

Abstract
Purpose
Malignant glioma (MG) is the most deadly primary brain cancer. Signaling though the PI3K/AKT/mTOR axis is activated in most MGs and therefore a potential therapeutic target. The mTOR inhibitor temsirolimus and the AKT inhibitor perifosine are each well‐tolerated as single agents but with limited activity reclinical data demonstrate synergistic anti‐tumor effects from combined treatment. Therefore, we initiated a phase I trial of combined therapy in recurrent MGs to determine safety and a recommended phase II dose.
Methods
Adults with recurrent MG, Karnofsky Performance Status ≥ 60 were enrolled, with no limit on the number of prior therapies. Temsirolimus dose was escalated using standard 3 + 3 design from 15 mg to 170 mg administered once weekly. Perifosine was fixed as a 600 mg load on day 1 followed by 100 mg nightly (single agent MTD) until dose level 7 when the load increased to 900 mg.
Results
We treated 35 patients with with glioblastoma (17) or other MGs (18; including nine anaplastic astrocytoma, nine anaplastic oligodendroglioma, one anaplastic oligoastrocytoma, and two low grade astrocytomas with radiographic transformation to MG). We observed five dose‐limiting toxicities (DLTs): one at dose level 3 (50mg temsirolimus), then two at dose level 7 expansion (170 mg temsirolimus), and then two more at dose level 6 expansion (170 mg temsirolimus). DLTs included thrombocytopenia (n = 3), intracerebral hemorrhage (n = 1) and lung infection (n = 1).
Conclusion
Combining the mTOR inhibitor temsirolimus dosed at 115 mg weekly and the AKT inhibitor perifosine dosed at 100 mg daily (following 600 mg load) is tolerable in heavily pretreated adults with recurrent MGs.
Introduction
Glioblastoma (GBM) is the most common primary brain tumor in adults. 1 Following initial therapy, nearly all patients eventually develop recurrent or progressive disease, and multiple clinical trials of novel agents failed to improve median survival of approximately 8 months. 2 , 3 , 4
Signaling through the phosphatidylinositol 3‐kinase (PI3K) axis (Figure1) is important in glioma biology. For example, approximately 70% of GBMs exhibit abnormally active PI3K signaling, 5 , 6 often resulting from loss or deactivating mutation of the tumor suppressor gene phosphatase and tensin homolog on chromosome 10 (PTEN). 7 Therefore, the PI3K/AKT/mTOR cascade is a compelling therapeutic target.
Figure 1.
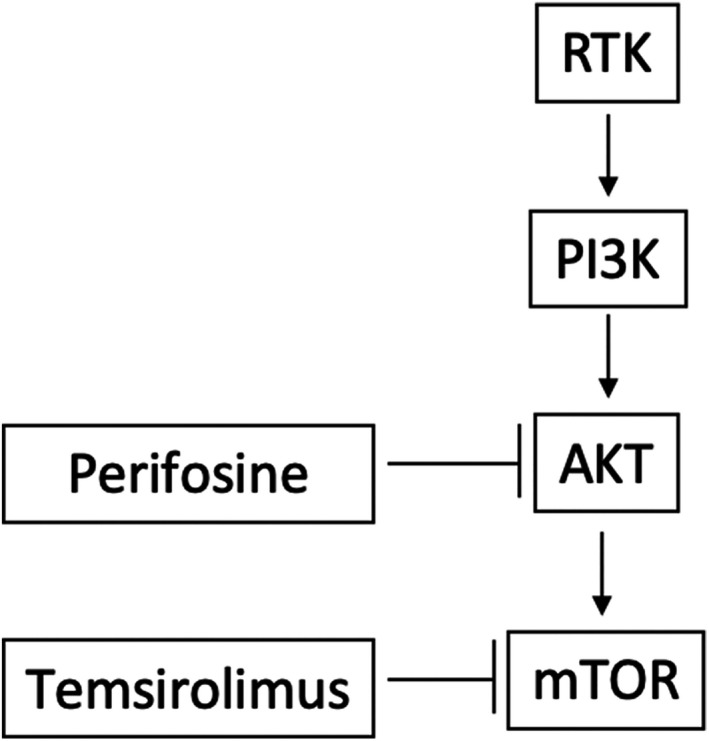
Signaling inhibition by perifosine and temsirolimus. A receptor tyrosine kinase (RTK) such as epidermal growth factor receptor activates phosphotidylinositol 3‐kinase (PI3K), leading to AKT activation, which activates mammalian Target of Rapamycin (mTOR). AKT and mTOR increase tumor cell invasion and proliferation, and reduce apoptosis. Perifosine inhibits AKT and temsirolimus inhibits mTOR. Arrows ending with pointed heads (→→) indicate activation and arrows ending in perpendicular lines (|) indicate inhibition.
However, mTOR inhibitors such as temsirolimus are generally ineffective as a single agents. 8 , 9 Possible explanations for their limited efficacy include the failure to inhibit AKT effectors independent of mTOR, induction of AKT activation by feedback loops as observed in other cancers, 10 , 11 , 12 and inadequate molecular selection. 12
Perifosine is an oral alkylphospholipid, a novel class of antineoplastics with distinct mechanisms and toxicities than other drugs. Topically applied miltefosine, another alkylphospholipids that occurs naturally, has efficacy against cutaneous metastatic cancer. However, it is intolerable when administered systemically. Accordingly, other alkylphospholipids with less systemic toxicity were synthesized, such as perifosine which inhibits AKT signaling in transformed glia upstream of mTOR. 13 , 14 , 15 , 16
Therefore, perifsoine has the theoretical advantage over temsirolimus or other mTOR inhibitors of avoiding feedback induced activation of AKT. However, despite inhibiting AKT, it leaves mTOR uninhibited. 16 Therefore, combined perifosine and temsirolimus is a compelling approach to shut down signaling through the PI3K axis both upstream of mTOR by perifosine and downstream by temsirolimus. In addition, perifosine inhibits other signal transduction cascades; these effects, such as on the RAS pathway, are also of interest and potentially therapeutic as RAS is activated in nearly all GBMs. 6 , 17
Preclinical experiments with genetically engineered mouse models of GBM demonstrate that combined temsirolimus + perifosine is synergistically efficacious. For example, temsirolimus inhibits mTOR (as evidenced by reducing pS6RP) and arrests tumor cell proliferation but does not cause any meaningful necrosis. Perifosine inhibits PI3K (as evidenced by reduced pAKT); however, perifosine causes regional necrosis in only a minority of tumors (approximately 40%) rather than pan‐tumor necrosis in all mice, and surviving tumor cells continue to proliferate. However, when combined, temsirolimus + perifosine synergistically induce massive intratumoral necrosis, and surviving cells undergo proliferation arrest. 18
As a necessary first step in the translation of our extensive preclinical data to humans, we conducted a human trial of perifosine alone before combination therapy studies because no toxicity or efficacy data existed for perifosine in glioma. As preclinical activity with perifosine alone was modest, we did not anticipate robust efficacy in humans, and indeed, 0 of the first 12 treated patients with recurrent GBMs responded. However, we intriguingly observed radiographic improvements in patients with other anaplastic gliomas. 19 In addition, with the exception of medically manageable gastrointestinal upset, temsirolimus and perifosine each have toxicities that are mainly nonoverlapping which could allow for the safe administration of both drugs concurrently. Therefore, we conducted a phase I human trial of temsirolimus combined with perifosine for recurrent GBM or other MG to determine the safety and tolerability of the regimen and to define a recommended phase II dose.
Patients and Methods
This was a prospective, open label, phase I trial that enrolled patients at Memorial Sloan Kettering Cancer Center and was sponsored by the National Cancer Institute (NCI), with drug supply from the Cancer Therapy Evaluation Program (local IRB #09‐058, NCT01051557, CTEP protocol #8249). All patients or legally authorized representatives signed an informed consent form that was approved by the Memorial Sloan Kettering Cancer Center Institutional Review Board prior to enrollment. The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and appropriate amendments.
Eligibility
Eligible subjects were least 18 years old with a Karnofsky Performance Status at least 60 with a diagnosis of recurrent/progressive GBM or other malignant glioma. Prior radiotherapy and temozolomide were mandatory, but there were otherwise no limitations on the number or types of prior therapies. Recovery from prior therapy following typical washout periods was required (6 weeks from radiotherapy and nitrosoureas, 4 weeks from temozolomide, 4 weeks from bevacizumab or other antiangiogenic therapy, 1 week from non‐cytotoxic agents, etc.), as were normal renal (defined as creatinine < 1.5 mg/dL), hepatic (defined as SGOT, SGPT, and bilirubin < 2 times the upper limit of normal), and bone marrow function (defined as WBC ≥ 2000/µL, ANC ≥ 1500/mm3, platelet count ≥ 100,000/mm3, and hemoglobin ≥ 10gm/dL). Total cholesterol ≤ 350 mg/dL and triglycerides ≤ 400 mg/dL were also required because temsirolimus can exacerbate or induce hyperlipidemia. Patients taking an enzyme inducing anti‐epileptic drug (EIAED, defined as phenytoin, fosphenytoin, phenobarbital, primidone, carbamazepine, and oxcarbamazepine) within 2 weeks before protocol therapy were excluded because of potential interactions on the hepatic p450 system, as is typical in trials of p450 drugs for patients with glioma. Women of child bearing potential and men who were unwilling to adhere to mandatory contraception were also excluded, as were pregnant or breast‐feeding women.
Treatment
All patients received both temsirolimus and perifosine. There was with no intrapatient dose escalation and no placebo was administered (Table1). Treatment was intended to continue until progressive disease per Macdonald criteria 20 by brain MRI every other cycle (progression defined as at least 25% increase in cross‐sectional area of enhancing tumor, or any new tumor, or neurologically worse, and corticosteroids stable or increased; partial response defined as at least 50% improvement confirmed at least 1 month later with stable or decreasing corticosteroids and neurologically stable or improved; this trial was initiated before the Response Assessment in Neuro‐Oncology, i.e., “RANO” criteria 21 were established), or until unacceptable toxicity which was evaluated using the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
Table 1.
Dose escalation scheme and patients treated per level
| Dose level | Temsirolimus in mg weekly | Perifosine load (day 1)/maintenance (starting day 2) in mg daily | Number of patients | Dose Limiting Toxicity |
|---|---|---|---|---|
| ‐6 | 5 | 150/50 | 0 | Not applicable |
| ‐5 | 10 | 150/50 | 0 | Not applicable |
| ‐4 | 15 | 150/50 | 0 | Not applicable |
| ‐3 | 15 | 150/100 | 0 | Not applicable |
| ‐2 | 15 | 300/100 | 0 | Not applicable |
| ‐1 | 15 | 450/100 | 0 | Not applicable |
| 1 (starting) | 15 | 600/100 | 4 1 | 1 (Prolonged grade 2 thrombocytopenia) |
| 2 | 25 | 600/100 | 3 | 0 |
| 3 | 50 | 600/100 | 6 | 0 |
| 4 | 75 | 600/100 | 3 | 0 |
| 5 | 115 | 600/100 | 7 1 | 0 |
| 6 | 170 | 600/100 | 6 |
2 (grade 3 thrombocytopenia; grade 2 intracerebral hemorrhage) |
| 7 | 170 | 900/100 | 6 | 2 (grade 4 thrombocytopenia; grade 2 lung infection) |
includes replacement patient at dose levels 1 and 5.
For the starting dose of perifosine (given orally), we used the recommended single‐agent phase II dose used in glioma 19 and other cancers consisting of a 600 mg load (in four divided doses of 250 mg each) on day 1 followed by 100 mg daily maintenance. We chose this regimen because we found this that it was relatively well‐tolerated in patients with high grade gliomas as we reported previously. 19 We also used this regimen because results of prior studies standardized treatment with a loading dose to achieve serum steady date (in the context of a half‐life exceeding 100 h) followed by daily treatment with 15%–20% of the loading dose 22 (17% in this study) as we described previously. 19 This starting dose of perifosine was the same for all patients except those at the highest dose levels (900 mg load, Table1).
We escalated the starting dose of temsirolimus on day 1 through a typical 3 x 3 design. In this manner, patients in the first cohort received temsirolimus 15 mg intravenously once weekly, and the highest prespecified dose level involved temsirolimus 170 mg weekly. Prior studies suggesting that doses above 25 mg would be intolerable when combined with other signal transduction inhibitors. 23 , 24 In addition, the FDA‐approved temsirolimus dose for renal cell carcinoma is 25 mg weekly. 25 Therefore, to be conservative, we started at 15 mg weekly with a plan to escalate in subsequent dose levels, or de‐escalate if overly toxic (Table1). Pharmacokinetic analyses were not conducted.
A cycle was defined as 28 days despite continuous treatment with weekly temsirolimus and daily perifosine. During cycle 1, patients were assessed for toxicity weekly both clinically and by laboratory evaluations. After cycle 1, assessments occurred biweekly. Finally, intracranial hemorrhages were observed in mice treated with combined temsirolimus and perifosine. 18 Therefore, a CT head or MRI brain was conducted during cycle 1 week 2 to evaluate for bleeding as a precaution with a resulting treatment hold/discontinuation if needed for patient safety.
Statistical analyses
A dose‐limiting toxicity (DLT) was defined as any of the following attributed as at least possibly related to temsirolimus, or perifosine, or the combination during the first 4‐week cycle of treatment: grade 3 thrombocytopenia, grade 4 anemia or neutropenia, unacceptable grade 2 non‐hematologic toxicity resulting in a treatment delay of> 7 days despite maximal medical treatment, grade 3 or 4 nonhematologic toxicity (except diarrhea and mucositis unless persisting despite maximal medical treatment), or failure to recover from toxicities to be eligible for retreatment within 14 days of last dose. Patients removed during cycle 1 for reasons other than toxicity were replaced.
The Maximum Tolerated Dose (MTD) was defined as the dose at which fewer than one‐third of patients experienced a DLT through a typical "3 + 3” dose escalation trial design. Cohorts treated at a dose level anticipated to be defined as the MTD would be expanded to accrue at least six patients. Finally, if the first dose level exceeded the MTD, then the dose would be de‐escalated in subsequent patients (Table1).
Efficacy was explored by progression‐free survival (PFS) and overall survival using the Kaplan–Meier method from start of treatment, as well as radiographic responses, 20 descriptively without prespecified goals or plans for comparison against historical controls.
Results
Patient characteristics
There were 35 patients enrolled (Table2), including two replaced for nonadherence to the protocol requirements and not evaluable for DLT (one patient was taking a contraindicated medication prescribed by her naturalist unbeknownst to investigators; one patient had a pre‐existing unrelated dental issue for which she decided to pursue treatment and hold drug for during cycle 1). Patients were recruited from February 2010 to November 2012. All 35 who received at least one dose of study treatment are, therefore, included in the toxicity analysis. Six patients were not evaluable for radiographic response because of discontinuation for nonadherence (n = 1) or toxicity during cycle 1 (n = 2, one each with grade 4 thrombocytopenia or grade 2 intracranial hemorrhage), or withdrawal of consent (n = 3) before reimaging. Four additional patients consented to trial but never received any study interventions and are excluded from all analyses.
Table 2.
Patient characteristics
| Characteristic | Evaluable Patients (n = 35) |
|---|---|
| Gender | |
| Men | 25 (76%) |
| Women | 10 (24%) |
| Age: median (range) | 52 (21–71) |
| Karnofsky Performance Status: median (range) | 80 (60–100) |
| Histology | |
| Glioblastoma | 17 (52%) |
| Anaplastic Astrocytoma | 9 (27%) |
| Anaplastic Oligodendroglioma | 6 (15%) |
| Anaplastic Mixed Glioma | 1 (3%) |
| Low‐grade Astrocytoma with radiographic evidence of transformation to high‐grade | 2 (3%) |
| Prior radiotherapy | 35 (100%) |
| Median number of prior chemotherapy regimens (range) | 2 (1–7) |
| Prior bevacizumab treatment | 19 (54%) |
| Isocitrate dehydrogenase (IDH) status | |
| Mutant | 3 |
| Wild‐type | 6 |
| Not available | 26 |
| 1P/19Q | |
| Codeleted | 7 |
| Neither deleted | 4 |
| Not available | 24 |
| MGMT promotor | |
| Methylated | 4 |
| Unmethylated | 12 |
| Not available | 19 |
Toxicity
There no DLTs at dose levels 1 or 2. At dose level 3 there was 1 DLT (prolonged grade 2 thrombocytopenia) but no additional DLTs were observed in three subsequent patients treated at level 3, nor any DLTs at dose levels 4‐6. However, at dose level 7, there were two DLTs among six subjects (grade 4 thrombocytopenia and grade 3 lung infection), resulting in de‐escalation for three additional subjects and expansion of dose level 6 among whom 2 DLTs were then observed (grade 3 thrombocytopenia and grade 2 intracerebral hemorrhage). In sum, 2/6 subjects experienced a DLT at dose level 6, and 2/6 also at level 7; therefore, the MTD was defined as dose level 5 at which no subjects (0/3 initially followed by 0/3 subsequent patients in an expansion group) experienced DLTs. Of the five patients who experienced DLT, two were on dexamethasone at the time.
Notable grade 3‐4 toxicities (Table 3) included lymphopenia and five cases of lung infections, including pneumocystis (jiroveci) pneumonia (PJP) in three. Of note, in the three patients with PJP, all three were on dexamethasone at the time and absolute lymphocyte counts were 0.4, 1.2, and 1.8 K/mcL. Prominent grade 1‐2 toxicities included intracranial hemorrhage (two grade 1, one grade 2), oral mucositis (16), hypertriglyceridemia (25), diarrhea (21), thrombocytopenia (31), leucopenia (18), nausea (9), pneumonitis (3), and thromboembolism (2). There were no treatment related deaths.
Table 3.
Grade 1‐4 toxicities per patient deemed possibly, probably, or definitely related to protocol therapy in patients who received at least one dose of drug (n = 35) using the NCI Common Terminology Criteria for Adverse Events version 4
| Toxicity |
Grade 1 N (%) |
Grade 2 N (%) |
Grade 3 N (%) |
Grade 4 N (%) |
|---|---|---|---|---|
| Alanine aminotransferase increased | 14 (40) | 9 (26) | 4 (11) | |
| Alkaline phosphatase increased | 8 (23) | 1 (3) | ||
| Alopecia | 1 (3) | |||
| Anemia | 5 (14) | 2 (6) | 2 (6) | |
| Anorexia | 2 (6) | |||
| Anxiety | 1 (3) | 1 (3) | ||
| Aspartate aminotransferase increased | 19 (54) | 2 (6) | 2 (6) | |
| Bilirubin increased | 6 (17) | 2 (6) | ||
| Cholesterol increased | 20 (57) | 12 (34) | ||
| Constipation | 6 (17) | |||
| Cough | 2 (6) | |||
| CPK increased | 1 (3) | |||
| Diarrhea | 15 (43) | 6 (17) | ||
| Dry skin | 1 (3) | |||
| Dysgeusia | 3 (9) | |||
| Dyspepsia | 3 (9) | 2 (6) | ||
| Dyspnea | 1 (3) | 2 (6) | ||
| Edema, limbs | 1 (3) | |||
| Epistaxis | 2 (6) | |||
| Erythema multiforme | 1 (3) | |||
| Fatigue | 12 (34) | 9 (26) | ||
| Fever | 1 (3) | |||
| Headache | 3 (9) | |||
| Hypercholesterolemia | 2 (6) | |||
| Hyperglycemia | 17 (49) | 6 (17) | 9 (26) | |
| Hyperkalemia | 3 (9) | 1 (3) | ||
| Hypernatremia | 11 (31) | |||
| Hypertriglyceridemia | 12 (34) | 13 (37) | 2 (6) | |
| Hypoalbuminemia | 28 (80) | |||
| Hypocalcemia | 12 (34) | 2 (6) | ||
| Hypoglycemia | 1 (3) | 1 (3) | ||
| Hypokalemia | 14 (43) | 2 (6) | ||
| Hypomagnesemia | 1 (3) | |||
| Hyponatremia | 12 (34) | |||
| Hypophosphatemia | 1 (3) | 14 (43) | 5 (14) | |
| INR increased | 3 (9) | |||
| Insomnia | 2 (6) | |||
| Intracranial hemorrhage | 2 (6) | 1 (3) | ||
| Leukopenia | 9 (26) | 9 (26) | 1 (3) | |
| Lung infection | 1 (3) | 1 (3) | 5 (14) | |
| Lymphopenia | 2 (6) | 11 (31) | ||
| Mucositis ‐ anal | 1 (3) | 1 (3) | ||
| Mucositis – oral | 9 (26) | 7 (20) | 2 (6) | |
| Myalgia | 1 (3) | |||
| Nausea | 6 (17) | 3 (9) | ||
| Neutropenia | 1 (3) | 5 (14) | 1 (3) | |
| Oral pain | 3 (9) | |||
| Paresthesia | 2 (6) | |||
| Pneumonitis | 1 (3) | |||
| Rash – acneiform | 17 (49) | 1 (3) | ||
| Rash – maculopapular | 6 (17) | 3 (9) | 1 (3) | |
| Rash – papulopustular | 2 (6) | |||
| Skin ulceration | 2 (6) | 1 (3) | ||
| Skin infection | 4 (11) | |||
| Thrombocytopenia | 27 (77) | 2 (6) | 1 (3) | |
| Thromboembolic event | 2 (6) | 2 (6) | ||
| Tooth infection | 1 (3) | 1 (3) | ||
| Upper respiratory infection | 1 (3) | |||
| Vomiting | 3 (9) | 3 (9) |
Exploratory evaluation of efficacy
Among the 29 evaluable for radiographic response, the best response was partial response in 1 (dose level 5) and stable disease in 13, and progressive disease in 15. Of note, there were two patients with formally designated stable disease who had reduction in tumor size by> 50% but discontinued treatment for toxicity at dose level 7 before a confirmatory MRI. Amongst all 35 patients, only two patients were still alive at the time of last follow‐up. Median OS was 10.4 months [95% CI (7.2, 16.7)], and median PFS was 2.7 months [95%CI: (1.8, 9.2)]. Median follow‐up amongst survivors was 8.9 months.
To explore efficacy further, we also evaluated the five bevacizumab‐naive subjects with GBM who were treated at or above the MTD (level 5, temsirolimus 115 mg weekly and perifosine 600mg on day 1 then 100 mg daily thereafter; Table 1). Among these five, there was one partial response durable for over 4.5 years. Intriguingly, next generation sequencing of the pretreatment archival tumor tissue in this patient demonstrated only PTEN loss; Isocitrate dehydrogenase (IDH) mutation was absent, and MGMT was unmethylated. He had undergone surgery demonstrating recurrence (rather than pseudoprogression), and prior treatment included failure of a PI3K inhibitor. There were also two patients each with stable and progressive disease as best response.
Discussion
In this trial we combined two drugs with mainly nonoverlapping toxicities designed to inhibit different activities within the PI3K/AKT/mTOR signal transduction cascade, perifosine which inhibits AKT and temsirolimus which inhibitors mTOR. Although each drug has minimal clinical efficacy in GBM as a single agent, combination therapy was synergistic in preclinical experiments. 18 However, no prior trial combined these agents together.
Toxicity was severe at the highest dose levels, with many patients experiencing common hematologic and treatable metabolic toxicities (i.e., hypophosphatemia, hypertriglyceridemia, hypercholesterolemia). However, a higher than expected incidence of other toxicities was seen (Table3). For example, three patients experienced intracerebral hemorrhage, although all were grade 1 or 2. In addition, five experienced lung infections, of which three were determined to be pneumocystis (jiroveci) pneumonia (PJP). PJP risk could have been exacerbated by lymphopenia; in addition, all three patients were receiving concurrent corticosteroids.
Our phase I results suggested that the MTD of the combination was temsirolimus 115 mg weekly with PRF loaded at 600 mg on day 1 (in 4 divided doses) followed by daily 100 mg thereafter. Notably, this is more than 4X the FDA‐approved dose of temsirolimus monotherapy for renal cell carcinoma (25 mg weekly).
It is unclear why higher temsirolimus doses were tolerable in this study, particularly when combined with another agent. We speculate that corticosteroids, a potential p450 stimulator given commonly to patients with brain tumors and in this trial administered concurrently in 17 (49%) subjects, could have contributed to increased tolerability. Others also found that temsirolimus 170 mg or 250 mg weekly is tolerable as a single agent in patients not taking or taking EIAEDs respectively. 8 , 9 , 26 However, pharmacokinetic analyses were not conducted to test the impact of corticosteroids or other concurrent medications on serum drug levels. Lack of pharmacokinetic analyses also limits our ability to draw any conclusions on the efficacy of the regimen as well.
In summary, this phase I trial declared an MTD of combined temsirolimus with perifosine, and responses were anecdotally observed, particularly at the higher dose levels. However, the large gap between temsirolimus doses (115mg weekly as the MTD and 170mg weekly as the next higher level) did not allow interrogation of intermediate levels between that may be as efficacious but more tolerable. Therefore, we are conducting a subsequent pilot study (NCT 02238496) combining temsirolimus at 140mg weekly with perifosine that also mandates PJP prophylaxis based on the results we reported here.
Conflict of Interest
TJK: research funding (outside the submitted work) from Merck, Ludwig, Eli‐Lilly. KSP: stock ownership (outside the submitted work) in Pfizer, Johnson and Johnson, Viking Therapeutics and Catalyst Biotech. EIP: none. IKM: research funding (outside the submitted work) from Agios, Amgen. CN: none. IG: none. LMD: none. LEA: became an employee of Novartis Oncology during the study. ECH: none. AO: none. MEL: consultant/speaking role (outside the submitted work) with Legacy Healthcare Services, Adgero, Amryt, Celldex, Debiopharm, Galderma, Johnson and Johnson, Novocure, Lindi, Merck Sharp and Dohme, BMS, Helsinn, Janssen, Menlo, Novartis, F. Hoffmann‐La Roche AG, AbbVie Inc, Boehringer Ingelheim, Allergan, Amgen, E.R. Squibb & Sons LLC, EMD Serono, AstraZeneca, Genentech, Leo, Seattle Genetics, Bayer, Lutris, Pierre Fabre, Paxman Coolers, Adjucare, Dignitana, Biotechspert, Teva, Parexel, OnQuality, Novartis, Harborside, Wiley, Azitra, NCODA and Takeda Millenium; research funding from Berg, Lutris, Paxman, Novocure, US Biotest, and Veloce. EL: none. ABL: received during the last year (all outside the submitted work): personal fees and nonfinancial support from Orbus, NW Biotherapeutics, Agios; grants, personal fees and nonfinancial support from Karyopharm, and AbbVie; grants and non‐financial support from Oncoceutics, Amgen, Millienium, Celldex, Novartis, Pfizer, Beigene, and VBI Vaccines, nonfinancial support from Tocagen, BMS; and personal fees from prIME Oncology, Sapience, Physicians' Education Resource/Chemotherapy Foundation Symposium, and from Bioclinica as an expert blinded independent reviewer of clinical and imaging data for a BMS‐sponsored clinical trial.
Funding Information
NCI‐Cancer Therapy Evaluation Program, NIH/NCI U01CA069856; Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of Memorial Sloan Kettering Cancer Center (MSKCC); Society for Memorial Sloan Kettering; Voices Against Brain Cancer; Brain Tumor Center of MSKCC; and AOI/Keryx Pharmaceuticals (as legacy to Aeterna Zentaris for development of perifosine). In addition, this work was supported by the MSKCC NIH/NCI Cancer Center Support Grant (P30CA008748).
Funding Statement
This work was funded by NIH/NCI Cancer Center Support Grant grant P30CA008748; NCI‐Cancer Therapy Evaluation Program grant NIH/NCI U01CA069856.
Contributor Information
Thomas J. Kaley, Email: kaleyt@mskcc.org.
Andrew B. Lassman, Email: ABL7@cumc.columbia.edu.
References
- 1. Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol 2013; 15 (Suppl 2):ii1–ii56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol 1999;17:2572–2578. [DOI] [PubMed] [Google Scholar]
- 3. Lamborn KR, Yung WK, Chang SM, et al. Progression‐free survival: an important end point in evaluating therapy for recurrent high‐grade gliomas. Neuro Oncol 2008;10:162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wick W, Gorlia T, Bendszus M, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med 2017;377:1954–1963. [DOI] [PubMed] [Google Scholar]
- 5. Holland EC, Celestino J, Dai C, et al. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 2000;25:55–57. [DOI] [PubMed] [Google Scholar]
- 6. Rajasekhar VK, Viale A, Socci ND, et al. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell 2003;12(4):889–901. [DOI] [PubMed] [Google Scholar]
- 7. Haas‐Kogan D, Shalev N, Wong M, et al. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol 1998;8:1195–1198. [DOI] [PubMed] [Google Scholar]
- 8. Chang SM, Wen P, Cloughesy T, et al. Phase II study of CCI‐779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. [DOI] [PubMed] [Google Scholar]
- 9. Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI‐779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol 2005;23:5294–5304. [DOI] [PubMed] [Google Scholar]
- 10. Sun SY, Rosenberg LM, Wang X, et al. Activation of Akt and eIF4E survival pathways by rapamycin‐mediated mammalian target of rapamycin inhibition. Cancer Res 2005;65:7052–8. [DOI] [PubMed] [Google Scholar]
- 11. O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 2006;66:1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN‐deficient glioblastoma. PLoS Med 2008;5:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pachioni Jde A, Magalhaes JG, Lima EJ, et al. Alkylphospholipids ‐ a promising class of chemotherapeutic agents with a broad pharmacological spectrum. J Pharm Pharm Sci 2013;16:742–759. [DOI] [PubMed] [Google Scholar]
- 14. Kondapaka SB, Singh SS, Dasmahapatra GP, et al. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther 2003;2:1093–1103. [PubMed] [Google Scholar]
- 15. Crul M, Rosing H, de Klerk GJ, et al. Phase I and pharmacological study of daily oral administration of perifosine (D‐21266) in patients with advanced solid tumours. Eur J Cancer 2002;38:1615–1621. [DOI] [PubMed] [Google Scholar]
- 16. Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res 2005;65:7429–7435. [DOI] [PubMed] [Google Scholar]
- 17. Guha A, Feldkamp MM, Lau N, et al. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene. 1997;15:2755–2765. [DOI] [PubMed] [Google Scholar]
- 18. Pitter KL, Galban CJ, Galban S, et al. Perifosine and CCI 779 co‐operate to induce cell death and decrease proliferation in PTEN‐intact and PTEN‐deficient PDGF‐driven murine glioblastoma. PloS One 2011;6:e14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaley TJ, Panageas KS, Mellinghoff IK, et al. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J Neurooncol 2019;144:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 1990;8:1277–1280. [DOI] [PubMed] [Google Scholar]
- 21. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high‐grade gliomas: response assessment in neuro‐oncology working group. J Clin Oncol 2010;28:1963–1972. [DOI] [PubMed] [Google Scholar]
- 22. Figg WD, Monga M, Headlee D, et al. A phase I and pharmacokinetic study of oral perifosine with different loading schedules in patients with refractory neoplasms. Cancer Chemother Pharmacol 2014;74:955–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wen PY, Chang SM, Lamborn KR, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04–02. Neuro Oncol 2014;16:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee EQ, Kuhn J, Lamborn KR, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05–02. Neuro Oncol 2012;14:1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. FDA . HIGHLIGHTS OF PRESCRIBING INFORMATION: TORISEL™ Kit (temsirolimus) injection, for intravenous infusion only. Initial U.S. approval 2007. 2017.
- 26. Chang SM, Kuhn J, Wen P, et al. Phase I/pharmacokinetic study of CCI‐779 in patients with recurrent malignant glioma on enzyme‐inducing antiepileptic drugs. Invest New Drugs 2004;22:427–435. [DOI] [PubMed] [Google Scholar]