

Abstract
Objective
Multiple sclerosis (MS) is a heterogenous, inflammatory disease of the central nervous system. Microbiota alterations in MS versus healthy controls (HC) are observed, but results are inconsistent. We studied diversity, enterotypes, and specific gut microbial taxa variation between MS and HC, and between MS subgroups.
Methods
Amplicon sequencing of the 16S ribosomal RNA V4 region (Illumina MiSeq) was used to evaluate alpha and beta diversity, enterotypes, and relative taxa abundances on stool samples. MS subgroups were based on phenotype, disease course modifiers, and treatment status. Results were controlled for recently identified confounders of microbiota composition.
Results
Ninety‐eight MS patients and 120 HC were included. Microbial richness was lower in interferon‐treated (RRMS_I, N = 24) and untreated relapsing–remitting MS during relapse (RRMS_R, N = 4) when compared to benign (BMS, N = 20; Z = −3.07, Pcorr = 0.032 and Z = −2.68, Pcorr = 0.055) and primary progressive MS (PPMS, N = 26; Z = −2.39, Pcorr = 0.062 and Z = −2.26, Pcorr = 0.071). HC (N = 120) and active untreated MS (RRMS_U, N = 24) showed intermediate microbial richness. Enterotypes were associated with clinical subgroups (N = 218, χ 2 = 36.10, P = 0.002), with Bacteroides 2 enterotype being more prevalent in RRMS_I. Butyricicoccus abundance was lower in PPMS than in RRMS_U (Z = −3.00, Pcorr = 0.014) and BMS (Z = −2.56, Pcorr = 0.031), lower in RRMS_I than in BMS (Z = −2.50, Pcorr = 0.034) and RRMS_U (Z = −2.91, Pcorr = 0.013), and inversely correlated with self‐reported physical symptoms (rho = −0.400, Pcorr = 0.001) and disease severity (rho = −0.223, P = 0.027).
Interpretation
These results emphasize the importance of phenotypic subcategorization in MS‐microbiome research, possibly explaining previous result heterogeneity, while showing the potential for specific microbiome‐based biomarkers for disease activity and severity.
Introduction
Multiple sclerosis (MS) is the most common chronic inflammatory disease of the central nervous system (CNS). 1 Patients experience a variety of physical and cognitive symptoms, including bowel dysfunction (>70% 2 ). Depending on relapses and/or magnetic resonance imaging (MRI) activity, MS is considered active or nonactive 3 . Relapses are presumably provoked by adaptive immune cells infiltrating the CNS, resulting in focal inflammation and myelin loss. 4 Disability accumulation independent of relapses is suggestive of progression, 3 mainly due to axonal degeneration. 5 Gene–environment interactions are thought to be involved. A modulating role of the gut microbiota has been proposed because of its potential to influence brain development/physiology, regulate immunity, 6 , 7 and increase the odds of murine CNS‐specific autoimmune disease. 8 , 9
Relapsing–remitting (RR)MS patients were repeatedly shown to have an altered microbiota composition from healthy controls (HC); however, the identified taxa differed between studies. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 Most studies were small scale and accounted for few confounders. 10 , 14 , 16 However, we recently identified >60 covariates affecting microbial composition, 17 among which stool consistency had the largest effect size on primary microbiome markers. 17 , 18 Although bowel dysfunction is prevalent in MS, this was never controlled for to date.
Controlling for such confounders, we conducted a cross‐sectional study in a cohort of MS patients and HC, including MS patients with a presumed high (active RRMS) and low degree of focal CNS inflammation (benign MS (BMS 19 , 20 ) and nonactive primary progressive (PP)MS). Subgroups were defined by MS phenotype, disease course modifiers, and treatment status. We hypothesized to detect a gut microbial community with higher pro‐inflammatory properties in MS subgroups with higher focal CNS inflammatory activity.
Materials and Methods
Study population
Patients with MS (2010 McDonald criteria 21 ) were recruited in the National MS Center (NMSC), Melsbroek (in‐ and outpatients) and in the University Hospital, Brussels (outpatients) according to group‐specific in‐ and exclusion criteria (Table 1). Five MS subgroups were predefined, including three subgroups with a presumed high degree of focal CNS inflammation: untreated active RRMS (RRMS_U); untreated RRMS with a relapse at the time of sampling (RRMS_R); and interferon (IFN) treated RRMS (RRMS_I). Relapse was defined as new neurological symptoms lasting ≥24 h, without fever or other triggers. 22 We selected one class of immune‐modulatory drugs (IMD) to obtain a homogenous treatment group. IFN‐beta was chosen because injectables are nonaggressive, spare the host’s systemic immunity (less bias by a direct immune effect on the gut microbiota as opposed to oral therapy), and limit contact with a hospital environment (less bias compared to regular visits for intravenous therapy). The two subgroups with a presumed low degree of focal CNS inflammation were progressive onset MS without relapses 2 years prior (PPMS) and RRMS with ≤3 on the Expanded Disability Status Scale (EDSS) after a disease duration of ≥15 years (BMS). 19 , 23 We excluded patients with secondary progressive MS, due to the difficulty to separate inflammation from neurodegeneration. HC were recruited among participants’ proxies, the HC database of the Metabolic Department in the University Hospital Brussels, and (para)medical staff in the same geographical regions as MS participants. The HC group was selected to represent age and sex distributions in the whole MS study population. Samples from the Flemish Gut Flora Project (FGFP 17 ), an independent, large, and diverse population study on the gut microbiome in Flanders, were added to this HC group and matched for age, sex, BMI, and the Bristol Stool Scale (BSS 24 ) against the entire MS population. This allowed to project our results on an independent and representative sample in the same geographical area. The ethics committee of the University Hospital, Brussels, and the local ethics committee of the NMSC, Melsbroek, approved the study (B.U.N. 143201317985), which is in compliance with the principles of the 2013 Declaration of Helsinki. All participants provided written informed consent.
Table 1.
In‐ and exclusion criteria. Untreated groups were untreated for at least 3 months before inclusion, irrespective of any treatment before this interval.
| Inclusion criteria |
| Untreated active RRMS (RRMS_U) |
| At least one clinical relapse during 2 years prior to screening, or at least one active, contrast‐enhancing lesion on brain MRI in the year prior to screening |
| EDSS < 7.0 |
| Untreated active RRMS during relapse (RRMS_R) |
| Able to perform first data collection prior to corticosteroid therapy |
| EDSS < 7.0 |
| Untreated BMS (BMS) |
| EDSS ≤ 3.0 at least 15 years after first MS symptoms 19 , 20 |
| IFN treated RRMS (RRMS_I) |
| At least 3 months of stable treatment with interferon beta |
| EDSS < 7.0 |
| Non‐active PPMS (PPMS) |
| No clinical relapse within 2 years prior to screening |
| EDSS < 7.0 |
| Exclusion criteria for MS patients |
| Secondary progressive MS patients |
| Disease‐modifying treatment other than IFN at screening |
| Glatiramer acetate treatment within 3 months prior to screening |
| Fingolimod or natalizumab treatment within 6 months prior to screening |
| Systemic corticosteroid use within 2 months prior to screening |
| Gastrointestinal conditions such as IBD |
| Antibiotic use in the 4 weeks prior to screening |
| Exclusion criteria for healthy controls |
| Systemic corticosteroid use within 2 months prior to screening |
| Gastrointestinal conditions such as IBD |
| Other diseases of the central nervous system |
| Antibiotic use in the 4 weeks prior to screening |
| Direct relation to or living with a participant in one of the MS groups |
BMS, benign MS; EDSS, expanded disease status scale; IBD, inflammatory bowel disease; IFN, interferon beta; PPMS, primary progressive MS; RRMS, relapsing–remitting MS.
Study design
During study setup, sample size calculation and power analysis could not be performed because effect size estimates for the gut microbiome were lacking. Following previous recommendations, 25 , 26 we aimed to include 30 patients per subgroup (and additional FGFP samples for HC). Potential confounders were assessed following FGFP protocols, 17 covering anthropometrics, general health, medication, dietary and bowel habits, lifestyle, fertility, and MS history. Participants noted date and time of stool sampling, time since last defecation, and scored stool consistency (BSS). Participants were further assessed using Age‐Related Multiple Sclerosis Severity score (ARMSS, 27 i.e. age‐corrected EDSS ranking), Brief‐H‐Neg Hopelessness scale, 28 Brief Illness Perception Questionnaire (IPQ‐K 29 ; MS only), three‐level EuroQol Five Dimensions’ questionnaire (EQ‐5D 30 ), Fatigue Scale for Motor and Cognitive Functions (FSMC), Hospital Anxiety and Depression Scale (HADS), Hauser Ambulation index (HAI), and Timed 25 Foot Walk Test (T25FW).
The primary objective was to explore whether gut microbiome diversity (alpha and beta), enterotype distribution, and microbial genus abundances were related to MS diagnosis (MS and MS subgroups vs. all HC, respectively) and/or differences in disease course (MS subgroups). Secondary objectives were to determine which genera abundances differed according to IFN treatment (RRMS_U vs. RRMS_I), and whether certain genera were associated with disease characteristics and clinical assessments.
Stool sample collection and processing
Fecal collection kits (including instructions following established protocols 17 , 18 ) were provided during consultation. Samples were stored at −20°C immediately after sampling, in the participants’ home freezer or at the NMSC, and transferred on dry ice to −80°C within 48 h. Extraction of microbial DNA from frozen aliquots (150–200 mg) was performed using an adapted Mobio PowerMicrobiome DNA/RNA isolation Kit‐based protocol. 17 , 18 Bacterial DNA was quantified via fluorometry (Qubit, Life Technologies). The hypervariable 4 region (V4) of the 16S rRNA gene was PCR amplified (515F/806R primer set 31 ), with Illumina sequencing adaptors and dual‐index barcodes to generate dual‐barcoded libraries. PCR amplicon quantification was carried out with Fragment Analyzer (Advanced Analytical Technologies) and sequencing on the Illumina MiSeq platform (MiSeq V2 sequencing kit), generating 250 bp paired‐end reads. Following demultiplexing of Illumina sequencing data, fastq sequences were merged with FLASH software version (v) 1.2.10. 32 Sequences with a quality score <25 (>90% of read length) were excluded from analysis (FASTX‐Toolkit v0.0.14, http://hannonlab.cshl.edu/fastx_toolkit/). Chimeric sequences were removed using the UCHIME algorithm in USEARCH v6.0.307. 33 Taxonomical sequence assignment was performed using the Ribosomal Database Project (RDP) classifier v2.12. 34 Phylum‐to‐genus matrices were created using custom Perl scripts. Samples were rarefied to 10,000 randomly selected reads.
Statistical analysis
Data analysis was performed in R (v3.3.1 35 ). After normality testing (Shapiro–Wilks test), demographics and clinical data were compared between groups with one‐way ANOVA or Wilcoxon rank‐sum and Kruskal–Wallis H tests, followed by a post hoc Dunn test (FSA R package 36 ). Microbiota richness was determined using observed richness (Sobs; i.e. total number of genera detected per sample), Pielou’s evenness index (i.e. expression of how evenly genera are distributed within one sample), and Simpson’s alpha diversity index (i.e. measure of alpha diversity including genus richness and evenness) using vegan 37 and phyloseq. 38 Group differences were tested with Wilcoxon rank‐sum or Kruskal–Wallis tests (with post hoc Dunn test). Beta diversity (i.e. difference in global microbiota composition between samples) was visualized using Principal Coordinate Analysis (PCoA) on genus‐level community composition (Bray–Curtis dissimilarity). The adonis function (vegan) was used to test for differences in community structure between groups, and betadisper (vegan) to verify whether differences were due to dissimilar dispersions among groups. Variables (N = 732) were filtered by report rate in the study population (≥10%). Next, when r > |0.8| using Pearson correlation (Hmisc 39 ), the variable with the lowest effect size on genus‐level community variation (adjusted R2, vegan’s capscale) was removed. Variables with the highest effect sizes in their category were retained. The 33 remaining metadata variables were used in further analysis. Forward stepwise distance‐based redundancy analysis (RDA, vegan’s ordiR2step) was performed to determine nonredundant microbiome covariates. FGFP individuals were excluded during RDA and correlation analysis with clinical variables, because not all clinical variables used in this study were also assessed in the FGFP cohort. In all other analyses, the FGFP individuals were analyzed alongside the original HC group, whenever the HC group was included. Enterotypes were determined with Dirichlet Multinomial Mixtures (DMM, DirichletMultinomial package 40 ), using Bayesian Information Criterion (BIC) to calculate the optimal number of clusters. 41 To increase accuracy, enterotyping was performed on a combined genus abundance matrix that included samples of this study and 2999 samples from the FGFP. 17 Clusters were named Bacteroides 1 (B1), Bacteroides 2 (B2), Prevotella (P), and Ruminococcaceae (R) based on enterotype‐discriminating predominant taxa. 42 B2 has been characterized by high Bacteroides and low Faecalibacterium abundances and lowered cell density. 42 Associations between enterotypes and groups were tested using pairwise chi‐square tests (chi.sq.post.hoc, fifer package 43 ). Differences in relative genera abundances were tested using Wilcoxon rank‐sum and Kruskal–Wallis tests (with post hoc Dunn test). Genus abundance and correlation analysis were restricted to genera with a mean relative abundance ≥0.0001 (mean ≥ 1 in 10,000 reads) and present in ≥20% of samples. Taxa unclassified at genus level were excluded. Where appropriate, analyses were corrected for multiple testing (Benjamini–Hochberg (BH) procedure, 44 FDR) resulting in corrected P‐values (Pcorr). A false discovery rate (FDR) of <0.1 was considered statistically significant. Multivariate analysis was performed using nested generalized linear models (GLM, glm function). Subgroup comparisons were controlled for stool consistency (BSS), age, sex, and BMI. This was not done for the MS versus HC comparison due to matching at baseline. Adjusted P‐values (Padj) were reported. Standardized regression coefficients were calculated (lm.beta function, QuantPsyc package 45 ). Associations between genera abundances and continuous metadata (with removal of HC for MS‐specific scores) were tested using Spearman correlations.
Results
Population
From February 2014 to October 2015, 118 MS patients and 30 HC were recruited and delivered stool samples. Data from 120 participants (98 MS and 22 HC) was retained for further analysis with 18.9% of samples having insufficient quality‐checked reads. Ninety‐eight samples from the FGFP were added to the HC pool. No significant differences were found in age, sex, BMI, and BSS between overall MS and HC (Table 2). However, age, EDSS, and disease duration differed among MS subgroups. Higher age and EDSS were seen in PPMS compared to RRMS_U (Dunn test, N = 120, Z = 2.74 and 4.86, Pcorr = 0.030 and <0.001) and RRMS_I (Z = 3.16 and 3.44, Pcorr = 0.016 and 0.002), and a higher age compared to RRMS_R (Z = 2.36, Pcorr = 0.061). BMS patients had lower EDSS than PPMS and RRMS_I (Z = −5.56 and 2.25, Pcorr < 0.001 and 0.062), and longer disease duration than PPMS and RRMS_U (Z = 2.79 and 4.02, Pcorr = 0.026 and <0.001). RRMS_I had longer disease duration than RRMS_U (Z = 2.63, Pcorr = 0.029).
Table 2.
Demographics of the participants included in microbiota analysis. Pcorr = P‐value corrected for multiple testing (Benjamini–Hochberg, FDR).
| HC total | MS total | P | HC | FGFP | BMS | RRMS_U | RRMS_I | RRMS_R | PPMS | Pcorr | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | 120 | 98 | 22 | 98 | 20 | 24 | 24 | 4 | 26 | ||
| Median/mean 4 age (y) (iqr/SD) | 49.0 (14.3) | 48.0 (13.8) | >0.05 1 | 54.5 (9.0) | 48.0 (14.0) | 48.7 (7.5) | 44.5 (10.5) | 44.0 (7.9) | 41.5 (5.1) | 51.8 (8.9) | <0.001 2 |
| Female (%) | 61.7 | 60.2 | >0.05 1 | 68.0 | 60.0 | 65.0 | 62.5 | 75.0 | 50.0 | 42.3 | >0.05 3 |
| Median BMI (iqr) | 23.7 (4.0) | 23.6 (4.9) | >0.05 1 | 24.9 (4.6) | 23.5 (4.1) | 23.5 (3.4) | 23.3 (4.3) | 23.9 (5.8) | 20.9 (4.6) | 24.5 (4.8) | >0.05 3 |
| Median EDSS (iqr) | – | 3.0 (3.5) | – | – | – | 2.25 (1.0) | 2.0 (1.1) | 3.0 (2.6) | 4.5 (1.9) | 6.0 (2.4) | <0.001 3 |
| Median disease duration (y) (iqr) | – | 12.5 (12.0) | – | – | – | 18.0 (7.0) | 6.0 (7.3) | 12.5 (12.3) | 11.0 (8.3) | 8.0 (11.3) | 0.001 3 |
| Median BSS (iqr) | 4 (1.0) | 4 (1.0) | >0.05 1 | 4 (0.8) | 4 (1.0) | 4 (1.0) | 4 (2.0) | 4 (2.0) | 3 (2.3) | 4 (1.0) | – |
| Inpatient setting or working in a hospital (%) 5 | 11 (9.2) | 25 (25.5) | – | 11 (9.2) | NA | 3 (14.3) | 4 (16.7) | 5 (21.7) | 1 (25.0) | 12 (46.2) | – |
HC, healthy controls; BMS, benign multiple sclerosis; FGFP, Flemish Gut Flora Project; RRMS_U, untreated active RRMS; RRMS_I, interferon‐beta‐treated RRMS; RRMS_R, RRMS during relapse; PPMS, primary progressive MS; BMI, body mass index; EDSS, expanded disease status scale; BSS, Bristol stool score; iqr, interquartile range; SD, standard deviation; NA, not available.
Wilcoxon rank sum test with continuity correction.
One‐way ANOVA.
Kruskal–Wallis H test.
Median (interquartile range) and mean (standard deviation) values, depending on results from the Shapiro–Wilk normality test.
No data available for the FGFP controls.
Alpha and beta diversity
Sobs varied greatly within our population (Kruskal–Wallis test, N = 218, χ 2 = 19.24, Pcorr = 0.007), with a downward trend from BMS to RRMS_U, RRMS_I, and RRMS_R (Fig. 1) (Table 3). Simpson’s alpha diversity index followed a similar trend. A comparison between the original HC group and the FGFP sample is made in Figure S1. There were no significant differences in Pielou’s index between subgroups (Kruskal–Wallis test, N = 218, Pcorr > 0.5), nor in Sobs, Simpson and Pielou indices between overall MS and HC (Wilcoxon rank‐sum test, N = 218, Pcorr> 0.5). There were no significant correlations with metadata.
Figure 1.
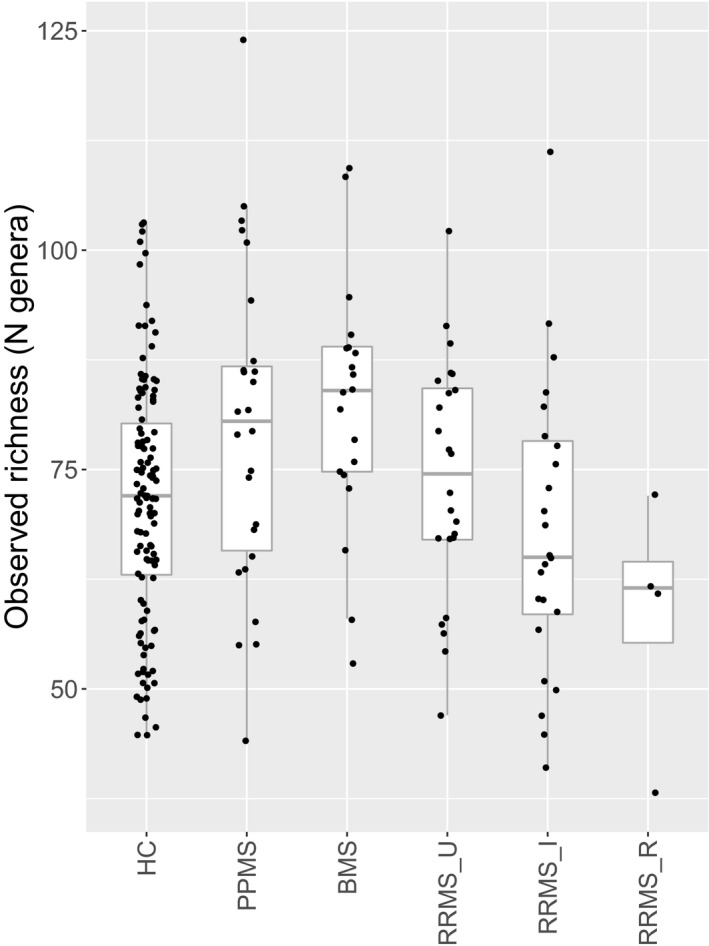
Boxplot of observed richness within patients (N = 98) and healthy controls (N = 120, including FGFP samples). Downward trend of observed richness within the relapsing–remitting patient population. The body of the boxplot represents the first and third quartiles of the distribution and median line. The whiskers extend to the last data point within 1.5 times the interquartile range. The outliers lie beyond. Individual data points (dots) may overlap. FGFP, Flemish Gut Flora Project. HC, healthy controls (N = 120). PPMS, primary progressive multiple sclerosis (N = 26). BMS, benign multiple sclerosis (N = 20). RRMS_U, untreated active RRMS (N = 24). RRMS_I, interferon‐beta‐treated RRMS (N = 24). RRMS_R, RRMS during relapse (N = 4). Kruskal–Wallis test (N = 218, χ 2 = 19.24, Pcorr = 0.007) with post hoc Dunn tests.
Table 3.
Differences in observed richness between subgroups. Only significant results are shown. Reported P‐values after Dunn test, with Benjamini–Hochberg corrected P‐values (Kruskal–Wallis test, N = 218, χ 2 = 19.24, Pcorr = 0.007).
| Comparison | N | Z‐score | Pcorr |
|---|---|---|---|
| BMS‐RRMS_I | 218 | 3.30 | 0.015 |
| BMS‐RRMS_R | 218 | 2.86 | 0.021 |
| BMS‐HC | 218 | 3.01 | 0.019 |
| PPMS‐RRMS_I | 218 | 2.55 | 0.040 |
| PPMS‐RRMS_R | 218 | 2.40 | 0.049 |
| PPMS‐HC | 218 | −2.09 | 0.092 |
HC, healthy controls (N = 120, including FGFP samples); PPMS, primary progressive multiple sclerosis (N = 26); BMS, benign multiple sclerosis (N = 20); RRMS_I, interferon‐beta‐treated RRMS (N = 24); RRMS_R, RRMS during a relapse (N = 4); P, uncorrected P‐value; Pcorr, P‐value corrected for multiple comparisons with Benjamini–Hochberg, FDR.
Global microbial composition differed between MS and HC (adonis test on beta diversity, N = 218, R2adj = 0.004, P = 0.027), and between subgroups (N = 218, R2adj = 0.016, P = 0.003). Excluding HC (N = 120), 2.4% of beta diversity variation was explained by allocation to an MS subgroup (N = 98, R2adj = 0.024, P = 0.012), largely driven by RRMS_I (N = 24). Subcutaneous IFN beta‐1‐a use (RDA, N = 120, R2adj = 0.024, Pcorr = 0.022), BSS (R2adj = 0.023, Pcorr = 0.022), subgroup (R2adj = 0.018, Pcorr = 0.092), weight (R2adj = 0.017, Pcorr = 0.022), inpatient recruitment (R2adj = 0.014, Pcorr = 0.056), mobility (25‐FWT; R2adj = 0.014, Pcorr = 0.056), sleep (R2adj = 0.012, Pcorr = 0.056), hopelessness (R2adj = 0.010, Pcorr = 0.090), and EQ‐5D (R2adj = 0.009, Pcorr = 0.092) were identified as covariates of microbiome variation. BSS had the highest nonredundant effect size on community variation (2.6%), followed by subcutaneous IFN beta‐1‐a use, weight, the first hopelessness question, daily amount of sleep, and the fifth EQ‐5D question.
Enterotypes
Enterotypes were differentially distributed in MS patients versus HC (Pearson’s chi‐square test for independence, N = 218, χ 2 = 8.77, P = 0.032), with MS containing the highest proportion of B2 and lowest proportion of B1 (Fig. 2); and among clinical subgroups (N = 218, χ 2 = 36.10, P = 0.002; Fig. 3). A comparison between the original HC group and the FGFP sample is made in Figure S2. B2 had a significantly different subgroup distribution compared to R and P (Post hoc pairwise chi‐square test, N = 218, χ 2 = 12.57 and 12.52, Pcorr = 0.047 for both). Among MS subgroups, most B2‐enterotyped individuals (N = 31) either belonged to RRMS_I (37.5%) or RRMS_R (18.8%), while P (N = 33) and R (N = 73) clusters consisted of more BMS (35.7% and 24.3%) and RRMS_U (42.9% and 24.3%).
Figure 2.
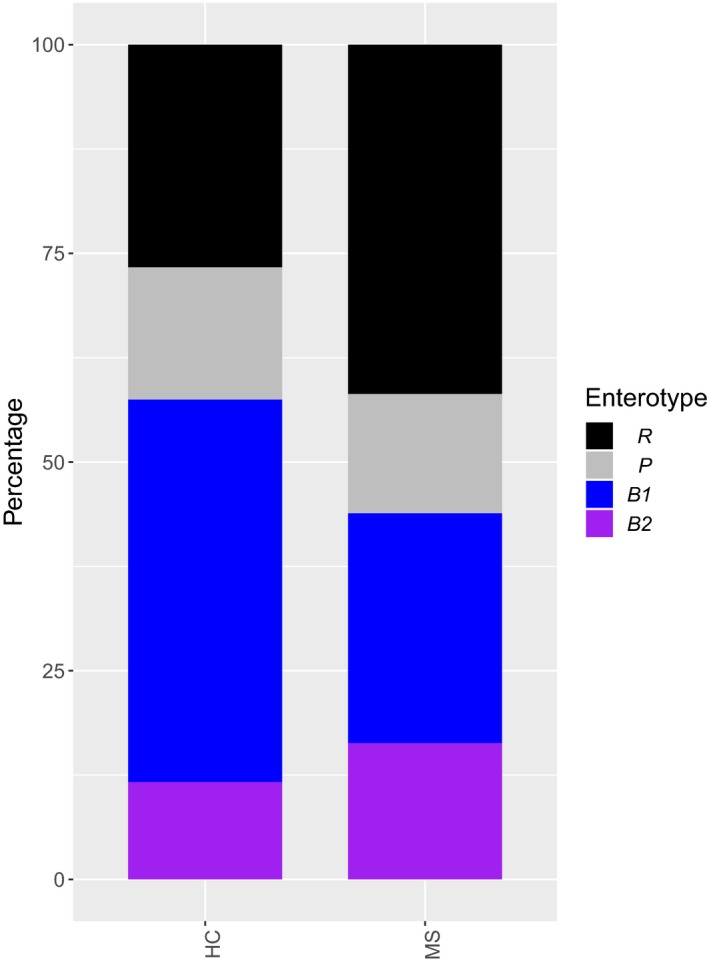
Bar plot illustrating the distribution of patient and control samples (MS vs. HC) over four enterotypes: Prevotella (P, N = 33), Ruminococcaceae (R, N = 73), Bacteroides 1 (B1, N = 81), and Bacteroides 2 (B2, N = 31). FGFP, Flemish Gut Flora Project. HC, healthy controls (N = 120, including FGFP samples). MS, multiple sclerosis (N = 98). Pearson’s chi‐square test for independence, N = 218, χ 2 = 36.10, P = 0.002 with pairwise chi‐square tests.
Figure 3.
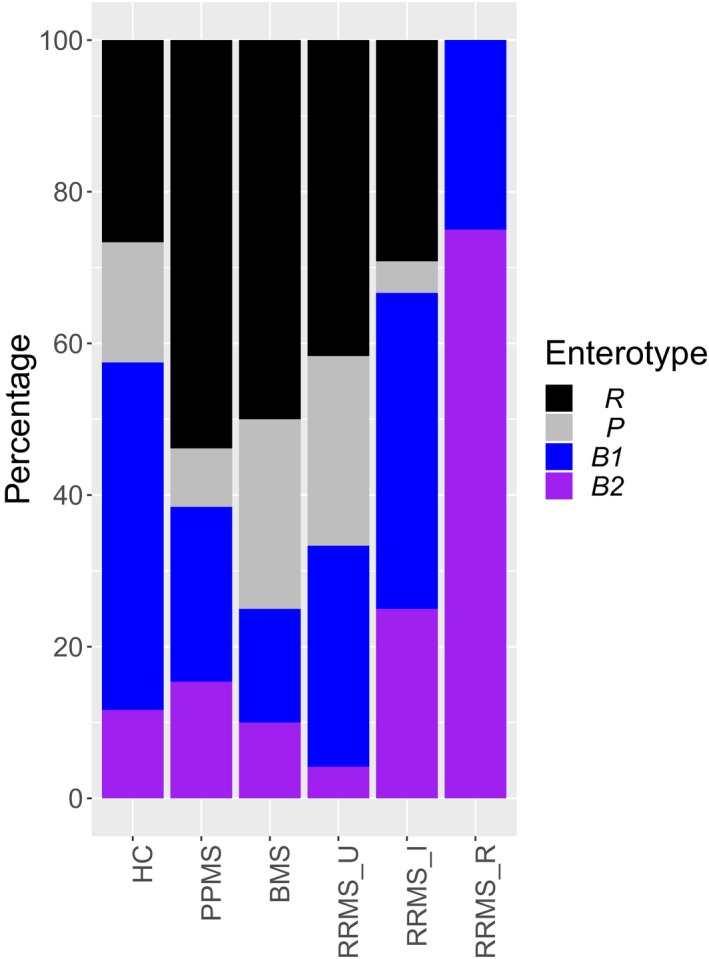
Bar plot illustrating the distribution of patient and control samples (comparison between subgroups) over four enterotypes: Prevotella (P, N = 33), Ruminococcaceae (R, N = 73), Bacteroides 1 (B1, N = 81), and Bacteroides 2 (B2, N = 31). FGFP, Flemish Gut Flora Project. HC, healthy controls (N = 120, including FGFP samples). PPMS, primary progressive multiple sclerosis (N = 26). BMS, benign multiple sclerosis (N = 20). RRMS_U, untreated active RRMS (N = 24). RRMS_I, interferon‐beta‐treated RRMS (N = 24). RRMS_R, RRMS during relapse (N = 4). Pearson’s chi‐square test for independence, N = 218, χ 2 = 36.10, P = 0.002 with pairwise chi‐square tests.
Reversely, we found significantly different enterotype distributions between RRMS_R and RRMS_U and HC (pairwise chi‐square test, N = 218, χ 2 = 14.73 and 12.47, Pcorr = 0.031 and 0.045), between HC and PPMS and BMS (N = 218, χ 2 = 8.63 and 7.83, Pcorr = 0.097 for both), between PPMS and RRMS_R (N = 218, χ 2 = 7.75, Pcorr = 0.097), between BMS and RRMS_I and RRMS_R (N = 218, χ 2 = 8.67 and 9.96, Pcorr = 0.097 and 0.095), and between RRMS_U and RRMS_I (N = 218, χ 2 = 8.20, Pcorr = 0.097). All but one RRMS_R patient had a B2 enterotype. The majority of RRMS_I had B1 (41.67%) or B2 (25%) enterotypes. Graphics showing the mean genera distribution within enterotypes as well as genus level hits per sample per (sub)group are in Figures [Link], [Link], [Link], [Link], [Link], [Link], [Link], [Link].
Compositional differences
Fourteen microbial genera were differentially abundant between MS and HC (Wilcoxon rank‐sum test, N = 218, Pcorr < 0.1) (Table 4). Seven were differentially abundant among subgroups (Kruskal–Wallis test, N = 218, Pcorr < 0.1) (Fig. 4). Post hoc Dunn test and nested GLM results are shown in Table 5. The small number of RRMS_R participants (N = 4) did not allow further statistical testing. Sporobacter, Clostridium cluster IV, and Ruminococcus were more abundant in the original HC group than in the FGFP sample (GLM, N = 120, Padj < 0.05) (Fig. S11). Median values for the relative abundances of OTU results in MS and HC groups are available in Tables S1 and S2.
Table 4.
Compositional differences between all MS patients and HC. Only significant results are shown. Reported P‐values after Wilcoxon rank‐sum test, with Benjamini–Hochberg corrected P‐values (Wilcoxon rank‐sum test, N = 218, Pcorr < 0.1).
| Genus | N | Effect size | Median MS | Median HC | P | Pcorr |
|---|---|---|---|---|---|---|
| Alistipes | 218 | −0.18 | 5.24 | 4.72 | 0.007 | 0.056 |
| Anaerotruncus | 218 | −0.16 | 0.69 | 0 | 0.016 | 0.080 |
| Butyricicoccus | 218 | −0.24 | 1.7 | 2.3 | <0.001 | 0.007 |
| Clostridium cluster IV | 218 | −0.35 | 4.14 | 3.18 | <0.001 | <0.001 |
| Faecalicoccus | 218 | −0.16 | 0 | 0 | 0.017 | 0.080 |
| Gemmiger | 218 | −0.30 | 2.71 | 3.73 | <0.001 | <0.001 |
| Intestinibacter | 218 | −0.21 | 0.69 | 1.79 | 0.002 | 0.029 |
| Lactobacillus | 218 | −0.18 | 0.69 | 0 | 0.009 | 0.065 |
| Methanobrevibacter | 218 | −0.20 | 1.61 | 0 | 0.004 | 0.037 |
| Olsenella | 218 | −0.19 | 0.69 | 0 | 0.006 | 0.049 |
| Parabacteroides | 218 | −0.15 | 4.56 | 4.12 | 0.022 | 0.097 |
| Roseburia | 218 | −0.17 | 6.36 | 6.71 | 0.014 | 0.078 |
| Ruminococcus | 218 | −0.17 | 4.86 | 4.52 | 0.014 | 0.078 |
| Sporobacter | 218 | −0.39 | 1.61 | 0.69 | <0.001 | <0.001 |
HC, healthy controls (N = 120, including FGFP samples); MS, multiple sclerosis (N = 98); Effect size (Z/sqrt(N)); Median MS, median abundance of genus (log1p transformed) for MS patients; Median HC, median abundances of genus (log1p transformed) for HC; P, uncorrected P‐value; Pcorr, P‐value corrected for multiple comparisons with Benjamini–Hochberg, FDR.
Figure 4.
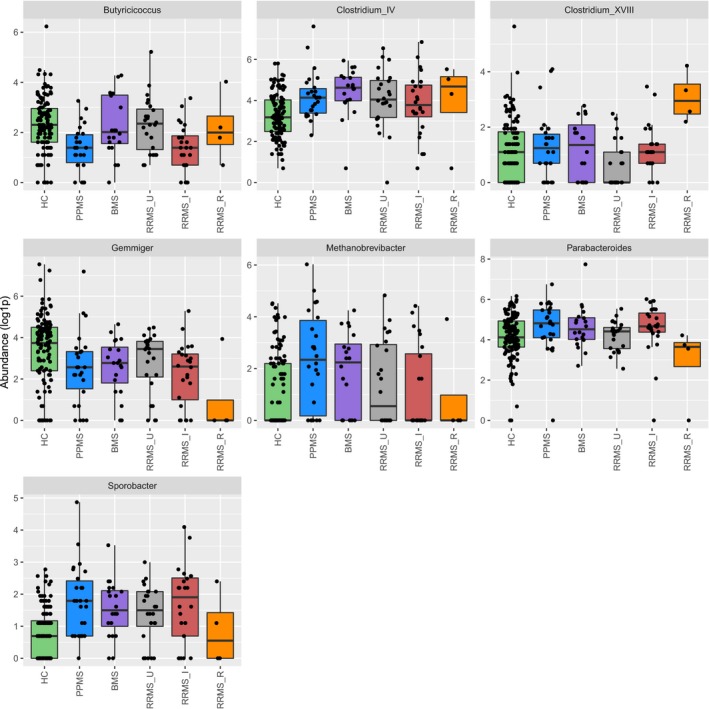
Boxplots illustrating the log1p abundances of Butyricicoccus, Clostridium cluster XVIII, Methanobrevibacter, Parabacteroides, Clostridium cluster IV, Gemmiger, and Sporobacter genera within the population. Abundances were log1p‐transformed for graphic purposes. Individual data points (dots) may overlap. HC, healthy controls (N = 120, including FGFP samples). PPMS, primary progressive multiple sclerosis (N = 26). BMS, benign multiple sclerosis (N = 20). RRMS_U, untreated active RRMS (N = 24). RRMS_I, interferon‐beta‐treated RRMS (N = 24). RRMS_R, RRMS during relapse (N = 4). Kruskal–Wallis test (N = 218, Pcorr < 0.1) with post hoc Dunn tests.
Table 5.
Multivariate analysis with nested generalized linear models taking into account Bristol stool score, age, sex, and BMI. Reported effect sizes and P‐values after Dunn and chi‐squared tests, with P‐values corrected for multiple comparisons with Benjamini–Hochberg.
| Genus | Comparison | N | Z‐score Dunn | Pcorr Dunn | SC | P | Pcorr |
|---|---|---|---|---|---|---|---|
| Butyricicoccus | PPMS–BMS | 46 | 2.56 | 0.03 | −1.83 | 0.020 | 0.030 |
| PPMS–RRMS_U | 50 | −2.99 | 0.01 | −2.94 | <0.001 | 0.003 | |
| PPMS–HC | 146 | 4.01 | <0.001 | 2.93 | <0.001 | <0.001 | |
| RRMS_I–BMS | 44 | 2.50 | 0.03 | −2.35 | 0.008 | 0.012 | |
| RRMS_I–RRMS_U | 48 | −2.91 | 0.01 | −2.54 | 0.002 | 0.004 | |
| RRMS_I–HC | 144 | −3.86 | <0.001 | 2.62 | <0.001 | 0.002 | |
| Clostridium cluster IV | BMS–HC | 140 | 4.32 | <0.001 | −3.26 | <0.001 | <0.001 |
| PPMS–HC | 146 | 3.33 | 0.006 | −1.92 | 0.002 | 0.005 | |
| RRMS_U–HC | 144 | −3.02 | 0.012 | −2.36 | <0.001 | 0.002 | |
| Clostridium cluster XVIII | RRMS_U–BMS | 44 | −2.11 | 0.058 | 1.86 | 0.023 | 0.030 |
| RRMS_U–PPMS | 50 | −2.80 | 0.037 | 2.01 | 0.008 | 0.012 | |
| RRMS_U–RRMS_I | 48 | −2.32 | 0.038 | 1.57 | 0.023 | 0.030 | |
| RRMS_U–HC | 144 | −2.82 | 0.024 | 2.20 | 0.004 | 0.007 | |
| Gemmiger | BMS–HC | 140 | −2.30 | 0.081 | 1.57 | 0.034 | 0.041 |
| PPMS–HC | 146 | −2.92 | 0.027 | −1.18 | 0.052 | 0.057 | |
| RRMS_I–HC | 144 | −3.39 | 0.011 | 2.14 | 0.002 | 0.004 | |
| Methanobrevibacter | PPMS–RRMS_I | 50 | 2.56 | 0.078 | −1.70 | 0.045 | 0.051 |
| PPMS–HC | 146 | −3.65 | 0.004 | −1.80 | 0.002 | 0.005 | |
| Parabacteroides | RRMS_I–RRMS_U | 48 | 2.00 | 0.098 | 0.89 | 0.236 | 0.236 |
| RRMS_I–HC | 144 | 2.44 | 0.004 | −1.55 | 0.064 | 0.067 | |
| Sporobacter | BMS–HC | 140 | 3.31 | 0.005 | −2.29 | 0.003 | 0.006 |
| PPMS–HC | 146 | 4.30 | <0.001 | −3.64 | <0.001 | <0.001 | |
| RRMS_I–HC | 144 | 3.75 | 0.001 | −3.80 | <0.001 | <0.001 | |
| RRMS_U–HC | 144 | 2.99 | 0.010 | −2.21 | <0.001 | 0.002 |
HC, healthy controls (N = 120, including FGFP samples); PPMS, primary progressive multiple sclerosis (N = 26); BMS, benign multiple sclerosis (N = 20); RRMS_U, untreated active RRMS (N = 24); RRMS_I, interferon‐beta‐treated RRMS (N = 24); RRMS_R, RRMS during a relapse (N = 4); SC, standard coefficient (beta); P, uncorrected P‐value; Pcorr, P‐value corrected for multiple comparisons with Benjamini–Hochberg, FDR.
Correlations with clinical variables
Butyricicoccus abundance inversely correlated with ARMSS (Spearman correlation test, N = 98, rho = −0.223, P = 0.027) (Fig. 5), and IPQ‐K5 (rho = −0.400, P = 0.001) (Fig. 6). One male RRMS_U participant with the shortest time since diagnosis among RRMS participants had the highest Butyricicoccus abundance. No correlations were found between genera and other clinical data.
Figure 5.
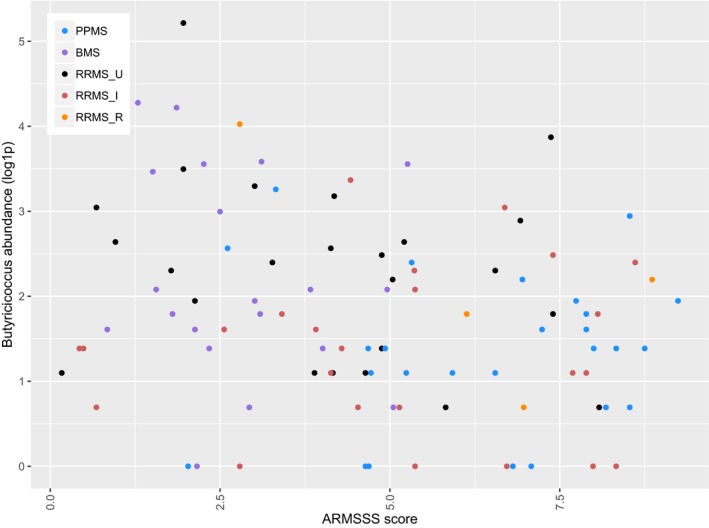
Scatterplot illustrating the abundance of Butyricicoccus (log1p‐transformed for graphical purposes) according to the ARMSS score of MS patients. ARMSS, age‐related multiple sclerosis severity. PPMS, primary progressive multiple sclerosis (N = 26). BMS, benign multiple sclerosis (N = 20). RRMS_U, untreated active RRMS (N = 24). RRMS_I, interferon‐beta‐treated RRMS (N = 24). RRMS_R, RRMS during relapse (N = 4). Spearman correlation test, N = 98, rho = −0.223, P = 0.027.
Figure 6.
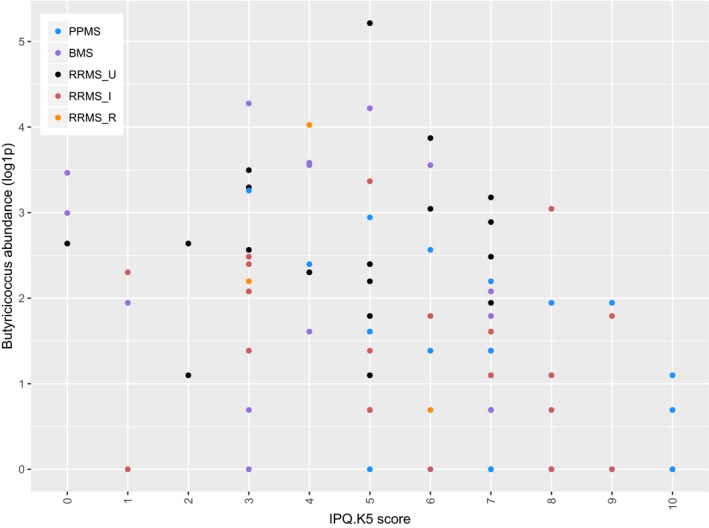
Scatterplot illustrating the abundance of Butyricicoccus according to the degree of physical symptoms patients report in the fifth question of the brief illness perception questionnaire (IPQ‐K5: 0 = none, 10 = multiple and serious symptoms). The outlier is explained in text. PPMS, primary progressive multiple sclerosis (N = 26). BMS, benign multiple sclerosis (N = 20). RRMS_U, untreated active RRMS (N = 24). RRMS_I, interferon‐beta‐treated RRMS (N = 24). RRMS_R, RRMS during relapse (N = 4). Spearman correlation test, N = 98, rho = −0.400, Pcorr = 0.001.
Discussion
No gut microbial diversity measure significantly differed between MS and HC. However, multiple microbiome readouts differed between MS subgroups, supporting our hypothesis of heterogeneity. First, Sobs was inversely related to the presumed degree of inflammatory disease activity in MS subgroups: highest in BMS and PPMS and lowest in RRMS_R. The lower Sobs in RRMS_I might be explained by the higher amount of CNS inflammation for which treatment was initiated. At the group level, BMS and PPMS patients had a higher richness than controls (including the FGFP sample). That Sobs in HC on average appears to be in between the Sobs in active (RRMS_I and RRMS_R) and nonactive MS groups (PPMS and BMS) is a novel finding, 17 , 18 which cannot be explained by major confounding factors (e.g. no differences in BSS to account for constipation). What the higher Sobs in PPMS and BMS patients when compared with HC exactly means, remains uncertain. A recent paper warned against basing health‐related conclusions on richness alone, as this could just be indicative of gut ecosystem age (i.e. the time the same ecosystem has been in place depending on the transit time). 46 Additionally, approximately 2.4% of interindividual microbiota compositional variation could be explained by allocation to a subgroup. Previous MS gut microbiota studies mainly focused on RRMS, 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 where the lack of a common observed signal is probably explained by the observed intergroup differences and distinct methodological issues.
Second, a high proportion of MS patients had a B2 enterotype. This was most pronounced in RRMS_I and RRMS_R patients, which suggests an association with higher presumed inflammatory activity. However, enterotype distributions of RRMS_U and BMS were not significantly different, even though they differ in inflammatory disease activity. The relationship between enterotypes and MS phenotypes, and the interplay between IFN and B2 remain to be elucidated. This is the first time that an enterotype is associated to (a specific subgroup of) MS. As this B2 enterotype was associated to Crohn’s disease, 42 our results warrant further research on overlapping host–microbiome interactions in immune‐mediated inflammatory diseases.
Third, we observed differences in microbial taxa. Results for Faecalicoccus, 47 Methanobrevibacter, 10 , 13 Ruminococcus, 16 and Gemmiger 47 confirm previous findings. While Alistipes and Anaerotroncus abundances reflect findings in a PPMS mouse model, 48 we are the first to find these differentially abundant in RRMS. Not in line with other studies, Lactobacillus, 14 Parabacteroides, 14 Sporobacter, 47 and Clostridium cluster IV 13 , 14 results are possibly explained by different population subtypes, since we also included less inflammatory phenotypes, or confounder control. Olsenella and Roseburia have been described as differentially abundant in other inflammatory conditions. 47 , 49 , 50 , 51
Butyricicoccus abundance was lower in MS than in the total HC group, as confirmed in a previous paper. 47 Between‐group analysis suggests an inverse relationship between Butyricicoccus abundance and presumed CNS inflammation in RRMS. Lower Butyricicoccus abundance in PPMS might be explained by the less prominent role of focal inflammatory disease activity. Butyricicoccus is a spore‐forming genus from the Clostridium cluster IV known to produce short‐chain fatty acids, 52 which can initiate anti‐inflammatory effects through regulatory T‐cell induction. 53 Through this mechanism the gut microbiota might contribute to MS pathogenesis. 54 , 55 Butyricicoccus and Clostridium cluster XVIII have never before been found differentially abundant within MS. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 While lower abundances of certain Clostridium cluster IV species have been reported, none were of the type known to induce regulatory T‐cells. 15 In line with our results, Butyricicoccus pullicaecorum was shown less abundant in IBD patients than in HC. 50 Lastly, Butyricicoccus abundance was inversely correlated with age‐corrected disease burden and self‐reported disability. Given also the fact that Butyricicoccus was found to be well tolerated for human administration, 56 these results highlight Butyricicoccus as a promising novel target in MS.
Strengths
Including MS subgroups with well‐defined disease characteristics and disease course modifiers enabled us to evaluate the gut microbiome in terms of higher or lower degree of presumed focal CNS inflammation. Restricting treatment to IFN‐beta allowed studying the gut microbiome in a homogenous treatment group. The RRMS_U group permitted to investigate the gut microbiome in MS without treatment effects. We controlled for the effect of constipation through BSS and other covariates known to affect gut microbial composition. 18
Limitations
First, categorization bypasses the natural spectrum of a heterogeneous disease, which may have resulted in errors of classification. Second, without MRI data on subclinical disease activity, the degree of focal inflammation may be underestimated in all groups. Third, despite attempts to increase the size of RRMS_R (N = 4), reported differences require replication in larger, independent study groups. Fourth, it is unclear whether microbial differences between the original HC and FGFP samples reflect natural diversity or sampling bias.
Author Contributions
TR performed recruitment and stool sampling in the University Hospital Brussels, aided in statistical analysis, and wrote the body of this manuscript. LD performed data quality checks, laboratory and statistical analyses under the guidance of MVC and JR, and aided in the writing process. MVC performed preprocessing of raw sequencing data for detailed analysis. AVR performed recruitment and stool sampling in the NMSC Melsbroek, and provided organizational support with MD. MD, JDK, GN, MJ, and JR prepared the outline of the study and provided insight.
Conflict of Interest
LD, BD, and JR have a patent A NEW INFLAMMATION ASSOCIATED, LOW CELL COUNT ENTEROTYPE (EP 17207770.3) issued to VIB VZW, KATHOLIEKE UNIVERSITEIT LEUVEN, K.U.LEUVEN R&D, and VRIJE UNIVERSITEIT BRUSSEL. MVC has a patent PCT/EP2018/084920 pending. MJ and JR have a patent EP 14184535.4—12.09.2014: Biological sampling and storage container, International patent application on 14.09.2015: PCT/EP2015/070977 issued to VIB, VUB, and LRD. There are no conflicts of interest for the other authors.
Supporting information
Figure S1. Boxplot of observed richness within the original healthy control samples (N = 22) and those from the FGFP (N = 98). The body of the boxplot represents the first and third quartiles of the distribution and median line. The whiskers extend to the last data point within 1.5 times the interquartile range. The outliers lie beyond. Individual data points (dots) may overlap. HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample.
Figure S2. Bar plot illustrating the distribution of four enterotypes within the original healthy control samples and those from the FGFP, Prevotella (P), Ruminococcaceae (R), Bacteroides 1 (B1), and Bacteroides 2 (B2). HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample.
Figure S3. Barplot showing the distribution of main discriminating genera in enterotypes in the whole study population (N = 218). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample.
Figure S4. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the healthy control group from the Flemish Gut Flora Project (HC_FGFP, N = 98). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S5. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the original healthy control group (HC_MICR, N = 22). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S6. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the benign MS group (BMS, N = 20). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S7. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the untreated active relapsing–remitting MS group (RRMS_U, N = 24). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S8. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the active relapsing–remitting MS group treated with interferon beta (RRMS_I, N = 24). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S9. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the active relapsing–remitting MS group recruited during a relapse (RRMS_R, N = 4). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S10. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the non‐active primary progressive MS group (PPMS, N = 26). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S11. Boxplots illustrating the log1p abundances of genera Bifidobacterium, Clostridium cluster IV, Clostridium cluster XIVa, Fusicatenibacter, Romboutsia, Ruminococcus, Sporobacter, Terrisporobacter, and Turicibacter within the healthy control population. Abundances were log1p‐transformed for graphic purposes. Individual data points (dots) may overlap. HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample. Kruskal–Wallis test (N = 120, Pcorr < 0.1) with post hoc Dunn tests.
Table S1. Relative abundance of bacterial genera found per MS subgroup (expressed per 10.000 reads). Taxa unclassified at genus level, genera with a mean relative abundance <1 in 10,000 reads and present in <20% of all samples were excluded. Median values are shown. The proportion (%) of samples per group containing the genus is mentioned between brackets.
Table S2. Relative abundance of bacterial genera found per healthy control group (expressed per 10,000 reads). Taxa unclassified at genus level, genera with a mean relative abundance <1 in 10,000 reads and present in <20% of all samples were excluded. Median values are shown. The proportion (%) of samples per group containing the genus is mentioned between brackets.
Acknowledgments
We thank Leen Rymenans, Chloë Verspecht, and other members of the Raes Lab for their helpful discussions. We are grateful for the support of Annick Van Merhaegen‐Wieleman, University Hospital Brussels, and Piet Eelen, NMSC Melsbroek, during recruitment. MJ is funded by a postdoctoral fellowship from Research Foundation—Flanders. The Raes lab is supported by the VIB, the Rega institute for Medical Research, KU Leuven, the FWO EOS program (30770923), FP7 METACARDIS (305312), H2020 SYSCID (733100), PIBD‐SET (668023), and IMMUNAID (779295). This work was funded by an FWO research grant (G038318N) to MD and JR.
Funding Information
MJ is funded by a postdoctoral fellowship from Research Foundation—Flanders. The Raes lab is supported by the VIB, the Rega institute for Medical Research, KU Leuven, the FWO EOS program (30770923), FP7 METACARDIS (305312), H2020 SYSCID (733100), PIBD‐SET (668023), and IMMUNAID (779295). This work was funded by an FWO research grant (G038318N) to MD and JR. Financial support for this project in the MS center was provided in 2015, 2016 and 2017, commissioned by the Belgian National MS society with the support of the Fund D.V., managed by the King Baudouin Foundation (2016‐C5812060‐206012).
Funding Statement
This work was funded by Fonds Wetenschappelijk Onderzoek grant G038318N; VIB grant ; the Rega institute for Medical Research grant ; KU Leuven grant ; the FWO EOS program grant 30770923; FP7 METACARDIS grant 305312; H2020 SYSCID grant 733100; PIBD‐SET grant 668023; IMMUNAID grant 779295.
Contributor Information
Marie D’hooghe, Email: marie.dhooghe@mscenter.be.
Jeroen Raes, Email: jeroen.raes@kuleuven.vib.be.
References
- 1. Reich DS, Lucchinetti CF, Calabresi PA. Multiple sclerosis. N Engl J Med 2018;378:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wiesel PH, Norton C, Glickman S, Kamm MA. Pathophysiology and management of bowel dysfunction in multiple sclerosis. Eur J Gastrol Hepatol 2001;13:441–448. [DOI] [PubMed] [Google Scholar]
- 3. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014;83:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med 2000;343:938–952. [DOI] [PubMed] [Google Scholar]
- 5. Tallantyre EC, Bo L, Al‐Rawashdeh O, et al. Clinico‐pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler 2010;16:406–411. [DOI] [PubMed] [Google Scholar]
- 6. Cox LM, Weiner HL. Microbiota signaling pathways that influence neurologic disease. Neurotherapeutics 2018;15:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. BMJ 2018;360:j5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berer K, Gerdes LA, Cekanaviciute E, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci 2017;114:10719–10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cekanaviciute E, Yoo BB, Runia TF, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci USA 2017;114:10713–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nat Commun 2016;7:12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cosorich I, Dalla‐Costa G, Sorini C, et al. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv 2017;3:e1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tremlett H, Fadrosh DW, Faruqi AA, et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol 2016;16:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tremlett H, Fadrosh DW, Faruqi AA, et al. Gut microbiota in early pediatric multiple sclerosis: a case‐control study. Eur J Neurol 2016;23:1308–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen J, Chia N, Kalari KR, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci Rep 2016;6:28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyake S, Kim S, Suda W, et al. Dysbiosis in the gut microbiota of patients with multiple sclerosis, with a striking depletion of species belonging to clostridia XIVa and IV clusters. PLoS One 2015;10:e0137429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cantarel BL, Waubant E, Chehoud C, et al. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J Invest Med 2015;63:729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falony G, Joossens M, Vieira‐Silva S, et al. Population‐level analysis of gut microbiome variation. Science 2016;352:560–564. [DOI] [PubMed] [Google Scholar]
- 18. Vandeputte D, Falony G, Vieira‐Silva S, et al. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016;65:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amato MP, Zipoli V, Goretti B, et al. Benign multiple sclerosis: cognitive, psychological and social aspects in a clinical cohort. J Neurol 2006;253:1054–1059. [DOI] [PubMed] [Google Scholar]
- 20. Calabrese M, Favaretto A, Poretto V, et al. Low degree of cortical pathology is associated with benign course of multiple sclerosis. Mult Scler 2013;19:904–911. [DOI] [PubMed] [Google Scholar]
- 21. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schumacher GA, Beebe G, Kibler RF, et al. Problems of experimental trials of therapy in multiple sclerosis: report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann N Y Acad Sci 1965;122:552–568. [DOI] [PubMed] [Google Scholar]
- 23. Calabrese M, Filippi M, Rovaris M, et al. Evidence for relative cortical sparing in benign multiple sclerosis: a longitudinal magnetic resonance imaging study. Mult Scler 2009;15:36–41. [DOI] [PubMed] [Google Scholar]
- 24. Lewis SJ, Heaton KW. Stool form scale as a useful guide to intestinal transit time. Scand J Gastroenterol 1997;32:920–924. [DOI] [PubMed] [Google Scholar]
- 25. Rumah KR, Linden J, Fischetti VA, Vartanian T. Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS One 2013;8:e76359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hertzog MA. Considerations in determining sample size for pilot studies. Res Nurs Health 2008;31:180–191. [DOI] [PubMed] [Google Scholar]
- 27. Manouchehrinia A, Westerlind H, Kingwell E, et al. Age related multiple sclerosis severity score: disability ranked by age. Mult Scler 2017;23:1938–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fraser L, Burnell M, Salter LC, et al. Identifying hopelessness in population research: a validation study of two brief measures of hopelessness. BMJ Open 2014;4:e005093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broadbent E, Petrie KJ, Main J, Weinman J. The brief illness perception questionnaire. J Psychosom Res 2006;60:631–637. [DOI] [PubMed] [Google Scholar]
- 30. Dakin H. Review of studies mapping from quality of life or clinical measures to EQ‐5D: an online database. Health Qual Life Outcomes 2013;11:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caporaso JG, Lauber CL, Walters WA, et al. Ultra‐high‐throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012;6:1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011;27:2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27:2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 2007;73:5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bates D, Chambers J, Dalgaard P, et al. R: a language and environment for statistical computing. 2013.
- 36. Ogle D. Simple fisheries stock analysis. R package version 0.8.14. 2017.
- 37. Oksanen J, Blanchet FG, Friendly M,et al. Vegan: Community Ecology Package. Version 2.4‐5. 2017.
- 38. McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 2013;8:e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Harrell FE Jr. Hmisc: Harrell miscellaneous. Version 4.0‐3. 2017.
- 40. Morgan M. DirichletMultinomial: Dirichlet‐multinomial mixture model machine learning for microbiome data. Version 1.18.0. 2017.
- 41. Holmes I, Harris K, Quince C. Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One 2012;7:e30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vandeputte D, Kathagen G, D'Hoe K, et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017;551:507–511. [DOI] [PubMed] [Google Scholar]
- 43. Fife D. fifer: a biostatisticians toolbox for various activities, including plotting, data cleanup, and data analysis. Version 1.1. 2017.
- 44. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B 1995;57:289–300. [Google Scholar]
- 45. Fletcher TD. QuantPsyc: quantative psychology tools. Version 1.5. 2015.
- 46. Falony G, Vieira‐Silva S, Raes J. Richness and ecosystem development across faecal snapshots of the gut microbiota. Nat Microbiol 2018;3:526–528. [DOI] [PubMed] [Google Scholar]
- 47. Forbes JD, Chen CY, Knox NC, et al. A comparative study of the gut microbiota in immune‐mediated inflammatory diseases‐does a common dysbiosis exist? Microbiome 2018;6:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carrillo‐Salinas FJ, Mestre L, Mecha M, et al. Gut dysbiosis and neuroimmune responses to brain infection with Theiler's murine encephalomyelitis virus. Sci Rep 2017;7:44377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vieira Colombo AP, Magalhaes CB, Hartenbach FA, et al. Periodontal‐disease‐associated biofilm: a reservoir for pathogens of medical importance. Microb Pathog 2016;94:27–34. [DOI] [PubMed] [Google Scholar]
- 50. Zakaria MN, Takeshita T, Shibata Y, et al. Microbial community in persistent apical periodontitis: a 16S rRNA gene clone library analysis. Int Endod J 2015;48:717–728. [DOI] [PubMed] [Google Scholar]
- 51. Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate‐producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014;63:1275–1283. [DOI] [PubMed] [Google Scholar]
- 52. Jeraldo P, Hernandez A, Nielsen HB, et al. Capturing one of the human Gut microbiome's most wanted: reconstructing the genome of a novel butyrate‐producing, clostridial scavenger from metagenomic sequence data. Front Microbiol 2016;7:783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Narushima S, Sugiura Y, Oshima K, et al. Characterization of the 17 strains of regulatory T cell‐inducing human‐derived Clostridia. Gut Microbes 2014;5:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mangalam A, Shahi SK, Luckey D, et al. Human gut‐derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep 2017;20:1269–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shahi SK, Freedman SN, Mangalam AK. Gut microbiome in multiple sclerosis: the players involved and the roles they play. Gut Microbes 2017;8:607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boesmans L, Valles‐Colomer M, Wang J, et al. Butyrate producers as potential next‐generation probiotics: safety assessment of the administration of Butyricicoccus pullicaecorum to healthy volunteers. mSystems 2018;3: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Boxplot of observed richness within the original healthy control samples (N = 22) and those from the FGFP (N = 98). The body of the boxplot represents the first and third quartiles of the distribution and median line. The whiskers extend to the last data point within 1.5 times the interquartile range. The outliers lie beyond. Individual data points (dots) may overlap. HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample.
Figure S2. Bar plot illustrating the distribution of four enterotypes within the original healthy control samples and those from the FGFP, Prevotella (P), Ruminococcaceae (R), Bacteroides 1 (B1), and Bacteroides 2 (B2). HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample.
Figure S3. Barplot showing the distribution of main discriminating genera in enterotypes in the whole study population (N = 218). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample.
Figure S4. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the healthy control group from the Flemish Gut Flora Project (HC_FGFP, N = 98). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S5. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the original healthy control group (HC_MICR, N = 22). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S6. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the benign MS group (BMS, N = 20). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S7. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the untreated active relapsing–remitting MS group (RRMS_U, N = 24). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S8. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the active relapsing–remitting MS group treated with interferon beta (RRMS_I, N = 24). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S9. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the active relapsing–remitting MS group recruited during a relapse (RRMS_R, N = 4). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S10. Barplot showing the distribution of the 61 most prevalent genera in samples from each subject in the non‐active primary progressive MS group (PPMS, N = 26). Other genera are combined in “Remainder.” The y‐axis shows the proportional abundance per sample. OTU, operational taxonomic unit.
Figure S11. Boxplots illustrating the log1p abundances of genera Bifidobacterium, Clostridium cluster IV, Clostridium cluster XIVa, Fusicatenibacter, Romboutsia, Ruminococcus, Sporobacter, Terrisporobacter, and Turicibacter within the healthy control population. Abundances were log1p‐transformed for graphic purposes. Individual data points (dots) may overlap. HC_FGFP, healthy controls from the Flemish Gut Flora Project. HC_MICR, original healthy control sample. Kruskal–Wallis test (N = 120, Pcorr < 0.1) with post hoc Dunn tests.
Table S1. Relative abundance of bacterial genera found per MS subgroup (expressed per 10.000 reads). Taxa unclassified at genus level, genera with a mean relative abundance <1 in 10,000 reads and present in <20% of all samples were excluded. Median values are shown. The proportion (%) of samples per group containing the genus is mentioned between brackets.
Table S2. Relative abundance of bacterial genera found per healthy control group (expressed per 10,000 reads). Taxa unclassified at genus level, genera with a mean relative abundance <1 in 10,000 reads and present in <20% of all samples were excluded. Median values are shown. The proportion (%) of samples per group containing the genus is mentioned between brackets.