

Abstract
Objective
To characterize the spectrum of neurologic involvement in Erdheim–Chester Disease (ECD), a treatable inflammatory neoplasm of histiocytes.
Methods
Sixty‐two patients with ECD were prospectively enrolled in a natural history study that facilitated collection of clinical, imaging, laboratory, neurophysiologic, and pathologic data.
Results
Ninety‐four percent of the patients had neurologic abnormalities on examination or imaging, and 22% had neurologic symptoms as the initial presentation of ECD. The most common neurologic findings were cognitive impairment, peripheral neuropathy, pyramidal tract signs, cranial nerve involvement, and cerebellar ataxia. Imaging revealed atrophy and demyelination along with focal lesions that were located throughout the nervous system, dura, and extra‐axial structures. The BRAF V600E variant correlated with cerebral atrophy. Brain pathology revealed lipid‐laden, phagocytic macrophages (histiocytes) accompanied by demyelination and axonal degeneration.
Interpretation
In patients with ECD, neurologic morbidity is common and contributes significantly to disability. Since neurologic symptoms can be the presenting feature of ECD and, given the mean delay in ECD diagnosis is 4.2 years, it is critical that neurologists consider of ECD and other histiocytosis in patients with inflammatory, infectious, or neoplastic‐appearing white matter. Furthermore, given the broad spectrum of neurologic involvement, neurologists have an important role in a team of specialists treating ECD patients.
Introduction
Erdheim–Chester disease (ECD) is a treatable histiocytic neoplasm frequently involving the brain. It is characterized by infiltration and accumulation of inflammatory foamy macrophages in multiple tissues leading to end‐organ dysfunction and failure through mass effect, tissue restriction, organ encasement, and by local and systemic inflammatory cytokines.1, 2, 3 The most commonly involved organs are bone, retroperitoneum, kidneys, brain, heart, skin, and lungs. The biopsy of affected tissue reveals foamy to epithelioid histiocytes that are CD1a–, CD68+, CD163+, factor XIIIa+, and S100±.4 Notably, this molecular signature is not specific to ECD, as it is also found in the macrophages of inflammatory conditions such as multiple sclerosis, sarcoid, and IgG4 disease. The treatment of ECD involves anti‐inflammatory or antineoplastic agents4 and vemurafenib is FDA‐approved for the treatment of ECD.5
To date, the neurologic features of ECD have been described in small series and retrospective studies.6, 7, 8, 9, 10 A meta‐analysis showed that approximately half of ECD patients have neurologic involvement, and these patients carry a poorer prognosis and may be refractory to first‐line treatments.11, 12 Given the therapeutic and prognostic implications, it is paramount to recognize ECD in neurologic patients, distinguish it from other inflammatory and oncologic disorders, and provide for timely diagnosis and treatment.
The goal of this study was to systematically characterize the spectrum of neurologic disease in ECD in a large cohort of patients enrolled in a longitudinal observational study.3
Methods
Standard protocol approvals, registrations, and patient consents
Patients were prospectively enrolled in the “Clinical and Basic Investigations into Erdheim‐Chester Disease” study (Protocol 11‐HG‐0207, clinicaltrials.gov identifier NCT01417520) at the National Human Genome Research Institute (NHGRI)8 and provided written informed consent. The NHGRI Institutional Review Board approved the study. Recruitment was primarily via physician referral or through the ECD Global Alliance. Inclusion required diagnosis of ECD based on clinical evaluation with histological confirmation. Seventy‐nine ECD patients were enrolled, and 62 patients were admitted for the first time to the NIH Clinical Center between October 2011 and September 2016. Seventeen patients were unable to travel to the NIH and were excluded from this analysis. ECD diagnosis was confirmed at the NIH using consensus criteria.4 Given the rarity of ECD, pre‐enrollment power calculations were not employed (the ECD Global Alliance estimates 359 patients worldwide, of which 191 are in the USA).13
Protocol 11‐HG‐0207 provides for the collection of tissue from confirmed ECD patients in the absence of clinical evaluation. Neurologic postmortem tissue was obtained from one additional patient.
Clinical evaluation
A multidisciplinary team focused on ECD composed of neurologists, ophthalmologists, geneticists, and endocrinologists performed a comprehensive evaluation.3 All patients were screened for neurologic comorbidities in a standardized manner. This included trauma, concussions, meningitis, encephalitis, surgery, vestibular disorders, other brain tumors (metastatic and primary), seizures, migraines, cerebrovascular ischemia, spinal injuries, toxic environmental exposure, and nerve entrapment syndromes. A complete general physical and neurologic examination – including assessment of mental status by a mini‐mental status examination (MMSE), cranial nerves, motor, sensory, coordination, reflexes, and gait – were performed on all participants.
Electrophysiologic investigation of the peripheral nervous system included nerve conduction studies of peroneal, tibial, and median nerves and limited EMGs of the lower extremities on 34 patients (selection limited by consent).
Formal neuropsychologic testing was obtained in 14 patients (limited by consent and availability of testing). A standardized battery was used to assess overall intelligence (Wechsler Reading and Wechsler Abbreviated Scale of Intelligence II), memory (Wechsler Memory test, digit span, Hopkins Verbal Learning Test), visuospatial functioning (Brief Visuospatial Memory Test, Rey Complex Figure), language function (Controlled Oral Word Association Test, Boston Naming Test), executive functioning (Symbol Digit Modality Test, Wisconsin Card Sorting Test, Paced Auditory Serial Addition Test), and mood (Frontal Systems Scale of Behavior, Beck Depression Inventory).
Imaging
MRIs of the brain, orbits, and pituitary (sellar and suprasellar regions) with and without gadolinium were obtained using a 1.5‐ or 3‐Tesla scanner (n = 58). Spinal imaging with gadolinium was obtained (n = 2). Three patients were unable to tolerate MRI or had contraindications to MRI imaging. Multiplanar T1‐weighted, T2‐weighted, and diffusion sequences were obtained using an MRI protocol developed for this study, and images were interpreted by one of three neuroradiologists experienced in ECD. The radiologists were provided clinical history and ECD diagnosis to facilitate interpretation within the context of this observational study. The written reports were used to generate frequencies of the imaging findings.
Subjects were considered to have atrophy if the report reflected generalized or lobar atrophy or ventricular enlargement. Intracranial tumors were defined as either measurable lesions or patchy, confluent, and large legions. Punctate changes (even if potentially related to ECD) were not considered tumor‐like.
Quantitative brain volumes were obtained in 15 patients and 15 age‐matched healthy controls using an unbiased voxel‐wise morphometric (VBM) approach using FSL (v5.0.11).14, 15, 16 Also, 3‐Tesla diffusion tensor imaging was obtained in 15 ECD patients and 15 age‐matched healthy controls, and voxel‐wise maps were created for fractional anisotropy, mean diffusivity, axial, and radial diffusivity using the TORTOISE and TBSS software packages.17 As these methods require a specific protocol for capturing and processing images, they could not be performed on the entire cohort.
Molecular studies and histology
DNA from CNS and non‐CNS biopsy tissues of 58 subjects was sequenced to evaluate for the presence of the BRAF V600E variant.3 Paraffin‐embedded sections prepared by referring institutions were evaluated by a hematopathologist experienced in histiocytic disorders.3 Unstained CNS tissue blocks were obtained from referring centers, and immunohistochemistry was performed at NIH (n = 5).
Statistical analysis
Clinical, imaging, and molecular data were analyzed using descriptive statistics, and BRAF versus atrophy comparisons were performed using a one‐tailed Fisher’s exact test. A one‐tailed test was used because the BRAF V600E variant is likely pathogenic and correlates with worse organ‐specific disease in untreated patients.2, 18, 19, 20 Diffusion tensor imaging and volumetric data were compared using the FSL Randomise tool (v5.0.11, 5000 permutations) – which conducts pairwise, permutation‐based inference on t‐statistic maps21, 22 – was used to identify clusters of voxels that differed between the healthy controls and patient group. The threshold for significance was P < 0.05, after correcting for multiple comparisons across space (FSL TFCE tool).14, 23, 24, 25
Results
Patient characteristics and therapy
We evaluated 62 patients (47 males and 15 females), all of whom met consensus criteria for a diagnosis of ECD.4 Mean age at enrollment was 54 years (range 22–74 years), mean age of symptom onset was 46 years (range 16–74 years), and the mean time to diagnosis was 4.2 years (range 0–24 years). Fifty‐four percent were positive for the targetable BRAFV600E variant.
Ninety‐four per cent of the patients had objective neurologic findings: abnormalities on examination or neuroimaging. Twenty‐two percent (14/62) of patients had an initial ECD presentation that was neurologic in nature, including cerebellar ataxia, focal weakness, gait imbalance, seizures, and Horner’s syndrome (Table S1). Bone pain (17%) and diabetes insipidus (25%) were the most common initial systemic presenting findings.3 Prior to reaching a conclusive ECD diagnosis, other considerations included autoimmune disease (27%), sarcoid (18%), IgG4 disease (17%), multiple sclerosis/neuromyelitis optica (15%), CNS malignancy (10%), and vasculitis (22%). Additionally, one patient had comorbid myasthenia gravis, and another had comorbid CNS lymphoma. One patient, initially considered to be CNS‐isolated ECD, had minimal asymptomatic perinephric fibrosis that facilitated the diagnosis.
Therapy prior to, or at, enrollment commonly included interferon α2b, anakinra, vemurafenib, imatinib, methotrexate, and cladribine (Table S1). Isolated cases were treated with natalizumab, cyclophosphamide, daclizumab, tocilizumab, dasatinib, 6‐mercoaptopurine with vincristine, vinblastine, or dabrafenib with trametinib. Clinical and radiologic neurologic improvement were seen with cladribine in a patient who had a brainstem lesion. The correlation of clinical improvement with specific therapies was limited since several subjects were receiving multiple agents for variable periods of time, sometimes prescribed for alternative diagnoses. Recent case reports suggest that MEK inhibitors may show promise for neurologic disease.26
Clinical features
We found involvement throughout the nervous system. The most common findings were subjective or objective cognitive difficulty (52%), cerebellar ataxia (46%), cranial neuropathy (61%), peripheral neuropathy (56%), pyramidal tract involvement (30%), and seizures (8%). Dysmetria was observed in 33% and dysdiadochokinesia in 26%. 44% of our cohort had oculomotor abnormalities resulting from lesions affecting the cerebellar peduncles, nuclei, nerve fibers, or extraocular muscles. Two patients had a myelopathy (confirmed on spinal imaging).
To assess peripheral nerve involvement, neurophysiologic testing was obtained in 34 patients. Fifty‐six percent of those patients have peripheral neuropathies. Electromyography revealed axonal neuropathy in all patients – peripheral demyelination was not seen. Deficits were equally distributed amongst polyneuropathies (15% sensorimotor; 6% isolated sensory), isolated mononeuropathies (12% peroneal; 12% median; 9% ulnar), and polyradiculopathies (9%). Contributing comorbidities included toxicity from therapies such as immunomodulators, chemotherapy and glucocorticoids (69%; 43/62), diabetes mellitus (14%), vitamin D deficiency (29%), and hypothyroidism (both central and primary, 28%). Symptomatically, subjects commonly complained of pain that reflected both tumor infiltration into bone marrow and neuropathic pain. In summary, neuropathies are common in ECD, but the etiologies may be multifactorial.
Elevated inflammatory markers were noted in some patients (ESR in 47%, CRP in 43% and borderline ANA in 23%; n = 62), but none of these patients met ACR diagnostic criteria for connective tissue disease or autoimmune thyroiditis seen. These results likely reflect the inflammatory nature of ECD.
Cognition
Cognitive impairment was unexpectedly common in our entire cohort. Fifty‐two percent of the subjects complained of disabling cognitive difficulties and 11% had an abnormal mini‐mental status examination. Neuropsychologic testing identified cognitive deficits in 64% (9 of 14) of studied subjects (see Table 1). One patient had dementia and eight had mild cognitive impairment. The most frequently affected domains were verbal fluency (COWAT; 6 patients), psychomotor slowing (FRSBE; 5 patients), executive dysfunction (SDMT, PASAT, WCST or Rey; 5 patients), and memory (WMS‐3, HVLT, VSMT; 5 patients). Two patients exhibited mood disorders (pseudobulbar affect and anxiety).
Table 1.
Summary of neuropsychologic testing results.
| # | WMS3 | HVLT | VSMT | COWAT | BNT | Peg | TMT | SDMT | WCST | FrBSE | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | X | X | |||||||||
| 5 | |||||||||||
| 7 | X | X | |||||||||
| 8 | X | X | X | X | REY | ||||||
| 12 | X | X | X | X | |||||||
| 13 | X | X | |||||||||
| 16 | X | ||||||||||
| 17 | X | X | X | ||||||||
| 20 | X | X | X | X | X | X | |||||
| 34 | |||||||||||
| 36 | |||||||||||
| 41 | X | X | X | X | X | PASAT | |||||
| 47 | X 1 | ||||||||||
| 60 | Significant dementia, MOCA 10/30, cannot complete battery | ||||||||||
x, more than 2 SD abnormal on any aspect of given test or one of its subsets.
BNT, Boston Naming Test; COWAT, Controlled Oral Word Association test; FrSBE, Frontal System Scale of Behavior Family‐rating Form; HVLT, Hopkins Verbal Learning Test; MOCA, Montreal Cognitive Assessment; PASAT, Paced Auditory Serial Addition Test (>2SD abnormal in indicated patient); Peg, Grooved Pegboard; Rey, Rey Complex Figure Task (abnormal in indicated patient); SDMT, Symbol Digit Modality Test; TMT, Trail Making Tests A & B; VSMT, Brief Visuospatial Memory Test; WCST, Wisconsin Card Sorting Test; WMS‐3, Wechsler Memory Scale, third edition.
Note: for patient #47, the abnormality was attributed to motor dysfunction rather than cognitive abnormality.
This cognitive dysfunction reflects the fact that ECD patients have reduced brain volumes compared to healthy controls as determined by quantitative brain volumetric analysis (n = 15 per group), with the ECD group being 2.7 mL smaller than the control group. The gray matter volume loss involved specific regions within the right frontal and parietal cortices (Fig. 3b). Congruently, analysis of brain MRI reports revealed that 28% of the entire cohort had age‐inappropriate atrophy (Fig. 1). The BRAF V600E variant correlated with the presence of atrophy on imaging (p = 0.047; odds ratio 3.5). Overall, symptomatic neurodegeneration is frequent in ECD.
Figure 3.
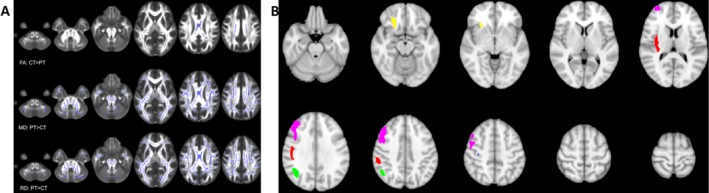
Quantitative MRI reveals demyelination and atrophy in ECD. (A) Purple demarcates white matter regions where fractional anisotropy (FA), mean diffusivity (MD), or radial diffusivity values were significantly different between patients (PT) and controls (CT) (P < 0.05). (B) Brain images were averaged for 15 ECD patients and 15 healthy controls, generating two image maps that were statistically compared on a voxel‐by‐voxel basis. Five clusters of gray matter volume loss were found in ECD patients compared to controls (P < 0.05) and are demarcated in various colors. These clusters are all located on the right hemisphere, within the middle frontal gyrus (1.356 mL/pink), insula and perirolandic cortex (0.699 mL/red), posterior‐parietal cortex (0.411 mL/green), orbitofrontal cortex (0.254 mL/yellow), and perirolandic cortex (0.004 mL/blue).
Figure 1.
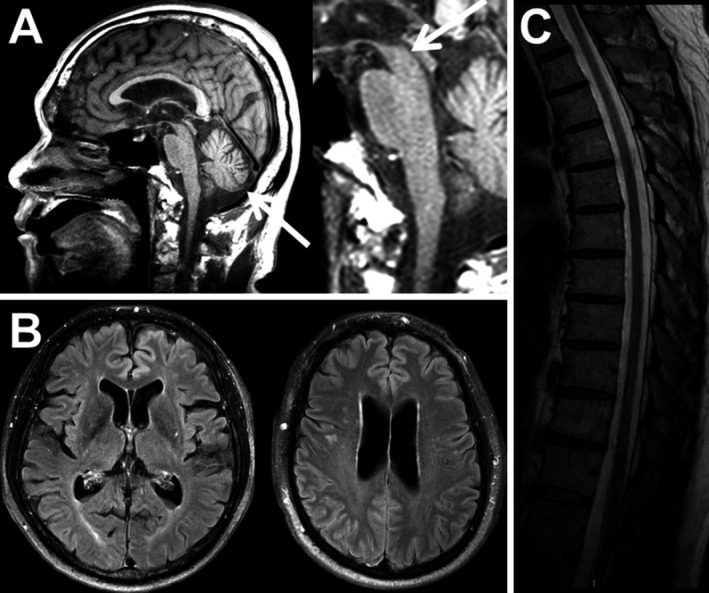
CNS neurodegeneration in Erdheim–Chester Disease. (A) Cerebellar and midbrain atrophy seen on FLAIR imaging. (B) Cerebral atrophy (FLAIR imaging). (C) Spinal cord atrophy (T2‐weighted image).
MRI lesions
Radiologic abnormalities involving the brain, orbits, or pituitary were noted in 75% of the cohort. Brain parenchyma (50%; 20/61), meninges (6%, 4/61), orbits (38%), and pituitary (26%) were most common (Fig. 2).
Figure 2.
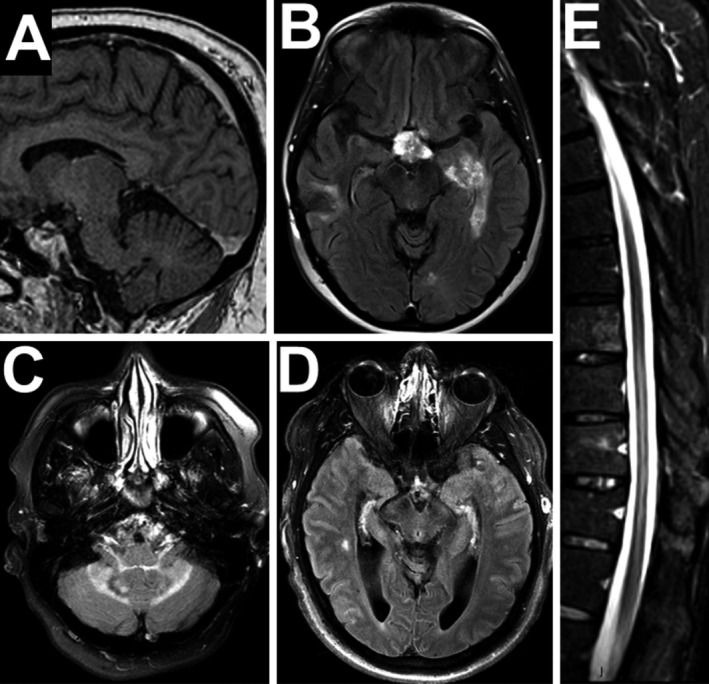
Examples of CNS parenchymal lesions in ECD. (A) Dural enhancing lesion on gadolinium‐enhanced T1 image. (B) Gadolinium‐enhanced FLAIR image revealing patchy enhancement within the left temporal lesion (right temporal resection is also seen) (C) Non‐enhancing cerebellar and peduncular lesion (D) Subtle bilateral temporal FLAIR lesions with cerebral atrophy. (E) Longitudinal spinal lesion and cord atrophy on short tau inversion recovery (STIR) imaging.
Parenchymal lesions were distributed throughout the brain: most frequently locations were the periventricular region (31%), pons (20%), and midbrain (16%) but lesions were seen in the frontal lobe, temporal lobe, occipital lobe, basal ganglia, and cerebellum. Enhancement was infrequent (18%) and typically heterogenous when present. Most of the lesions were tumor‐like (67%; 20/31) but punctate abnormalities unexplained by age or vascular disease were also seen (potentially ECD‐related). The tumor‐like lesions were frequently ill‐defined or patchy (55%) and not always discrete or measurable. Lesion‐associated edema was seen in only one patient. These findings suggest that neuro‐ECD is a multifocal disease.
To better understand parenchymal disease, quantitative diffusion tractography was performed in 15 patients and 15 age‐matched healthy controls. Compared to controls, ECD patients exhibited increased radial diffusion of water molecules (Fig. 3). These results indicate disruption of myelin integrity, and suggest ECD may involve CNS demyelination (see pathology section).27
Orbital lesions were common (38%) and caused proptosis in 22% of patients. The most frequent location was intraconal (22%) but 5% of the patients had tumor infiltration of the extraocular musculature and 9% had extraconal lesions. Five percent of patients had optic nerve encasement or infiltration but chiasmal compression was not seen. Pituitary involvement typically manifest as nodules, stalk thickening (20%) or empty sella (6%). Meningeal disease was dural in location (no leptomeningeal disease was seen), rarely enhancing, and did not compress the brain parenchyma.
Pathology
Biopsy tissue of CNS lesions revealed lipid‐laden (foamy) macrophages (Fig. 4) positive for CD68 (100%, 5/5) and negative for CD1a (100%), accompanied by gliosis (80%) and Touton giant cell formation (100%). Cells were variably positive for CD163 (60%) and S100 (40%). The ECD histiocytes were surrounded by demyelination (Fig. 4D) and scant axonal loss on neurofilament staining, consistent with the tractography findings. It should be noted that these findings may not be specific for ECD (see Discussion).
Figure 4.
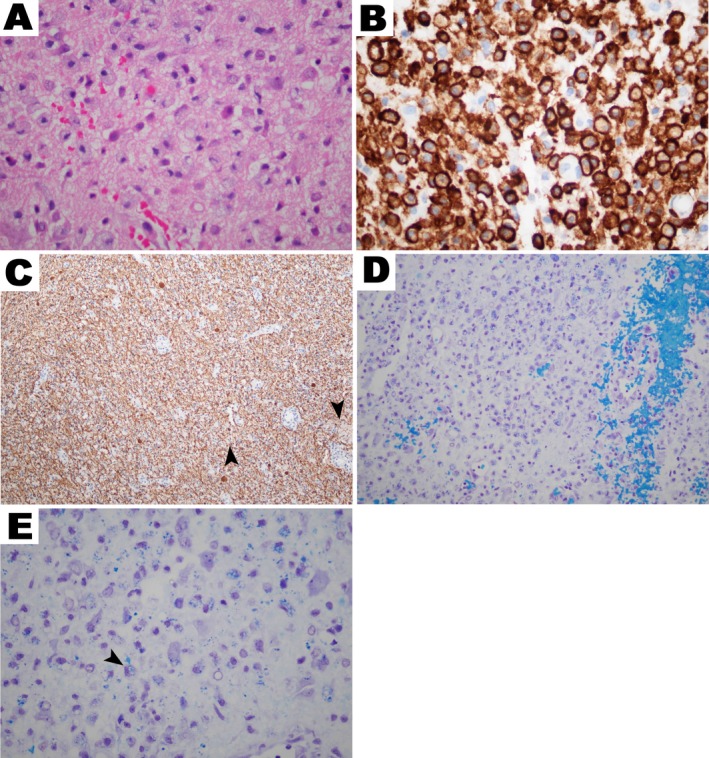
Histology of ECD brain lesions. Brain biopsy of an ECD lesions revealing (A) lipid‐laden histiocytes on H&E stain. (B) CD163 + reactivity on histiocytes (brown). (C) Neurofilament staining (brown) demonstrating axonal neurodegeneration in the vicinity of cerebral ECD histiocytes (arrowhead is an example). (D) Area of demyelination surrounding histiocytes (myelin is stained blue). (E) ECD macrophage phagocytosing PAS + myelin fragments (arrowhead for examination).
Additionally, two patients with confirmed ECD had brain pathology suggesting comorbid disease including B‐cell lymphoma and Langerhans cell histiocytosis (CD68+, CD1a+, S100+). Autopsy tissue was available from a patient whose brain contained multiple well‐circumscribed, but not encapsulated, fibrohistiocytic infiltrates involving the parenchyma, dural venous sinuses and the neurohypophysis.
Discussion
Erdheim–Chester disease is a rare, life‐threatening disorder characterized by macrophage (histiocyte) infiltration in multiple organs including the brain.4, 28, 29 Since these histiocytes can be clonal30 and contain disease‐causing variants in proliferative signaling pathways,31, 32 the World Health Organization classifies ECD as an inflammatory neoplasm.33
Our results derive from a large observational cohort study on ECD and therapeutic decisions were made by referring physicians (ethical considerations preclude studying treatment‐naïve ECD). This study design does not permit conclusions regarding therapeutic efficacy or disease mechanisms, and we cannot exclude the potential impact of treatment on our findings.
Our data suggest that neurologic disease is more prevalent than previously reported, likely a consequence of our prospective study design. Also, in contrast to initial reports suggesting that intracranial ECD is posterior‐fossa predominant, we found broad neurologic involvement throughout the brain, spinal cord, meninges, orbits, and pituitary. Extra‐axial mass effects can cause significant neurologic morbidity in this population. Examples include tumor infiltration in orbital vault and musculature causing visual symptoms or carotid bulb lesions resulting in dysautonomia.
We found cognitive impairment (and sometimes dementia) was common in ECD patients. Cognitive difficulty was assessed both clinically and by neuropsychologic testing, suggesting impairment in 50–60% of the cohort. Moreover, the BRAF V600E variant – which may be pathogenic and is an emerging risk factor for aggressive ECD2, 18, 19, 20 – statistically correlates with poor cognitive outcomes. This concurs with 28% of the cohort exhibiting cerebral atrophy on MRI interpretation. Since the interpreting radiologist was not blinded and clinical judgment may confound the results, we also assessed brain volumes using a fully automated method to quantify regional brain volumes. The statistical approach used directly compares two cohorts (ECD vs. age‐matched healthy controls) to each other but does not yield individual‐level data or prevalence information. ECD brain volumes were smaller than age‐matched healthy controls by 2.7 mL, and the volume loss was concentrated within the right frontal and parietal cortices. The regional selectivity and BRAF variant association data raise the hypothesis that impaired cognition might be pathophysiologically related to ECD. Proving this hypothesis would require identifying mechanisms underlying neurodegeneration in ECD, a subject for further investigation.
Electrophysiologic studies revealed neuropathies in half of our patients, which was purely axonal in nature. The lack of demyelination and rheumatologic comorbidity argues against an inflammatory etiology. Although direct perineural ECD tumor invasion is possible, complications of ECD2 (e.g., hypothyroidism or pituitary abnormalities), treatment (e.g., glucocorticoid, immunosuppressant, or antineoplastic therapies), or diabetes (which, in some cases, may have been induced by corticosteroid therapy) likely contribute to the neuropathy. Nonetheless, it behooves the practicing clinician to remain vigilant for possible comorbid neuropathy while treating ECD patients.
Neuro‐ECD is frequently mistaken for diseases such as progressive multiple sclerosis, neurosarcoid, CNS vasculitis, IgG4 disease, or adrenoleukodystrophy. It typically affects middle‐aged adults (rarely children) and has a progressive course. Transient improvement can sometimes be seen with glucocorticoids. ECD should be a consideration in patients who respond poorly to lymphocyte‐directed immunotherapy (such as natalizumab, cyclophosphamide, rituximab, or ocrelizumab). Also, since the systemic manifestations of ECD may be asymptomatic or otherwise not readily apparent,3 a thorough search for extra‐neurologic involvement (including recurrent evaluations over time) can facilitate diagnosis.
Radiologically, ECD lesions can mimic sarcoid, lymphoma, atypical multiple sclerosis, astrocytoma, and leukoencephalopathy. ECD lesions are multifocal, variably sized and demarcated, infrequently enhancing, and rarely cause significant mass effect or elicit surrounding edema. Dural, orbital, pituitary, or osteosclerotic lesions, when present, should be biopsied to facilitate the identification of ECD.
The brain pathology of ECD mimics inflammatory CNS disorders with the presence of lipid‐laden macrophages, demyelination, and relative axonal preservation. Although histologic staining helps differentiate ECD from other histiocytic disorders,2, 4 these markers are identical between ECD histiocytes and the reactive macrophages found in MS or sarcoid. Giant cells are not specific to ECD and can be a feature of sarcoid. Thus, routine clinical histologic analysis does not differentiate ECD from inflammatory disorders such as multiple sclerosis, sarcoid, vasculitis, or IgG4 disease. The assessment of clonality and identification of BRAF and MAP‐kinase pathway variants have the potential to distinguish the neoplastic histiocytes in ECD from reactive macrophages in multiple sclerosis.3, 30
BRAF and MEK inhibitors are effective in treating systemic ECD, and investigations are underway to assess the efficacy of these agents in modulating CNS disease (see also clinicaltrials.gov).5, 34 While vemurafenib is FDA approved for patients bearing the BRAF V600E variant, cobimetinib may be effective in BRAF‐negative patients who have other MAP kinase pathway variants.35 Such studies promise a changing landscape in our understanding and treatment of ECD, and patients are best served at specialized referral centers. A list is maintained by the ECD Global Alliance at erdheim‐chester.org.
In conclusion, neurologists play a critical role in identifying and monitoring ECD and other histiocytic disorders, because patients may present with neurologic symptoms, isolated neurologic disease occurs, and neurologic involvement portends a poorer prognosis.36, 37, 38 Furthermore, identification of ECD can spare patients morbidity associated with immunotherapies directed toward other diseases and ineffective therapies.39, 40 Ultimately, it is important for neurologists to understand histiocytic diseases because the associated dysfunction might respond to appropriate treatment.26, 41
Conflict of Interest
All authors report no conflicts of interest relevant to this work.
Author Contributions
LCB analyzed the data and wrote the manuscript; KOB, WG, JIEV, and RHD designed the study, analyzed the data and wrote the manuscript; NO, TL, BG, FHS, AN, and CT analyzed the data and wrote the manuscript; AM analyzed the data.
Supporting information
Table S1. Patient Characteristics
Acknowledgments
Sarah Kranick, Minal Bhanushali, Kavya Mathur, Gail Tarlton, Edmund Fitzgibbons, and NIH Clinical Center neuroradiology faculty for assisting with clinical data collection. The Clinical Center staff provided outstanding patient care. We thank the ECD patients and their families for participation in this study and the Erdheim‐Chester Disease Global Alliance (ECDGA) remains an invaluable resource for both us and ECD patients. This study was entirely funded by an intramural grant to the National Human Genome Research Institute, NIH.
Funding Information
This study was entirely funded by an intramural grant to the National Human Genome Research Institute, NIH.
Funding Statement
This work was funded by National Human Genome Research Institute grant .
References
- 1. Chester W. über Lipoidgranulomatose. Virchows Arch Path Anat 1930;279:561–602. [Google Scholar]
- 2. Haroche J, Cohen‐Aubart F, Charlotte F, et al. The histiocytosis Erdheim‐Chester disease is an inflammatory myeloid neoplasm. Expert Rev Clin Immunol 2015;11:1033–1042. [DOI] [PubMed] [Google Scholar]
- 3. Estrada‐Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim‐Chester disease: an observational cohort study. Blood Adv 2017;1:357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim‐Chester disease. Blood 2014;124:483–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oneal PA, Kwitkowski V, Luo L, et al. FDA approval summary: vemurafenib for the treatment of patients with Erdheim‐Chester Disease with the BRAFV600 mutation. Oncologist 2018;23:1520–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trikamji B, Mishra S. Neurological manifestation of Erdheim Chester disease: a series of 3 patients. Neurol India 2017;65:392–393. [DOI] [PubMed] [Google Scholar]
- 7. Rouco I, Arostegui J, Canovas A, et al. Neurological manifestations in Erdheim‐Chester disease: two case reports. Neurologia 2016;31:426–428. [DOI] [PubMed] [Google Scholar]
- 8. Selvarajah JR, Rodrigues MG, Ali S. Histiocytosis for the neurologist: a case of Erdheim‐Chester disease. Pract Neurol 2012;12:319–323. [DOI] [PubMed] [Google Scholar]
- 9. Brodkin CL, Wszolek ZK. Neurologic presentation of Erdheim‐Chester disease. Neurol Neurochir Pol 2006;40:397–403. [PubMed] [Google Scholar]
- 10. Wright RA, Hermann RC, Parisi JE. Neurological manifestations of Erdheim‐Chester disease. J Neurol Neurosurg Psychiatry 1999;66:72–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cives M, Simone V, Rizzo FM, et al. Erdheim‐Chester disease: a systematic review. Crit Rev Oncol Hematol 2015;95:1–11. [DOI] [PubMed] [Google Scholar]
- 12. Arnaud L, Hervier B, Neel A, et al. CNS involvement and treatment with interferon‐alpha are independent prognostic factors in Erdheim‐Chester Disease: a multicenter survival analysis of 53 patients. Blood 2011;117:2778–2782. [DOI] [PubMed] [Google Scholar]
- 13. Erdheim‐Chester Global Alliance . Available at: http://www.erdheim-chester.org.
- 14. Douaud G, Smith S, Jenkinson M, et al. Anatomically related grey and white matter abnormalities in adolescent‐onset schizophrenia. Brain 2007;130:2375–2386. [DOI] [PubMed] [Google Scholar]
- 15. Good CD, Johnsrude IS, Ashburner J, et al. A voxel‐based morphometric study of ageing in 465 normal adult human brains. Neuroimage 2001;14:21–36. [DOI] [PubMed] [Google Scholar]
- 16. Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 2004;23(Suppl 1):S208–S219. [DOI] [PubMed] [Google Scholar]
- 17. Pierpaoli C. Quantitative brain MRI. Top Magn Reson Imaging 2010;21:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cavalli G, Biavasco R, Borgiani B, Dagna L. Oncogene‐induced senescence as a new mechanism of disease: the paradigm of Erdheim‐Chester Disease. Front Immunol 2014;5:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen‐Aubart F, Guerin M, Poupel L, et al. Hypoalphalipoproteinemia and BRAF(V600E) mutation are major predictors of aortic infiltration in the Erdheim‐Chester Disease. Arterioscler Thromb Vasc Biol 2018;38:1913–1925. [DOI] [PubMed] [Google Scholar]
- 20. Chasset F, Barete S, Charlotte F, et al. Cutaneous manifestations of Erdheim‐Chester disease (ECD): clinical, pathological, and molecular features in a monocentric series of 40 patients. J Am Acad Dermatol 2016;74:513–520. [DOI] [PubMed] [Google Scholar]
- 21. Nichols TE, Holmes AP. Nonparametric permutation tests for functional neuroimaging: a primer with examples. Hum Brain Mapp 2002;15:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Winkler AM, Ridgway GR, Webster MA, et al. Permutation inference for the general linear model. Neuroimage 2014;92:381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Smith SM, Jenkinson M, Johansen‐Berg H, et al. Tract‐based spatial statistics: voxelwise analysis of multi‐subject diffusion data. Neuroimage 2006;31:1487–1505. [DOI] [PubMed] [Google Scholar]
- 24. Andersson JLR, Jenkinson M, Smith S. Non‐linear optimisation. FMRIB Technical Report TR07JA1. 2007. Available at: http://www.fmrib.ox.ac.uk/datasets/techrep/tr07ja1/tr07ja1.pdf. Accessed May 16, 2017.
- 25. Andersson JLR, Jenkinson M, Smith S. Non‐linear registration, aka Spatial normalisation. FMRIB Technical Report TR07JA2. 2007. Available at: http://www.fmrib.ox.ac.uk/datasets/techrep/tr07ja2/tr07ja2.pdf. Accessed May 16, 2017.
- 26. Euskirchen P, Haroche J, Emile JF, et al. Complete remission of critical neurohistiocytosis by vemurafenib. Neurol Neuroimmunol Neuroinflamm 2015;2:e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alexander AL, Lee JE, Lazar M, Field AS. Diffusion tensor imaging of the brain. Neurotherapeutics 2007;4:316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verdalles U, Goicoechea M, Garcia de Vinuesa S, et al. Erdheim‐Chester disease: a rare cause of renal failure. Nephrol Dial Transplant 2007;22:1776–1777. [DOI] [PubMed] [Google Scholar]
- 29. Conrad M, Haroche J, Charlotte F, et al. A 75‐year‐old woman admitted to the ICU with respiratory failure. Chest 2012;142:1063–1067. [DOI] [PubMed] [Google Scholar]
- 30. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Emile JF, Charlotte F, Amoura Z, Haroche J. BRAF mutations in Erdheim‐Chester disease. J Clin Oncol 2013;31:398. [DOI] [PubMed] [Google Scholar]
- 32. Emile JF, Diamond EL, Helias‐Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim‐Chester disease. Blood 2014;124:3016–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tzoulis C, Schwarzlmuller T, Gjerde IO, et al. Excellent response of intramedullary Erdheim‐Chester disease to vemurafenib: a case report. BMC Res Notes 2015;8:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Diamond EL, Durham BH, Ulaner GA, et al. Efficacy of MEK inhibition in patients with histiocytic neoplasms. Nature 2019;567:521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deiva K, Mahlaoui N, Beaudonnet F, et al. CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology 2012;78:1150–1156. [DOI] [PubMed] [Google Scholar]
- 37. Cohen‐Aubart F, Maksud P, Saadoun D, et al. Variability in the efficacy of the IL1 receptor antagonist anakinra for treating Erdheim‐Chester disease. Blood 2016;127:1509–1512. [DOI] [PubMed] [Google Scholar]
- 38. Haroche J, Amoura Z, Trad SG, et al. Variability in the efficacy of interferon‐alpha in Erdheim‐Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006;54:3330–3336. [DOI] [PubMed] [Google Scholar]
- 39. Mascalchi M, Nencini P, Nistri M, et al. Failure of radiation therapy for brain involvement in Erdheim Chester disease. J Neurooncol 2002;59:169–172. [DOI] [PubMed] [Google Scholar]
- 40. Rushing EJ, Bouffard JP, Neal CJ, et al. Erdheim‐Chester disease mimicking a primary brain tumor. J Neurosurg 2004;100:1115–1118. [DOI] [PubMed] [Google Scholar]
- 41. Haroche J, Cohen‐Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAF(V600E)‐mutated Erdheim‐Chester disease. J Clin Oncol 2015;33:411–418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Patient Characteristics