

Abstract
We report four patients from two families who presented attacks of childhood‐onset episodic ataxia associated with pathogenic mutations in the FGF14 gene. Attacks were triggered by fever, lasted several days, and had variable frequencies. Nystagmus and/or postural tremor and/or learning disabilities were noticed in individuals harboring FGF14 mutation with or without episodic ataxia. These cases and literature data delineate the FGF14‐mutation‐related episodic ataxia phenotype: wide range of age at onset (from childhood to adulthood), variable durations and frequencies, triggering factors including fever, and association to chronic symptoms. We propose to add FGF14‐related episodic ataxia to the list of primary episodic ataxia as Episodic Ataxia type 9.
Introduction
Hereditary cerebellar ataxias constitute a large, heterogeneous group of neurological diseases presenting as a cerebellar syndrome, variably combining gait alteration, limb incoordination, dysarthria, and eye movement abnormalities. FGF14 mutations have been identified as a rare cause of autosomal dominant spinocerebellar ataxia type 27 (SCA27). SCA27 is characterized by gait and limb ataxia, dysarthria, and nystagmus; tremor is often associated and may be the presenting manifestation. Other symptoms, including orofacial dyskinesias, psychiatric manifestations, cognitive delay, or parkinsonism are common. Symptom onset occurs early in life nevertheless the disease course is usually very slow and motor function is maintained through life in most of the patients. 1 More recently, animal model studies and genome‐wide association studies suggested that FGF14 may be a risk factor for various neuropsychiatric diseases including depression addiction and schizophrenia, as well as neurodegenerative diseases, 2 , 3 FGF14 encodes fibroblast growth factor 14 highly expressed in the brain and especially in Purkinje cells, where it interacts with voltage‐gated Na+ channels to regulate neuronal excitability. FGF14 also regulates synaptic transmission from granule cells to Purkinje cells and plays a role in synaptic plasticity and neurogenesis in the hippocampus. 3 , 4 The occurrence of episodic ataxia (EA) has been occasionally associated with FGF14 mutations. 5 , 6 , 7 , 8 , 9 Here, we report four patients from two families harboring FGF14 mutations with a peculiar phenotype including the variable combination of fever‐triggered EA and permanent mild neurological symptoms.
Material and Methods
Standard protocol approvals, registrations and patient consents
The study was carried out in accordance with the Declaration of Helsinki and was approved by the local ethical committee. Written informed consent was obtained from the patient's legal representatives.
Genetic analysis
Patient IV‐10 underwent Sanger sequencing for CACNA1A, KCN1A, PRRT2, and FGF14. Index case from family B was analyzed using a panel of genes known to be associated with episodes of ataxia (CACNA1A, KCNA1, CACNB4, SLC1A3, FGF14, SLC2A1, ATP1A3, PRRT2). Sanger sequencing was used to confirm the variant in this patient and to test the relatives of patient IV‐10.
Results
Family A
Patient IV‐10 was the index case of a large family previously briefly published (Fig. 1A). 10 Pregnancy and delivery were uneventful. Psychomotor achievements were normal (head control acquired at 3 months, independent walking at 15 months, first sentences at 18 months). He was diagnosed with hyperopia at 20 months.
Figure 1.
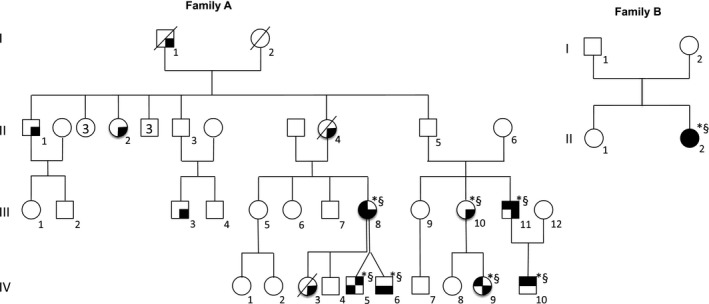
Pedigrees of families A and B. Black upper right quadrant: episodic ataxia; black upper left quadrant: nystagmus; black lower right quadrant: tremor; black lower left quadrant: developmental delay/learning difficulties; *: mutation carrier; §: clinical examination performed by a neurologist, a neuro pediatrician or a geneticist.
At 24 months, a multidirectional nystagmus was noticed. Shortly after, the patient exhibited a first episode of vertigo with nausea, vomiting, and unsteady gait during a febrile illness; it lasted two days then completely resolved. Attacks of ataxia during fever recurred three to four times a year. The last episode occurred at age 4, consisting of severe ataxia with an inability to walk and dizziness for almost a week. Between the attacks, clinical examination only disclosed multidirectional nystagmus without a tremor. At age 5, clinical examination, academic achievements and cognitive assessment were normal.
Patient III‐11 was patient IV‐10's father. At 8 years he exhibited an episode of vertigo with inability to walk and dizziness, lasting one week and triggered by a febrile illness; the second episode of gait unsteadiness occurred at 31 years during a period of intense stress. Nystagmus was diagnosed in infancy. He reported having tremor of the upper limbs (UL) from childhood, without functional impairment, and was never referred for this symptom. He succeeded in university studies. Clinical examination at 37 years disclosed horizontal nystagmus and mild UL rest tremor.
Patient IV‐5 was the second cousin of patient IV‐10. He was born at 36 weeks of gestation, from a dizygotic twin pregnancy. Birth parameters were normal. At 3 months, he exhibited two accesses of breath‐holding spells. He experienced a first episode of acute ataxia at 2.8 years, after a febrile illness; it lasted 2 days, and then the patient regained prior neurological function. After this event, the mother noticed monthly paroxysmal attacks of gait disorders. They were never observed by a physician, they were described by the mother as gait unsteadiness with bizarre posture of the left limb, triggered by fatigue or intercurrent illness, lasting several hours and resolving spontaneously. These episodes did not recur after the age of 5.5. The patient walked unaided at 14 months; language delay and behavioral disturbances (poor attention, sleep difficulties) were noticed from the age of 36 months. Clinical examination at 5 years disclosed very discrete action tremor. At last follow‐up (6.5 years), neurological examination showed coordination disabilities without tremor. The boy exhibited gross and fine motor skills difficulties (unable to get dressed or cut his food alone, unable to ride a bicycle, very poor graphism abilities). He could associate a few words but not form full sentences. He benefited from language and psychomotor rehabilitation from age 3. Due to learning difficulties, he underwent a special educational program.
Patient IV‐6 was the twin brother of patient IV‐5. Birth parameters were normal. He managed to walk alone at 14 months; language was delayed. At 6.5 years of age, he exhibited gross and fine motor skills difficulties, discrete UL postural tremor with coordination difficulties, language disabilities related to expressive dysphasia, poor attention span, but displayed no ataxia and was less severely affected than his brother. He also benefited from language and psychomotor rehabilitation and special educational program. He never experienced paroxysmal neurological episode.
Their mother (Patient III‐8) reported UL tremor from childhood without functional impairment, and she never sought medical advice concerning this tremor. She never exhibited paroxysmal neurological episode. She experienced learning disabilities and reached the middle school level. Clinical examination at 45 years disclosed mild symmetric postural tremor and nystagmus.
Patient IV‐9 had a normal psychomotor development. Minutes‐lasting episodes of spontaneously resolving lower limb hypertonia were reported between 7 and 9 months of age. Nystagmus was reported from the first months of life, and UL postural tremor was noticed during the second year, associated with fine motor skills disturbances.
Her mother (Patient III‐10) exhibited UL postural tremor from childhood.
Genetic analysis identified the variant NM_004115.3:c.439G>T leading to a stop mutation p.Glu147Ter in FGF14 gene in the index case and patients III‐11, IV‐5, IV‐6, III‐8, IV‐9, and III‐10. Other family members were reported having hand tremor from childhood, but genetic testing was not performed (Fig. 1A).
Family B
Patient II‐2 exhibited delayed motor and language skills (Fig. 1B). Vertical nystagmus was noticed at one year, and UL tremor was noticed soon after. At 2 years she exhibited a first access of severe ataxia with inability to walk and sit unaided, triggered by a febrile illness; after one week she regained her previous neurological status. Such episodes, always triggered by fever, recurred twice a year until the last follow‐up (6.3 years). Clinical examination then disclosed nystagmus, discrete UL postural and action tremor, and coordination difficulties. Mild learning difficulties were reported, and she benefited from language rehabilitation. She was described as very smiley and communicative with her peers.
Genetic analysis identified the variant NM_004115.3:c.486_487del leading to a premature stop codon p.Tyr162Ter in FGF14 gene. Due to parental child neglect, she was placed in foster care from the age of 7 months, and the parents were not available for analysis.
Discussion
We report four patients harboring a dominant FGF14 mutation who presented prolonged episodes of seldom‐recurring, fever‐triggered EA with childhood onset (Table 1). EA are rare genetic disorders characterized by brief recurrent attacks of ataxia, sometimes associated with other ictal symptoms (tremor, dysarthria, vomiting, vertigo, headache) and typically a normal interictal neurological examination. Triggering factors in episodic ataxia include physical and psychological stress, startle, sudden movements, fatigue, caffeine, alcohol, or fever. 11 , 12 , 13 Eight genetically distinct subtypes have been described (Table 2).
Table 1.
Main clinical features of the patients harboring FGF14 mutation.
| Family | Family A | Family B | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient | IV‐10 | III‐11 | IV‐5 | IV‐6 | III‐8 | IV‐9 | III‐10 | patient I‐2 | |
| Sex/age at last follow‐up (years) | M/5 | M/37 | M/6.5 | M/6.5 | F/45 | F/3 | F/NK | F/6.3 | |
| Acute episodes of ataxia | Yes | Yes | Yes | No | No | No | No | Yes | |
| Age at onset (years) | 2 | 8 | 2.8 | 2 | |||||
| Triggering factor | Febrile illness | Febrile illness, stress | Febrile illness | Febrile illness | |||||
| Duration | 2–7 days | 7 days | 2 days | 7 days | |||||
| Total number |
8 No recurrence after age 4 |
2 | 1 | Twice a year until last follo‐up | |||||
| Symptoms | Vertigo, ataxia, nausea, vomiting, occacional confusion | Dizziness, ataxia | Ataxia | Severe ataxia | |||||
| Other paroxysmal signs | No | No | 2 access of breath holding spell at 3 months; between 2.8 and 5.5 years, daily access of gait unsteadiness with bizarre posture of the left limb lasting several hours | No | No | Brief episodes of lower limb hypertonia lasting some minutes between 7 and 9 months of age | No | No | |
| Permanent symptoms | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Tremor (age at onset) | No | UL postural tremor (from childhood) | No | UL postural tremor (from childhood) | UL postural tremor (from childhood) | UL postural tremor (from 2.5 years) | UL postural tremor (from childhood) | UL postural tremor (from infancy) | |
| Nystagmus (age at onset) | Yes (from 24 months) | Yes (from infancy) | No | No | Yes | Yes (from infancy) | No | Yes (from infancy) | |
| Permanent ataxia | No | No | No | No | No | No | No | No | |
| Learning difficulties | No | No | Yes | Yes | Yes | No | NK | Yes | |
| Psychomotor development | Normal | Normal | Language delay | Language delay | Normal | Normal | NK | Global delay | |
| Paraclinical investigations (age) | Blood electrolytes, lactate, aminoacid, ammonemia, acylcarnitine dosage, urinary organic acid screening, cerebrospinal fluid analysis, auditory and vestibular assessment, brain MRI: normal (2 years) | None |
Auditory and vestibular assessment, CGH‐array, FMR1 gene analysis, brain MRI normal (3.3 years) |
None | None | None | None |
EEG, brain MRI, ophthalmological assessment normal (1.2 years) |
|
UL, upper limbs; M, male; F, female; NK, not known.
Table 2.
Cardinal features of the various subtypes of EA. Clinical features of childhood‐onset EA related to FGF14 mutation are emphasized in bold characters.
| EA type (gene) |
EA1 (KCNA1) |
EA2 (CACNA1A) |
EA3 (12q42, no gene) |
EA4 (No gene‐ |
EA5 (CACNB4) |
EA6 (SLC1A3) |
EA7 (No gene) |
EA8 (UBR4) |
EA9 (FGF14) |
|---|---|---|---|---|---|---|---|---|---|
| Age at onset (years) | Childhood | Childhood or adolescence | 1–40 | 20–50 | 20–60 | From infancy to adulthood | <20 | Infancy | From childhood to adulthood |
| Attacks duration | Seconds to minutes | Hours to days | Minutes to hours | Brief | Hours | Hours to days | Hours to days | Minutes to hours | Seconds to several days |
| Triggering factors | Movement, stress, fatigue, caffeine, alcohol | Exertion, fatigue, stress | Stress, fatigue, sudden changes in head position | Changes in head position, fatigue, environmental motion | Fever, stress, heat, smoking | Excitement, exercise | Anxiety, cold, stress | Fever, stress, physical activity | |
| Interictal manifestations | Myokymia | Nystagmus | Myokymia | Nystag‐mus, abnormal smooth pursuit | Nystag‐mus, ataxia | Nystagmus, ataxia | None | Nystag‐mus, ataxia, myoky‐mia, tremor | Nystagmus, upper limb postural and action tremor |
| Additional features/atypical manifestations | Epileptic seizures, myokimia, prolonged attacks,rigidity and cataplexy, deafness, distal weakness | Other forms of episodic neurological syndroms, permanent ataxia,developmental delay,cerebellar atrophy | Epileptic seizures | Epileptic seizures | None | Cognitive deficit,hemiplegic migraine | None | None | Develop‐mental delay, Learning disabilitiesP Paroxysmal dyskinesia |
| Follow‐up | Improve‐ment with age | Improvement after acetozo‐lamide | Poor therapeutic response | Poor therapeutic response | Poor therapeutic response | Variable response to acetazo‐lamide | Poor therapeutic response | Poor therapeutic response | Variable Improvement with age |
NK, not known.
EA associated with FGF14 mutations have been previously reported in 8 patients from 5 families 5 , 6 , 7 , 8 , 9 , 14 (Table 3). Literature analysis and the precise clinical description of our four patients allow better delineation of the phenotype of EA related to autosomal dominant FGF14 mutations: (i) wide range of age at onset (early childhood such as in our patients, to adulthood), (ii) variable triggering factors, especially fever (7/10 patients with available data), (iii) variable duration with long‐lasting attacks not uncommon (often mimicking febrile cerebellitis), (iv) variable frequency (ranging from limited number of episodes with spontaneous improvement before the teens to weekly access), (v) associated to chronic upper limb tremor and/or nystagmus, (vi) pharmacological responsiveness still unknown (improvement after acetazolamide treatment in 2 patients). 9 (vii) genetic heterogeneity including point mutations and gene deletions. The FGF14 mutation was also identified in family members exhibiting a milder phenotype (isolated nystagmus or upper limb postural tremor with childhood‐onset, without EA). Patients IV‐5 and IV‐9 exhibited paroxysmal manifestations suggesting paroxysmal dyskinesia, as previously described in one case 15 ; some of these episodes spontaneously resolved in the first years of life, which is unusual compared to other forms of PD. 13
Table 3.
Main clinical features of patients with paroxysmal neurological manifestations (episodic ataxia, paroxysmal dyskinesia) harboring FGF14 mutation previously reported in the literature.
| Patient |
Choquet Patient 1 |
Choquet Patient 2 |
Choquet Patient 3 |
Amado Patient 1 & 2 |
Choi |
Coebergh patient 1 |
Schesny | Shimojima | |
|---|---|---|---|---|---|---|---|---|---|
| Chromosomal abnormality/FGF14 gene mutation |
NM_004115:c.211_212insA/p.(Ile71Asnfs*27) |
Deletion of 424 kb on chromosome 13q33.1 including FGF14 | NM_175929:c.31A>/p.(Thr11Ala) | Deletion of 202 kb on chromosome 13q33.1 including exons 1‐4 of FGF14 |
NM_175929:c.208+1G>A |
Translocation t(13;21)(q32;q22.3) disrupting FGF14 | |||
| Sex/age at last follow‐up (years) | M/31 | M/NK | F/NK | F/NK | M/46 | M/6 | M/twenties | M/6 | |
| Acute episodes | Episodic ataxia | Episodic ataxia | Episodic ataxia | Episodic ataxia | Episodic ataxia | Episodic ataxia | Episodic ataxia | Paroxysmal dyskinesia | |
| Age at onset | 26 years | NK | NK | Childhood | 39 years | Childhood | Twenties | 8 months | |
| Triggering factor | None | Fatigue exercise | NK | Fever | NK | Fever | High emotional stress levels, physical activity, certain body positions (e.g., bending forward), and caffeine intake | Crying | |
| Duration | NK | 20 min | NK | NK | Hours | NK | Min to hours | 5 min | |
| Frequency (or total number) | NK | 4 times per week | Rare | NK | NK | (3 episodes) | 4 times per month | Several per week | |
| Symptoms | Incoor‐dination, unsteady gait, vertical oscillopsy, dysarthria, headache | Dysarthria, unsteady gait, and diplopia | Vertigo dysatrhia | Ataxia | Dizziness, headache | Ataxia | Intense vertigo, dizziness, nausea, dysphagia, diplopia | Attacks of choreic movements | |
| Treatment | Acetazo‐lamide discontinued due to adverse effects | No | No | No | Response to acetazolamide | No | Improvement after acetazolamide | Valproic acid and phenobarbital non effective | |
| Other paroxysmal signs | Attacks of right upper limb dystonia | No | Head‐ache | No | No | No | No | Breath holding spells from 9 months | |
| Permanent symptoms | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | |
| Tremor | Yes (From age 29) | No | No | Yes | No | No | UL postural tremor | ||
| Nystagmus | Yes (From age 29) | Yes | Yes | Yes | Yes | Yes | Yes | ||
| Ataxia | Yes (From age 29) | No | No | Yes | No | Yes (From age 2) | Slight | ||
| Learning difficulties | NK | NK | NK | Yes | No | Yes | No | Yes (mental al disability) | |
| Psycho‐motor develop‐ment | NK | NK | NK | NK | Normal | Delayed | Delayed | Mental deficiency | |
NK, not known; UL, upper limb.
FGF14 regulates the Cav2.1 presynaptic channels by modulating the current and the vesicular recycling. 4 EA could result from this dysregulation, at least partially. FGF14 also directly regulates the function of Nav1.2 and Nav1.6 channels at the axon initial segment. NaV1.2 is the main channel subtype implicated in the mediation of fever‐induced neuronal hyperexcitability. 16 This could explain why fever is a triggering factor in FGF14‐mutated patients.
Some genes involved in progressive ataxia may also be responsible for episodic neurological manifestations: recently, permanent or even progressive neurological signs including ataxia, combined with various types of other episodic syndromes have been reported in patients with the so‐called « episodic ataxia» (linked to CACNA1A, PRRT2, SLC2A1). 11 , 13 , 17 FGF14‐associated phenotypes emphasize this overlap between progressive and episodic ataxias, with phenotypic continuum encompassing chronic (SCA27) and paroxysmal ataxia. Learning difficulties in most of our patients and in previously published cases strongly enlarge the implication of FGF14 mutation in developmental disabilities 6 , 7 , 15 and is concordant with the putative role of FGF14 in behavioral, cognitive, and psychiatric disturbances. 2 , 3
We propose that fever‐triggered EA associated with upper limb tremor in the patients and/or in relatives suggest FGF14 involvement; genetic analysis (targeted gene or panel gene testing and if negative, gene deletion testing) establishes the diagnosis. We propose to add FGF14‐related EA to the list of primary EAs as “type 9 episodic ataxia.”
Conflicts of Interest
The authors declare that they have no conflict of interest related to the research covered in the article.
Acknowledgments
We acknowledge patients and their families for participating in the study. We thank Dr Lagavulin for his helpful discussions.
This study was supported by Clinical Research Hospital Program from the French Ministry of Health, year 2010 (Project number 14‐12).
As the corresponding author, Agathe Roubertie takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding Information
This study was supported by Clinical Research Hospital Program from the French Ministry of Health, year 2010 (Project number 14‐12).
Funding Statement
This work was funded by Clinical Research Hospital Program from the French Ministry of Health grant 14‐12.
References
- 1. Groth CL, Berman BD. Spinocerebellar Ataxia 27: a review and characterization of an evolving phenotype. Tremor Other Hyperkinet Mov (N Y) 2018;8:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoxha E, Marcinnò A, Montarolo F, et al. Emerging roles of Fgf14 in behavioral control. Behav Brain Res 2019;356:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Di Re J, Wadsworth PA, Laezza F. Intracellular fibroblast growth factor 14: emerging risk factor for brain disorders. Front Cell Neurosci 2017;11:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yan H, Pablo JL, Pitt GS. FGF14 regulates presynaptic Ca2+ channels and synaptic transmission. Cell Rep 2013;4:66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Choquet K, La Piana R, Brais B. A novel frameshift mutation in FGF14 causes an autosomal dominant episodic ataxia. Neurogenetics 2015;16:233–236. [DOI] [PubMed] [Google Scholar]
- 6. Coebergh JA, Fransen van de Putte DE, Snoeck IN, et al. A new variable phenotype in spinocerebellar ataxia 27 (SCA 27) caused by a deletion in the FGF14 gene. Eur J Paediatr Neurol 2014;18:413–415. [DOI] [PubMed] [Google Scholar]
- 7. Amado A, Blanco MO, Repáraz‐Andrade A. Spinocerebellar Ataxia 27: clinical phenotype of twin sisters with FGF14 deletion. Neuropediatrics 2017;48:131–131. [DOI] [PubMed] [Google Scholar]
- 8. Choi K‐D, Kim J‐S, Kim H‐J, et al. Genetic variants associated with episodic ataxia in Korea. Sci Rep 2017;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schesny M, Joncourt F, Tarnutzer AA. Acetazolamide‐responsive episodic ataxia linked to novel splice site variant in FGF14 gene. Cerebellum 2019;18:649–653. [DOI] [PubMed] [Google Scholar]
- 10. Humbertclaude V, Krams B, Nogue E, et al. Benign paroxysmal torticollis, benign paroxysmal vertigo, and benign tonic upward gaze are not benign disorders. Dev Med Child Neurol 2018;55:1358. [DOI] [PubMed] [Google Scholar]
- 11. Méneret A, Roze E. Paroxysmal movement disorders: an update. Rev Neurol (Paris) 2016;172:433–445. [DOI] [PubMed] [Google Scholar]
- 12. McGovern EM, Roze E, Counihan TJ. The expanding spectrum of paroxysmal movement disorders. Curr Opin Neurol 2018;31:491–497. [DOI] [PubMed] [Google Scholar]
- 13. De Gusmao CM, Silveira‐Moriyama L. Paroxysmal movement disorders ‐ practical update on diagnosis and management. Expert Rev Neurother 2019;19:807–822. [DOI] [PubMed] [Google Scholar]
- 14. Pablo JL, Pitt GS. FGF14 is a regulator of KCNQ2/3 channels. Proc Natl Acad Sci USA 2017;114:154–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shimojima K, Okumura A, Natsume J, et al. Spinocerebellar ataxias type 27 derived from a disruption of the fibroblast growth factor 14 gene with mimicking phenotype of paroxysmal non‐kinesigenic dyskinesia. Brain Dev 2012;34:230–233. [DOI] [PubMed] [Google Scholar]
- 16. Ye M, Yang J, Tian C, et al. Differential roles of NaV1.2 and NaV1.6 in regulating neuronal excitability at febrile temperature and distinct contributions to febrile seizures. Sci Rep 2018;8:753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Humbertclaude V, Riant F, Krams B, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol 2020;62:330–337. [DOI] [PubMed] [Google Scholar]