

Abstract
Mutations in the ATPase family 3‐like gene (AFG3L2) have been linked to autosomal‐dominant spinocerebellar ataxia type 28 and autosomal recessive spastic ataxia‐neuropathy syndrome. Here, we describe the case of a child carrying bi‐allelic mutations in AFG3L2 and presenting with ictal paroxysmal episodes associated with neuroimaging suggestive of basal ganglia involvement. Studies in skin fibroblasts showed a significant reduction of AFG3L2 expression. The relatively mild clinical presentation and the benign course, in spite of severe neuroimaging features, distinguish this case from data reported in the literature, and therefore expand the spectrum of neurological and neuroradiological features associated with AFG3L2 mutations.
INTRODUCTION
The ATPase family 3‐like gene (AFG3L2) encodes an ATP‐dependent proteolytic complex of the mitochondrial inner membrane that degrades misfolded proteins and regulates ribosome assembly.1, 2 AFG3L2 forms either homo‐ or hetero‐oligomeric complexes with SPG7, another m‐AAA protease whose mutations lead to a relatively common form of spastic ataxia.3 The SPG7‐AFG3L2 complex is involved in several pathways crucial for mitochondrial function, including mitochondrial protein quality control and homeostasis. Its impairment can lead to dysfunctions in mitochondrial protein synthesis, respiration, mitochondrial integrity and networking, axonal transport, as well as dysfunctions in Ca2+ flux and Ca2+‐induced cell death.4 Mutations in AFG3L2 have been associated with both autosomal dominant spinocerebellar ataxia type 28 (SCA28) and autosomal recessive spastic ataxia‐neuropathy syndrome (SPAX5).5 The clinical phenotype associated with variations in AFG3L2 was recently expanded with the description of two brothers showing oculomotor apraxia, extrapyramidal features and myoclonic epilepsy.1
The relationships found between different AFG3L2 mutations and distinct forms of the disease, different levels of severity and neuropathological correlates in mice indicate that these variants impact differentially on m‐AAA protease structure and activity. However, a lack of structural information has limited our understanding of the molecular mechanisms linking specific mutations to altered enzymatic activity and, ultimately, disease features.6
We here describe bi‐allelic mutations in AFG3L2 in a further child who showed ictal episodes of loss of balance and neuroimaging features suggestive of a mitochondrial disorder.
METHODS
This study was approved by the Tuscany Regional Pediatric Ethics Committee. All the procedures complied with the Helsinki Declaration of 1975. Total DNA purification, genetic studies and muscle, and skin punch biopsies were performed after parental written informed consent had been obtained.
Routine morphology, histochemical stains for oxidative metabolism, and spectrophotometric determination of respiratory chain (RC) complexes in muscle homogenate, and whole mtDNA sequencing were performed according to standard protocols. In the family trio, we performed a targeted multigene resequencing panel (SureSelect, Agilent, Santa Clara, CA) including 273 genes related to hereditary ataxias (Table S1) followed by whole‐exome sequencing (WES). using an Illumina (San Diego, CA) NextSeq500 platform, as reported elsewhere.7 The Ingenuity Variant Analysis suite (Qiagen, Hilden, Germany) was used for variant annotation according to an in‐house validated pipeline; the impact of mutations on AFG3L2 was determined in silico using eleven prediction tools.7 Gene variants were confirmed by Sanger sequencing, and segregation studies were performed in the healthy parents and sister.
Western blot (WB) analysis of cultured skin fibroblasts derived from the proband and from healthy controls was performed using the following antibodies: rabbit polyclonal anti‐AFG3L2 (Proteintech, Rosemont, IL), mouse monoclonal anti‐SPG7 (ThermoFisher Scientific, Waltham, MA), mouse monoclonal anti‐GAPDH (Abcam, Cambridge, UK), and rabbit polyclonal anti‐β‐tubulin (Cell Signaling Technology, Danvers, MA).
Structural changes in the mitochondrial network were evaluated by fluorescence microscopy after live staining of patient fibroblasts using MitoTracker Red (Invitrogen, Carlsbad, CA). Cells were cultured either in regular medium or with 5mM galactose in place of glucose as the primary source of energy to enhance mitochondrial metabolism. A treatment with the uncoupling agent carbonyl cyanide p‐(trifluoromethoxy) phenylhydrazone (FCCP) (4 h, 20 µmol/L) followed by 4 h in the initial medium was used to highlight the cells’ capacity to recover a normal mitochondrial network.
RESULTS
We describe the case of a 12‐year‐old boy with atypical history of episodic gait and speech disturbances. The proband is the first child of unrelated and apparently healthy parents. However, closer investigation of the family history disclosed symptoms suggestive of poor motor coordination (not better specified) in the 49‐year‐old father when he was a teenager. This man works as a puppeteer, and he occasionally complains of vague and diffuse muscle pain that he attributes to his work and frequent travels.
The proband had uneventful prenatal and perinatal periods and his psychomotor development was normal except for a history of clumsiness from infancy. At the age of 2 years, during a fever (38°C), he showed the first episode of a sudden loss of speech with psychomotor agitation, hypersalivation, pale extremities, loss of vision and incontinence. The episode resolved spontaneously within a few minutes without consequences; the parents did not seek medical attention, considering the event related to the high fever. Over the following five years, these symptoms recurred with a relapsing‐remitting course in which the acute episodes, triggered by high fever or mild exercise, were also accompanied by difficulty in movement planning and control, balance problems and reduced manual dexterity. During episodes, the child experienced apparent loss of consciousness, sweating, and occasionally dyspnea. Following the attack, he showed slurred speech, ataxic gait, and felt tired. An electroencephalogram (EEG), CT scan, and cardiac examinations performed occasionally in the emergency room were normal. Similar paroxysmal episodes were also triggered by heightened emotion; these lasted a few minutes during which the boy experienced impaired consciousness. Examined elsewhere, the child was deemed to be affected by a phobic anxiety disorder. Because of his clumsiness, he has never been able to engage in sports or in active play with his peers. Nonetheless, on several occasions, he has acted as puppeteer’s assistant during his father’s shows without experiencing any difficulty. Most recently, the child experienced an additional episode while playing in his first football match. During the game, he suddenly became pale, sweaty and unable to speak; he also presented unsteady gait. This episode lasted over thirty minutes without neurological consequences, and afterward the child had no recollection of it. An electrocardiogram and EEG monitoring near the episode and during exercise were unremarkable. Based on these features, we hypothesized this child suffered from a possible paroxysmal disorder with episodic ataxia and proposed the family to start 250 mg/die acetazolamide treatment. Therapy was associated with a reduction in frequency and intensity of these manifestations.
At the latest neurological examination (age 12), we observed clumsiness when running and jumping but not frank muscle weakness. Gait ataxia and trunk ataxia were seen but the examination was otherwise normal. The boy’s SARA score was 2/40 (gait). Ophthalmological, amino acid and organic acids in blood and urinary metabolic screening tests were all normal. Neurological examination of both parents was unremarkable. Cardiac examination and EEG were again normal.
Brain MRI in the proband at age 8 showed, bilaterally, the following findings: rounded T2‐weighted hyperintense signals in the mesial anterior region of the globi pallidi and in the region of the substantia nigra pars reticulate. T*‐weighted images showed punctiform hypointense signals in the anteroinferior portion of the globi pallidi with high magnetic susceptibility. Moreover, MRI showed enlarged fissures in comparison with normal foliae in the region of the superior vermis (Fig. 1). MR‐spectroscopy was normal. Neuroimaging studies at age 12 were unchanged. Muscle biopsy showed subsarcolemmal rims in some fibers on cytochrome C oxidase and succinate dehydrogenase stains. Biochemically, we observed normal RC activities.
Figure 1.
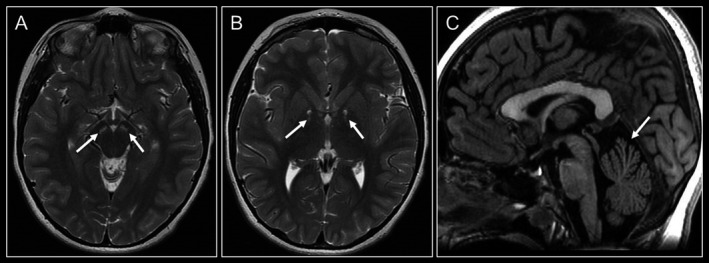
Brain MRI scans at the age of 8 years. (A‐B) Axial T2‐weighted imaging revealing, bilaterally, rounded T2‐weighted hyperintense signals in the region of the substantia nigra pars reticulata (A) and in the mesial anterior region of the globi pallidi (B) (arrows). (C) Sagittal T1‐weighted image shows enlarged fissures in comparison with normal foliae in the region of the superior vermis (arrow).
Target resequencing panel and WES studies identified the c.634dupG/p.Val212Glyfs*4 on the paternal allele and c.2167G> A/p.Val723Met on the maternal allele in AFG3L2 (NM_006796.2) (Fig 2A). We did not find other predictably or possibly pathogenic variants segregating in this family and with clinical significance. Whereas p.Val212Glyfs*4 is absent in gnomAD (https://gnomad.broadinstitute.org), p.Val723Met is present with a low frequency (1.78 E‐4) in the database where it is scored as predictably deleterious in silico. WB analysis demonstrated a significant reduction of AFG3L2 expression in cultured skin fibroblasts (Fig 2B). Structural changes in the mitochondrial network were seen upon the use of galactose as metabolic stressor: unlike control fibroblasts, the patient’s cells presented a predominantly fragmented mitochondrial reticulum, suggestive of a metabolic dysfunction under stress conditions (Fig. 3).
Figure 2.
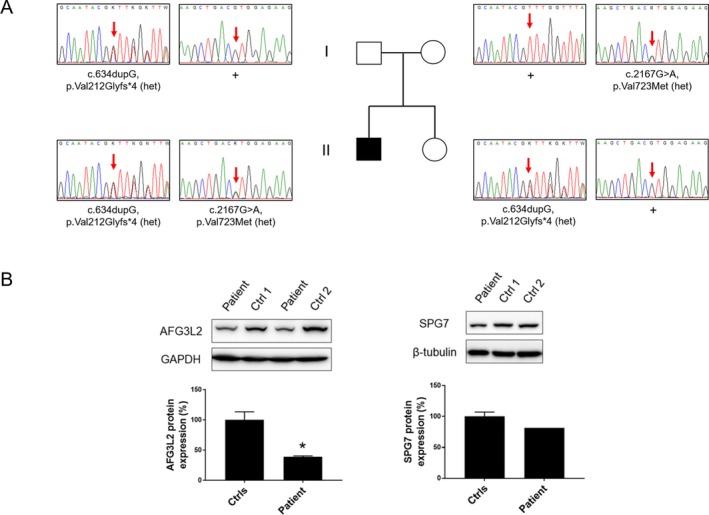
Genetic findings and Western blot analysis. (A) Pedigree of the family and electropherograms showing the segregation of the variants in the family members. (B) Western blot analysis revealed a significant reduction of AFG3L2 expression (left panel), whereas paraplegin levels were comparable with controls (right panel). Statistical analysis was performed using Student’s t‐test. *P < 0.05.
Figure 3.
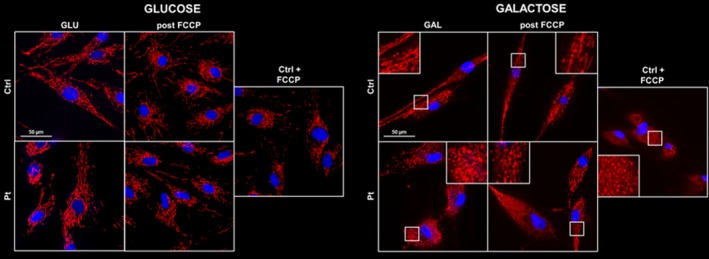
Mitochondrial network analyses in patient’s skin fibroblasts. Cells were cultured in regular glucose medium (left panel) or in galactose medium (right panel). In standard growth conditions, no differences in the mitochondrial network were detected between patient and control cells (left side). When patient cells were grown under stress conditions, they were clearly unable to recover a normal mitochondrial network after FCCP treatment, showing mostly a fragmented profile (right side). Control cells treated for 4 h with 20µM FCCP were used as evidence of mitochondrial fragmentation. Hoechst 33342 staining was used to visualize cell nuclei. Images were obtained using a Nikon Ti2‐E inverted microscope at 60 × magnification. Scale bar, 50 µm.
DISCUSSION
Heterozygous mutations in AFG3L2 cause SCA28 (MIM 610246), whereas bi‐allelic mutations in the same gene have been found in SPAX5;8 this latter condition is characterized by early‐onset spasticity resulting in significantly impaired ambulation, cerebellar ataxia, oculomotor apraxia, dystonia, and myoclonic epilepsy (MIM 614487). The mechanisms by which different mutations in AFG3L2 lead to distinct neurological phenotypes constitute an important, open question in the field. Recently, Puchades and colleagues6 described distinct biochemical impacts of different mutations in the m‐AAA AGF3L2 protease, suggesting that these differences might constitute the molecular basis for the different neurodegenerative conditions associated with mutations in AFG3L2. We here describe the case of a child with SPAX5 in whom severe MRI features suggesting a mitochondrial disorder are difficult to reconcile with his milder, fluctuating clinical course. Whilst the p.Val212Glyfs*4 predicts a severe impact on protein function, and can lead to nonsense‐mediated decay, the pathogenicity of the missense p.Val723Met mutation is less obvious, possibly on account of its localization in the proteolytic domain, which is essential for protein function and also the site where most SCA28 mutations reside.9 Remarkably, p.Val723Met was recently deemed disease causative8 when present in compound heterozygosity with the previously reported p.Tyr616Cys,5 a mutation already functionally characterized by others.10 SinceAFG3L2 homozygous missense mutations (i.e. p.Tyr616Cys) were shown to impair both the activity and formation of AFG3L2 homo‐oligomers and to affect the proper oligomerization with paraplegin5 without reduction of protein levels,10 we are tempted to speculate that deficits in the correct assembling of homo‐ and hetero‐oligomers, as seen in severe SPAX5 phenotypes, can have more dramatic effects rather than the simple reduction of the protein expression as observed in our patient. In line with previous reports,5, 8 we demonstrated that the combination of variants in our proband led to enhanced oxidative stress and mitochondrial network dysfunction when cells were opportunely stressed.
More than 30 different AFG3L2 disease‐causing mutations have thus far been reported in over 80 SCA28 patients,11, 12 whereas few SPAX5 families have been described.1, 6, 13 Interestingly, previously described patients with bi‐allelic mutations in AFG3L2 (Table 1) have severe phenotypes, including early‐onset spastic ataxia‐neuropathy syndrome or severe developmental delay and microcephaly, and often present progressive myoclonic epilepsy.1, 13 However, a recent report of a milder phenotype consisting of slowly progressive ataxia and associated with the p.Val723Met mutation8 suggests that this variant can be less aggressive, as seen in our patient.
Table 1.
Description of the main clinical, electroencephalographic, brain imaging, and genetic features recorded in SPAX5 cases harboring bi‐allelic mutations in AFG3L2.
| Pierson et al., 20115 | Eskandrani et al., 20171 | Muona et al., 201513 | Tunc et al., 20198 | This case | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Age at onset | 2 years | 8 months | 8 months | 8 months | 8 months | 8 months | 10 years | 10 years | 20 years | 2 years |
| Sex | M | F | M | F | M | F | M | F | M | M |
| Clinical features at last examination | Progressive motor degeneration, dysarthria, dysphagia | Severe development‐al delay. Microcephaly | Severe development‐al delay. Microcephaly | Severe development‐al delay. Microcephaly | Severe development‐al delay. Microcephaly | Severe development‐al delay. Microcephaly | SCA 28, spastic ataxia and severe myoclonic epilepsy | Progressively worsening myoclonus and ataxia | Limb and gait ataxia, dysarthria | Episodes triggered by exercise |
| Epilepsy | + | + | + | + | + | + | + | + | ‐ | ‐ |
| Electroencephalogram | Diffuse disorganization, F‐C spikes/waves | Hypsarrhyth‐mia | Hypsarrhyth‐mia | Consistent with myoclonic seizures | Hypsarrhyth‐mia | Hypsarrhyth‐mia | Data not available | Data not available |
Unremarkable alpha‐EEG |
Normal |
| Brain MRI | Moderate cerebellar atrophy | Bilateral involvement of putamina and caudate nuclei | Bilateral involvement of putamina | Bilateral involvement of putamina | Bilateral involvement of putamina |
Bilateral involvement of putamina and caudate nuclei |
Data not available | Data not available | Cerebellar atrophy | Bilateral involvement of globi pallidi, pars reticulata of the substantia nigra |
| AFG3L2 mutation |
c.1847G.A/ pTyr616Cy (homozygous) |
c.1714G> A/ p.Ala572Thr (homozygous) |
c.1714G> A/ p.Ala572Thr (homozygous) |
c.1714G> A/ p.Ala572Thr (homozygous) |
c.1714G> A/ p.Ala572Thr (homozygous) |
c.1714G> A/ p.Ala572Thr (homozygous) |
c.1875G> A/ p.Met625Ile (homozygous) |
c.1875G> A/ p.Met625Ile (homozygous) |
c.2167G> A/ p.Val723Met; c.1847A> G/ p.Tyr616Cys (comppund heterozyg.) |
c.634dupG/ p.Val212Glyfs*4; c.2167G> A/ p.Val723Met (compound heterozygous) |
Bold typesetting indicate features observed in the patient described in this work.
A final consideration emerges upon revisiting brain imaging data of patients with recessive AFG3L2 mutations. When available, brain MRI revealed multiple abnormalities, including symmetrical hyperintense signals in the basal ganglia, mostly the putamina and caudate nuclei, associated with severe and rapid neurodegeneration. Cerebellar atrophy seems common, but can be mild or barely detectable.1 Neuroimaging in two patients with p.Val723Met6 showed cerebellar atrophy, whereas the basal ganglia were spared. The new case of SPAX5 described in the present report corroborates the impression that a wide spectrum of features, including iron deposition in the basal ganglia, can be attributed to m‐AAA protease defects. Taken together our data suggest that some missense variants may have hypomorphic effects. If this is the case, our patient’s relatively mild clinical phenotype might be associated with abnormal mitochondrial morphology and inefficient oxidative phosphorylation activity, but the actual cause of the neuronal loss is not known.
Conflicts of interest
None.
Supporting information
Table S1. Targeted resequencing panel gene list used in this study.
Acknowledgments
We thank Catherine J. Wrenn for expert editorial assistance and useful discussion. This study was partially supported by the Italian Ministry of Health‐Ricerca Finalizzata RF‐2016‐02361610 (to FMS), and the E‐RARE‐3 Joint Transnational Call grant “Preparing therapies for autosomal recessive ataxias” (PREPARE) (MoH; project 3398 to FMS).
Funding information
This study was partially supported by the Italian Ministry of Health‐Ricerca Finalizzata RF‐2016‐02361610 (to FMS), and the E‐RARE‐3 Joint Transnational Call grant “Preparing therapies for autosomal recessive ataxias” (PREPARE) (MoH; project 3398 to FMS).
Funding Statement
This work was funded by Italian Ministry of Health grant Ricerca Corrente 5X1000.
References
- 1. Eskandrani A, Al Hashem A, Ali ES, et al. Recessive AFG3L2 mutation causes progressive microcephaly, early onset seizures, spasticity, and basal ganglia involvement. Pediatr Neurol 2017;71:24–28. [DOI] [PubMed] [Google Scholar]
- 2. Edener U, Wöllner J, Hehr U, et al. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four‐generation family with a novel AFG3L2 mutation. Eur J Hum Genet 2010;18:965–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Gassen KL, van der Heijden CD, de Bot ST, et al. Genotype‐phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain 2012;135:2994–3004. [DOI] [PubMed] [Google Scholar]
- 4. Patron M, Sprenger HG, Langer T. m‐AAA proteases, mitochondrial calcium homeostasis and neurodegeneration. Cell Res 2018;28:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pierson TM, Adams D, Bonn F, et al. Whole‐exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia‐neuropathy syndrome linked to mitochondrial m‐AAA proteases. PLoS Genet 2011;7:e1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Puchades C, Ding B, Song A, et al. Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol Cell 2019;75(5):1073–1085.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lieto M, Riso V, Galatolo D, et al. The complex phenotype of spinocerebellar ataxia type 48 in eight unrelated Italian families. Eur J Neurol. 2020;27:498–505. 10.1111/ene.14094. [DOI] [PubMed] [Google Scholar]
- 8. Tunc S, Dulovic‐Mahlow M, Baumann H, et al. Spinocerebellar ataxia type 28 ‐ phenotypic and molecular characterization of a family with heterozygous and compound‐heterozygous mutations in AFG3L2. Cerebellum 2019;18:817–822. [DOI] [PubMed] [Google Scholar]
- 9. Charif M, Roubertie A, Salime S, et al. A novel mutation of AFG3L2 might cause dominant optic atrophy in patients with mild intellectual disability. Front Genet 2015;6:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tulli S, Del Bondio A, Baderna V, et al. Pathogenic variants in the AFG3L2 proteolytic domain cause SCA28 through haploinsufficiency and proteostatic stress‐driven OMA1 activation. J Med Genet 2019;56:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Di Bella D, Lazzaro F, Brusco A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet 2010;42:313–321. [DOI] [PubMed] [Google Scholar]
- 12. Szpisjak L, Nemeth VL, Szepfalusi N, et al. Neurocognitive characterization of an SCA28 family caused by a novel AFG3L2 gene mutation. Cerebellum 2017;16:979–985. [DOI] [PubMed] [Google Scholar]
- 13. Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Targeted resequencing panel gene list used in this study.