

Abstract
Objectives
We ascertained the prevalence of ictal arrhythmias to explain the high rate of sudden unexpected death in epilepsy (SUDEP) in Dravet syndrome (DS).
Methods
We selected cases with clinical DS, ≥6 years, SCN1A mutation, and ≥1 seizure/week. Home‐based ECG recordings were performed for 20 days continuously. Cases were matched for age and sex to two epilepsy controls with no DS and ≥1 major motor seizure during video‐EEG. We determined the prevalence of peri‐ictal asystole, bradycardia, QTc changes, and effects of convulsive seizures (CS) on heart rate, heart rate variability (HRV), and PR/QRS. Generalized estimating equations were used to account for multiple seizures within subjects, seizure type, and sleep/wakefulness.
Results
We included 59 cases. Ictal recordings were obtained in 45 cases and compared to 90 controls. We analyzed 547 seizures in DS (300 CS) and 169 in controls (120 CS). No asystole occurred. Postictal bradycardia was more common in controls (n = 11, 6.5%) than cases (n = 4, 0.7%; P = 0.002). Peri‐ictal QTc‐lengthening (≥60ms) occurred more frequently in DS (n = 64, 12%) than controls (n = 8, 4.7%, P = 0.048); pathologically prolonged QTc was rare (once in each group). In DS, interictal HRV was lower compared to controls (RMSSD P = 0.029); peri‐ictal values did not differ between the groups. Prolonged QRS/PR was rare and more common in controls (QRS: one vs. none; PR: three vs. one).
Interpretation
We did not identify major arrhythmias in DS which can directly explain high SUDEP rates. Peri‐ictal QTc‐lengthening was, however, more common in DS. This may reflect unstable repolarization and an increased propensity for arrhythmias.
Introduction
Dravet syndrome (DS; OMIM #607208) is a severe childhood‐onset developmental epileptic encephalopathy, with multiple seizures types. People with DS face a substantial risk of early epilepsy‐related death, of up to 15% by age 20 years. 1 , 2 Sudden unexpected death in epilepsy (SUDEP) is common, accounting for nearly half of all deaths 2 and affecting one in 107 individuals per year. 1 The underlying pathophysiology of SUDEP is poorly understood, but likely heterogeneous and multifactorial. 3 It is a seizure‐related event and a high frequency of convulsive seizures (CS) is the major risk factor. 3
In >70% of people with DS, heterozygous loss‐of‐function mutations are found in the SCN1A gene, encoding the α‐subunit of the voltage‐gated sodium channel Nav1.1 in the mammalian brain and heart. 4 , 5 , 6 Other so called “cardiocerebral channelopathies,” are known to have features related to both organs which may lead to sudden death – for example, mutations in SCN5A have been linked to epilepsy and long QT syndrome. 7 , 8 Mouse models support the strong association between DS and SUDEP. 9 , 10 , 11 Reports of postictal bradycardia 12 , 13 , 14 and ventricular fibrillation 13 progressing to fatal asystole in DS mice suggest that the mutation not only alters cortical excitability but also increases the propensity to arrhythmias. 12 , 13 , 14 Interictal data in people with DS showed increased QT dispersion and decreased heart rate variability (HRV), 15 , 16 , 17 , 18 which may increase risk of lethal ventricular dysrhythmias. 19 , 20 We prospectively performed long‐term ECG recordings in DS cases and compared them to historical epilepsy controls to find an explanation for the high SUDEP rate. We determined the prevalence of seizure‐induced arrhythmias and repolarization/conduction abnormalities and assessed peri‐ictal HR and HRV.
Methods
Subject and seizure selection
We selected individuals with clinical DS and confirmed pathogenic SCN1A mutation. Other inclusion criteria were: (1) ≥6 years of age, (2) ≥1 seizure per week averaged over the previous year (all types except absences or myoclonic seizures), and (3) no history of self‐harm. Cases were recruited from participating centers in the Netherlands (SEIN, Heemstede and Zwolle; Kempenhaeghe, Heeze), Germany (University Hospital of Bonn), and the United Kingdom (Great Ormond Street Hospital for Children, London).
All cases had two baseline 12‐lead ECGs (standard and Brugada 21 lead placement) and wore a 3‐lead ECG device continuously for 20 days (nECG MINDER, Nuubo®, Madrid, Spain; sampling frequency 250Hz and including a tri‐axial accelerometer). The device was attached to a comfortable vest with textile electrodes, allowing for normal daily activities; only during showering the device was not recording. Data were recorded per 24 h (then the device had to be charged and a new recording file was created when the device was turned back on). Carers and participants were asked to record seizure details (e.g., seizure time and type) and the time the person got out and into bed. All continuous data were saved on a microSD card and could be downloaded at the end of the recording period. All ECG data was inspected visually (page by page; each page was a 10‐s window). During inspection the corresponding tachogram was visible below the ECG. Reported seizures were identified based on diary notes, accelerometry, and HR profiles. A HR increase of ≥10% was required to recognize seizures. We additionally included “unreported seizures” with HR patterns resembling a reported CS of that person if: (1) HR changes did not coincide with a sudden body position change (as indicated by the triaxial accelerometer) and (2) seizures in this person were known to be sometimes missed (e.g., found in a postictal state).
Historical controls were selected from a video‐EEG database. A 1‐lead ECG was measured during EEG recording. Two controls were selected for each case, as fewer seizures were expected in controls due to shorter recordings. Inclusion criteria were: (1) ≥6 years of age, (2) definite diagnosis of epilepsy, (3) no suspicion of DS, (4) ≥1 major motor seizure (CS (i.e., focal to bilateral or generalized tonic‐clonic), generalized or focal motor seizures), and (5) postictal recording of >5 min. Controls were frequency matched to mean age and sex ratio of the Dravet group, as these variables may affect cardiac function. Clinical seizures (except absences and myoclonic jerks) which resulted in a HR increase of ≥10% were included.
The following data were obtained from the medical records: sex, age, epilepsy duration, seizure frequency, seizure cluster (yes/no), occurrence (nocturnal/diurnal/both), anti‐seizure medications (ASM), vagal nerve stimulation (yes/no), and etiology. Additionally, for DS, we recorded SCN1A mutation type, age of onset developmental delay, and family history of febrile seizures, epilepsy, and sudden cardiac death.
The study protocol was independently approved by the local Medical Ethics Committee of each participating center. A written informed consent was obtained from the participants or assent from parents or legal guardians in case of minors or those with learning disability. The study was registered at ClinicalTrials.gov (NCT02415686).
Electrocardiographic analysis
Baseline 12‐lead ECG recordings of DS cases were evaluated by one experienced cardiologist (HLT). The ECG of all seizures in cases and controls were assessed manually for abnormalities from 1 min before onset to 5 min after end of seizure (by SS and, in case of uncertainty, by HLT).
All ECG data were imported into MATLAB to enable assessment in the same viewer, with the same measuring tools. HR changes were used to determine the timing of seizure onset and end. We noted peri‐ictal timepoints: T1 just before seizure onset, T2 immediately after seizure end, T3 2 min, and T4 5 min after seizure end. When an ECG abnormality was identified, the cardiologist consulted and if deemed necessary referral for further cardiac evaluation arranged.
Analysis part 1: Bradyarrhythmias and QTc‐intervals
The main study parameters were postictal asystole (sinus arrest of ≥3s) and bradycardia (<2nd HR percentile for age; average of three consecutive RR intervals; female 6–8 years 68 bpm, 8–12 years 58 bpm, 12–16 years 54 bpm; male 6–8 years 62 bpm, 8–12 years 55 bpm, 12–16 years 48 bpm; >16 years 50 bpm 21 ). Bradycardia and asystole detection were done manually (SS).
We manually measured QT intervals and RR intervals at time points T1–4. QT intervals and RR intervals were averaged from three successive ECG complexes. Four correction formulas (Bazett, Fridericia, Hodges, and Framingham) were used to calculate QTc intervals. All formulas are known to over‐ or undercorrect QT intervals. 23 To reduce bias error of putatively pathologic intervals, we considered only those on which Bazett and at least one other formula were abnormal. Table 1 shows the QTc parameters that were determined for each seizure. QTc‐lengthening and ‐shortening of ≥60 ms was determined by subtracting QTc at T2–4 from the preictal value (T1). We noted the occurrence of clinically significant prolonged QTc (defined as: ≤13 years ≥ 460 ms, males > 13 years ≥ 470 ms and females > 13 years ≥ 480 ms), shortened QTc (≤340 ms), and marked prolongation (≥500 ms) and shortening (≤300 ms) at every time point. 23
Table 1.
Overview of QTc parameters that were determined for each seizure.
| QTc parameters | |
|---|---|
| Marked prolongation | Marked shortening |
| ≥500 ms | ≤300 ms |
| Clinically significant prolonged | Clinically significant shortened |
| ≥460 ms ≤ 13 years | ≤340 ms |
| ≥470 ms > 13 years male | |
| ≥480 ms > 13 years female | |
| Lengthening of ≥ 60 ms | Shortening of ≥ 60 ms |
| T2 versus T1 | T2 versus T1 |
| T3 versus T1 | T3 versus T1 |
| T4 versus T1 | T4 versus T1 |
T1, time of seizure onset; T2, seizure end; T3, two minutes after seizure end; T4, five minutes after seizure end.
Analysis part 2: Peri‐ictal heart rates, heart rate variability, QRS‐width, and PR‐interval in convulsive seizures
This analysis was only done in CS. For most cases, one of the three leads had good recording quality and could be used for all subsequent analyses. In case of prominent ECG artifacts, all leads were visually inspected to determine the least affected lead (could vary between and within seizures).
Consecutive RR intervals were automatically determined by one of the two peak detection methods. First, the Pan–Tompkins QRS detection algorithm was used and the results were visually inspected. 25 If there were quality concerns a second detection method based on the Hilbert–Huang transform was applied. 25 Additional filtering was applied in individual cases depending on signal quality (e.g., removal of low frequencies as only R peaks are needed).
Peri‐ictal HR was determined automatically in all CS at T1–T4 by averaging 10 RR intervals. In case a HR was aberrant, it was checked and measured manually (e.g., all extreme values <50 bpm or >180 bpm or inexplicable values like higher HR at T2 than at T1).
We estimated HRV from 1‐minute windows during three periods: pre‐ictal, postictal, and resting state. We visually inspected the HR segments and selected the most optimal QRS detection method. Segments with insufficient quality of ECG were excluded from further analysis. RR intervals > 3 SD from the average were considered artefacts; these beats and their corresponding interbeat intervals were excluded from analysis. The windows were only analyzed if the summed RR intervals ≥ 50s. The windows were selected as follows:
The preictal minute was the minute immediately before T1.
The post‐ictal minute was selected between T3 and T4, depending on the ECG quality. A 1‐minute sliding window was moved along the postictal ECG with 1 s step (starting at T3) until an adequate window was reached (summed RR intervals ≥ 50 s).
The resting‐state minute required an awake state in a lying position (between 5 and 15 min after onset of supine rest as determined by video inspection (controls) or diary and accelerometry (cases)) and that no seizure had occurred in the previous hour and no CS in the previous 6 h. 27
The following HRV measures were estimated: average RR interval, root mean square of successive differences of RR intervals (RMSSD), the standard deviation of RR intervals (SDNN), and the percentage of consecutive RR intervals differing by ≥50 ms (pNN50).
QRS‐widths and PR‐intervals were measured manually at T1 and T4. We noted whether these values were pathologically prolonged (>98th percentile for age: QRS‐width female 6–8 years 95 ms, 8–12 years 99 ms, 12–16 years 106 ms; male 6–8 years 98 ms, 8–12 years 103 ms, 12–16 years 111ms; all > 16 years 120 ms; PR‐interval female 6–8 years 156 ms, 8–12 years 163 ms, 12–16 years 176 ms; male 6–8 years 160 ms, 8–12 years 174 ms, 12–16 years 178 ms; all > 16 years 200 ms). 22
Statistical analysis
Clinical characteristics were described with means, SDs, medians, (interquartile) ranges, frequencies, and percentages, and compared between groups using the two‐sided unpaired t‐test or Mann–Whitney U test for continuous variables and chi‐square for categorical data. To determine whether DS was independently associated with the occurrence of bradycardia, asystole, and QTc variables, we compared these to the controls using generalized estimating equation (GEE) logistic models (analysis part 1). Preictal and postictal HR and HRV variables in DS were compared to controls using GEE linear models (analysis part 2). GEE models were applied to correct for within‐person correlation, seizure onset from sleep or wakefulness, and (in analysis part 1 only) for the presence of CS (yes/no). Resting‐state HRV variables were compared using unpaired t‐tests or Mann–Whitney U test. For visual comparison, boxplots were constructed for HRs at T1‐4 of CS for both groups. Normality tests were performed, and logarithmic transformation was applied to right‐skewed‐dependent variables. Outliers were defined as values that lie more than three SD from the mean. The Holm–Bonferroni method was used to correct for multiple comparisons within the different data sets; adjusted p‐values are shown. All tests were two‐tailed and a value of P < 0.05 was considered significant. Statistical analyses were performed with IBM SPSS Statistics 24 (IBM Corp. Armonk, NY).
Results
Subjects
Fifty‐nine people were included from June 2015 to January 2018 (48 in the Netherlands, nine in Germany and two in the UK; Fig. 1). No seizures were recorded in eight cases and in six the study was terminated prematurely prior to any seizure thus leaving 45 cases with ictal ECG recordings. Mean age was 19 years (±10 years) and 23 were female (51%; Table 2). In 90 controls, the mean age was 20 years (±9 years) and 46 were female (51%). Most controls were refractory to treatment (n = 82, 91%) and 25 controls had a learning disability (28%). A total of 22 different ASM were used between the groups and most were using more than one ASM (DS n = 41 (91%) and controls n = 57 (63%)). Table s1 shows the SCN1A variants.
Figure 1.
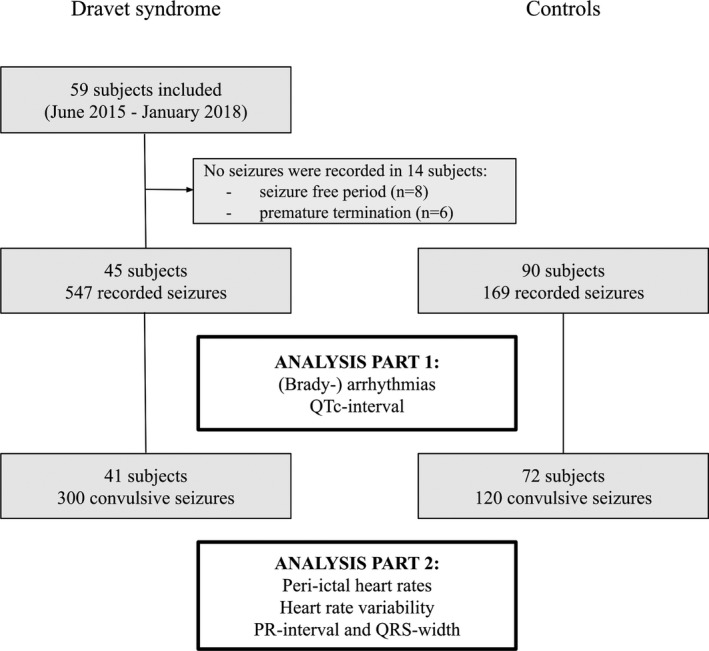
Study flowchart.
Table 2.
Clinical characteristics of Dravet syndrome cases and historical epilepsy controls.
| Characteristics | Dravet syndrome (n = 45) | Controls (n = 90) | P‐value |
|---|---|---|---|
| Sex, n (%) female | 23 (51) | 46 (51) | 1 |
| Age, years mean (SD) | 19 (10) | 20 (9.4) | 0.54 |
| Epilepsy duration, years mean (SD) | 19 (12) | 9 (6.5) | <0.001 |
| Seizure frequency per month, median (IQR) | 12 (8–25) | 8 (3–30) | 0.09 |
| Seizures predominantly, n (%) 1 | 0.001 | ||
| Nocturnal | 26 (58) | 26 (29) | |
| Diurnal | 4 (9) | 31 (34) | |
| Both | 15 (33) | 33 (37) | |
| Number of ASM, median (range) | 3 (0–4) | 2 (0–4) | <0.001 |
| ASM type | |||
| Valproic acid | 35 (78) | 27 (30) | <0.001 |
| Clobazam | 28 (62) | 19 (21) | <0.001 |
| Stiripentol | 19 (42) | 1 (1) | <0.001 |
| Topiramate | 14 (31) | 3 (3) | <0.001 |
| Lamotrigine | 3 (7) | 28 (31) | 0.001 |
| Carbamazepine | 2 (4) | 24 (27) | 0.002 |
| Oxcarbazepine | 2 (4) | 21 (23) | 0.006 |
| Lacosamide | 0 | 8 (9) | 0.039 |
| Drug tapering, n (%) | NA | 35 (39) | NA |
| VNS, n (%) | 8 (18) | 3 (3.3) | 0.012 |
| MRI abnormalities, n (%) | 8 (18) | 39 (43) | 0.001 |
| History cardiac illness, n (%) 2 | 0 | 2 (2.2) | NA |
| Epilepsy etiology, n | NA | ||
| Structural | 0 | 50 | |
| Genetic | 45 | 13 3 | |
| Infectious | 0 | 1 | |
| Metabolic | 0 | 0 | |
| Immune | 0 | 1 | |
| Unknown | 0 | 25 | |
| Learning disability, n (%) | 19 (42) | 25 (28) | NA |
| SCN1A variant type, n 4 | Not assessed | NA | |
| Missense | 15 | ||
| Splice site | 2 | ||
| Nonsense | 12 | ||
| Small frameshift deletions | 11 | ||
| Small frameshift duplications | 3 | ||
| Gross deletions | 2 | ||
| Gross duplications | 1 | ||
| Unknown | 2 | ||
ASM, antiseizure medication; IQR, interquartile range; NA, not applicable; VNS, vagal nerve stimulator.
Reported by cases/caregivers.
One control with an atrial septum defect type II and one with bigeminy/trigeminy.
Six controls had a generalized epilepsy syndrome with a presumed genetic etiology (e.g., juvenile myoclonic epilepsy), five a genetic cause of a focal epilepsy/encephalopathy (DEPDC5, GRIN1, SLC6A5, PCDH19, and trisomy 13), one Doose syndrome and one blepharophimosis‐mental retardation syndrome (BMRS).
One case had a missense variant type and a small frame shift deletion, and two cases had both a small frameshift deletion and insertion.
Seizures
Continuous ECG during a total of 19,174 h was recorded in the 45 cases (mean 426 h per case; Fig. 2). A total of 547 seizures were captured, resulting in a median of seven seizures per person (range 1–69). Seizure types recorded in cases were: 300 CS (55%), 33 tonic (6%), 12 focal seizures with impaired awareness (2.2%), nine focal motor seizures (1.6%), seven hemiclonic (1.3%), one clonic (0.2%), 41 unknown type (7.5%), and 144 unreported seizures (26%; Table 2). Of all 547 seizures, 77 arisen from wakefulness (14%) and 470 from sleep (86%). Most unreported seizures were from sleep (135, 94%). None of the recorded seizures in cases occurred during a period of illness or fever.
Figure 2.
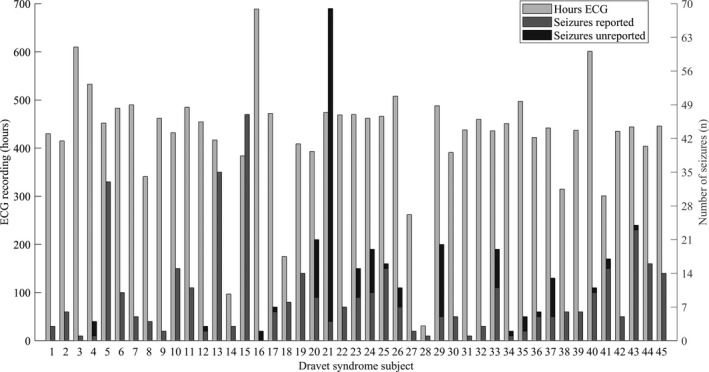
Hours ECG, number of recorded seizures, and proportion unreported seizures for each Dravet syndrome case.
In the 90 controls, 169 seizures were recorded with a median of one seizure per person (range 1–8). Seizure types recorded were: 120 CS (71%), 29 tonic seizures (17%), 18 focal seizures with impaired awareness (11%), and two hemiclonic seizures (1.2%). Of the 169 seizures, 67 were from wakefulness (40%) and 102 from sleep (60%).
Electrocardiographic findings
Baseline 12‐lead ECG
In five people with DS, abnormalities were found in the baseline ECG: atrial rhythm (n = 2), right axis deviation (n = 1), ST‐segment abnormalities in all leads (n = 1), and negative T‐waves in V1‐4 (n = 1) leading to two referrals for further cardiac evaluation (ST‐segment abnormalities and negative T‐waves). No signs of cardiomyopathy were found in the case with negative T‐waves.
Analysis part 1: Peri‐ictal bradyarrhythmias and QTc‐intervals
Asystole was not seen. Bradycardia was more common in seizures of controls (11 seizures in eight subjects, 6.5% of seizures) compared to cases (four seizures in two subjects, 0.7% of seizures; P = 0.002; Table 3). In the controls, five of these 11 seizures were CS and six were tonic. In cases, all four were tonic seizures.
Table 3.
Peri‐ictal electrocardiographic findings in the Dravet syndrome and historical epilepsy control group.
|
Dravet syndrome n = 45 subjects |
Controls n = 90 subjects |
P‐value | 95% CI of OR | |
|---|---|---|---|---|
| Seizure types, n seizures (%) | ||||
| Total number of seizures | 547 | 169 | NA | NA |
| Convulsive | 300 (55) | 120 (71) | NA | NA |
| Tonic | 33 (6) | 29 (17) | NA | NA |
| Focal impaired awareness | 12 (2.2) | 18 (11) | NA | NA |
| Focal motor | 9 (1.6) | 0 | NA | NA |
| Hemiclonic | 7 (1.3) | 2 (1.2) | NA | NA |
| Clonic | 1 (0.2) | 0 | NA | NA |
| Unknown type reported | 41 (7.5) | 0 | NA | NA |
| Unreported | 144 (6) | 0 | NA | NA |
| Peri‐ictal ECG variables, n seizures (n people; %) | ||||
| Bradycardia, n seizures (n subjects; %) | 4 (2; 0.7) | 11 (8; 6.5) | 0.002 | 1.2 to 5.3 |
| Prolonged QTc, n seizures (n subjects; %) | 1 (1; 0.2) | 1 (1; 0.6) | 0.7 1 | −0.99 to 2.8 |
| T1 | 0 | 0 | ||
| T2 | 0 | 1 | ||
| T3 | 1 | 0 | ||
| T4 | 1 | 0 | ||
| Shortened QTc, n seizures (n subjects; %) | 31 (12; 5.7) | 12 (12; 7.1) | 0.82 1 | −0.72 to 0.92 |
| T1 | 17 | 5 | ||
| T2 | 5 | 4 | ||
| T3 | 10 | 3 | ||
| T4 | 5 | 2 | ||
| Ictal QTc‐lengthening, ≥60 ms compared to T1, n seizures (n subjects; %) | 64 (23; 12) | 8 (8; 4.7) | 0.048 1 | −1.7 to −0.21 |
| T2 | 53 | 4 | ||
| T3 | 4 | 1 | ||
| T4 | 9 | 3 | ||
| Ictal QTc‐shortening, ≥60 ms compared to T1, n seizures (n subjects; %) | 15 (7; 2.7) | 13 (11; 7.7) | 0.39 1 | −0.26 to 2 |
| T2 | 12 | 10 | ||
| T3 | 3 | 5 | ||
| T4 | 2 | 4 | ||
CI, confidence interval; OR, odds ratio; T1, time of seizure onset; T2, seizure end; T3, 2 min after seizure end; T4, 5 min after seizure end.
The Holm–Bonferroni method was used to correct for the multiple comparisons of the QTc‐interval; corrected p‐values and original CIs are shown. Generalized estimating equations were used to correct for within‐subject correlation, seizure onset from sleep or wakefulness and seizure type (convulsive seizure yes or no). QTc changes can occur at multiple time points within seizures.
Peri‐ictal QTc‐lengthening of ≥60 ms was more common in cases (64 seizures in 23 subjects, 12% of seizures) than controls (eight seizures in eight subjects, 4.7% of seizures; P = 0.048 (P = 0.012*4)). Within the Dravet group, the median age did not differ between those with QTc‐lengthening (n = 23; median 14 years, IQR 12–21) and those without (n = 22; median 18 years, IQR 14–25; P = 0.12). Anti‐seizure medications and the presence of ictal QTc‐lengthening of ≥60 ms or bradycardia, for cases and controls are provided in Table S2. No difference was found in the number of seizures with ictal QTc‐shortening of ≥60 ms between cases (15 seizures in seven subjects, 2.7%) and controls (13 seizures in 11 subjects, 9.5%; P = 0.39 (P = 0.13*3)). The occurrence of prolonged (cases n = 1, 0.2% vs. controls n = 1, 0.6%; P = 0.7 (P = 0.35*2)) and shortened QTc (cases n = 31, 5.7% vs. controls n = 12, 7.1%; P = 0.82 (P = 0.82*1)) also did not differ between the groups. Marked prolongation and shortening did not occur in either group.
Analysis part 2: Peri‐ictal heart rates, heart rate variability, QRS‐width, and PR‐interval in convulsive seizures
In 41 cases, 300 CS and in 72 controls, 120 CS were analyzed. In cases, 56 CS (19%) were from wakefulness and 244 (81%) from sleep and in the controls 49 CS (42%) from wakefulness and 71 from sleep (59%). Mean age in this selection of the Dravet group was 19 years (±11 years) and 20 were female (49%). In controls, the mean age was 21 years (±9.3 years) and 35 were female (49%).
At T1, there was a trend of higher mean HR in DS (81 bpm ± 19) compared to controls (76 bpm ± 17, P = 0.092; Fig. 3). At the other time points, mean HR did not differ between cases and controls: T2 154 bpm (±23) versus 151 bpm (±20; P = 0.75); T3 113 bpm (±24) versus 118 bpm (±22; P = 0.92); T4 106 bpm (±22) versus 110 bpm (±21; P = 0.39; corrected for within‐person correlation and onset sleep/wakefulness). An example of a continuous HR curve during a CS in a case is shown in Figure 4.
Figure 3.
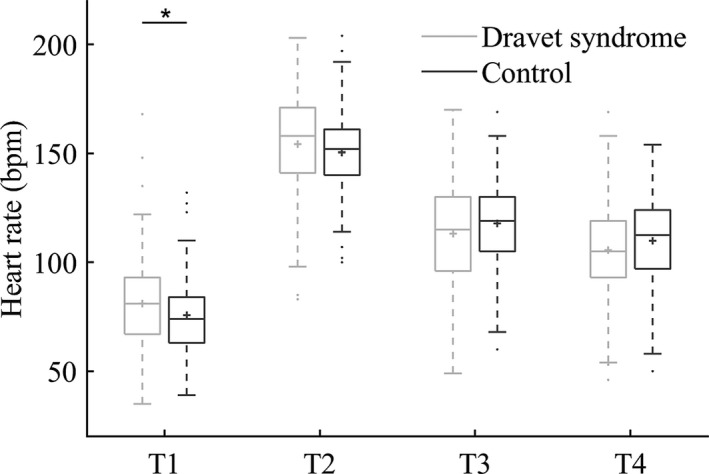
Box plots of peri‐ictal heart rates in convulsive seizures of the Dravet syndrome and historical epilepsy control groups. T1 = time of seizure onset; T2 = seizure end; T3 = two minutes after T2; T4 = 5 min after T2; * significance P < 0.05. Median (solid line), mean (plus sign), interquartile interval (box), minimum, maximum (whiskers, 1.5 IQR), and suspected outliers (dots) are shown. Generalized estimating equation linear models were used to compare heart rates between the groups, correcting for within‐subject correlation and seizure onset from sleep or wakefulness.
Figure 4.
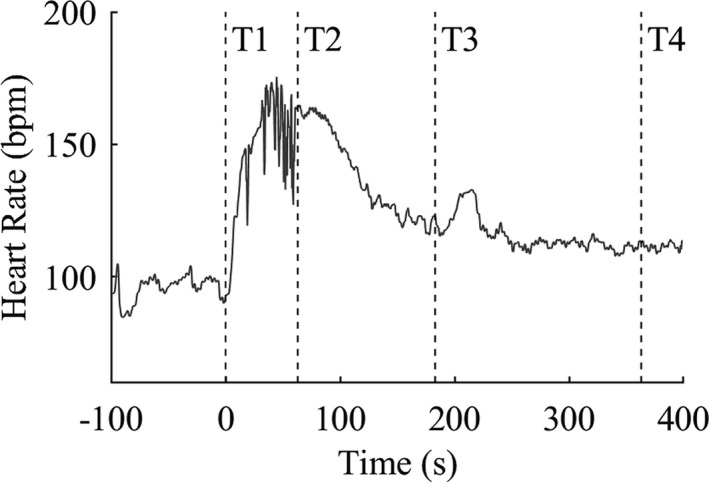
Heart rate curve during a convulsive seizure of a 14‐year‐old girl with Dravet syndrome from 1 min prior to seizure onset to 5 min after seizure end. T1 = time of seizure onset; T2 = seizure end; T3 = 2 min after T2; T4 = 5 min after T2.
The median resting‐state HRV variable RMSSD was lower in Dravet (37 ms) than in controls (51 ms; P = 0.029). Other resting‐state HRV variables were not significantly lower in DS: SDNN was 40 ms in Dravet and 55 ms in controls (P = 0.052) and pNN50 was 16 ms in Dravet and 32 ms in controls (P = 0.06). The Mean resting‐state HR was higher in cases (RR 740 ms) than in controls (RR 884 ms; P < 0.001). Mean HR and HRV variables of the pre‐ and post‐ictal minute did not differ between groups (Table 4).
Table 4.
Heart rate variability in rest and before and after convulsive seizures in the Dravet syndrome and historical epilepsy control groups.
| HRV variables (ms) |
Dravet syndrome n = 41 |
Controls n = 72 |
P‐value | 95% CI |
|---|---|---|---|---|
| Awake rest | CI of difference | |||
| Number of people | 41 | 66 | NA | NA |
| RR interval, mean (SD) | 740 (140) | 884 (175) | <0.001 | 80 to 208 |
| RMSSD, median (IQR) 1 | 37 (20–58) | 51 (33–76) | 0.029 | −15 to 26 |
| SDNN, median (IQR) 1 | 40 (23–60) | 55 (37–68) | 0.052 | −10 to 22 |
| pNN50, median (IQR) | 16 (1.2–42) | 32 (12–52) | 0.06 | 0.14 to 19 |
| Pre‐ictal | CI of OR | |||
| Number of seizures | 285 | 100 | NA | NA |
| RR interval, mean (SD) | 769 (201) | 821 (202) | 0.18 | 2 to 168 |
| RMSSD, median (IQR) 1 | 46 (21–107) | 44 (26–81) | 1 | −0.1 to 0.09 |
| SDNN, median (IQR) 1 | 46 (26–99) | 51 (31–73) | 1 | −0.07 to 0.08 |
| pNN50, median (IQR) 2 | 22 (2.4–58) | 20 (4.9–51) | 0.99 | −11 to 11 |
| Postictal | ||||
| Number of seizures | 288 | 117 | NA | NA |
| RR interval, mean (SD) | 582 (151) | 530 (105) | 0.34 | −98 to 6 |
| RMSSD, median (IQR) 1 | 21 (8.8–66) | 17 (6.3–52) | 0.8 | −0.24 to 0.07 |
| SDNN, median (IQR) 1 | 29 (15–59) | 27 (14–50) | 0.68 | −0.13 to 0.09 |
| pNN50, median (IQR) 2 | 3.2 (0–31) | 1.7 (0–16) | 0.72 | −12 to 5 |
The Holm–Bonferroni method was used to correct for multiple comparisons within each epoch. Corrected p‐values and original CIs are shown. Resting‐state HRV variables were compared using two‐sided unpaired t‐tests or Mann–Whitney U test. Peri‐ictal HRV variables were compared using generalized estimating equation linear models, correcting for within‐person correlation and seizure onset from sleep or wakefulness.
CI, confidence interval; OR, odds ratio; HRV, heart rate variability; IQR, interquartile range; NA, not applicable; pNN50, proportion of pairs of successive RR intervals that differ ≥50 ms; RMSSD, root mean square of successive differences of RR intervals; SDNN, standard deviation of RR intervals.
Logarithmic transformation was applied to RMSSD and SDNN.
pNN50 variable was treated as normal distribution in the model as no distribution type fitted original or transformed data.
Peri‐ictal prolonged QRS did not occur in the CS of cases while in controls one mild prolongation was seen (at T1, 2 ms above normal limit). Prolonged PR was also more common in controls (n = 3) than in DS (n = 1). The case had a mildly prolonged PR interval in one CS at T1 (1 ms above normal limit). The first control had prolonged PR in one CS at T4 (3 ms above normal limit), the second subject in one of six CS at T1 and T4 (by 17 ms and 6 ms), and the third control in one CS at T1 and T4 (by 3 ms and 7 ms).
Discussion
We prospectively recorded peri‐ictal ECG of 547 seizures in a cohort of 45 people with DS and did not identify actionable major arrhythmias. Peri‐ictal QTc‐lengthening of ≥60 ms was, however, more prevalent in DS compared to controls, occurring in over half of the cases. In line with previous reports, interictal HRV was lower in DS compared to controls.
Strengths and limitations
The strength of our study is that despite DS being a rare disease, a substantial cohort was recruited. This, combined with the long duration of ECG recordings, resulted in a large overall number of recorded seizures in the home setting. Another strength is that all ECG data were assessed manually for abnormalities. Our Dravet cohort might be an enriched selection, as SUDEP in DS mostly affects young children. 2 The reported SUDEP cases in DS are, however, likely to be biased toward the young, as improved genetic testing and awareness over the last decades has enabled increased early diagnosis. A younger cohort might have exhibited a higher SUDEP risk, but the high frequency of CS alone places them at high risk for SUDEP. 28 , 29 The age of cases with and without QTc‐lengthening did not differ. The age factor therefore does not appear to influence conclusions of this study.
Seizure diaries are known to be unreliable. 30 Some seizures, and thus potential ictal arrhythmias, within our cohort may therefore have been missed. To overcome this, the complete recordings were meticulously inspected for likely CS, and many unreported seizures were identified. These unreported seizures were not included in the second part of the analysis to ensure only definite CS were involved. Ideally, controls should resemble the Dravet phenotype, with refractory epilepsy and learning disabilities, and be recorded prospectively at home to avoid contrasts in physiological state (e.g., sleep deprivation or drug tapering during clinical stay). Equal recording methods would also enable blinding for analysis, which was not possible due to different data sources. The study burden would, however, be of concern in view of the young age and behavioral problems. The burden would not be outweighed by evidence for increased propensity for arrhythmias, as in DS. The controls here, however, were predominantly people with refractory epilepsy and learning disabilities were present in a considerable proportion – strengthening the power of our study population and supporting the validity of our results.
Cardiac function in Dravet syndrome
Peri‐ictal QTc‐lengthening of ≥60 ms in DS was up to four times more common than in other epilepsy syndromes. 24 , 31 , 32 The lengthening may result from a less stable cardiac repolarization in DS that increases the propensity for malignant tachyarrhythmias. 33 Peri‐ictal QTc‐lengthening was mostly brief and often resolved right after seizure end. Peri‐ictal respiratory dysfunction is common in people with DS. 14 Studies in healthy people showed that hypoxia and hypercapnia can prolong QTc. 34 Results of studies comparing QTc changes in seizures with and without an SpO2 drop of <90% were conflicting: one found more QTc‐shortening and lengthening in the desaturation group, 35 while the other found no differences. 32 A recent larger study, using a mixed‐effect model, that, unlike previous studies, included SpO2 as a continuous variable and HR and peri‐ictal phase as covariates, found that peri‐ictal QTc changes strongly correlated with SpO2. 36 The QTc‐lengthening found in our cohort may thus be explained by ictal hypoxemia rather than unstable repolarization due to the SCN1A mutation. Alternatively, QT changes may be related to contrasts in ASM profiles. With this cohort size and high rates and heterogeneity of ASM polytherapy, we were insufficiently powered to assess the effect of separate ASM types on the study outcomes.
Prolongation of QTc can provoke potentially lethal tachyarrhythmias, 33 but whether peri‐ictal QTc‐prolongation relates to SUDEP is unclear. Only two SUDEP and four near SUDEP cases in which tachyarrhythmias played a role have been reported. 37 , 38 , 39 A prospective study of out‐of‐hospital cardiac arrests due to ECG‐documented ventricular tachycardia or fibrillation showed a threefold increased risk of these arrhythmias in people with epilepsy compared to the general population. 40 Most arrhythmias were not seizure‐related and occurred in the context of preexisting or acute heart disease, but some were unexplained and classified as (near) SUDEP. 41 Sudden cardiac arrest and SUDEP may thus partially overlap.
The lower resting‐state HRV in DS compared to controls confirms previous findings. 15 , 16 , 17 , 18 Resting‐state HR was, however, higher in DS than controls, which was also seen as a trend in the other studies. 15 , 16 , 17 HR increases are correlated with decreases in HRV. 18 , 26 Lower resting‐state HRV in Dravet may thus cohere with a higher resting‐state HR. Decreased HRV has been proposed as a SUDEP biomarker as it may lower the propensity to ictal arrhythmias, but direct evidence linking HRV to SUDEP is still lacking. Postictal bradycardia was unexpectedly more common in controls than in DS. Mouse model studies have reported episodes of bradycardia postically prior to death, 12 , 13 , 14 but also during nonfatal seizures. 14 Ictal bradycardia only occurred if there was apnea or severely decreased breath amplitude of Scn1aR1407X/+ mice. 14 The role of cardiac dysfunction in SUDEP in people with DS may thus not be as prominent as previous experimental evidence suggested and most likely occurs in response to respiratory dysfunction. Mice with selective knockout of SCN1A in brain interneurons only, and not in cardiac myocytes only, can also experience seizures and die spontaneously. 42 Single‐cell electrophysiology experiments of human‐ and mice‐derived cardiac myocytes did find increased sodium currents and spontaneous contraction rates in DS compared to control cells. 12 , 43 It may seem counterintuitive that reduced sodium channels (due to SCN1A haploinsufficiency) leads to these findings. This may, however, reflect compensatory overexpression of other sodium channels (i.e., Nav1.5 encoded by SCN5A), 12 , 43 which would be in line with the QTc‐lengthening we observed. It is not clear, however, how these electrophysiological studies of mice and single mutated cells translate to human cardiac function. Phenotype variability (i.e., types, locations, and mosaicism) is known to affect developmental outcome and epilepsy severity in DS 3 , 44 , 45 and may also apply to cardiac function.
Clinical implications
Our analysis did not suggest any major peri‐ictal cardiac arrhythmias which directly explain high SUDEP rates in DS. Postictal QTc‐lengthening in the DS cohort is more likely explained by respiratory dysfunction rather than unstable repolarization due to the SCN1A mutation. Prospective data to determine whether QTc lengthening and decreased HRV can predict SUDEP risk in DS is warranted. A 10‐year follow‐up of our cohort and additional analyses in case some succumbed to SUDEP will be performed. An important factor underlying SUDEP risk in DS seems to be epilepsy severity. As CS frequency is the most important risk factor, optimizing seizure control and nocturnal supervision, particularly in view of the substantial number of unreported CS in our cohort, are the most effective preventative measures. 28 , 46
Conflict of Interest
RS reports personal fees from UCB Pharma, EISAI, Cyberonics, Bial, Desitin, and LivaNova, RS is member of the editorial board of Epilepsy and Behavior and Epilepsia Open. JWS reports personal fees from UCB and Zogenix, grants from UCB, Eisai, UCB, GW Pharma. JWS's current position is endowed by the Epilepsy Society, he is a member of the Editorial Board of the Lancet Neurology and receives research support from the Marvin Weil Epilepsy Research Fund. JHC reports grants from GW Pharma, Zogenix, Marinius, Vitaflo, Nutricia, and National Institute for Health Research Biomedical Centre at Great Ormond Street Hospital for Children NHS Foundation trust. SS reports personal fees from UK Epilepsy Society. BG reports personal fees from GW Pharmaceuticals, Zogenix, and OVID/Takeda. RDT reports personal fees from UCB, GSK, Theravance, Novartis and Medtronic and grants from Nuts OHRA Foundation, Medtronic, AC Thomson Foundation and The Netherlands Organisation for Health Research and Development (ZonMW). RDT is a member of the editorial board of Seizure, Epilepsia and Clinical Autonomic Research. The remaining authors have no conflicts of interest relevant to this research.
Ethical publication statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1. SCN1A variants of the 45 subjects in the Dravet syndrome group.
Table S2. Antiseizure medication types and the presence of peri‐ictal QTc prolongation ≥60 ms and postictal bradycardia in ≥1 of the recorded seizures, of Dravet syndrome cases, and historical epilepsy controls.
Acknowledgments
This work was funded by the Dutch Epilepsy Foundation (project number 15‐10) and supported by Christelijke Vereniging voor de Verpleging van Lijders aan Epilepsie, The Netherlands. We thank all participants and their families. We thank Myra de Groot, of the Dravet syndrome foundation Netherlands/Flandres for her invaluable support and Amy Muggeridge, research coordinator Young Epilepsy UK, for her assistance. We thank Professor Job van der Palen, from University of Twente, for his help with the statistical analysis. This work was partly carried out at UCLH/UCL Comprehensive Biomedical Research Centre, which receives a proportion of funding from the UK Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
Funding Information
This work was funded by the Dutch Epilepsy Foundation (project number 15‐10) and supported by Christelijke Vereniging voor de Verpleging van Lijders aan Epilepsie, The Netherlands. This work was partly carried out at UCLH/UCL Comprehensive Biomedical Research Centre, which receives a proportion of funding from the UK Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
Funding Statement
This work was funded by Dutch Epilepsy Foundation grant ; Christelijke Vereniging voor de Verpleging van Lijders aan Epilepsie grant .
References
- 1. Cooper MS, McIntosh A, Crompton DE, et al. Mortality in Dravet syndrome. Epilepsy Res 2016;128:43–47. [DOI] [PubMed] [Google Scholar]
- 2. Shmuely S, Sisodiya SM, Gunning WB, et al. Mortality in Dravet syndrome: a review. Epilepsy Behav 2016;64(Pt A):69–74. [DOI] [PubMed] [Google Scholar]
- 3. Depienne C, Trouillard O, Saint‐Martin C, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet 2009;46:183–191. [DOI] [PubMed] [Google Scholar]
- 4. Djemie T, Weckhuysen S, von Spiczak S, et al. Pitfalls in genetic testing: the story of missed SCN1A mutations. Mol Genet Genomic Med 2016;4:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marini C, Mei D, Temudo T, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia 2007;48:1678–1685. [DOI] [PubMed] [Google Scholar]
- 6. Bagnall RD, Crompton DE, Petrovski S, et al. Exome‐based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol 2016;79:522–534. [DOI] [PubMed] [Google Scholar]
- 7. Friedman D, Kannan K, Faustin A, et al. Cardiac arrhythmia and neuroexcitability gene variants in resected brain tissue from patients with sudden unexpected death in epilepsy (SUDEP). NPJ Genom Med 2018;3:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leu C, Balestrini S, Maher B, et al. Genome‐wide polygenic burden of rare deleterious variants in sudden unexpected death in epilepsy. EBioMedicine 2015;2:1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tu E, Bagnall RD, Duflou J, Semsarian C. Post‐mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol 2011;21:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldman AM, Behr ER, Semsarian C, et al. Sudden unexpected death in epilepsy genetics: molecular diagnostics and prevention. Epilepsia 2016;57:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goldman AM, Glasscock E, Yoo J, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Science Translational Medicine 2009;1(2):2ra6–2ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Auerbach DS, Jones J, Clawson BC, et al. Altered cardiac electrophysiology and SUDEP in a model of Dravet syndrome. PLoS One 2013;8:e77843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kalume F, Westenbroek RE, Cheah CS, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 2013;123:1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim Y, Bravo E, Thirnbeck CK, et al. Severe peri‐ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 2018;128:1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Delogu AB, Spinelli A, Battaglia D, et al. Electrical and autonomic cardiac function in patients with Dravet syndrome. Epilepsia 2011;52(Suppl 2):55–58. [DOI] [PubMed] [Google Scholar]
- 16. Ergul Y, Ekici B, Tatli B, et al. QT and P wave dispersion and HR variability in patients with Dravet syndrome. Acta Neurol Belg 2013;113:161–166. [DOI] [PubMed] [Google Scholar]
- 17. Lyu SY, Nam SO, Lee YJ, et al. Longitudinal change of cardiac electrical and autonomic function and potential risk factors in children with Dravet syndrome. Epilepsy Res 2019;152:11–17. [DOI] [PubMed] [Google Scholar]
- 18. Myers KA, Bello‐Espinosa LE, Symonds JD, et al. Heart rate variability in epilepsy: a potential biomarker of sudden unexpected death in epilepsy risk. Epilepsia 2018;59:1372–1380. [DOI] [PubMed] [Google Scholar]
- 19. Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 1996;93(5):1043–1065. [PubMed] [Google Scholar]
- 20. Elming H, Holm E, Jun L, et al. The prognostic value of the QT interval and QT interval dispersion in all‐cause and cardiac mortality and morbidity in a population of Danish citizens. Eur Heart J 1998;19:1391–1400. [DOI] [PubMed] [Google Scholar]
- 21. Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc Res 2005;67:367–378. [DOI] [PubMed] [Google Scholar]
- 22. Rijnbeek PR, Witsenburg M, Schrama E, et al. New normal limits for the paediatric electrocardiogram. Eur Heart J 2001;22:702–711. [DOI] [PubMed] [Google Scholar]
- 23. Aytemir K, Maarouf N, Gallagher MM, et al. Comparison of formulae for heart rate correction of QT interval in exercise electrocardiograms. Pacing Clin Electrophysiol 1999;22:1397–1401. [DOI] [PubMed] [Google Scholar]
- 24. Moseley BD, Wirrell EC, Nickels K, et al. Electrocardiographic and oximetric changes during partial complex and generalized seizures. Epilepsy research. 2011;95:237–245. [DOI] [PubMed] [Google Scholar]
- 25. Pan J, Tompkins WJ. A real‐time QRS detection algorithm. IEEE Trans Biomed Eng 1985;32:230–236. [DOI] [PubMed] [Google Scholar]
- 26. Tavares C, Martins RC, Laranjo S, Rocha I. Computational tools for assessing cardiovascular variability. EMBS/IEEE 2011. 10.1109/ENBENG.2011.6026082, E‐ISBN : 978‐1‐4577‐0521‐2. [DOI]
- 27. Toth V, Hejjel L, Fogarasi A, et al. Periictal heart rate variability analysis suggests long‐term postictal autonomic disturbance in epilepsy. Eur J Neurol 2010;17:780–787. [DOI] [PubMed] [Google Scholar]
- 28. Harden C, Tomson T, Gloss D, et al. Practice guideline summary: Sudden unexpected death in epilepsy incidence rates and risk factors: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 2017;88:1674–1680. [DOI] [PubMed] [Google Scholar]
- 29. Hesdorffer DC, Tomson T, Benn E, et al. Combined analysis of risk factors for SUDEP. Epilepsia. 2011;52:1150–1159. [DOI] [PubMed] [Google Scholar]
- 30. Elger CE, Hoppe C. Diagnostic challenges in epilepsy: seizure under‐reporting and seizure detection. Lancet Neurol 2018;17:279–288. [DOI] [PubMed] [Google Scholar]
- 31. Brotherstone R, Blackhall B, McLellan A. Lengthening of corrected QT during epileptic seizures. Epilepsia. 2010;51:221–232. [DOI] [PubMed] [Google Scholar]
- 32. Moseley BD, Britton JW. Peri‐ictal QTc changes are not associated with hypoxemia. Epilepsy Res 2014;108:982–985. [DOI] [PubMed] [Google Scholar]
- 33. Johnson JN, Ackerman MJ. QTc: how long is too long? Br J Sports Med 2009;43:657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roche F, Reynaud C, Pichot V, et al. Effect of acute hypoxia on QT rate dependence and corrected QT interval in healthy subjects. Am J Cardiol 2003;91:916–919. [DOI] [PubMed] [Google Scholar]
- 35. Seyal M, Pascual F, Lee CY, et al. Seizure‐related cardiac repolarization abnormalities are associated with ictal hypoxemia. Epilepsia 2011;52:2105–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Goldenholz DM, Kuhn A, Austermuehle A, et al. Long‐term monitoring of cardiorespiratory patterns in drug‐resistant epilepsy. Epilepsia 2017;58:77–84. [DOI] [PubMed] [Google Scholar]
- 37. van der Lende M, Surges R, Sander JW, Thijs RD. Cardiac arrhythmias during or after epileptic seizures. J Neurol Neurosurg Psychiatry 2016;87:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Seo MH, Sung WY. A case of near‐sudden unexpected death in epilepsy due to ventricular fibrillation. Open Access Emerg Med 2019;11:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuttab HI, Harris EA, Tataris KL, et al. Cardiac arrhythmia following an epileptic seizure. Clin Pract Cases Emerg Med 2019;3:354–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bardai A, Lamberts RJ, Blom MT, et al. Epilepsy is a risk factor for sudden cardiac arrest in the general population. PLoS One 2012;7:e42749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lamberts RJ, Blom MT, Wassenaar M, et al. Sudden cardiac arrest in people with epilepsy in the community: circumstances and risk factors. Neurology 2015;85:212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheah CS, Yu FH, Westenbroek RE, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 2012;109:14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frasier CR, Zhang H, Offord J, et al. Channelopathy as a SUDEP biomarker in Dravet syndrome patient‐derived cardiac myocytes. Stem Cell Reports 2018;11:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Lange IM, Koudijs MJ, van 't Slotet al. Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia. 2018;59:690–703. [DOI] [PubMed] [Google Scholar]
- 45. Meng H, Xu HQ, Yu L, et al. The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum Mutat 2015;36:573–580. [DOI] [PubMed] [Google Scholar]
- 46. van der Lende M, Hesdorffer DC, Sander JW, Thijs RD. Nocturnal supervision and SUDEP risk at different epilepsy care settings. Neurology 2018;91:e1508–e1518. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. SCN1A variants of the 45 subjects in the Dravet syndrome group.
Table S2. Antiseizure medication types and the presence of peri‐ictal QTc prolongation ≥60 ms and postictal bradycardia in ≥1 of the recorded seizures, of Dravet syndrome cases, and historical epilepsy controls.