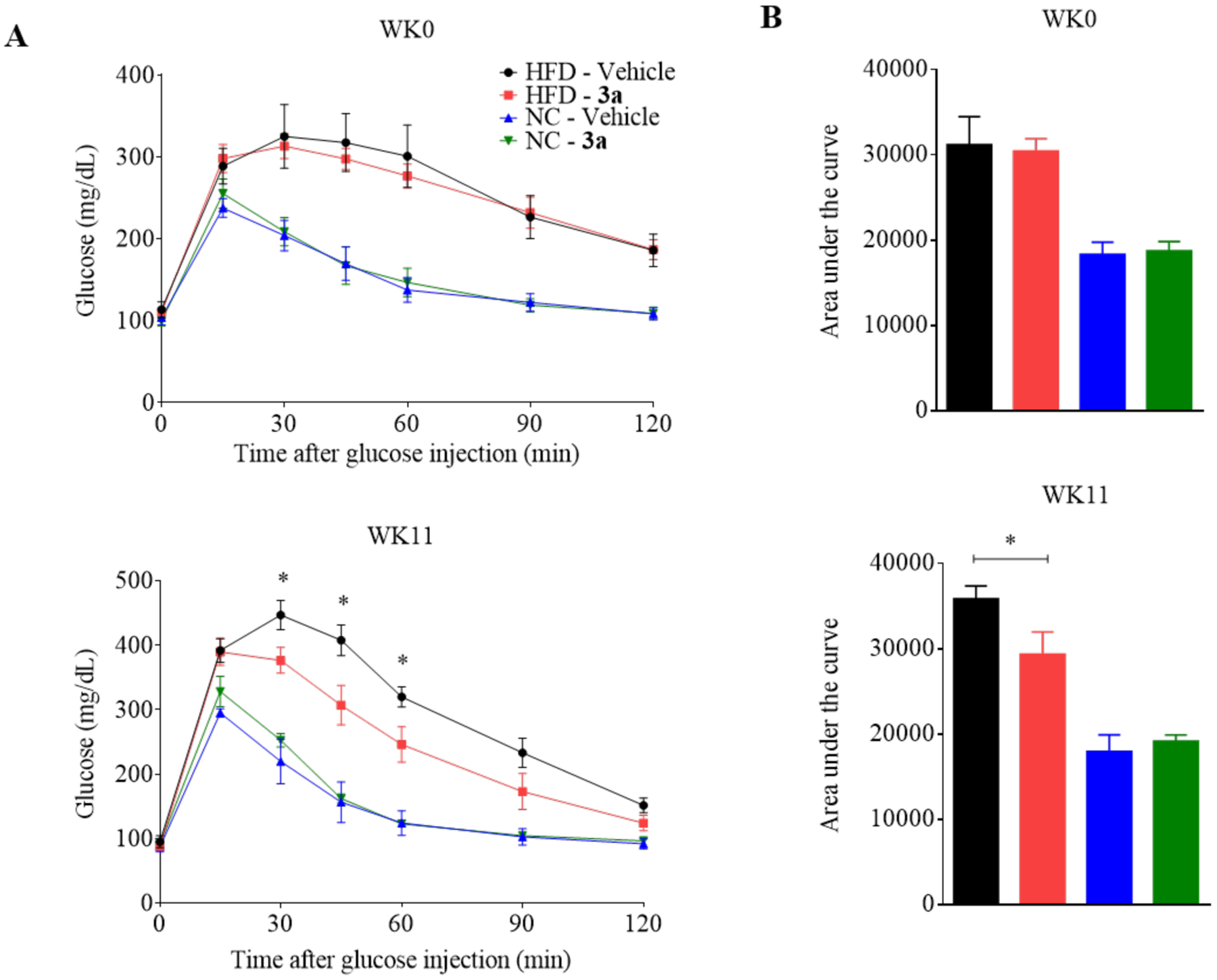
Improvement of glucose tolerance by compound 3a in C57BL/6J mice fed with a high fat diet. (A) Intraperitoneal glucose tolerance test (IPGTT) was carried out on the mice fed with the high fat diet (HFD) and normal chow diet (NC) at the start of treatment (WK0) and after 11 weeks (WK11) of treatment. The mice received intraperitoneal injection of 40 mg/kg compound 3a or vehicle (corn oil) every two days. (B) The area under the curve (AUC, min*mg/dL) for the IPGTT. Data represents mean ± SEM, n = 5 per group. *p < 0.05 as compared to the vehicle group.
Figure 6.
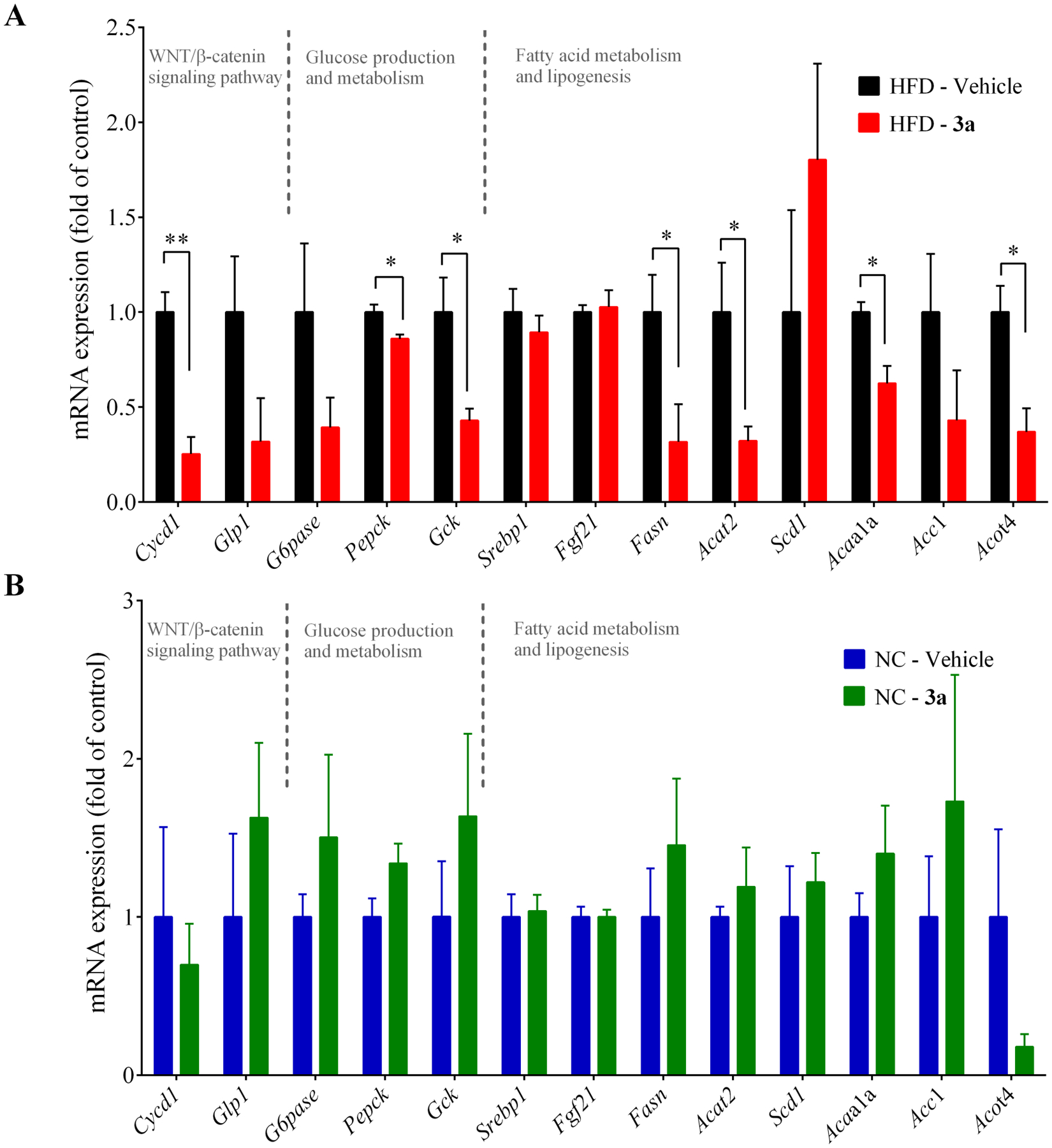
Effects of compound 3a on the hepatic expression of select Wnt/β-catenin pathway target genes and those involved in energy metabolism in mice. (A) The mRNA expression of select genes in the mice fed with a high fat diet. (B) The mRNA expression of select genes in the mice fed with normal chow diet. The mice received i.p. injection of 40 mg/kg compound 3a or vehicle (corn oil) every two days for 11 weeks. Data represents mean ± SEM, n = 5 per group. * p < 0.05, ** p < 0.01.
To further assess the efficacy of compound 3a against the metabolic disorders induced by the high fat diet in mice, we conducted additional measurements. The body weight and the liver weight adjusted by the body weight were increased in the high fat diet-fed mice. However, the increases were suppressed by the treatment of compound 3a. Of note, the treatment caused no effects in the normal chow diet-fed mice (Figure 7). Consistently, in our hepatic histological examination, compound 3a treatment drastically reduced the hepatic lipid accumulation induced by the high fat diet. In addition, the hepatic triglyceride content and the serum cholesterol level were reduced by the treatment of compound 3a in the high fat diet-fed mice. Importantly, the mice that received compound 3a treatment did not exhibit any significant toxicity as indicated by the blood chemical measurements of liver and kidney function and by the histological examination (Table 2, Figure S5). Taken together, the effects of compound 3a were pronounced in the mice with metabolic disorders induced by the high fat diet. Inhibition of the Wnt signaling pathway appeared to mediate the efficacy of compound 3a against the metabolic disorders induced by the high fat diet because the results phenocopied those seen in genetic knockdown of Axin, Ctnnb1 (β-catenin) and Tcf7l2 in the mice fed a high fat diet.11,46,47
Figure 7.
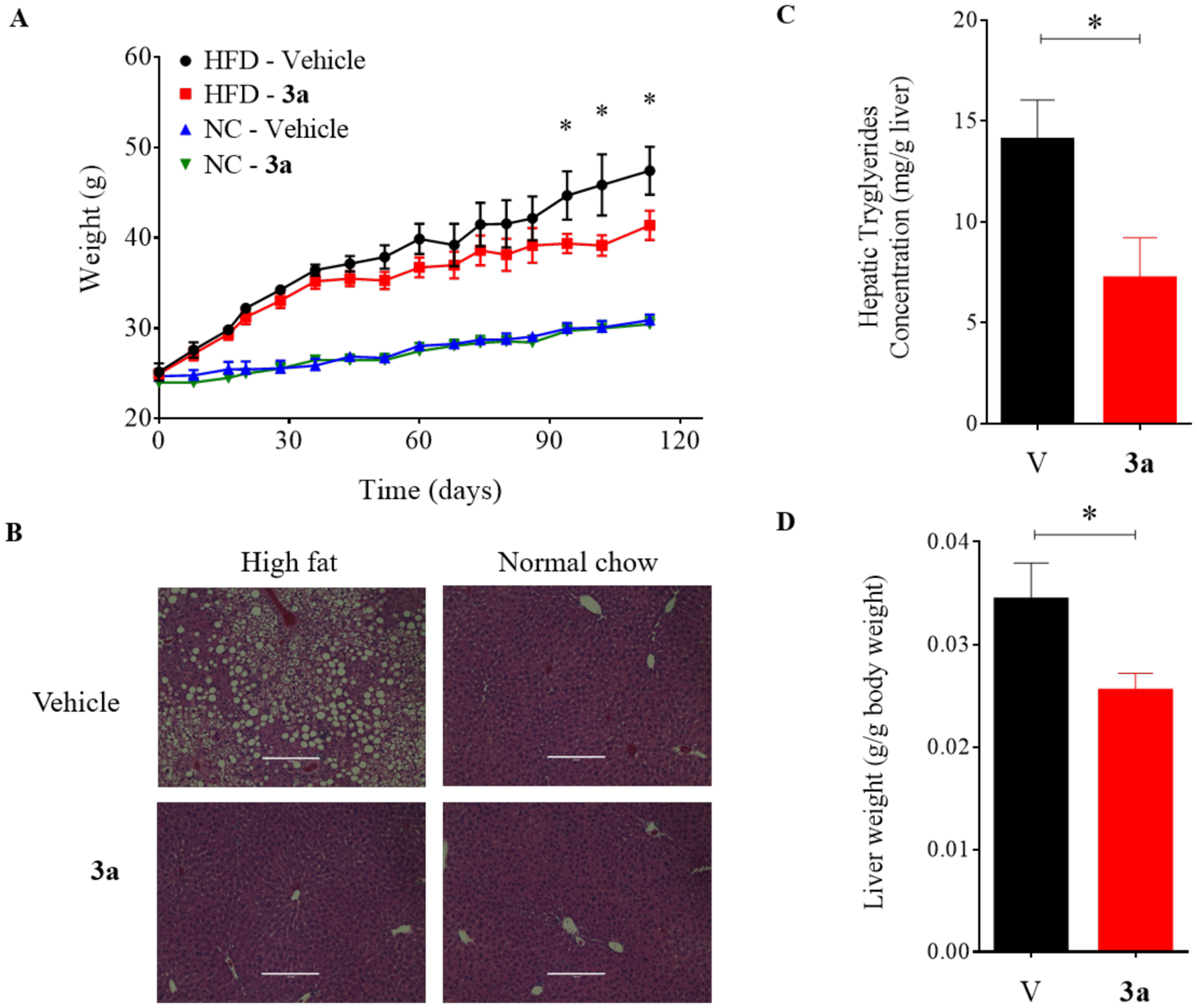
Effects of compound 3a treatment on body weight gain and hepatic lipid accumulation in C57BL/6J mice. (A) Body weight gain in high fat diet and normal chow group mice treated with 40 mg/kg compound 3a or vehicle (corn oil). (B) Hematoxylin and eosin staining for liver tissue samples. (C) Hepatic triglyceride content and (D) the ratio of liver/body weight in high fat diet-fed mice. The mice received i.p. injection of 40 mg/kg compound 3a or vehicle (corn oil) every two days for 11 weeks. V= vehicle, 3a = Compound 3a. * p < 0.05.
Table 2.
Serum Biochemical Parameters in Mice Received Compound 3a or Vehicle
| Physiologic parameter | Unit | High fat diet (n=10) | Normal chow (n=10) | ||||
|---|---|---|---|---|---|---|---|
| vehicle | 3a | p-value | vehicle | 3a | p-value | ||
| ALT | U/L | 63.3 ± 26.9 | 21.0 ± 1.9 | NS | 20.0 ± 5.1 | 12.8 ± 1.6 | NS |
| Alkaline Phosphatase | U/L | 58.3 ± 3.8 | 51.0 ± 4.6 | NS | 66.5 ± 1.8 | 57.5 ± 1.1 | NS |
| AST | U/L | 131 ± 15.6 | 94.8 ± 17.2 | NS | 119 ± 28.3 | 74.5 ± 6.25 | NS |
| Total bilirubin | mg/dL | 0.20 ± 0.0 | 0.20 ± 0.03 | NS | 0.20 ± 0.0 | 0.20 ± 0.0 | NS |
| Cholesterol | mg/dL | 316 ± 25.0 | 223 ± 18.1 | 0.003 | 183 ± 16.6 | 194 ± 13.7 | NS |
| Creatinine Jaffe | mg/dL | 0.30 ± 0.02 | 0.20 ± 0.02 | NS | 0.20 ± 0.01 | 0.2 ± 0.01 | NS |
| LDH | U/L | 469 ± 67 | 313 ± 40.8 | <0.001 | 324 ± 63.5 | 190 ± 20.2 | <0.001 |
| Triglycerides | mg/dL | 93.5 ± 12.1 | 85.3 ± 7.7 | NS | 69.0 ± 2.5 | 55.0 ± 4.8 | NS |
| BUN | mg/dL | 17.3 ± 0.5 | 14.8 ± 0.3 | NS | 19.0 ± 1.8 | 21.8 ± 0.5 | NS |
| Uric Acid | mg/dL | 6.30 ± 0.5 | 4.80 ± 0.6 | NS | 4.00 ± 0.2 | 3.80 ± 0.3 | NS |
The mice were fed either a high-fat diet or normal chow diet. Data analysis was performed using ANOVA and Tukey’s post-hoc test. The values are expressed as mean ± SE. NS, not significant; ALT, Alanine Aminotransferase; AST, Aspartate aminotransferase; LDH, Lactate dehydrogenase; BUN, Blood urea nitrogen.
CONCLUSION
We have designed, synthesized, and characterized a novel class of triazole-based compounds as novel inhibitors of the Wnt/β-catenin signaling pathway. One inhibitor of the class, compound 3a, potently inhibits Wnt/β-catenin signaling pathway and elicits favorable efficacy against the metabolic disorders induced by high fat diet. Our results have indicated that the new inhibitors stabilized the cellular level of Axin, fortifying the β-catenin destruction complex to attenuate β-catenin cytoplasmic level and suppress the transcription of Wnt/β-catenin pathway target genes. The identification of direct protein target of compound 3a is currently underway in our group using biotinylated chemical affinity chromatography, proteomics, and other biochemical methods. Based on the parent scaffold for these new compounds, further modification can be made to generate potent analogues with even improved drug properties that may be applied to the treatment of metabolic disorders, such as fatty liver, diabetes, and obesity, and other diseases, such as cancers, with aberrant regulation of Wnt/β-catenin pathway.
EXPERIMENTAL SECTION
1. Chemical Analysis.
All chemicals were obtained from commercial suppliers and used without further purification. Analytical thin layer chromatography was visualized by ultraviolet light at 256 nM. 1H NMR spectra were recorded on a Varian (400 MHz) spectrometer. Data are presented as follows: chemical shift (in ppm on the δ scale relative to δ = 0.00 ppm for the protons in tetramethylsilane (TMS), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant (J/Hz). 13C NMR spectra were recorded at 100 MHz, and all chemical shifts values are reported in ppm on the δ scale with an internal reference of δ 77.0 or 39.0 for CDCl3 or DMSO-d6, respectively. The purities of title compounds were determined by analytic HPLC, performed on an Agilent 1100 instrument and a reverse-phase column (Waters XTerrra RP18, 5 μM, 4.6 × 250 mm). All compounds were eluted with 45% CH3CN/55% H 2O (0.1% TFA) over 20 min with a detection at 260 nm and a flow rate at 1.0 mL/min. All tested compounds were >95% purity. Yields were not optimized. Compounds 1a-n and potassium trifluoroborate derivatives were commercially available.
General Procedure A: Synthesis of Compounds 2a-2p.
To a mixture of substituted aniline 1a-1n (20 mmol) in concentrated HCl (8.0 mL) was added a solution of NaNO2 (1.4 g, 21 mmol) in H2O (6.0 mL) dropwise at 0 °C. The mixture was stirred at 0 °C for 1 h followed by the addition of a solution of NaN3 (1.3 g, 20 mmol) in H2O (6.0 mL) at 0 °C. The resulting mixture was stirred for another 1 h, extracted with diethyl ether (Et2O) for three times. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. To the resulting residue was added ethyl 3-oxobutanoate, or ethyl 3-oxopentanoate, or ethyl 4-methyl-3-oxopentanoate (22 mmol) and a solution of NaOEt (2.04 g, 30 mmol) in EtOH (120 mL) at room temperature. The mixture was stirred at 80 °C overnight, cooled and concentrated. To the resulting residue was added HCl (1 N, 250 mL) and the resulting precipitate was collected by filtration and washed with water to offer the crude product that was recrystallized from EtOH to give 2a-2p (25–83%).
5-Methyl-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxylic acid (2a).
The title compound was synthesized according to General Procedure A (55%): 1H-NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 7.57–7.52 (m, 2H), 7.46–7.44 (m, 2H), 2.32 (s, 3H), 2.00 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 163.0, 140.1, 136.5, 135.5, 134.6, 131.7, 131.2, 127.8, 127.6, 17.1, 9.6.
5-Methyl-1-(m-tolyl)-1H-1,2,3-triazole-4-carboxylic acid (2b).
The title compound was synthesized according to General Procedure A (50%): 1H-NMR (400 MHz, DMSO-d6) δ 13.47 (s, 1H), 7.85 (t, J = 8.0 Hz, 1H), 7.78–7.73 (m, 3H), 2.83 (s, 3H), 2.75 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 163.0, 140.0, 139.3, 136.9, 135.7, 131.2, 130.0, 126.4, 123.0, 21.3, 10.3.
5-Methyl-1-(p-tolyl)-1H-1,2,3-triazole-4-carboxylic acid (2c).
The title compound was synthesized according to General Procedure A (46%): 1H-NMR (400 MHz, DMSO-d6) δ 13.08 (s, 1H), 7.42–7.38 (m, 4H), 2.41 (s, 3H), 2.36 (s, 3H); 13C-NMR (100 MHz, DMSO-d 6) δ 163.1, 140.3, 136.8, 133.3, 130.5, 125.7, 123.2, 21.2, 10.1.
1-(2-Fluorophenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2d).
The title compound was synthesized according to General Procedure A (60%). 1H-NMR (400 MHz, DMSO-d6) δ 13.24 (s, 1H), 7.74–7.68 (m, 2H), 7.64–7.56 (m, 1H), 7.52–7.45 (m, 1H), 2.40 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 162.8, 157.5, 155.0, 140.8, 136.7, 133.5, 133.5, 129.5, 126.1, 123.2, 123.0, 117.6, 117.4, 9.5.
1-(2-Bromophenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2e).
The title compound was synthesized according to General Procedure A (80%): 1H-NMR (400 MHz, DMSO-d6) δ 13.26 (s, 1H), 7.98–7.96 (m, 1H), 7.73–7.62 (m, 3H), 2.34 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 167.6, 145.3, 141.3, 139.3, 138.8, 138.1, 134.9, 134.4, 125.9, 14.4.
1-(2-Cyanophenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2f).
The title compound was synthesized according to General Procedure A (71%): 1H-NMR (400 MHz, DMSO-d6) δ 13.37 (s, 1H), 8.23–8.21 (d, J = 6.8 Hz, 1H), 8.05–8.01 (t, J = 7.2 Hz), 7.92–7.87 (m, 2H), 2.47 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 162.7, 140.6, 136.9, 136.8, 135.3, 134.9, 132.0, 128.8, 115.6, 110.5, 9.8.
1-(2-Methoxyphenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2g).
The title compound was synthesized according to General Procedure A (80%): 1H-NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 7.66–7.62 (t, J = 8.0 Hz, 1H), 7.49–7.47 (d, J = 8.0 Hz, 1H), 7.35–7.33 (d, J = 8.0 Hz, 1H), 7.20–7.16 (t, J = 8.0 Hz, 1H), 3.80 (s, 3H), 2.31 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 163.0, 154.1, 140.8, 136.3, 132.7, 128.9, 123.9, 121.3, 113.3, 56.4, 9.5.
1-(2-Carbamoylphenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2h).
The title compound was synthesized according to General Procedure A (65%): 1H-NMR (400 MHz, DMSO-d6) δ 13.10 (s, 1H), 7.93 (s, 1H), 7.70–7.68 (m, 1H), 7.67–7.62 (m, 2H), 7.55–7.52 (m, 1H), 7.38 (s, 1H), 2.38 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 167.6, 163.2, 140.8, 136.0, 135.9, 131.4, 131.0, 129.1, 128.5, 129.1, 128.5, 128.3, 9.9.
1-(2-Acetylphenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2i).
The title compound was synthesized according to General Procedure A (40%): 1H-NMR (400 MHz, DMSO-d6) δ 8.02–7.99 (m, 1H), 7.77–7.73 (m, 2H), 7.61–7.59 (m, 1H), 2.32 (s, 3H), 2.26 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 199.0, 163.0, 140.7, 136.5, 136.3, 133.3, 132.6, 131.5, 130.4, 128.8, 29.3, 9.8.
5-Methyl-1-(2-morpholinophenyl)-1H-1,2,3-triazole-4-carboxylic acid (2j).
The title compound was synthesized according to General Procedure A (83%): 1H-NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 7.63–7.59 (t, J = 8.0 Hz, 1H), 7.44–7.42 (d, J =8.0 Hz, 1H), 7.33–7.31 (d, J = 8.0 Hz, 1H), 7.30–7.26 (t, J = 8.0 Hz, 1H), 3.42 (br s, 4H), 2.63 (br s, 4H), 2.38 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 162.9, 148.0, 140.5, 136.7, 132.3, 129.2, 129.1, 124.0, 120.9, 66.6, 51.4, 10.0.
1-([1,1’-Biphenyl]-2-yl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2k).
The title compound was synthesized according to General Procedure A (65%): 1H-NMR (400 MHz, DMSO-d6) δ 7.72–7.68 (m, 1H), 7.63–7.61 (m, 2H), 7.53–7.51 (m, 1H), 7.26–7.21 (m, 3H), 7.02–6.96 (m, 2H), 1.97 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 164.2, 140.1, 138.9, 137.9, 137.5, 133.5, 131.5, 131.4, 129.3, 129.0, 128.8, 128.4, 128.3, 9.5.
1-(2,3-Dimethylphenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2l).
The title compound was synthesized according to General Procedure A (70%): 1H-NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 7.46–7.45 (m, 1H), 7.36–7.32 (m, 1H), 7.26–7.25 (m, 1H), 2.36 (s, 3H), 2.30 (s, 3H), 1.81 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 167.8, 144.9, 143.9, 141.2, 139.3, 138.9, 137.0, 131.6, 130.1, 24.9, 18.7, 14.3.
1-(2,4-Dimethylphenyl)-5-methyl-1H-1,2,3-triazole-4-carboxylic acid (2m).
The title compound was synthesized according to General Procedure A (73%): 1H-NMR (400 MHz, DMSO-d6) δ 13.01 (s, 1H), 7.28–7.24 (m, 2H), 7.20–7.18 (m, 1H), 2.34 (s, 3H), 2.26 (s, 3H), 1.88 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 163.0, 140.9, 140.1, 136.4, 135.1, 132.2, 132.1, 128.0, 127.5, 21.1, 17.0, 9.6.
5-Methyl-1-(naphthalen-1-yl)-1H-1,2,3-triazole-4-carboxylic acid (2n).
The title compound was synthesized according to General Procedure A (72%): 1H-NMR (400 MHz, DMSO-d6) δ 13.15 (s, 1H), 8.27–8.25 (d, J = 8.0 Hz, 1H), 8.16–8.14 (d, J = 8.0 Hz, 1H), 7.80–7.30 (m, 2H), 7.69–7.65 (t, J = 8.0 Hz, 1H), 7.62–7.58 (t, J = 8.0 Hz, 1H), 7.14–7.12 (d, J = 8.0 Hz, 1H), 2.31 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 163.0, 141.1, 136.7, 134.1, 131.5, 129.2, 128.9, 128.8, 127.9, 127.7, 126.2, 126.0, 122.0, 9.7.
5-Ethyl-1-(2-fluorophenyl)-1H-1,2,3-triazole-4-carboxylic acid (2o).
The title compound was synthesized according to General Procedure A (80%): 1H-NMR (400 MHz, DMSO-d6) δ 13.31 (s, 1H), 7.77–7.74 (m, 2H), 7.64–7.59 (t, J = 8.8 Hz, 1H), 7.52–7.48 (t, J = 8.0 Hz, 1H), 2.84–2.78 (q, J = 7.2 Hz, 2H), 1.02–0.98 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 167.4, 162.6, 160.1, 150.5, 140.9, 138.6, 138.5, 134.6, 130.9, 130.8, 122.3, 122.1, 21.6, 17.6.
1-(2-Fluorophenyl)-5-isopropyl-1H-1,2,3-triazole-4-carboxylic acid (2p).
The title compound was synthesized according to General Procedure A (25%): 1H-NMR (400 MHz, DMSO-d6) δ 13.01 (s, 1H), 7.77–7.74 (m, 2H), 7.64–7.60 (t, J = 8.0 Hz, 1H), 7.52–7.48 (t, J = 8.0 Hz, 1H), 3.24–3.20 (m, 1H), 1.23–1.21 (d, J = 6.8 Hz, 6H); 13C-NMR (100 MHz, DMSO-d6) δ 167.6, 162.8, 160.3, 153.5, 140.6, 138.7, 138.6, 135.0. 130.8, 122.2, 122.0, 29.5, 24.6.
General Procedure B: Synthesis of Compounds 3a-3r.
To a solution of 2-aminoquinoline or naphthalen-2-amine or 6-bromoquinolin-2-amine (0.5 mmol) and 2a-2p (0.675 mmol) in DCE (2 mL) was added PyCIU (0.775 mmol) and DIPEA (2.33 mmol). The mixture was stirred at 80 °C overnight, then cooled and concentrated. To the resulting residue was added ethyl acetate (30 mL), and the mixture was washed with brine, dried over Na2SO4 and concentrated. The crude material was purified by column chromatography (hexane/AcOEt, v/v = 4/1 to 2/1) to give products 3a-3r (47–69%).
5-Methyl-N-(quinolin-2-yl)-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3a).
The title compound was synthesized according to General Procedure B (60%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 8.49–8.47 (d, J = 8.8 Hz, 1H), 8.42–8.40 (d, J = 8.8 Hz, 1H), 8.00–7.98 (d, J = 8.0 Hz, 1H), 7.91–7.89 (d, J = 8.0 Hz, 1H), 7.69 (t, J = 8.0 Hz, 1H), 7.61–7.56 (m, 3H), 7.51–7.50 (m, 2H), 2.45 (s, 3H), 2.04 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 160.0, 150.8, 146.8, 139.5, 139.1, 137.6, 135.5, 134.4, 131.8, 131.4, 130.7, 128.3, 127.8, 127.7, 127.7, 126.4, 125.8, 114.7, 17.2, 9.4. HRMS (ESI): m/z [M + H]+ calcd for C20H18N5O, 344.1511, found, 344.1510. HPLC: tR = 8.75 min, 98.2%.
5-Methyl-N-(quinolin-2-yl)-1-(m-tolyl)-1H-1,2,3-triazole-4-carboxamide (3b).
The title compound was synthesized according to General Procedure B (65%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.92 (s, 1H), 8.57 (d, J = 8.0 Hz, 1H), 8.22 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.70 (t, J = 7.2, 7.6 Hz, 1H), 7.47 (d, J = 7.2 Hz, 2H), 7.38 (d, J = 8.0 Hz, 1H), 7.33 (s, 1H), 7.27 (d, J = 5.6 Hz, 1H), 2.71 (s, 3H), 2.48 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.0, 150.6, 146.9, 140.1, 138.4, 138.1, 137.9, 135.3, 130.9, 129.9, 129.4, 127.8, 127.5, 126.4, 125.9, 125.1, 122.2, 114.2, 21.3, 9.9; HRMS (ESI): calcd. for C20H18N5O [M + H]+ 344.1511, found 344.1508. HPLC: t R = 6.51 min, 100%.
5-Methyl-N-(quinolin-2-yl)-1-(p-tolyl)-1H-1,2,3-triazole-4-carboxamide (3c).
The title compound was synthesized according to General Procedure B (68%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 8.55 (d, J = 8.0 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.68 (t, J = 7.6, 8.0 Hz, 1H), 7.45 (t, J = 6.8, 8.0 Hz, 1H), 7.43–7.35 (m, 4H), 2.68 (s, 3H), 2.46 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.0, 150.6, 146.9, 140.4, 138.4, 138.1, 137.9, 132.9, 130.2, 129.9, 127.8, 127.5, 126.4, 125.1, 125.0, 114.2, 21.3, 9.9; HRMS (ESI): m/z [M + H]+ calcd for C20H18N5O, 344.1511; found, 344.1511. HPLC: tR = 6.40 min, 99.9%.
1-(2-Fluorophenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3d).
The title compound was synthesized according to General Procedure B (50%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.25 (s, 1H), 8.46–8.44 (m, 2H), 7.97–7.54 (m, 9H), 2.52 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 159.8, 157.5, 155.0, 150.8, 146.8, 140.2, 139.1, 137.7, 133.7, 130.6, 129.5, 128.2, 127.7, 126.4, 126.2, 125.8, 123.0, 117.7, 117.5, 114.7, 9.3. HRMS (ESI): m/z [M + H]+ calcd for C19H15FN5O, 348.1261; found, 348.1252. HPLC: tR = 9.53 min, 99.9%.
1-(2-Bromophenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3e).
The title compound was synthesized according to General Procedure B (68%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.26 (s, 1H), 8.48–8.46 (d, J = 8.0 Hz, 1H), 8.41–8.39 (d, J = 8.0 Hz, 1H), 8.02–8.99 (t, J = 8.8 Hz, 2H), 7.91–7.89 (d, J = 8.0 Hz, 1H), 7.77–7.67 (m, 4H), 7.57–7.53 (d, J = 8.0 Hz, 1H), 2.46 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 164.6, 155.5, 151.6, 144.7, 143.9, 142.3, 139.2, 138.9, 138.3, 135.4, 135.0, 134.5, 133.0, 132.5, 131.2, 130.6, 125.9, 119.6, 14.2. HRMS (ESI): m/z [M + H]+ calcd for C 19H15BrN5O2, 408.0460; found, 408.0458. HPLC: tR = 8.21 min, 99.9%.
1-(2-Cyanophenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3f).
The title compound was synthesized according to General Procedure B (57%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.49–8.46 (d, J = 8.8 Hz, 1H), 8.40–8.38 (d, J = 8.8 Hz, 1H), 8.27–8.25 (d, J = 8.0 Hz, 1H), 8.08–8.04 (t, J = 8.0 Hz, 1H), 7.99–7.96 (t, 2H), 7.94–7.90 (t, J =8.0 Hz, 2H), 7.79–7.75 (t, J = 8.0 Hz, 1H), 7.57–7.53 (t, J = 8.0 Hz, 1H), 2.60 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 159.7, 150.8, 146.8, 140.1, 139.1, 138.0, 136.6, 135.4, 135.1, 132.1, 130.7, 128.8, 128.3, 127.8, 126.4, 125.8, 115.6, 114.9, 110.5, 9.6. HRMS (ESI): m/z [M + H]+ calcd for C20H15N6O, 355.1307; found 355.1304. HPLC: tR = 7.45 min, 99.8%.
1-(2-Methoxyphenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3g).
The title compound was synthesized according to General Procedure B (48%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.61–8.51 (m, 2H), 8.09 (m, 1H), 8.01 (m, 1H), 7.88 (m, 1H), 7.78 (m, 1H), 7.66 (m, 2H), 7.52–7.47 (m, 1H), 7.35–7.32 (m, 1H), 3.97–3.94 (q, J = 4 Hz, 3H), 2.56–2.53 (q, J = 4 Hz, 3H); 13C-NMR (100 MHz, DMSO-d 6) δ 160.0, 154.1, 150.9, 146.2, 140.2, 139.1, 137.3, 132.9, 130.6, 128.9, 128.2, 127.7, 126.4, 125.7, 123.7, 121.4, 114.7, 113.4, 56.5, 9.4. HRMS (ESI): m/z [M + H]+ calcd for C 20H18N5O2, 360.1460; found, 360.1460. HPLC: tR = 8.88 min, 99.8%.
1-(2-Carbamoylphenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3h).
The title compound was synthesized according to General Procedure B (49%, a pale yellow solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.48–8.46 (d, J = 8.8 Hz, 1H), 8.42–8.39 (d, J = 8.8 Hz, 1H), 8.06 (s, 1H), 7.99–7.97 (d, J = 8.0 Hz, 1H), 7.90–7.88 (d, J = 8.0 Hz, 1H), 7.80–7.74 (m, 4H), 7.68–7.66 (m, 1H), 7.57–7.53 (t, J = 8.0 Hz, 1H), 7.49 (s, 1H), 2.50 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 172.3, 164.8, 155.6, 151.6, 145.1, 143.9, 141.7, 139.7, 137.6, 136.2, 135.9, 135.4, 133.9, 133.3, 133.0, 132.5, 131.2, 130.5, 119.4, 14.4. HRMS (ESI): m/z [M + H]+ calcd for C20H17N6O2, 373.1413; found, 373.1413. HPLC: tR = 2.72 min, 95.0%.
1-(2-Acetylphenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3i).
The title compound was synthesized according to General Procedure B (48%, a brown solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.37 (s, 1H), 8.65–8.63 (d, J = 8.8 Hz, 1H), 8.58–8.56 (d, J = 8.8 Hz, 1H), 8.30–8.28 (d, J = 7.2 Hz, 1H), 8.16–8.14 (d, J = 8.0 Hz, 1H), 8.07–8.01 (m, 3H), 7.95–7.89 (m, 2H), 7.74–7.70 (t, J = 7.2 Hz, 1H), 2.67 (s, 6H); 13C-NMR (100 MHz, DMSO-d6) δ 203.7, 164.7, 155.6, 151.6, 144.9, 143.9, 142.3, 140.9, 138.2, 137.1, 136.4, 135.4, 133.6, 133.0, 132.5, 131.2, 130.5, 119.5, 34.1, 14.3. HRMS (ESI): m/z [M + H]+ calcd for C21H18N5O2, 372.1460; found, 372.1455. HPLC: tR = 7.39 min, 99.2%.
5-Methyl-1-(2-morpholinophenyl)-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3j).
The title compound was synthesized according to General Procedure B (48%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.20 (s, 1H), 8.48–8.46 (d, J = 8.8 Hz, 1H), 8.43–8.40 (d, J = 8.8 Hz, 1H), 7.99–7.97 (d, J = 8.4 Hz, 1H), 7.90–7.88 (d, J = 8.4 Hz, 1H), 7.78–7.74 (t, J = 8.0 Hz, 1H), 7.66–7.62 (t, J = 8.0 Hz, 1H), 7.57–7.53 (t, J = 8.0 Hz, 1H), 7.50–7.48 (d, J = 8.0 Hz, 1H), 7.37–7.33 (t, J = 8.0 Hz, 1H), 7.31–7.29 (d, J = 8.0 Hz, 1H), 3.45 (br s, 4H), 2.67 (br s, 4H), 2.50 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 159.9, 150.8, 146.8, 139.8, 139.1, 137.8, 132.4, 130.6, 129.3, 129.0, 128.3, 127.7, 126.4, 125.7, 124.1, 121.0, 114.7, 66.6, 51.4, 9.7. HRMS (ESI): m/z [M + H]+ calcd for C 23H23N6O2, 415.1882; found, 415.1873. HPLC: tR = 10.48 min, 99.6%.
1-([1,1’-Biphenyl]-2-yl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3k).
The title compound was synthesized according to General Procedure B (58%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 8.45–8.43 (d, J = 8.0 Hz, 1H), 8.33–8.31 (d, J = 8.0 Hz, 1H), 7.98–7.96 (d, J = 8.0 Hz, 1H), 7.89–7.87 (d, J = 8.0 Hz, 1H), 7.81–7.69 (m, 5H), 7.56–7.52 (t, J = 7.2 Hz, 1H), 7.34–7.32 (m, 3H), 7.09–7.07 (m, 2H), 2.16 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 159.7, 150.7, 146.8, 139.6, 139.1, 137.4, 137.2, 132.8, 131.9, 131.5, 130.6, 129.5, 129.2, 128.7, 128.5, 128.2, 127.7, 126.4, 125.8, 114.7, 9.28. HRMS (ESI): m/z [M + H]+ calcd for C25H20N5O, 406.1668; found, 406.1665. HPLC: t R = 11.17 min, 99.9%.
1-(2,3-Dimethylphenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3l).
The title compound was synthesized according to General Procedure B (67%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 8.57–8.55 (d, J = 8.4 Hz, 1H), 8.23–8.21 (d, J = 8.4 Hz, 1H), 7.93–7.91 (d, J = 8.8 Hz, 1H), 7.81–7.79 (d, J = 8.4 Hz, 1H), 7.71–7.67 (t, J = 8.0 Hz, 1H), 7.49–7.45 (t, J = 7.2 Hz, 1H), 7.40–7.38 (d, J = 7.2 Hz, 1H), 7.31–7.27 (t, J = 8.0 Hz, 1H), 7.12–7.10 (d, J = 8.0 Hz, 1H), 2.50 (s, 3H), 2.39 (s, 3H), 1.91 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.1, 150.6, 146.9, 139.1, 139.0, 138.4, 137.7, 134.3, 134.0, 132.2, 129.9, 127.8, 127.5, 126.5, 126.4, 125.1, 124.8, 114.2, 20.3, 14.0, 9.3. HRMS (ESI): m/z [M + H]+ calcd for C21H20N5O, 358.1668; found, 358.1670. HPLC: tR = 11.23 min, 99.9%.
1-(2,4-Dimethylphenyl)-5-methyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3m).
The title compound was synthesized according to General Procedure B (69%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.94 (s, 1H), 8.57–8.55 (d, J = 8.8 Hz, 1H), 8.23–8.21 (d, J = 8.8 Hz, 1H), 7.93–7.91 (d, J = 8.0 Hz, 1H), 7.82–7.80 (d, J = 8.0 Hz, 1H), 7.71–7.67 (t, J = 8.0 Hz, 1H), 7.49–7.45 (t, J = 8.0 Hz, 1H), 7.26–7.24 (d, J = 8.0 Hz, 1H), 7.20–7.18 (d, J = 8.0 Hz, 1H), 7.15–7.13 (d, J = 8.0 Hz, 1H), 2.51 (s, 3H), 2.44 (s, 3H), 2.03 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.1, 150.6, 141.1, 138.9, 138.4, 137.7, 135.1, 132.1, 131.7, 129.9, 127.8, 127.7, 127.5, 126.9, 126.4, 125.1, 114.2, 21.5, 17.1, 9.3. HRMS (ESI): m/z [M + H]+ calcd for C21H20N5O 358.1668, found 358.1661. HPLC: tR = 17.12 min, 99.9%.
5-Methyl-1-(naphthalen-1-yl)-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3n).
The title compound was synthesized according to General Procedure B (47%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.76–8.69 (m, 2H), 8.58–8.56 (d, J = 8.0 Hz, 1H), 8.46–8.44 (d, J = 8.0 Hz, 1H), 8.26–8.24 (d, J = 8.0 Hz, 1H), 8.19–8.17 (d, J = 8.0 Hz, 1H), 8.10 (s, 1H), 8.07–8.10 (m, 2H), 7.97–7.95 (m, 1H), 7.92–7.89 (t, J = 6.4 Hz, 1H), 7.82 (t, J = 6.4 Hz, 1H), 7.49–7.47 (d, J = 8.0 Hz, 1H), 2.67 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 160.0, 150.9, 146.8, 140.4, 139.2,137.8, 134.1, 131.7, 131.4, 130.7, 129.2, 128.9, 128.8, 128.3, 127.8, 127.7, 126.4, 126.3, 126.0, 125.8, 122.1, 114.7, 9.5. HRMS (ESI): m/z [M + H]+ calcd for C23H18N5O, 380.1511; found, 380.1512. HPLC: tR = 9.54 min, 97.4 %.
5-Ethyl-1-(2-fluorophenyl)-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3o).
The title compound was synthesized according to General Procedure B (60%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.49–8.46 (d, J = 8.8 Hz, 1H), 8.42–8.40 (d, J = 8.8 Hz, 1H), 8.00–7.98 (d, J = 8.8 Hz, 1H), 7.91–7.89 (d, J = 8.8 Hz, 1H), 7.84–7.75 (m, 3H), 7.69–7.65 (t, J = 8.0 Hz, 1H), 7.57–7.53 (m, 2H), 2.96–2.91 (q, J = 7.2 Hz, 2H), 1.11–1.08 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6) δ 164.4, 162.6, 160.1, 155.6, 151.6, 149.9, 143.9, 142.1, 138.8, 138.7, 135.4, 134.6, 133.0, 132.5, 131.2, 131.0, 130.9, 130.5, 122.4, 122.2, 119.5, 21.6, 17.5. HRMS (ESI): m/z [M + H]+ calcd for C 20H17FN5O, 362.1417; found, 362.1415. HPLC: tR = 14.04 min, 99.7%.
1-(2-Fluorophenyl)-5-isopropyl-N-(quinolin-2-yl)-1H-1,2,3-triazole-4-carboxamide (3p).
The title compound was synthesized according to General Procedure B (69%, a white solid): 1H-NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.49–8.47 (d, J = 8.8 Hz, 1H), 8.43–8.41 (d, J = 8.8 Hz, 1H), 8.00–7.98 (d, J = 8.8 Hz, 1H), 7.91–7.89 (d, J = 8.8 Hz, 1H), 7.84–7.75 (m, 3H), 7.69–7.65 (t, J = 8.0 Hz, 1H), 7.57–7.53 (m, 2H), 3.24–3.20 (m, 1H), 1.34–1.32 (d, J = 7.2 Hz, 6H); 13C-NMR (100 MHz, DMSO-d6) δ 164.3, 162.8, 160.3, 155.6, 153.1, 151.6, 143.9, 142.1, 138.9, 138.8, 135.4, 134.9, 133.0, 132.5, 131.2, 131.0, 130.5, 128.4, 128.3, 122.3, 122.1, 119.5, 29.7, 24.8. HRMS (ESI): m/z [M + H]+ calcd for C 21H19FN5O, 376.1574; found, 376.1581. HPLC: tR = 10.69 min, 99.0%.
5-Methyl-N-(naphthalen-2-yl)-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3q).
The title compound was synthesized according to General Procedure B (57%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 8.47 (s, 1H), 7.87–7.81 (m, 3H), 7.67–7.64 (dd, J 1 = 1.6 Hz, J2 = 8.8 Hz, 1H), 7.53–7.39 (m, 5H), 7.26 (s, 1H), 2.53 (s, 3H), 2.09 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 159.4, 138.4, 138.1, 135.5, 135.2, 134.3, 133.9, 131.5, 130.8, 130.7, 128.8, 127.7, 127.6, 127.2, 127.1, 126.5, 125.0, 119.8, 116.4, 17.2, 9.2. HRMS (ESI): m/z [M + H]+ calcd for C21H19N4O, 343.1559; found, 343.1561. HPLC: tR = 11.64 min, 98.6%.
N-(6-Bromoquinolin-2-yl)-5-methyl-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3r).
The title compound was synthesized according to General Procedure B (60%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 8.59 (d, J = 8.4 Hz, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.95 (s, 1H), 7.80–7.75 (m, 2H), 7.51–7.49 (m,1H), 7.45–7.40 (m, 2H), 7.26 (s, 1H), 2.51 (s, 3H), 2.08 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.0, 150.8, 139.0, 137.8, 135.5, 134.2, 133.4, 131.5, 130.9, 129.5, 129.3, 127.1, 118.7, 115.0, 17.2, 9.3; HRMS (ESI): m/z [M + H]+ calcd for C20H17BrN5O, 422.0616; found, 422.0621. HPLC: t R = 8.14 min, 98.2%.
General Procedure C: Synthesis of Compounds 3s-3u.
In a conical shaped microwave vial was added 3r (0.169 mmol), potassium trifluoroborate derivatives (0.338 mmol), 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos, 0.034 mmol), cesium carbonate (0.507 mmol), and palladium (II) acetate (0.017 mmol), THF (0.5 mL) and water (0.05 mL). The reaction mixture was sealed and stirred at room temperature for 10 min. Once a clear solution was obtained, the vial was heated to 80 °C for 15 min followed by 145 °C for 45 min. After cooling, the reaction mixture was diluted with DCM and dried over Na2SO4. The solution was filtered, concentrated and purified by column chromatography (5–15% MeOH in DCM) to give compounds 3s-3u (55–73%).
5-Methyl-N-(6-(morpholinomethyl)quinolin-2-yl)-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3s).
The title compound was synthesized according to General Procedure C (73%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.91 (s, 1H), 8.54 (d, J = 8.8 Hz, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.71–7.69 (m, 2H), 7.49–7.47 (m, 1H), 7.43–7.39 (m, 2H), 7.25 (d, J = 7.2 H, 1H), 3.74–3.67 (m, 6H), 2.50–2.51 (m, 7H), 2.06 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 160.0, 150.5, 146.4, 138.8, 138.2, 137.7, 135.5, 134.2, 131.5, 131.4, 130.9, 127.8, 127.4, 127.1, 126.1, 114.3, 66.8, 63.1, 53.6, 17.2, 9.3; HRMS (ESI): m/z [M + H]+ calcd for C25H27N6O2, 443.2195; found, 443.2189. HPLC: tR = 1.96 min, 97.2%.
5-Methyl-N-(6-(piperidin-1-ylmethyl)quinolin-2-yl)-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3t).
The title compound was synthesized according to General Procedure C (55%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.89 (s, 1H), 8.51 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H), 7.68–7.66 (m, 2H), 7.47–7.46 (m, 1H), 7.42–7.36 (m, 2H), 7.24 (d, J = 7.6 Hz, 1H), 3.61 (s, 2H), 2.49 (s, 3H), 2.41–2.40 (m, 4H), 2.05 (s, 3H), 1.58–1.57 (m, 4H), 1.43–1.41 (m, 2H); 13C-NMR (100 MHz, CDCl3) δ 159.9, 150.3, 146.3, 138.8, 138.2, 137.8, 135.8, 135.5, 134.3, 131.7, 131.5, 130.8, 127.5, 127.2, 127.1, 126.1, 114.1, 63.6, 54.6, 25.9, 24.3, 17.2, 9.3; HRMS (ESI): m/z [M + H]+ calcd for C 26H29N6O, 441.2403; found, 441.2417. HPLC: tR = 2.05 min, 100%.
5-Methyl-N-(6-(thiomorpholinomethyl)quinolin-2-yl)-1-(o-tolyl)-1H-1,2,3-triazole-4-carboxamide (3u).
The title compound was synthesized according to General Procedure C (70%, a white solid): 1H-NMR (400 MHz, CDCl3) δ 9.90 (s, 1H), 8.53 (d, J = 8.4 Hz, 1H), 8.16 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.68–7.66 (m, 2H), 7.50–7.47 (m, 1H), 7.43–7.37 (m, 2H), 7.25 (d, J = 7.6 Hz, 1H), 3.66 (s, 2H), 2.74–2.69 (m, 8H), 2.50 (s, 3H), 2.06 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 159.9, 150.4, 146.4, 138.8, 138.1, 137.8, 135.5, 135.1, 134.2, 131.5, 131.3, 130.9, 127.8, 127.1, 127.1, 126.1, 114.2, 63.4, 55.0, 28.0, 17.2, 9.3; HRMS (ESI): m/z [M + H]+ calcd for C25H27N6OS, 459.1967; found, 459.1955. HPLC: tR = 2.12 min, 99.9%.
2. Wnt-HEK293 Luciferase Reporter Assay in a 1536-Well Plate Format.
Stably transfected Wnt-HEK293 cells were generated by transfection of HEK293 cells with TCF based M50 Super 8x TOPFlash plasmid along with a pLenti-GFP-Puro empty plasmid (Addgene). The clones carrying the TOPFlash and GFP constructs were screened with 2 μg/ml puromycin for a period of 3 weeks. Stably expressing clones were validated with prototypical Wnt activators (WNT3a and LiCl) and inhibitor (XAV939). For our screening studies, cells were dispensed at 2000/well in 4 μL of the Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum into white-solid 1,536-well plates (Greiner Bio-One North America Inc., Monroe, NC) using flying reagent dispenser (FRD, Aurora Discovery, San Diego, CA). The assay plates were incubated for overnight at 37 °C. Then 23 nL of test compounds were transferred to the assay plates using pintool station (Wako, San Diego, CA), followed by the addition of 1 μL of 200 ng/mL recombinant human Wnt-3a (R&D Systems Inc., Minneapolis, MN) using an FRD to all the wells except in the top half portions of the first four columns which received the control assay medium instead. The assay plates were incubated for 24 h at 37 °C. The cell viability was determined as described below. For luciferase reporter assay, 4 μL/well ONE-Glo reagent (Promega Corporation) was added using an FRD and luminescence signal was measured through ViewLux plate reader after 30 min incubation at room temperature. Data were expressed as relative fluorescence units (cell viability assay) and relative luminescence units (luciferase reporter assay).
3. Confirmatory Luciferase Reporter Assay in a 24-Well Plate Format.
Plasmids M50 Super 8x TOPFlash and FOPflash were gifts from Randall Moon (Addgene plasmid # 12456 and 12457).48 Renilla luciferase was used as a control for the gene reporter assays. Human pcDNA3-wild type and pcDNA3-S33Y β-catenin were gifts from Eric Fearon (Addgene plasmid # 19286 and 16828).49 HEK293 cells were transfected with the plasmids for 16 h using lipofectamine 2000 (Qiagen). Compounds were incubated with the transfected cells for additional 24 h and luciferase was measured using Promega’s Dual-Luciferase Reporter Assay System according to the manufacturer’s directives. Calculations were carried out by using the ratio of the TOPFlash or FOPFlash luciferase adjusted by the Renilla luciferase.
4. Determination of Cell Viability.
The effects of compounds on cell viability were determined under two incubation conditions. First, the cell viability under the condition for luciferase reporter assay was measured. The assay 1536-well plates were incubated with a compound for 24 h at 37 °C. Then 1 μL/well CellTiter-Fluor reagent (Promega Corporation, Madison, WI) was added using an FRD and fluorescence signal was measured through ViewLux plate reader (Perkin Elmer, Boston, MA) after 30 min incubation at 37 °C. Second, the cytotoxic effects of select compoundswere assessed in unstimulated normal HEK293 cells with inherently low background Wnt pathway activity. The cells were plated at a density of 3.5 × 105/well in 24-well plates for 24 h and then incubated with each compound for a much longer incubation time of 72 h relative to that for the reporter assay. Cell viability was determined by using CellTiter-Fluor reagent according to manufacturer’s instruction.
5. Immunocytochemistry Analysis.
HEK293 cells were seeded on slides coated with poly-D-lysine. In the following day, the cells were treated with compounds with or without Wnt activation by 20 mM LiCl or Wnt3a conditioned medium. Then the cells were washed once with PBS and fixed with 10% formalin (Sigma) at room temperature for 20 min. The cells were permeabilized with methanol for 5 min and washed three times with PBS. Blocking solution containing 5% bovine serum albumin in PBS was added to the slides for 1 h. Primary antibodies were incubated with the slide overnight and then secondary antibodies were added for additional 2 h. Further wash step was carried out and DAPI was incubated with the slide for 30 min. The slides were washed twice and mounted prior to microscopy analysis.
6. Cellular Fractionation and Western Blot Analysis.
Lysis of the cells were carried out using lysis buffer (10 mM HEPES, 100 μM EDTA, 10 mM KCl, 0.5% NP40, protease and phosphatase inhibitor cocktails).50 Cells were lysed for 20 min, vortexed for 10 seconds and centrifuged at 13,000 x g for 15 min. Tissue lysis was carried out using RIPA Lysis Buffer System (Santa Cruz Biotechnology, #sc-24948) and its fractionation was carried out strictly as published by Wieckowski et al.51
Proteins were quantitated with bicinchoninic acid assay. The adjusted protein concentrations were resolved on 10% SDS-PAGE gel. This was transferred to nitrocellulose membrane and western blot was carried out using antibodies specific for the proteins as followed: β-actin (Abcam) , p-catenin (#9561), Axin (#2087) were purchased from Cell Signaling, and β-catenin (sc-133240), GSK-3α/β (sc-7291) were from Santa Cruz Biotechnology.
Overexpression pcDNA3 plasmid vectors for Flag tagged human beta-catenin and beta-catenin S33Y were gifts from Eric Fearon (Addgene plasmid #19286).49 All siRNAs were purchased from Sigma-Adrich.
7. Quantitative PCR.
Total RNA was extracted from cells or tissues using Trizol reagent (Thermo Fisher) following the manufacturer’s instructions. Reverse transcription was carried out to obtain the complementary DNA, which was further used to determine the gene expression using quantitative PCR. The human primers used in our studies are outlined in Supplementary table 1 (Table S1) and those of the mouse have been previous published.11
8. Immunoprecipitation.
Following experiments, cells were washed in PBS and scraped into new Eppendorf tubes. Cell lysis was carried out with IP lysis buffer (25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol; pH 7.4) for 20 min. The lysates were centrifuged at 13,000 × g for 15 min and the supernatant was collected and pellet discarded. Protein A/G plus beads (Santa Cruz biotechnology) were added to preclear the lysate for 1 h. The lysate was centrifuge at 1,000 x g for 1 min and the supernatant was transferred to a new Eppendorf tube. Protein concentration was determined with the BCA assay and 2 mg total protein was used for the immunoprecipitation experiment using the indicated antibodies overnight. The protein beads were then added to the lysates and incubated for 12 h. The samples were centrifuge at 1,000 × g for 1 min and further washed with lysis buffer three times. Laemmli sample buffer was added to the final pelleted beads. The sample was heated at 95 °C for 5 min, then vortexed and centrifuge for additional 1 min. The supernatant was immunoblotted for the indicated proteins.
9. RNAi Mediated Knockdown.
All knockdown studies were carried out with lipofectamine RNAimax (Thermofisher) for RNAi transfection according to manufacturer’s instructions. Small interfering RNAs were purchased from Sigma-Aldrich. Cells were transfected for 36 h and treated with compounds for additional 24 h prior to harvest unless otherwise indicated.
10. Surface Plasmon Resonance (SPR) Analysis.
SPR assays were performed and analyzed by the University of Maryland School of Medicine Biosensor Core Facility. GST tagged human GSK3β (#14–306-D) was purchased from Millipore Sigma.
11. Nile Red Staining.
Huh7 cells were seeded on coverslips coated with rat Collagen I overnight. The cells were then treated with varying concentrations of the compounds for 36 h and together with 200 μM sodium oleate for additional 12 h. The Hanks’ Balanced Salt solution (HBSS) was used to wash the cells once and incubated with the cells along with 1 μM Nile red for 10 min at 37 °C in the dark. Excess Nile red was washed off with HBSS buffer and the cells were imaged with a fluorescence microscope using 552/636 nm (excitation/emission) lamp settings.
12. Animals.
The animal experiments received prior approval from and followed the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Maryland Baltimore. All the mice were housed in 12-h light/ 12-h dark cycle with food and water provided ad libitum. For the efficacy studies, mice were either fed a standard normal chow diet or high fat diet (45% kcal fat) purchased from Harlan Laboratories (Indianapolis, IN). All experiments utilized the C57BL/6J mice purchased from the Jackson Laboratories (Bar Harbor, ME).
13. Mouse Efficacy and Pharmacokinetic Study Design.
For the efficacy study, four groups of 8 week-old male mice weighing 24–27 g were used for the experiments. Two groups of mice were fed high fat diet obtained from Harlan Laboratories (#TD.08811) containing 45% kcal fat initially for 6 weeks, then in combination with intraperitoneal injection of 40 mg/kg compound 3a or vehicle (corn oil) every two days until the end of the experiments. The other two groups received normal chow diet throughout the study with or without 40 mg/kg compound 3a. Intraperitoneal glucose tolerance test (IPGTT) was performed before and at the end of compound/vehicle treatment. Mice were sacrificed one week after the second glucose tolerance test and about 24 h following the final dose. For the pharmacokinetic evaluations, compound 3a was dissolved at 1 mg/ml with 10% Kolliphor EL and 10% Polyethylene Glycol in saline. Ten mice were separated into two groups for intravenous and oral administration. Compound 3a was given at 10 mg/kg to the mice and blood samples were collected via tail vein at various time points and analyzed by LC/MS/MS.
14. Intraperitoneal Glucose Tolerance Test.
Mice were fasted for 6–8 h in new cages without food. Following this, the mice were injected intraperitoneally glucose solution (2 g/kg) in saline. Glucometer was used to measure blood glucose levels from tail vein in a time dependent manner.
15. Triglyceride Assay.
Serum triglyceride level and liver triglyceride content were quantitated using Triglyceride Quantitation Assay Kit (Abcam, #ab65336) following the instructions from the manufacturer.
16. Hematoxylin and Eosin Staining.
Tissue embedding and hematoxylin and eosin staining was carried out by the University of Maryland School of Medicine Pathology Core Facility.
17. Serum Biochemical Analysis.
Comprehensive clinical biochemical assays to determine metabolic status, liver and renal function were carried out by VRL™-Maryland (Gaithersburg, MD).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the National Institute of General Medical Sciences of the US National Institutes of Health (NIH) for research support [R01GM099742] to Y.S. Dr. Yan Shu is a co-founder for and owns equity in Optivia Biotechnology. We also thank the Chinese National Natural Science Foundation [#21702073] to W.Y. and FX, and HuaiAn Science and Technology Bureau [HAC201706] to W.Y. and F.X. This work was supported in part by the Intramural Research Program of the National Center for Advancing Translational Sciences (NCATS).
ABBREVIATION USED
- APC
Adenomatous polyposis coli
- GSK3β
Glycogen synthase kinase
- LEF/TCF
T-cell factor/lymphoid enhancer factor
- TCF7L2
transcription factor 7 like 2
- CK
Casein kinase
- CBP
CREB-binding protein
- FDA
food and drug administration LiCl – lithium chloride
- IPGTT
Intraperitoneal glucose tolerance test
- HFD
high fat diet
- NC
normal chow diet
- AUC
Area under the curve
- K D
dissociation constant
- IC50
half-maximal inhibitory concentration
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publication websites.
1H and 13C NMR spectra of new compounds and molecular formula strings and additional data.
REFERENCES
- (1).Clevers H; Nusse R Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- (2).Valenta T; Hausmann G; Basler K The many faces and functions of β-catenin. EMBO J. 2012, 31, 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Oh DY; Olefsky JM Medicine. Wnt fans the flames in obesity. Science 2010, 329, 397–398. [DOI] [PubMed] [Google Scholar]
- (4).Bordonaro M Role of Wnt signaling in the development of type 2 diabetes. Vitam. Horm 2009, 80, 563–581. [DOI] [PubMed] [Google Scholar]
- (5).Schinner S Wnt-signalling and the metabolic syndrome. Horm. Metab. Res 2009, 41, 159–163. [DOI] [PubMed] [Google Scholar]
- (6).Grant SF; Thorleifsson G; Reynisdottir I; Benediktsson R; Manolescu A; Sainz J; Helgason A; Stefansson H; Emilsson V; Helgadottir A; Styrkarsdottir U; Magnusson KP; Walters GB; Palsdottir E; Jonsdottir T; Gudmundsdottir T; Gylfason A; Saemundsdottir J; Wilensky RL; Reilly MP; Rader DJ; Bagger Y; Christiansen C; Gudnason V; Sigurdsson G; Thorsteinsdottir U; Gulcher JR; Kong A; Stefansson K Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet 2006, 38, 320–323. [DOI] [PubMed] [Google Scholar]
- (7).Gaulton KJ; Nammo T; Pasquali L; Simon JM; Giresi PG; Fogarty MP; Panhuis TM; Mieczkowski P; Secchi A; Bosco D; Berney T; Montanya E; Mohlke KL; Lieb JD; Ferrer J A map of open chromatin in human pancreatic islets. Nat. Genet 2010, 42, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stitzel ML; Sethupathy P; Pearson DS; Chines PS; Song L; Erdos MR; Welch R; Parker SC; Boyle AP; Scott LJ; NISC Comparative Sequencing Program, Margulies EH; Boehnke M; Furey TS; Crawford GE; Collins FS Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab. 2010, 12, 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lyssenko V; Lupi R; Marchetti P; Del Guerra S; Orho-Melander M; Almgren P; Sjögren M; Ling C; Eriksson KF; Lethagen AL; Mancarella R; Berglund G; Tuomi T; Nilsson P; Del Prato S; Groop L Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Invest 2007, 117, 2155–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Savic D; Ye H; Aneas I; Park SY; Bell GI; Nobrega MA Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. 2011, 21, 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Yang H; Li Q; Lee JH; Shu Y Reduction in Tcf7l2 expression decreases diabetic susceptibility in mice. Int. J. Biol. Sci 2012, 8, 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ip W; Shao W; Chiang YT; Jin T The Wnt signaling pathway effector TCF7L2 is upregulated by insulin and represses hepatic gluconeogenesis. Am. J. Physiol. Endocrinol. Metab 2012, 303, E1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shao W; Wang D; Chiang YT; Ip W; Zhu L; Xu F; Columbus J; Belsham DD; Irwin DM; Zhang H; Wen X; Wang Q; Jin T The Wnt signaling pathway effector TCF7L2 controls gut and brain proglucagon gene expression and glucose homeostasis. Diabetes 2013, 62, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Boj SF; van Es JH; Huch M; Li VS; José A; Hatzis P; Mokry M; Haegebarth A; van den Born M; Chambon P; Voshol P; Dor Y; Cuppen E; Fillat C; Clevers H Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 2012, 151, 1595–1607. [DOI] [PubMed] [Google Scholar]
- (15).Thompson MD; Moghe A; Cornuet P; Marino R; Tian J; Wang P; Ma X; Abrams M; Locker J; Monga SP; Nejak-Bowen K β-Catenin regulation of farnesoid X receptor signaling and bile acid metabolism during murine cholestasis. Hepatology 2018, 67, 955–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Popov VB; Jornayvaz FR; Akgul EO; Kanda S; Jurczak MJ; Zhang D; Abudukadier A; Majumdar SK; Guigni B; Petersen KF; Manchem VP; Bhanot S; Shulman GI; Samuel VT Second-generation antisense oligonucleotides against β-catenin protect mice against diet-induced hepatic steatosis and hepatic and peripheral insulin resistance. FASEB J. 2016, 30, 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kahn M Can we safely target the WNT pathway? Nat. Rev. Drug Discov 2014, 13, 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Polakis P Drugging Wnt signalling in cancer. EMBO J. 2012, 31, 2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zimmerman ZF; Moon RT; Chien AJ Targeting Wnt pathways in disease. Cold Spring Harb. Perspect. Biol 2012, 4, a008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wodarz A; Nusse R Mechanisms of Wnt signaling in development. Annu. Rev. Cell Dev. Biol 1998, 14, 59–88. [DOI] [PubMed] [Google Scholar]
- (21).Ohishi K; Toume K; Arai MA; Koyano T; Kowithayakorn T; Mizoguchi T; Itoh M; Ishibashi M 9-Hydroxycanthin-6-one, a β-carboline alkaloid from eurycoma longifolia, is the first Wnt signal inhibitor through activation of glycogen synthase kinase 3β without depending on casein kinase 1α. J. Nat. Prod 2015, 78, 1139–1146. [DOI] [PubMed] [Google Scholar]
- (22).Lu D; Choi MY; Yu J; Castro JE; Kipps TJ; Carson DA Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 13253–13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hao J; Ao A; Zhou L; Murphy CK; Frist AY; Keel JJ; Thorne CA; Kim K; Lee E; Hong CC Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gwak J; Hwang SG; Park HS; Choi SR; Park SH; Kim H; Ha NC; Bae SJ; Han JK; Kim DE; Cho JW; Oh S Small molecule-based disruption of the Axin/β-catenin protein complex regulates mesenchymal stem cell differentiation. Cell Res. 2012, 22, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huang SM; Mishina YM; Liu S; Cheung A; Stegmeier F; Michaud GA; Charlat O; Wiellette E; Zhang Y; Wiessner S; Hild M; Shi X; Wilson CJ; Mickanin C; Myer V; Fazal A; Tomlinson R; Serluca F; Shao W; Cheng H; Shultz M; Rau C; Schirle M; Schlegl J; Ghidelli S; Fawell S; Lu C; Curtis D; Kirschner MW; Lengauer C; Finan PM; Tallarico JA; Bouwmeester T; Porter JA; Bauer A; Cong F Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [DOI] [PubMed] [Google Scholar]
- (26).Henderson WR Jr.; Chi EY; Ye X; Nguyen C; Tien YT; Zhou B; Borok Z; Knight DA; Kahn M Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 14309–14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Toume K; Kamiya K; Arai MA; Mori N; Sadhu SK; Ahmed F; Ishibashi M Xylogranin B: a potent Wnt signal inhibitory limonoid from Xylocarpus granatum. Org. Lett 2013, 15, 6106–6109. [DOI] [PubMed] [Google Scholar]
- (28).Lepourcelet M; Chen YN; France DS; Wang HS; Crews P; Petersen F; Bruseo C; Wood AW; Shivdasani RA Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell 2004, 5, 91–102. [DOI] [PubMed] [Google Scholar]
- (29).Park HY; Toume K; Arai MA; Sadhu SK; Ahmed F; Ishibashi M Calotropin: a cardenolide from calotropis gigantea that inhibits Wnt signaling by increasing casein kinase 1α in colon cancer cells. Chembiochem 2014, 15, 872–878. [DOI] [PubMed] [Google Scholar]
- (30).García-Reyes B; Witt L; Jansen B; Karasu E; Gehring T; Leban J; Henne-Bruns D; Pichlo C; Brunstein E; Baumann U; Wesseler F; Rathmer B; Schade D; Peifer C; Knippschild U Discovery of inhibitor of Wnt Production 2 (IWP-2) and related compounds as selective ATP-competitive inhibitors of Casein Kinase 1 (CK1) δ/ε. J. Med. Chem 2018, 61, 4087–4102. [DOI] [PubMed] [Google Scholar]
- (31).Cha PH; Cho YH; Lee SK; Lee J; Jeong WJ; Moon BS; Yun JH; Yang JS; Choi S; Yoon J; Kim HY; Kim MY; Kaduwal S; Lee W; Min do S; Kim H; Han G; Choi KY Small-molecule binding of the axin RGS domain promotesβ-catenin and Ras degradation. Nat. Chem. Biol 2016, 12, 593–600. [DOI] [PubMed] [Google Scholar]
- (32).Liu J; Pan S; Hsieh MH; Ng N; Sun F; Wang T; Kasibhatla S; Schuller AG; Li AG; Cheng D; Li J; Tompkins C; Pferdekamper A; Steffy A; Cheng J; Kowal C; Phung V; Guo G; Wang Y; Graham MP; Flynn S; Brenner JC; Li C; Villarroel MC; Schultz PG; Wu X; McNamara P; Sellers WR; Petruzzelli L; Boral AL; Seidel HM; McLaughlin ME; Che J; Carey TE; Vanasse G; Harris JL Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 20224–20229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Madan B; Ke Z; Harmston N; Ho SY; Frois AO; Alam J; Jeyaraj DA; Pendharkar V; Ghosh K; Virshup IH; Manoharan V; Ong EH; Sangthongpitag K; Hill J; Petretto E; Keller TH; Lee MA; Matter A; Virshup DM; Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 2016, 35, 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mo ML; Li MR; Chen Z; Liu XW; Sheng Q; Zhou HM Inhibition of the Wnt palmitoyltransferase porcupine suppresses cell growth and downregulates the Wnt/β-catenin pathway in gastric cancer. Oncol. Lett 2013, 5, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lu W; Lin C; Roberts MJ; Waud WR; Piazza GA; Li Y Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS One 2011, 6, e29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Thorne CA; Hanson AJ; Schneider J; Tahinci E; Orton D; Cselenyi CS; Jernigan KK; Meyers KC; Hang BI; Waterson AG; Kim K; Melancon B; Ghidu VP; Sulikowski GA; LaFleur B; Salic A; Lee LA; Miller DM 3rd; Lee E Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol 2010, 6, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Shinoda K; Ohyama K; Hasegawa Y; Chang HY; Ogura M; Sato A; Hong H; Hosono T; Sharp LZ; Scheel DW; Graham M; Ishihama Y; Kajimura S Phosphoproteomics identifies CK2 as a negative regulator of beige adipocyte thermogenesis and energy expenditure. Cell Metab. 2015, 22, 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rossi M; Ruiz de Azua I; Barella LF; Sakamoto W; Zhu L; Cui Y; Lu H; Rebholz H; Matschinsky FM; Doliba NM; Butcher AJ; Tobin AB; Wess J CK2 acts as a potent negative regulator of receptor-mediated insulin release in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E6818–6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhong L; Ding Y; Bandyopadhyay G; Waaler J; Börgeson E; Smith S; Zhang M; Phillips SA; Mahooti S; Mahata SK; Shao J; Krauss S; Chi NW The PARsylation activity of tankyrase in adipose tissue modulates systemic glucose metabolism in mice. Diabetologia 2016, 59, 582–591. [DOI] [PubMed] [Google Scholar]
- (40).Smith TC; Kinkel AW; Gryczko CM; Goulet JR Absorption of pyrvinium pamoate. Clin. Pharmacol. Ther 1976, 19, 802–806. [DOI] [PubMed] [Google Scholar]
- (41).Zhu W; Groh M; Haupenthal J; Hartmann RW A detective story in drug discovery: elucidation of a screening artifact reveals polymeric carboxylic acids as potent inhibitors of RNA polymerase. Chemistry 2013, 19, 8397–8400. [DOI] [PubMed] [Google Scholar]
- (42).Baell JB; Holloway GA New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (43).Martino-Echarri E; Brocardo MG; Mills KM; Henderson BR Tankyrase inhibitors stimulate the ability of tankyrases to bind axin and drive assembly of β-catenin degradation-competent axin puncta. PLoS One 2016, 11, e0150484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Rowan AJ; Lamlum H; Ilyas M; Wheeler J; Straub J; Papadopoulou A; Bicknell D; Bodmer WF; Tomlinson IP APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 3352–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).de La Coste A; Romagnolo B; Billuart P; Renard CA; Buendia MA; Soubrane O; Fabre M; Chelly J; Beldjord C; Kahn A; Perret C Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 8847–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang YL; Guo H; Zhang CS; Lin SY; Yin Z; Peng Y; Luo H; Shi Y; Lian G; Zhang C; Li M; Ye Z; Ye J; Han J; Li P; Wu JW; Lin SC AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 2013, 18, 546–555. [DOI] [PubMed] [Google Scholar]
- (47).Behari J; Li H; Liu S; Stefanovic-Racic M; Alonso L; O’Donnell CP; Shiva S; Singamsetty S; Watanabe Y; Singh VP; Liu Q β-catenin links hepatic metabolic zonation with lipid metabolism and diet-induced obesity in mice. Am. J. Pathol 2014, 184, 3284–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Veeman MT; Slusarski DC; Kaykas A; Louie SH; Moon RT Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol 2003, 13, 680–685. [DOI] [PubMed] [Google Scholar]
- (49).Kolligs FT; Hu G; Dang CV; Fearon ER Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol. Cell. Biol 1999, 19, 5696–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Schreiber E; Harshman K; Kemler I; Malipiero U; Schaffner W; Fontana A Astrocytes and glioblastoma cells express novel octamer-DNA binding proteins distinct from the ubiquitous Oct-1 and B cell type Oct-2 proteins. Nucleic Acids Res. 1990, 18, 5495–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Wieckowski MR; Giorgi C; Lebiedzinska M; Duszynski J; Pinton P Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc 2009, 4, 1582–1590. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.