

Abstract
The resurgence of haploidentical stem cell transplantation (HaploSCT) over the last decade is one of the most important advances in the field of hematopoietic stem cell transplantation (HSCT). The modified platforms of T cell depletion either ex vivo (CD34+ selection, ‘mega-dose’ of purified CD34+ cells, or selective depletion of T-cells) or newer platforms of in vivo depletion of T cells, with either post-transplant high-dose cyclophosphamide (PTCy) or intensified immune suppression, have contributed to better outcomes, with survival that is similar to HLA-match donor transplantation. Further efforts are on the way to control viral reactivation using modified T cells, improve immunologic reconstitution and decreased relapse rate post-transplant using donor-derived cellular therapy products like genetically modified donor lymphocytes or NK cells. Improvements in treatment-related mortality have allowed extension of the use of haploidentical donor transplants to hemoglobinopathies like thalassemia or sickle cell disease, and possible development as platform for immunotherapy for solid tumors. Moreover, combining HSCT from related donors with solid organ transplants could allow early taper of immunosuppression for recipients of solid organ transplants and hopefully prevent organ rejection in this setting. This symposium summarizes some of the most important recent advances in this field of haploidentical transplantation and provides a glimpse in the future of fast growing field.
Keywords: Haploidentical transplantation, post-transplant cyclophosphamide, T-cell depletion, cellular therapy, immunologic reconstitution
INTRODUCTION
Haploidentical transplantation continues to expand around the world owing primarily to the application of post-transplantation cyclophophosphamide (PTCy), a cheap and easy to use alkylating agent, which was found to successfully control intense alloreactive reactions seen in this setting.1 This step forward in donor availability for hematopoietic stem cell transplant recipients is more important in the developing countries, where most patients do not have an HLA matched donor for transplantation, and the use of unrelated donors is either not available or cost prohibitive. Thus haploidentical transplants fill an enormous void and will likely become the predominant type of transplant, at least in the developing world. Availability of such donors will likely continue to drive expansion of allogeneic stem cell transplant in the coming years.2 Parallel to this expansion, the number of papers reporting results with this type of transplant is also exponentially going up3. A reflection of such an increased interest in this field is also the Haploidentical Transplantation Symposium, organized before the beginning of American Society of Hematology Meeting, now at its fourth edition.
This proceeding from this annual meeting summarizes recent developments in haploidentical presented at the Fourth Symposium on Haploidentical Transplantation, HAPLO2016, held in San Diego, California. The meeting is structured in 3 parts dedicated to Biology of Transplantation, Clinical Applications, and Cellular Therapy and Immunologic Reconstitution.
Dr. Richard Jones delivered the keynote presentation this year with on an overview of the development of port-transplant cyclophosphamide (PTCy) over the last 50 years. This was started early at Johns Hopkins in the 1960’s when Santos and Owens explored potential new bone marrow transplant (BMT) conditioning agents by evaluating the immunosuppressive properties of the known anticancer agents. Nearly 50 years ago, the group found cyclophosphamide (Cy) to be the most immunosuppressive, and accordingly developed it as a replacement for total body irradiation (TBI)4. At the same time, Santos and Owens also found that Cy, when used at high-doses on days 2–5 after transplantation, was highly effective in preventing alloreactivity in mice5,6. However, because of concerns at the time that high-dose Cy given after BMT might damage transplanted hematopoietic stem cells (HSCs), the clinical trials that arose from these preclinical studies actually used low-dose Cy as graft versus host disease (GVHD) prophylaxis.7
Subsequently, laboratory as well as corroborating clinical data showed that HSCs are resistant to high-dose Cy8. HSCs highly express aldehyde dehydrogenase 1 (ALDH1), the body’s primary means of inactivating Cy, while lymphocytes generally express low levels8. The primary function of ALDH1, also known as retinaldehyde dehydrogenase, is the biosynthesis of retinoic acid, which is required for the growth and differentiation of HSCs. Cy was actually rationally designed as an inactive prodrug that would selectively target cancer cells expressing phosphamidase capable of cleaving Cy’s phosphoramide bond, releasing phosphoramide mustard; normal cells that were thought to express less phosphamidase would be relatively spared. Although an inactive prodrug, this hypothetical mechanism of action was ultimately proven to be incorrect. It took nearly a quarter century after Cy was FDA-approved in 1959, for Hilton and Colvin at Hopkins to dissect its actual metabolic pathway. The prodrug actually undergoes bioactivation by liver P450 enzymes to its active transport congeners, 4-hydroxyCy and aldophosphamide, which exist in tautomeric equilibrium. ALDH1 actually inactivates Cy by serendipity, through oxidation of the active metabolic aldehyde intermediate aldophosphamide to the inactive carboxylic acid carboxyphosphamide8. The success of high-dose Cy without HSC rescue as treatment for aplastic anemia and other autoimmune diseases at Hopkins9 was further proof of its potent immunosuppressive, but HSC-sparing, properties.
This laboratory and clinical evidence of HSC resistance to Cy led to re-exploring it as GVHD prophylaxis, but this time at the high doses as Santos and Owens found were most effective in animal models,5,6 but were unwilling to test clinically. High-dose Cy given early after BMT was again shown to effectively prevent alloreactivity (GVHD and graft rejection) and spare HSCs, allowing successful mismatched BMT in mice.10 Translational clinical trials based on these preclinical studies showed that haploidentical related BMT using PTCy produced outcomes similar to those seen with HLA-matched BMT.11 Moreover, the degree of HLA disparity did not influence outcome as long as the donor was at least haploidentical,12 and outcomes were excellent even in patients in their 70’s13. A national multi-institutional clinical trial through the BMT Clinical Trials Network (CTN) confirmed the safety and effectiveness of haploidentical BMT using PTCy14. Multiple studies, including large registry analyses, have now shown that that haploidentical BMT with PTCy produces results similar to those seen with matched sibling and unrelated transplants.15–18
In addition to controlling haploidentical alloreactivity, PTCy is associated with excellent immune reconstitution and a low incidence of severe opportunistic infections.11 The timing of the PTCy appears to contribute to its selectivity toward alloreactive T cells. Early after allogeneic BMT, both donor and host alloreactive T cells are maximally activated and proliferative, while T cells specific for infectious agents are quiescent and thus less sensitive to Cy-mediated cytotoxicity. Recent data demonstrated that memory lymphocytes, like other cells with substantial proliferative capacity, also highly express ALDH1 and are thus relatively resistant to Cy; these Cy-resistant cells undoubtedly also contribute to the favorable immune reconstitution after PTCy.19
Allograft T cell depletion (TCD) can also limit GVHD after haploidentical related BMT. Although success with this approach has been reported in children,20 most trials in adults have been associated with relatively high non-relapse mortality (NRM) rates primarily as a result of slow immunologic reconstitution and infectious complications.21 Ciurea et al. reported the first direct comparison between TCD and PTCy for haploidentical related BMT.22 In a retrospective analysis of two cohorts of haploidentical BMT patients treated at MD Anderson Cancer Center using the same conditioning regimen, PTCy produced statistically superior actuarial overall and progression-free survivals when compared to TCD. Although both approaches were associated with low rates of GVHD, PTCy was associated with significantly improved immune reconstitution and lower NRM, predominately because of fewer infectious deaths, suggesting clearly that complete depletion of T cells from the graft should be avoided.17
In contrast to PTCy, TCD shows no selectivity toward alloreactive T cells, also eliminating T cells reactive against infectious agents as well as memory T cells. Thus, functional immune reconstitution after TCD can adequately develop in children and young adults from the thymic output of naïve T cells, but not in older adults (with thymic involution) who rely predominately on the peripheral expansion of memory T cells.23 Transplantation with another alternative donor source, unrelated umbilical cord blood, also produces excellent results in children and young adults. However, immune reconstitution and infectious complications have also been concerns in older adults transplanted with these products that are similarly deficient in memory T cells.23 A randomized trial comparing unrelated umbilical cord blood and haploidentical related transplantation in adults lacking matched donors is currently in progress through the BMT CTN. Regardless of the results of this trial, PTCy now allows safe and effective mismatched donor BMT such that no patient in need of allogeneic BMT should ever be denied the procedure for lack of a matched donor option. Moreover, the successful development of PTCy would not have been possible without the integrated work of a team of laboratory and clinical scientists with broad expertise in transplantation biology, pharmacology, hematopoiesis, and immunology.
Biology of Transplantation
Dr. Qing Ma discussed the use of post-transplant bendamustine (PTBen) instead of PTCy for prevention of GVHD in murine models. Since the main effector cells in GVHD are T lymphocytes, various approaches to modify T cell mediated alloreactivity and immune modulator agents are being explored for novel GVHD therapeutics. Given the emerging body of work suggests that PTCy is an effective strategy for GVHD prevention and similar role in selective killing of proliferating cells as “classical alkylating agents”, Bendamustine may be an appealing alternative alkylating reagent that can prevent GVHD after hematopoietic stem cell transplantation (HSCT), especially in patients with lymphoma.
Herein, Dr. Ma et al. report that bendamustine can inhibit human T cell proliferation without affecting regulatory T cells (Tregs). This could significantly reduce primary human T cell proliferation upon activation in mixed lymphocyte reactions (MLR). At 20 μM concentration, bendamustine can induce cell cycle arrest and apoptosis in human alloreactive T cells. While the frequency of dividing cells was greatly decreased for total CD4+ and CD8+ T cells, bendamustine treatment preserved CD4+Foxp3+ Treg in vitro. This was used in xenogeneic GVHD mouse model to compare the efficacy of PTBen vs. PTCy in vivo. At seven weeks post-transplant, the investigators found 90% of PTBen-treated recipients survived in comparison to 15% of PBS control mice (P=0.0067). Whereas control recipients had severe GVHD in the intestine, PTBen-treated mice exhibited only mild changes. On day 7 post-transplant, there was a significantly lower number human CD4+ and CD8+ T cells in PTBen-treated mice compared to control recipients. While PTBen treatment eliminated alloreactive T cells in mice, Tregs were spared and remained intact. Furthermore, PTBen appears to be more potent in eliminating alloreactive T cells, has less myelosuppressive effect and increased survival when compared with PTCy in a xeno-GVHD mouse model.
Dr. Dean Lee discussed current and future expansion methods for NK cells. He described the recent understanding of natural killer (NK) cells anti-leukemic response through recognition of stressed-self, resulting in lysis of leukemic cells by release of granzyme/perforin and signaling through FasL/TRAIL.24,25 In addition, he described the important role of NK cells in activating the adaptive immune system by liberating tumor antigens and establishing a pro-inflammatory microenvironment that enables dendritic cell maturation and antigen presentation, B cell maturation and class switching, and T cell (CD4, CD8, and NKT) recruitment and activation.26
In support of their critical role in HSCT, both quantitative and qualitative NK cell recovery after transplant are associated with improved outcomes, including decreased GVHD, reduced relapse, and improved overall survival.27 However, approaches aimed at enhancing NK cell number and function through adoptive transfer have not been successful until methods became available to purify NK cells and effectively eliminate T cells from the infusion product to avoid the confounding role of GVHD caused by the T cells. The first evidence of clinical benefit was demonstrated by Miller et al., wherein NK cells from haploidentical donors were infused after lymphodepleting chemotherapy, enabling the small numbers of NK cells attainable by apheresis and T cell depletion to proliferate in a permissive environment in vivo and thereby attain sufficient number to exert an anti-tumor effect.28 Further clinical investigation showed that the degree of lymphodepletion was positively associated with in vivo expansion, and this in turn was associated with clinical response.29
Based on this approach, Dr. Lee’s group designed a clinical trial to infuse NK cells from a haploidentical family member (obtained by apheresis and T cell depletion) during the pre-transplant period of a matched-donor allotransplant in patients with myeloid leukemia. NK cells were administered after busulfan/fludarabine conditioning, were allowed to exert an anti-leukemia effect for five days, which was then followed by ATG and the stem cell infusion. It was found that relapse-free survival was associated with the dose of NK cells infused.30 However, achieving these doses with the apheresis/depletion method was inconsistent and was only sufficient to deliver one infusion at approximately 1x107/kg dose. If future studies were to investigate higher doses or multiple doses, a reliable method of ex vivo expansion was necessary.
Previously, the group had developed a method to generate large numbers of NK cells utilizing a genetically-modified K562 cell line and demonstrated the utility of this platform for expanding NK cells from normal donors,31 patients,32,33 cord blood,34 and embryonic/pluripotent stem cells35. Importantly, this approach leads to NK cells with extended telomeres to avoid proliferative senescence, and produces NK cells with both high cytotoxicity and high cytokine secretion.31 The same group then established a master cell bank of the feeder cells36 to enable expansion of clinical-grade NK cells using this approach.
This enabled the conduct of several trials to test whether increased numbers of hyper-functional NK cells would improve on the results seen in prior NK cell studies. First, the MD Anderson group initiated a dose-escalation study to deliver the maximum number of NK cells tolerated after FLAG chemotherapy for relapsed/refractory leukemia, which is ongoing (NCT01787474). The first patient treated achieved a prolonged complete remission of over 4 months, despite coming into the trial in 3rd relapse with 93% marrow blasts after over 10 cycles of high-dose therapy including 5 induction/re-induction failures and a haploidentical stem cell transplant. Building on our group’s prior evidence for NK cell benefit in allogeneic transplantation, a study was initiated to further increase the number of haploidentical NK cells delivered during matched donor allogeneic transplantation, which finished accrual at the highest dose level without toxicity and is completing long-term patient follow-up (NCT01823198).
The MD Anderson group previously demonstrated feasibility of a reduced-toxicity HaploSCT regimen for myeloid leukemia utilizing PTCy for GvHD prophylaxis37. To maximize the benefit of expanded NK cells, a dose escalation trial was initiated based on this HaploSCT regimen that infused multiple doses of expanded NK cells from the same donor before and after the stem cell infusion (NCT01904136). Compared to patients treated on the original regimen, those who received NK cells at day +7 had improved NK cell function at day +30, had fewer CMV and BK virus reactivations, and may have improved overall survival compared with a retrospective group of similar patients (not yet statistically significant likely because of small numbers in the Phase I trial).38
Lastly, Dr. Lee discussed his phase I trial of locoregional infusions of expanded autologous NK cells for pediatric patients with relapsed 4th ventricle brain tumors (medulloblastoma, ependymoma, or atypical teratoid/rhabdoid tumor) (NCT01823198). Because of the small volume of the 4th ventricle and proximity to the brainstem, frequent infusions of small numbers of cells in a 2mL infusion volume were performed. This trial also continues to enroll patients.
Overall, the results of these early trials are encouraging. All the NK cell infusions were well tolerated with promising evidence of long-lasting immune effects and disease responses, and similar positive results of NK cell adoptive immunotherapy are being reported by other groups. Continued improvements in the NK cell manufacturing process are being pursued using a method for disrupting the feeder cells into exosome-like plasma-membrane particles.39 Advances are being made in applying unique phenotypic and/or functional NK cell subtypes, such as adaptive NK cells40 and memory-like NK cells.41 It is now believed that, with novel expansion methods to obtain high does of NK cells, NK cell therapy if becoming a mainstream approach for treatment of various tumor types.
Dr. Yair Reisner discussed how transplantation of T cell–depleted BM (TCD-BM) with non-myeloablative conditioning (NMA) can be associated with minimal toxicity and low risk of GVHD. This could offer an attractive therapeutic option for patients with nonmalignant hematologic disorders or can be used to induce immune tolerance to subsequent organ transplantation. However, overcoming TCD-BM rejection after reduced conditioning intensity remains a challenge. Dr. Reisner’s group previously demonstrated that this barrier could be overcome by using donor-derived naïve CD8+ T cells if they are expanded against third-party alloantigens under culture conditions, inducing a central memory phenotype. Thus, such anti-third party CD8+ T cells (Tcm) exhibit marked veto activity and reduced risk for GVHD due to the lower frequency of anti-host clones achieved during the culture period42. In the present study, his group tested the feasibility of generating veto Tcm by stimulation against well-defined peptides including viral antigens. For proof of concept, they used OT1 mice that express a transgenic (Tg) TCR designed to recognize ovalbumin (OVA) residues 257–264 in the context of H2Kb MHC-I. Prior to harvest of OT1 CD8+ T cells, mice were immunized twice with OVA-peptide. Mice were sacrificed 7 to 14 days after immunization, their spleens and lymph nodes removed and crushed, and magnetic bead sorting utilized to isolate the memory cells (CD8+CD44+). The resulting population was subjected to third-party stimulation by co-culture with irradiated splenocytes generated from spleens of OVA-expressing mice under cytokine deprivation. HIL-15 (10ng/ml) was added to the culture 60 hours after culture initiation to induce the cells to express a Tcm like phenotype, as previously described for anti-third party Tcm generated from WT naïve CD8+ T cells (WT Tcm).43
When tested in vivo, the OT-1 Tcm (H2Kb) were able to enhance engraftment of allogeneic T cell depleted BMT (H2b) in sub-lethally irradiated (5.5 Gy TBI) Balb/c recipients (H2Kd), in analogy to the chimerism induced by WT Tcm.43 This initial successful experiment was then followed by an experiment, which more closely resembled the human setting, in which the CD8+CD44+ cells were isolated from WT C57BL/6 OVA-immunized mice and subsequently introduced to co-culture with irradiated splenocytes generated from spleens of OVA-expressing mice. Results showed that Tcm grown from a starting population of CD8+CD44+ cells was also able to achieve marked donor chimerism when administered with mega-dose of TCD alloHSCT in sub-lethally irradiated Balb/c recipients. Notably, infusion of 5x106 Tcm into sub-lethally irradiated mice (5.5. GY TBI) without BMT, did not cause any GVHD as measured by weight loss, in contrast to CD8+CD44+ T cells used to generate the Tcm.43 These results demonstrate that CD8+CD44+ derived from memory CD8+ T cells by expansion against cognate peptides exhibit markedly reduced risk for GVHD compared to freshly isolated memory cells, while retaining their veto activity and inducing tolerance.
Finally, based on these proof of concept studies, Dr. Reisner’s group were able to translate this approach and generate human anti-viral CD8+ veto cells with central memory phenotype. Thus, CD8+CD45RO+ memory T cells were selected by depletion of CD4+CD56+CD45RA+ cells from peripheral blood mononuclear cells of normal donors and then co-cultured with donor dendritic cells pulsed with a viral peptide mixture of 3 prominent viruses (EBV, CMV and adenovirus). In three large-scale experiments using leukapheresis preparations of normal CMV and EBV positive donors, with GMP grade reagents, more than 1x109 Tcm could be attained by the end of 9 days of culture (average expansion of Tcm =13.5±4 fold) with greater than 90% purity of CD45RO+CD62L+CD8+ T cells. The harvested anti-viral Tcm exhibited more than 3 log depletion of alloreactivity, compared to fresh CD8+ T cells, as measured by limit dilution analysis of cytotoxic T cell precursors against host type target cells. Dr. Reisner concluded that potent veto CD8+ Tcm can be generated from the memory pool of donors positive for viral reactivity by stimulation against viral antigens. Such veto Tcm could be most attractive for TCD-BM HaploSCT, as they can enable engraftment following NMA conditioning and at the same time provide anti-viral protection.43
Dr. Jeffery Miller discussed immune activation strategies to prevent relapse including IL-15, tri-specific killer cells engagers and adaptive NK cells with immune memory. In clinical trials involving patients with refractory acute myelogenous leukemia at the University of Minnesota, Dr. Miller’s group used a lymphodepleting regimen with high-dose cyclophosphamide and fludarabine (Hi-Cy/Flu) followed by infusion of adoptively transferred HLA-haploidentical NK cells from related donors.28 This preparative regimen uniformly resulted in lymphopenia and a surge in endogenous serum IL-15 (up to 200 pg/mL). IL-2 was also administered with the intent of further expanding NK cells in vivo. In a cumulative experience, 40 patients have been treated with this regimen, and in a recent trial IL-2 diphtheria toxin (IL2DT) was added to test the hypothesis that IL2DT would selectively deplete IL-2 receptor (CD25hi) expressing regulatory T cells (Treg).29 Among the 15 patients treated with this regimen, 10 had detectable donor NK cells at day 7 (median 68% donor DNA). At day 14, 27% had successfully expanded NK cells in vivo, with median absolute donor - derived NK cell counts of 1000 cells/μL blood. These results improved upon their previous rate of 10% in vivo NK cell expansion observed with the same regimen but without Treg depletion. Augmented lymphocyte and Treg depletion with IL2DT resulted in 53% patients attaining complete remission, significantly better compared to strategies without IL2DT (CR rate 10%, P=0.02). These outcomes suggest that the NK cells themselves played a role in the antileukemia response over and above the activity of the high-dose chemotherapy preparative regimen. Patients achieving remission also had a significantly higher proportion of circulating donor NK cells, further suggesting that persistence and expansion is required to observe clinical efficacy. Therefore, limitations of NK cell therapy include the need for: 1) better activation without inducing exhaustion or other suppressive mechanisms, 2) functional memory, and 3) better specificity. Newer cytokines are now available; IL-15 is the homeostatic receptor controlling NK cell development, proliferation, and activation.44 Recombinant human IL-15 (rhIL-15) is a cytokine and growth factor capable of expanding and activating T cells and NK cells. By broad consensus, the NCI Immunotherapy Workshop (2007) ranked IL-15 as the #1 agent with “high potential for immunotherapy.” The NCI Biological Resource Branch has manufactured E. coli-expressed rhIL-15, and this was the first IL-15 product tested at the University of Minnesota. Based on preclinical, non-human primate and early phase clinical trial data at the NCI45, systemic administration of rhIL-15 by daily intravenous bolus was tested in adoptive NK cell schema. The clinical MTD of rhIL-15 using both intravenous and subcutaneous dosing when used to promote NK cell adoptive transfer have been established. There may be limitations in addition to short half-life. High levels of free rhIL-15 may decrease circulating IL-15 Receptor-alpha (IL-15Rα), yielding negative feedback and limiting IL-15 trans-presentation, the physiologic mechanism by which IL-15 activates the immune system neighboring NK or CD8+ T cells which express common IL-2Rα receptors.
Dr. Millers’ group has “first in human” clinical data on a novel IL-15/IL-15Rα-Fc construct, ALT-803 (Altor Biosciences, Miramar, FL), designed to have a prolonged serum half-life allowing for intermittent weekly dosing. ALT-803 is a soluble complex consisting of two protein subunits of a human IL-15 variant associated with high affinity to a dimeric human IL-15 receptor α (IL-15Rα) sushi domain/human IgG1 Fc fusion protein. The IL-15 variant is a 114 aa polypeptide comprising the mature human IL-15 cytokine sequence with an Asn to Asp substitution at position 72 of helix C (N72D). Aside from the N72D substitution, all of the protein sequences are human. ALT-803 has a prolonged serum half-life in preclinical animal models and has a 4-fold increase in biologic activity greater than wild-type IL-15.46,47
The University of Minnesota in collaboration with Washington University have completed a first-in-human phase 1 trial of ALT-803 in patients who relapsed after alloHCT. While safe dosing up to 10 mcg/kg intravenous or subcutaneous were tested, 6 mcg/kg SC was selected in a relapse-prevention trial because: 1) Current data shows favorable PK with this SC dose, 2) Known biologic activity of this dose to proliferate NK cells and CD8+ T cells in vivo, and 3) When comparing intravenous to subcutaneous dosing, the peak concentration decreases eliminating cytokine release and fever seen 5–6 hours after each dose of IV ALT-803.
Dr. Miller further discussed on better NK cell functional memory as terminal maturation of CD56dim NK cells is associated with acquisition of CD57. Rather than being an immunosenescence marker, CD57 acquisition represents a shift toward greater effector function, including increased CD16 signaling (Fc receptor responsible for triggering antibody-dependent cellular cytotoxicity) and enhanced cytotoxicity.40 Cytomegalovirus (CMV) infection is uniquely associated with expansion of CD57+ NK cells expressing the activating receptor NKG2C.48 This group has reported that in vivo expanded of CD57+NKG2C+ NK cells (referred to as adaptive NK cells) persist for over one year and are directly associated with reduced leukemia relapse after reduced intensity HSCT40.
Ex vivo expansion to enrich the subset of cells with the adaptive NK cell phenotype represents a new strategy to obtain high numbers of NK cells with enhanced effector function for use in adoptive transfer to treat cancer patients. Clinical trials will start in 2017 with their first generation adaptive NK cell product. Also Dr. Miller discussed the need for better NK cell specificity, as with several tumor-targeted antibody strategies have been proposed to enhance NK cell activity or targeting. These approaches are intended to interrupt NK cell inhibition, provide co-stimulation, or to enhance targeting through CD16. Each of these strategies has the potential to augment the therapeutic benefit of NK cells. The Fc receptor CD16 is present on most peripheral blood NK.49 Upon recognition of antibody-coated tumor cells, CD16 delivers potent activating signals to NK cells, leading to target elimination through direct killing and cytokine production. In addition to monoclonal antibodies, Dr. Miller’s group focused on a platform using bi-specific killer engagers (BiKE) constructed with a single-chain Fv against CD16 and a single-chain Fv (scFv) against a tumor-associated antigen. They initially developed a CD16/33 BiKE to target myeloid malignancies (AML and myelodysplastic syndrome).50
One of the most remarkable properties of this drug is its potent signaling. In refractory AML, CD16/33 BiKE overcomes inhibitory KIR signaling,51 leading to potent killing and production of cytokines by NK cells.52 However, the BiKE does not sustain NK cell survival or deliver a proliferative signal to NK cells. Therefore, a third function to BiKE was added by inserting IL-15 between the two scFv components. Their tri-specific hybrid drug (CD16/IL15/CD33 TriKE) binds NK cells, myeloid CD33+ targets and generates an IL-15 proliferative and survival response in vitro and in vivo.53
Dr. Peter Lang presented preliminary results on anti-CD19-based immunotherapy post HaploSCT in children with immunomagnetic depletion of TCRαβ+ T cells and CD19+ B cells, and how it can improve immune recovery without increasing the risk of GvHD.54 Herewith, sufficient numbers of potentially anti-leukemic effector cells expressing the Fc low-affinity receptor FcγRIIIa (type III receptor for IgG; CD16), like NK cells and γδ T cells, can be achieved already within the first 30 days post-transplant, preceding the recovery of αβ T cells.54 Those donor derived effector cell populations, which are not influenced by pharmacological immune suppression, can form a basis for further post-transplant immunotherapeutic strategies. Since B-lineage acute lymphoblastic leukemia (ALL) is the most common malignancy in childhood, Dr. Lang’s group focuses on 2 approaches which target the CD 19 antigen: 1) A chimerized SDIE-modified CD19 antibody (4G7SDIE mAb), for treatment of minimal residual disease (MRD) or for prophylactic use, and 2) A bispecific single-chain antibody construct (BiTE) blinatumomab for treatment of hematopoietic relapse. 4G7SDIE mAb exerted significant lysis of several leukemic cell lines and primary ALL blasts via NK cells and γδ T cells from healthy donors or transplanted patients in vitro. A correlation of CD19 expression on leukemic blasts and 4G7SDIE-mediated lysis underlined the importance of sufficient antigen sites on target cells.55 Moreover, 4G7SDIE mAb was administered prior or post stem cell transplantation in a cohort of 14 MRD-positive patients, suffering from primary refractory and/or relapsed CD19-positive B-lineage ALL at very high risk for relapse, to reduce or eradicate residual leukemic cells and prevent recurrence. Ten out of 14 patients responded to the treatment with a reduction of MRD ≥ 1 log. The side effect profile of 4G7SDIE infusion was very low, and only intermittent increased temperature and fever, headache and nausea were observed. Neither gastrointestinal nor neurotoxic events nor GvHD were documented.55 In another group of 8 patients at extremely high risk for relapse who had received ≥2nd transplant due to subsequent relapses, 4G7SDIE mAb infusions were given as prophylaxis every 4 weeks over 24 months post-transplant. A favorable 2-year EFS of 60% was observed. Thus, this antibody format may be preferably used (1) In a patient with MRD, (2) In patients who may not be able to tolerate severe side effects, and (3) As a prophylactic treatment in patients without detectable MRD but at high risk for leukemic relapse.
For treatment of full-blown relapse after HSCT, 9 patients were treated with blinatumomab. All patients had a 1st (n=1) or subsequent (2nd-4th, n=8) post-transplant relapse and did not respond sufficiently to previous chemotherapy. Four out of 9 patients achieved a complete remission after the 1st cycle of treatment. In other 2 patients, who were refractory to the 1st cycle of blinatumomab at a dose of 5 μg/m²/day, chemotherapy was administered after blinatumomab treatment to stop leukemia progression. Remission was finally achieved by the 2nd cycle of blinatumomab, which was administered at a higher dose of 15 μg/m²/day.56 It is remarkable that an additional chemotherapy after initial blinatumomab courses resulted in a secondary response. The synergic effect of conventional chemotherapy to reduce blast load and then re-attempt blinatumomab treatment might be a reasonable therapeutic strategy in non-responders. In patients with poor bone marrow function, the use of stem cell boosts from the previous donor might enable such additional chemotherapy and may contribute to relevant T-cell response. Apart from already described, typical side effects of blinatumomab applications, no GvHD occurred despite intensive stimulation of the donor-derived immune system. In a currently ongoing clinical trial with a larger patient cohort suffering from post-transplant relapse, similar response rates have been observed. Further investigations have to address the questions if extramedullary non-CNS and non-gonadal relapses at atypical sites, which might be a limiting factor, and if additional use of checkpoint inhibitors can increase response rates.
Dr. Lang concluded that in HaploSCT with ex vivo manipulated grafts, post-transplant immune therapies can provide long lasting pharmacological immune suppression. Especially in case of CD19+ leukemia, antibody-based approaches are already available for clinical use and may help to reduce the risk of disease recurrence.
Clinical Applications
Dr. Stefan Ciurea presented a brief overview of HSCT outcomes for patients with ALL in 1st complete remission (CR1) with HLA matched donors as well as outcomes from 2 recent analyses on patients with ALL with haploidentical donors, one from the newly formed Haploidentical Transplantation Research Consortium (HIT-RC) and one from the European Group for Blood and Marrow Transplantation (EBMT).
Approximately 6,590 new cases with ALL were diagnosed in 2016, with a median age at presentation of 14 years. The outcomes in children are significantly better, with 5-year overall survival (OS) of approximately 80% vs. approximately 40% in adults, reflecting different biology between these 2 groups. Approximately 60% of patients will eventually relapse and the median survival after relapse is less than 10 months.57,58 There is a controversial role for HSCT in CR1 patients with ALL. However, several meta-analyses showed benefit for HSCT in CR1 for Ph− and Ph+ ALL patients in adults.59–61
Kako et al. analyzed outcomes of 649 patients with Ph− ALL in CR1 reported to the Japanese Transplantation Society Registry between 1993 and 2007. Four hundred and eight patients had HSCT, while 241 received chemotherapy on 2 contemporaneous clinical trials. The primary endpoint was 10-year survival. This group found better survival with HSCT from matched sibling donors in CR1 compared with chemotherapy alone (48.3% vs. 32.6%), benefit seen for all subgroups of patients (standard-risk, high-risk, lower age and higher age groups). The authors concluded that HSCT in CR1 for Ph-ALL is superior to chemotherapy.60 A second meta-analysis was performed by Gupta et al., who retrospectively analyzed results of 2,962 patients with HSCT vs. autologous HSCT vs. chemotherapy for Ph-ALL patients in CR1.61 This group found that patients undergoing HSCT had lower relapse rate but higher treatment-related mortality (TRM) with better OS in the HSCT arm (OR 0.87, p=0.0003). HSCT appeared to be better for patients younger than 35 years (OR 0.79, p=0.0003); however, no survival benefit was seen for patients 35 years or older (OR 1.01, p=0.9). The 5-year survival was 55% vs. 45.1% for patients younger than 35 years and 39.2% vs. 37.2% for patients age 35 years or older, in favor of HSCT vs. chemotherapy, respectively. The survival curves crossed early (before 2 years) and differences became progressively different with a trend favoring HSCT with time.61
For Ph+ ALL patients, results from a recent SWOG phase II prospective multicenter trial (conducted in 37 centers) were published in abstract format by Ravandi and colleagues at ASH 2015. Ninety-seven patients with newly diagnoses Ph+ ALL age 18–60 years were randomized to received HyperCVAD plus dasatinib followed by dasatinib maintenance vs. HSCT in CR1 followed by dasatinib maintenance for up to 5 years. Forty-one patients (42.3%) received HSCT (35/41 with myeloablative, TBI-based conditioning). The median age was 44 years, and median follow-up of survivors was 26 months. The 12- month relapse-free survival (RFS) and OS were 71% and 87%, respectively for HSCT arm, superior for HSCT compared with maintenance dasatinib only [RFS (p=0.03) and OS (p=0.07)].62 Although longer follow-up is needed, this clearly suggests superiority of early allogeneic transplantation for Ph+ ALL in CR1.
Regarding HaploSCT, Wang et al. from China recently reported results of a phase III prospective multicenter study conducted between 2010–2013 using haploidentical vs. HLA matched sibling donor (MSD) transplants for Ph− high-risk ALL. The patients were biologically randomized to receive either HSCT from a MSD (N=83) or a haploidentical donor (N=103). The median follow-up was 3 years. The median age for MSD transplants was 38 years vs. 26 years for haploidentical donors (P<0.001). The 3-year RFS was 68% vs. 64% for haploidentical vs. MSD HSCT, respectively, while the NRM was 13% vs. 11% (p=0.84), and relapse was 18% vs. 24% (p=0.3). There were no significant differences in rate of acute and chronic GVHD.63
Experiences with HaploSCT for patients with ALL in the United States and Europe have been recently summarized in 2 retrospective analyses performed in the US and Europe, which showed similar results. In the US analysis, outcomes of 109 patients with a median age of 32 years were analyzed. Only 29% were in CR1, 33% in second complete remission (CR2) and the rest were beyond CR1/CR2 or had HaploSCT as a second transplant (N=13), 26 % of patients were MRD+, 32% had WBC >30,000/uL at presentation for B-cell ALL and 40% >100,000/uL for T-cell ALL patients. The hematopoietic stem cell transplant-comorbidity index (HCT-CI) was 0–1 in 52% of the patients, 54% had a peripheral blood graft, 49% with a sibling, 34% with a parent and 17% with a child donor, 64% of patients received a myeloablative conditioning regimen.64 The median follow-up was 13 months. The cumulative incidence (CI) of acute grade II-IV and III-IV GVHD at day 100 post-transplant were 32% and 11%, respectively, whereas the CI of chronic GVHD was 32%. The 1-year NRM, relapse rate and RFS for all patients were 21%, 27% and 51%, respectively. The 3-year RFS for patients in CR 1 was 52%. In this analysis, the only factor associated with better RFS in MVA was the use of a myeloablative conditioning regimen compared with the NMA conditioning with Flu/Cy/TBI regimen64.
In the corresponding European analysis from the EBMT, 208 patients were analyzed.65 The median age was also 32 years, the median follow-up for survivors was 16 months. When compared with the US study, only 52% of patients had PTCy-based GVHD prophylaxis, the rest received ATG-based GVHD prophylaxis. Forty-four percent of the patients were in CR1 and 57% had a peripheral blood graft. The CI of grade II-IV and III-IV acute GVHD were 31% and 11%, respectively, and the CI of chronic GVHD was 29%. NRM and relapse incidence were 32% and 37%, respectively and OS, RFS and GRFS were 33%, 31% and 26%, respectively. Patients in CR1 had 52% OS, 47% RFS and 40% GVHD-free, relapse-free survival (GRFS). Factors associated with better RFS in this analysis were being in CR1 vs. other (HR 0.51, p<0.01), Karnopsky performance status (KPS) ≥90% (HR 0.58, p=0.05), while the use of ATG-based GVHD prophylaxis was associated with worse NRM and RFS (HR 1.03, p=0.03 for RFS). Factors associated with worse GvHD-free, relapse-free survival (GRFS) were KPS<90 (HR 2.22, p=0.02), female donor to male recipient (HR 1.53, p=0.05), and the use of PB when compared with BM graft (HR1.56, p=0.02).65
Dr. Ciurea concluded that according to multiple analyses of patients with ALL, transplant outcomes with haploidentical donors show encouraging results. PTCy-based GVHD prophylaxis is associated with better survival in recipients of HaploSCT. At present, only a minority of transplant eligible ALL patients are transplanted in CR1, although transplant outcomes for HaploSCT in CR1 appear similar with HLA matched donor transplants. Future studies will evaluate HaploSCT outcomes compared with matched unrelated donor transplants in a matched cohort analysis.
Dr. Suradej Hongeng discussed his group’s experience with HaploSCT for patients with severe thalassemia using a pre-transplant immunosuppressive (PTIS) program consisting of fludarabine (Flu) in combination with dexamethasone (Dxm) to suppress patients’ immune system and facilitate engraftment of donor cells, followed by a reduced-toxicity conditioning regimen. In this study peripheral blood rather than bone marrow graft was used to be able to obtain a larger number of target CD34+ progenitor cells in the graft (10 × 106 CD34+ cells/kg recipient body weight). Dr. Hongeng reported results of 31 consecutive patients with severe thalassemia (both homozygous β-thalassemia and β-thalassemia/hemoglobin E diseases), who had onset of transfusion dependence (225 mL/kg/year) during the first 3 years of age, pre-transfusion hemoglobin level ≤7 gm/dL, and hepatosplenomegaly. Donors were selected from parents without clinical hemoglobinopaties, who had the highest degree of HLA matches of A, B, C, DRB1 and DQB1 alleles. If degree of allele mismatch between the patient and both parents was similar, the mother was considered a preferred donor. Data on donor-specific anti-HLA antibodies (DSA) were obtained and analyzed but was not used for a donor selection consideration and patients with DSA positivity did not received desensitization treatment before transplant. The PTIS consisted of Flu 40 mg/m2/day IV together with 5 days of Dxm 25 mg/m2/day IV administered in 2 courses on day −68 to −64 and −40 to −36. Patients then received ‘early’ antithymocyte globulin (Thymoglobulin; Sanofi-Genzyme Canada, Ontario, CA, USA) 1.5 mg/kg/day on day −12 to −10. The conditioning chemotherapy consisted of Flu 35 mg/m2/day IV over 60 minutes once daily on day −8 to −3, and IV busulfan (Bu) 130 mg/m2 IV over 3 hours once daily on day −8 to −5. The GVHD prophylaxis consisted of Cy 50 mg/kg/day on day +3 and +4, tacrolimus or sirolimus and mycophenolate mofetil (MMF) at 15 mg/kg orally twice daily for 60 days starting on day 5.66
In Dr. Hongeng’s initial report, 31 patients (4 patients with homozygous β-thalassemia and 27 patients with β-thalassemia/hemoglobin E disease) with median age of 10 years (range: 2–20) were treated with this PTIS and HaploSCT program. Eleven and 20 patients received peripheral blood stem cell graft from father and mother, respectively. Fifteen, 8, 7 and 1 patient received a 5/10, 6/10, 7/10 and 8/10 HLA allele-matched graft, respectively. On weekly outpatient evaluation, none of the patients developed complications or side effects during or following the PTIS treatment phase. All patients were admitted to the inpatient unit on day −13 before starting the conditioning regimen. The median CD34+ cell dose was 11.6 × 106 cells/kg body weight (range: 4.0–19.0 × 106). The median time to neutrophil and platelet engraftment was 14 days (range: 11–18) and 30 days (range: 20–45), respectively. Three patients had high antibody titers (>1:3000) before transplant while 2 patients developed primary graft failure. Grade 1–2 mucositis was common (n=11) but grade 3 was seen in only 3 patients. Five patients developed mild to moderate venoocclusive disease, all resolved with supportive care measures. Infections reported were: CMV reactivation (n=6), BK-virus cystitis (n=4), adenovirus cystitis (n=2), herpes zoster (n=1), and gram-negative septicemia (n=2). But all infections resolved with appropriate therapies. Nine patients developed grade II acute GVHD and 1 patient had grade IV acute GVHD. Five patients developed limited-chronic GVHD. All surviving patients who developed GVHD were able to discontinue systemic immunosuppression within 6–12 months after transplant. With median follow up of surviving patients of 12 months (range: 7–33), 2-year OS and event-free survival (EFS) were 95% (95% CI 69.5–99.3%) and 94% (95% CI 76.6–98.4%), respectively (Figure 2).66 In the updated analysis, a total of 67 thalassemia patients were treated on this protocol. The 2-year OS and EFS were 94% (95% CI; 82.2–97.9%) and 94% (95%CI; 82.2–97.9%), respectively.
Dr. Hongeng concluded that outcomes after HaploSCT in patients with severe thalassemia are promising. The rapid and durable engraftment is associated with low incidence of serious complications and with low risk for severe acute GVHD. Additionally, the program can be adapted and used for treatment of patients with sickle cell anemia and other non-malignant diseases such as severe aplastic anemia.
Dr. Parameswaran Hari discussed data on HaploSCT in multiple myeloma (MM). He started his argument that a variety of novel immunotherapy approaches (i.e. vaccines, immune check point inhibitors, and reengineered T cells) are being explored in active clinical trials, whereas HSCT represents the most established immunotherapeutic strategy available for MM. The evidence for the existence of a sustained graft-versus-myeloma (GVM) effect67,68 is well established but excessive TRM has prohibited wide adoption of this modality. Similarly, it has been observed that chronic GVHD correlates with protection from relapse,68,69 donor derived lymphocytes can eliminate residual or relapsing disease70 and lower relapse rates observed in recipients of T-cell replete allografts compared with T-depleted/syngeneic grafts. A variety of NMA and RIC regimens have been developed to address the TRM issue,71,72 but lower TRM seemed to be offset by increased relapse risk73 with discordant results in clinical trials. Notably, all of these data are with fully HLA matched donors. PTCy-based GVHD prophylaxis in recent years has made haploidentical donor grafts much safer and led to universal donor availability even in ethnic minorities and older individuals.13 While prospective data are not available, Dr. Hari’s group examined the limited data available from the Center for International Blood and Marrow Transplant Research (CIBMTR). Forty-one patients with median age of 55 years (range 36–73) received haploidentical grafts with PTCy- based GVHD prophylaxis. HCT-CI ≥3 in 41%. NMA (FluCyTBI) conditioning regimen was used in majority of the patients (71%), while 50% received marrow grafts. In the setting of advanced myeloma, TRM was 8% but relapse rate was high at 72% in the first year and 2 years- OS was 48%. Castagna et al. reported the results of a retrospective multi-center study in abstract form at ASH 2016.74 Data were reported on 29 patients receiving haploidentical grafts for relapsed/refractory myeloma between 2011 −2015 following either MAC (n=15) or RIC regimens (n=14) with PTCy-based GVHD prophylaxis. Although 93% had relapsed after a prior autologous transplantation and 92% had previous lenalidomide and bortezomib exposure, only 62% of the patients were chemotherapy sensitive prior to transplant. Marrow grafts were used in 60% of the patients. TRM was at 15% while the risk of relapse or progression was 39%, 18-month PFS and OS were 34% and 68%, respectively. CI of grade II-IV acute GVHD was 38%.
Dr. Hari concluded that with these data in multiply relapsed patients with MM, PTCy-based HaploSCT could be considered a feasible form of immunotherapy in relapsed high-risk patients who achieve a remission after salvage regimens. However with a significantly high relapse rate, HaploSCT could be considered a platform for further cellular/non-cellular immune manipulation. A variety of approaches including donor derived NK cells, myeloma-specific T cells, marrow infiltrating lymphocytes, checkpoint inhibitors, and immunomodulatory drugs can be considered to induce myeloma-specific immunity.
Dr. Samuel Strober presented data on combined solid organ and hematopoietic stem cell transplantation. He started by describing the goals of tolerance induction in organ transplant patients including elimination of lifelong need for immunosuppressive drugs as well as preventing graft loss due to rejection. In his work, twenty-eight patients with HLA matched and 19 with haploidentical donor HSCT were conditioned with total lymphoid irradiation (TLI) and rabbit ATG during the first 11 days after kidney transplantation. Immediately after TLI, HLA matched recipients received at least 4x106 CD34+ cells/kg purified on a Miltenyi column, and a low dose of 1 x106 CD3+ T cells/kg. Donor cells were collected from G-CSF mobilized peripheral blood and cryopreserved about 6 weeks before kidney transplantation. Twenty-one recipients were given 1 month of MMF and 9–12 months of cyclosporine before discontinuation of both drugs. There was no evidence of subsequent rejection during a follow-up period of up to 8 years except in 1 case after 4 years. Five patients did not meet drug withdrawal criteria, and 2 are in taper, none have lost kidney grafts with up to 11 years of observation. Of the 21 patients, 7 have been stable mixed chimeras for up to 8 years, and 14 lost donor chimerism in the second year despite maintenance of excellent graft function off drugs.
Haplotype matched recipients were given escalating doses of donor T cells up to 100 x106/kg and persistent chimerism for at least 1 year has been achieved in 5 patients despite the discontinuation of MMF and the taper of tacrolimus to a blood level of 3–5ng/ml (minimal effective dose; MED). These patients were withdrawn from tacrolimus during the second year to determine whether chimerism persists, and rejection was prevented for at least one year after complete withdrawal. He added that his group is currently determining the fraction of mismatched patients given the high doses of donor T cells who can maintain a stable chimerism and graft function on the MED of tacrolimus versus complete immunosuppressive drug withdrawal.
Dr. Ephraim Fuchs presented data on HaploHCT for solid tumors. He started with the proposal that HSCT can cure poor risk acute or chronic leukemia via a “graft-versus-leukemia” (GVL) effect mediated by donor T cells reactive against disparate major or minor histocompatibility antigens expressed by the malignant cells. Due to the expression of histocompatibility antigens by all parenchymal cells, this favorable effect is associated an immunologic attack on normal tissues called “GVHD”. This association is not absolute and it is possible to achieve a curative GVL effect without GVHD in case of absence of inflammatory stimuli, when donor T cells remain confined to the hematopoietic compartment and limit their destruction to cells of lymphohematopoietic origin.75
Dr. Fuchs added that success of this procedure in treating chemotherapy unresponsive hematologic malignancies has created interest in using HSCT to treat metastatic solid tumors. However, with the exception of one report for patients with kidney cancer in which 10 of 19 patients responded,76 results of HSCT to treat solid tumors have been largely disappointing. A recent review of HSCT for solid tumors in Europe showed only 6 long-term disease-free survivors out of 61 patients (9%) owing to a 21% NRM and a 70% incidence of relapse.77 Interestingly, 3 of the 6 long-term survivors experienced graft rejection, including 1 patient with pancreatic carcinoma, 1 patient with metastatic renal carcinoma, and 1 patient with hepatic cholangiocarcinoma. The patient with metastatic renal cell carcinoma underwent a second transplant and engrafted followed by the development of GVHD.
Dr. Fuchs admitted that it is hard to justify HSCT for solid tumors when the rate of NRM (21%) is more than double the long-term survival rate (9%). Furthermore, simple logic dictates that it should not be possible to cure a metastatic solid tumor by a GVL reaction that is directed solely against ubiquitously expressed histocompatibility antigens. If the antigens being targeted for destruction by donor T cells are not differentially expressed by normal versus malignant tissues, then it simply is not possible to destroy the cancer without destroying the rest of the host tissues. It is possible then that tumor responses in patients who experienced graft rejection were initiated by the donor’s immune system but then carried to completion by tumor-specific T cells of host origin.78,79 A priori reasoning combined with clinical observations lead to 2 potential strategies to achieve anti-tumor effects in solid tumor patients through the infusion of allogeneic lymphocytes: 1) Infusion of transiently engrafting lymphocytes to revive a tumor-specific T cell response of host origin; and 2) Immunizing donors against tumor-specific antigens, then infusing tumor-specific T cells as part of or after HSCT.
The use of transiently engrafting lymphocytes to stimulate anti-tumor immunity began in 1960 with a report of a melanoma patient who achieved a disease response following infusion of lymphocytes from a second melanoma patient who had a spontaneous regression of disease.80 Several reports followed (reviewed in81), but without a detailed understanding of alloreactivity or significant mechanistic insights, interest in non-engrafting allogeneic cell therapy waned. Dr. Fuchs’ group interest in non-engrafting cell therapy was piqued by the observation of clinical responses of hematologic malignancies despite graft rejection after HaploSCT.82,83 This observation prompted animal experiments, which revealed a novel cooperation between alloreactive donor CD4+ T cells and host CD8+ T cells in mediating anti-tumor effects against either hematologic or solid cancers.84 A model to account for this cooperation is provided in Figure 1. The steps in this cooperation are as follows: 1) The infused alloreactive CD4+ T cell recognizes HLA Class II alloantigen on a recipient antigen-presenting cell (APC); 2) The CD4+ T cell is activated, upregulates CD40 ligand (CD154), ligates CD40 on the APC, and this triggers the activation of the APC and upregulation of CD7085; 3) CD70 on the APC ligates CD27 on an exhausted, tumor-specific recipient CD8+ T cell86; and 4) The revived CD8+ T cell migrates to and kills leukemia cells expressing the target antigen.87 This model hypothesizes that tolerance in endogenous, tumor-specific CD4+ T cells results in CD8+ T cell exhaustion and that the immune response to either solid or hematologic cancers can be revived by providing an exogenous source of T cell help. The group plans to test this prediction in a clinical trial of CD8 depleted, HLA mismatched donor lymphocyte infusion (DLI) after induction chemotherapy for patients with myelodysplastic syndrome after hypomethylating agent failure or for patients with secondary acute myeloid leukemia. However, transiently engrafting, HLA-haploidentical DLI could also be applied to patients with solid tumors and tumor-infiltrating lymphocytes.
Figure 1.
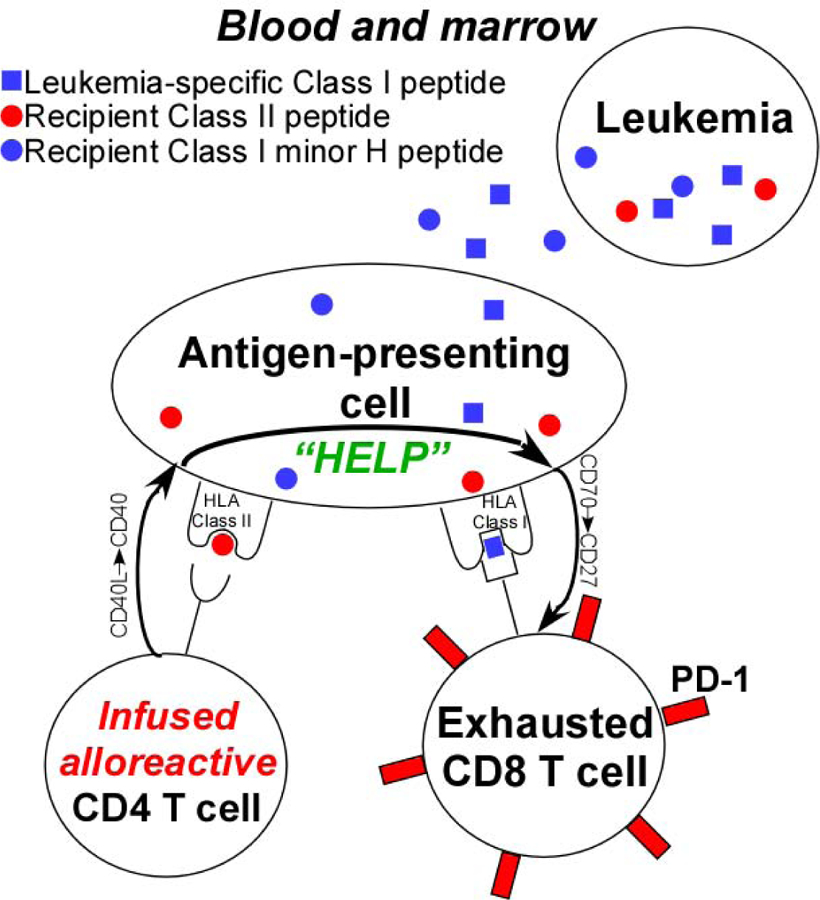
Model to account for the anti-tumor effect of transiently engrafting, HLA-mismatched donor lymphocyte infusion.
The second proposed method of using HLA-haploidentical T cells to treat solid tumors is to vaccinate donors against tumor antigens prior to HaploSCT. For this method to be effective, the antigen being targeted must either be expressed uniquely by the tumor (a tumor-specific antigen) or expressed by both the tumor and a tissue type that is dispensable (for example, prostate or ovary). In the category of tumor-specific antigens, it may be possible to target either viral antigens expressed by virally induced cancers or tumor neo-antigens, the antigens that arise from somatic mutations in cancer cells. While it is relatively easy to identify expressed somatic mutations in cancer cells by a combination of whole genome sequencing of tumor versus normal tissue and RNA sequencing in the tumor tissue, the methods for identifying which of the mutations encode immunogenic peptides are in their infancy, and it is also not clear that it is ethical to vaccinate donors against tumor neo-antigens. In contrast, virus-induced malignancies, in particular malignancies associated with the human papillomavirus (HPV), may provide an ideal scenario to test the hypothesis that donor vaccination can augment anti-tumor immunity in a transplant recipient. HPV encodes 2 proteins, E6 and E7, which are required for cellular transformation and are potentially immunogenic in humans. Moreover, autologous T cells targeting E6 and E7 have been expanded ex vivo and have induced clinical responses in 3 of 9 patients with metastatic cervical cancer, including 2 durable complete responses.88 A clinical trial is planned in which patients with recurrent or metastatic, HPV-associated oropharyngeal cancer will receive allogeneic HSCT, including HaploSCT, from donors vaccinated against the E6 and E7 antigens of HPV serotypes HPV16 and HPV18, which are responsible for >90% of such cancers.
Cell Therapy and Immunologic Reconstitution
Dr. Jianhua Yu discussed arming natural killer cells with chimeric antigen receptors for treatment of relapsed hematological malignancies after HSCT. One of the challenges in the CAR field involves finding a proper tumor-associated antigen to target. Dr. Yu’s group focused on CS1 in multiple myeloma (MM), as it is a cell surface glycoprotein highly expressed on tumor cells in more than 95% of all MM patients with low expression on NK cells, plasma cells, and a subset of T cells but not on other normal cells.89,90 Dr. Yu described their experience in CS1-specific viral construct and transduced primary NK cells and the human NK cell line (NK-92) with this construct. In vitro, CS1-CAR NK cells displayed enhanced cytotoxicity and IFN-γ production when co-cultured with MM cell lines. This occurred in a CS1-dependent manner because CS1 negative MM cell lines did not trigger these CAR effects unless CS1 was ectopically overexpressed in these lines. Ex vivo, CS1-CAR NK cells also showed enhanced activities when responding to patient MM tumor cells.91 In an aggressive orthotopic MM xenograft mouse model, adoptive transfer of CS1-CAR NK cells efficiently suppressed the growth of xenografted MM cells and also significantly prolonged survival of MM-bearing mice.91 In addition to treating MM with the CS1-CAR NK cells this approach is being investigated in acute myeloid leukemia (AML) by targeting AML-specific tumor antigens, where enhanced in vitro and in vivo efficacy has also been observed.
Dr. Richard Maziarz talked about the role of inducible suicidal switch in cellular immunotherapy. He discussed the recent advances with gene modified T cells and natural killer cells and how cellular immune therapy has expanded to autologous or off-the-shelf allogeneic products. He explained the extensive applications including allogeneic hematopoietic stem cell transplantation, monoclonal antibodies, bi-specific antibody constructs, antibody toxin conjugate, cytokines, fusion proteins, dendritic cell and peptide vaccines, antigen specific HLA restricted T -cell lines, natural killer cells, cytokine activated killer cells, chimeric antigen receptor T (CAR-T) cells and exogenous T-cell receptor transduced T cells.92 Some of these therapies are nonspecific but other, receptor specific therapy can be complicated with systemic inflammatory conditions due to cytokine release and as well, to “on” and “off” target directed toxicities when dealing with adoptively transferred T-cell cellular therapy.
Dr. Maziarz further discussed the adverse effects of adoptively transferred therapies including cytokine release syndrome (CRS), macrophage activation syndrome, tumor lysis syndrome, B cell aplasia with subsequent hypogammaglobinemia, stress cardiomyopathy and cardiac dysfunction, respiratory distress syndrome, neurologic toxicity, renal and/or hepatic failure and disseminated intravascular coagulation. CRS has been most recently under scrutiny and new grading systems have been developed to guide intervention with the goal of ameliorating the complicated clinical circumstance.92 Interestingly, awareness of CRS as a complication not just of CAR-T application, but that it represents an adoptively transferred T-cell disease is emerging with a recent report of a patient who developed CRS after HaploSCT for multiply relapsed Hodgkin lymphoma.93,94 These T-cell associated complications can be unpredictable and severe and recently, due to mortality associated with cerebral edema, a CAR-T trial for relapse refractory adult acute lymphoblastic leukemia was halted.
The first clear demonstration of the efficacy of adoptive T cell therapy is highlighted by donor leukocyte infusions (DLI) used in the treatment of relapse after allogeneic HCT.95 The utilization of post-transplant DLI has been associated with control of relapsed malignancies as well as reinstitution of sustained remission states. It is also associated with bone marrow aplasia and GVHD. Bonini’s group96,97 reported the first DLI that were gene modified with a suicide gene. They utilized herpes virus associated thymidine kinase (TK), a gene that would be inactive in normal human cells but would confer susceptibility to exogenously administered ganciclovir. This TK-modified DLI could eradicate the abnormal clone of CMML in patients with relapsed disease after HSCT. At the same time, it was demonstrated that in patients who developed acute GVHD, reversal of GVHD could be accomplished by ex vivo administration of intravenous ganciclovir. A follow up study also demonstrated that these adoptively transferred T cell products can have long term functional persistence.98
Other suicide gene strategies have emerged. One strategy utilizes a gene therapy to transduce host cells that will allow the co-expression of surface receptors that can be targets of established antibody therapy, such as expression of CD20 which could be a target for anti-CD20 antibody99 or the epidermal growth factor receptor (EGFR) that would allow the induction of cell death by treatment with the pro-drug Erlotinib, which can bind to the EGFR and lead to the death of the transduced cells.100 Another interesting strategy that is under investigation for clinical exploitation is the generation of a trans-gene of activated Caspase 9. This was first reported by the Baylor group101 where a lentiviral vector incorporating a fusion gene with the activation components of the caspase 9 gene with a surface FK binding protein with coordinate expression of the surface marker for CD19. After integration into T cells, in the setting of IL-2 exposure, high levels of Caspase 9 would be expressed. In the absence of a target agent, the cells would continue to proliferate. However, if one exposes cells to Rimiducid (AP1903), an agent that would be inert when there is no target molecule availability, one would then get high affinity Rimiducid binding and activation of the caspase pathway leading to death of the cells. Advantages are that the kinetics were shown to be rapid and dose responsive within nano-molar concentrations that could be achieved in vivo after IV administration of the rimiducid. The investigators demonstrated the successful T-cell responses in 5 patients treated with DLI transduced with the caspase gene construct. Importantly, one could detect iCasp9-transduced T cells in the blood and they could demonstrate that treatment with Rimiducid was able to reverse the clinical complication in patients, who developed GVHD. This strategy has now been applied to a large number of patients on clinical trials and in detailed immune reconstitution studies, sustained expression of the transgene within the transduced cells could be identified.102
No randomized trials exist comparing suicide gene therapies at present. However, we anticipate that these strategies will be increasingly utilized as more adoptively transferred T-cell therapies are implemented into our clinical arena. At the end of his talk, Dr. Maziarz concluded that using these strategies will likely contribute to amelioration of complications and thus ultimately improve the outcomes of patients undergoing a variety of adoptive immunotherapies and as such, will increase the application to a greater patient population.
Dr. Domenico Mavilio described NK cell immune-reconstitution (IR) after HaploSCT with both clinical and therapeutic implications. Dr. Mavilio started with the notion that kinetic and quality of IR determines the degree of GVL effect, incidence of tumor relapse, onset and grade of GVHD and control of life-threatening opportunistic infections.103 Donor-derived and alloreactive NK cells play a key role, as they are important effectors able to kill non-self-dangerous targets such as viral-infected or tumor-transformed cells and produce pro-inflammatory mediators such as interferon γ (IFN- γ) upon activation and in the absence of a prior sensitization to specific antigens. NK cell recognition of allogenic cells relies on large family of inhibitory NK cell receptors (iNKRs) including Killer cell immunoglobulin-like receptors (KIRs) and C-type lectins such as CD94/NKG2A that recognize different and specific alleles of “self” major histocompatibility complex of class I (MHC-I) expressed only on autologous cells (missing self-hypothesis).104
Alloreactive donor-derived NK cells early immune reconstitute after HaploSCT (within the first month from transplantation and soon after neutrophil and monocyte IRs), eliminate recipient immune cells surviving conditioning (i.e. avoid graft reject), kill recipient antigen presenting cells (APCs) presenting host antigens to donor T cells (i.e. avoid the onset of GVHD) and clearance residual malignant cells in the recipient (i.e. induce GvL). This knowledge made it possible to develop adoptive NK cell transfer therapies to cure both solid and hematologic tumors.105,106 Similar to what Dr. Mavilio’ s group observed with B and T cells,107,108 from all patients receiving T cell-replete (TCR) HaploSCT with RIC followed by PTCy, showed a complete donor-dependent chimerism for NK cells after a month from transplant. NK cells that are commonly identified within the CD3neg/CD19neg/CD14neg lymphocytes according to the expression of CD56 and CD16.
In healthy subjects, the largest subset of circulating and cytotoxic NK cells (up to 90%) expresses a CD56dim/CD16pos (CD56dim) phenotype. The second circulating subset of CD56brigh/CD16dim-neg (CD56bright) NK cells (up to 10%−15%) is able to secrete high amount of pro-inflammatory cytokines while displaying poor cytotoxicity.109 Dr. Mavilio’ s preliminary data showed that donor-derived NK cells start to reconstitute approximately around the 2nd week after HaploSCT and the first subset to be detected is an unconventional NK cell population characterized by a CD56dim/CD16neg phenotype (uCD56dim NK cells). This neglected NK cell population is presented at very low frequency under homeostatic condition and only recently a study reported that its frequency increases in pediatric patients affected by acute lymphoblastic leukemia.110 Very little is known about the homeostasis and the origin of these uCD56dim NK cells and even the nomenclature of these lymphocytes is being debated as they have been also termed CD56low/CD16low NK cells.110 Also, another group claimed that uCD56dim arise from conventional CD56dim (cCD56dim) NK cell subset as a consequence of cell activation that induces the metalloproteinase-17 (ADAM-17)-mediated shedding of CD16 from cell surface.111 Nonetheless, uCD56dim NK cells represent the first NK cell subset detectable in the early phase of IR in HaploSCT, when Dr. Mavilio’ s group could not find at all any expansion of cCD56dim NK cell subset. Donor-derived uCD56dim NK cell are bona fide NK cell expressing several NKRs such as NKp30, NKG2D, CD94, NKG2A, NKG2C and low levels of KIRs. His group also observed that uCD56dim NK cells are highly cytotoxic in healthy donors (even more than cCD56dim NK cells). In contrast, the same subset purified from recipients undergone HaploSCT is highly anergic and defective in killing the 721.221 cell lines expressing their putative ligands HLA-E. This impairment is associated with the high expression of CD94/NKG2A on donor-derived uCD56dim NK cells. Currently, Dr. Mavilio’ s group is determining the impact of this subset in NK cell homeostasis/ontogenesis and in the clinical outcome of HaploSCT as uCD56dim NK cells can be potentially targeted to improve the prognosis of patients affected by hematologic malignancies undergone HSCT.
Dr. Denis-Claude Roy updated the group on IR after HaploSCT with photodepletion. He started with a comparison of in vivo or ex vivo T cell depletion strategies that are necessary to overcome the major histocompatibility complex disparity between donor and recipient that would normally lead to GVHD. However, these strategies are also responsible for delayed IR, and are therefore at increased risk of infectious complications and disease relapse. In an attempt to overcome this reconstitution issue, the group of Dr. Roy et al. have developed a photodynamic approach that virtually eliminates from leukapheresis donor peripheral blood cells, those donor cells with the ability to recognize the patient and cause lethal GVHD.112–114 This strategy relies on a photosensitizing agent, dibromorhodamine that is retained in donor cells that are activated ex vivo upon exposure to host antigens. Light illumination in the visible spectrum eliminates these host-recognizing cells, leaving out resting T cells, both of naïve and memory phenotype, to fight infection and cancer. These allodepleted T cells can then be administered after T cell depleted transplantation to accelerate T cell immune reconstitution (ATIR).115 This strategy has been tested in a Phase I cell-dose escalation clinical trial and is currently undergoing evaluation in a Phase II trial.116
The ex vivo cell photodepletion procedure specifically eliminates anti-host cells and spares T cells with the ability to proliferate in response to third party antigens and to CD3/CD28 stimulation. The Phase I study showed that a cell dose of 3.2x105 to 2.0x106 CD3+ cells/kg afforded a high survival rate (67%) without any treatment-related mortality.114,117 Not surprisingly, there was a trend for earlier T cell reconstitution in those patients who received a higher T cell dose. However, even patients without detectable T cells in their peripheral blood could mobilize CD3, CD4 and CD8 T cells in the few patients who developed rising titers of Epstein-Barr virus (EBV). Interestingly, EBV titers decreased following ATIR cell administration alone in most patients. Patient cohorts receiving the higher T ATIR cell doses also showed improved T cell reconstitution with a high proportion of naïve T cells, when compared to patients receiving lower ATIR cell doses. The preservation of naïve, T cells who have the ability to react to novel infectious agents, as well as memory T cells, may explain the most favorable results observed at the high ATIR cell doses. No GVHD immunoprophylaxis was used in any of these patients and no patient developed grade III or IV GVHD.(Manuscript in preparation)
A Phase II clinical trial (NCT01794299) was performed internationally in 23 patients aged 21 to 64 (median 41) year-old with acute myeloid (n=16) or acute lymphoblastic (n=7) leukemia in first or second complete remission. All patients received a CD34+ selected stem cell graft followed 4 weeks later by ATIR infusion at a single 2 million CD3+ cells/kg cell dose. All patients demonstrated rapid engraftment, reaching 0.5 x 109 neutrophils/L at a median of 12 days (range: 8–34) and 20 x 109 platelets/L at a median of 12 days (range: 9–35). No patients experienced early or late graft failure. The majority of patients had an adverse cytogenetic risk profile118 and a high disease-risk index.119 NK cells recovered early post-transplant. Similarly, B cells were present from only a few weeks after ATIR infusion onwards, rapidly followed by an increase in immunoglobulin levels. T cells increased in peripheral blood at approximately 3 months after ATIR infusion. Interestingly, no mortality occurred in the first 3 months post-transplant, suggesting that, while T cell levels were low in the first week post-ATIR, these cells could be recruited upon proper antigenic expression to fight infections. Again, no grade III-IV GVDH was observed in these patients after ATIR infusion. These most interesting results demonstrate that it is possible to provide patients with photo-depleted haploidentical donor T cells without causing GVHD while maintaining their immunologic potential. A Phase III haploidentical transplantation clinical trial comparing CD34 depleted transplant followed by ATIR infusion to T-replete transplantation with post-transplantation cyclophosphamide will be initiated at the end of 2017 (NCT02999854).
In the last talk, Dr. Chiara Bonini discussed T cell dynamics after HaploSCT. She started her presentation describing how HaploSCT can offer a chance for cure to patients affected by high-risk hematological malignancies who lack a conventional donor. She added that HaploSCT offers a unique model system to unravel, in humans, the mechanisms governing the generation and maintenance of immunological memory. Her group have longitudinally tracked the generation of antigen-experienced T cell subsets, including effectors (TE), effector memory (TEM), central memory (TCM), and memory stem T cells (TSCM) from naïve T lymphocytes (TN) infused after HaploSCT with PTCy. The observation was that donor-derived TSCM cells are highly enriched early after HSCT and showed, at the antigen-specific and clonal level, that TSCM lymphocytes can differentiate directly from naïve precursors infused within the graft. In vivo fate mapping by TCR clone typing revealed that TSCM have the potential to differentiate in all other antigen-experienced T cell subsets.120
To further investigate and compare the long-term persistence ability of human memory T cell subsets, her group analyzed samples from patients affected by acute myeloid leukemia, undergoing gene therapy in the context of T-cell depleted HaploSCT. Within this clinical trial, patients received donor T lymphocytes genetically modified by a retroviral vector to express the Thymidine Kinase (TK)-suicide gene, 20–40 days after T-cell depleted HaploSCT.121 Engineered T cells were traced in 10 patients alive and well up to 14 years after treatment. It was showed that, in the presence of a broad and resting immune system, engineered lymphocytes are still detectable in peripheral blood at low but stable levels in all patients. Longitudinal analysis of viral specific engineered cells indicates that antigen recognition is dominant for T cell in vivo expansion. By sequencing retroviral integration sites, TCRα and TCRβ clonal markers on sorted T cell subsets, they could trace T-cell clone types from infused cellular products to late follow-up. They also observed that dominant T cell clones, detected long-term, preferentially originate from infused TSCM cells, thus confirming the long-term persistence ability of this T cell subset,98 and suggesting the value of genetically engineered TSCM cells in cancer immunotherapy.
In conclusion, exciting new developments in the field of haploidentical transplantation have demonstrated clinical outcomes resembling outcomes of HLA matched donor transplants. Application of haploidentical transplants to non-malignant disease will likely expand the possibility to apply this live-saving procedure to recipients without HLA matched donors. In addition, combining haploidentical transplants with solid organ transplants from the same donor could favorably impact organ rejection and allow patients to discontinue early immunosuppression. Understanding immunologic reconstitution post-transplant will allow administration of cellular therapy products to fasten recovery of the immune system and hopefully provide increase anti-tumor effect, to improve safety and efficacy of allogeneic transplantation not only from haploidentical donors, but also for transplants from other HLA matched and mismatched donor sources.
ACKNOWLEDGMENTS
The organizers thank Bellicum Pharmaceuticals, Kiadis Pharma, Miltenyi Biotec, Astellas Pharma, CytoSen Therapeutics for their support in organizing the meeting.
Financial disclosure statement: D.C.R. has received research and travel support from Kiadis Pharma. S.O.C. served as advisory board member for Spectrum Pharmaceuticals, received research support from Miltenyi Biotec and has equity/leadership in CytoSen Therapeutics. Y.R. serves as a consultant and shareholder of Cell Source Ltd., which supported part of this work. S.S. is a co-founder of a company that is planning multi-center clinical trials of tolerance induction to combined kidney and hematopoietic cell transplants. D.A.L. has equity/leadership in CytoSen Therapetics and equity in Courier Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Passweg JR, Baldomero H, Bader P, et al. Use of haploidentical stem cell transplantation continues to increase: the 2015 European Society for Blood and Marrow Transplant activity survey report. Bone marrow transplantation 2017;52:811–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gratwohl A, Pasquini MC, Aljurf M, et al. One million haemopoietic stem-cell transplants: a retrospective observational study. The Lancet Haematology 2015;2:e91–100. [DOI] [PubMed] [Google Scholar]
- 3.Devine SM. Haploidentical Hematopoietic Cell Transplantation Using Post-Transplantation Cyclophosphamide: Does Graft Source Matter? Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017;35:2984–6. [DOI] [PubMed] [Google Scholar]
- 4.Owens AH Jr., Santos GW. The induction of graft versus host disease in mice treated with cyclophosphamide. The Journal of experimental medicine 1968;128:277–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos GW, Owens AH. Production of graft-versus-host disease in the rat and its treatment with cytotoxic agents. Nature 1966;210:139–40. [DOI] [PubMed] [Google Scholar]
- 6.Owens AH Jr., Santos GW. The effect of cytotoxic drugs on graft-versus-host disease in mice. Transplantation 1971;11:378–82. [DOI] [PubMed] [Google Scholar]
- 7.Santos GWTP, Brookmeyer R, Saral R, Beschorner WE, Bias WB, Braine HG, Burns WH, Farmer ER, Hess AD, Kaizer H, Mellits D, Sensenbrenner LL, Stuart R, Yeager AM. Cyclosporine plus methylprednisolone versus cyclophosphamide plus methylprednisolone as prophylaxis for graft-versus-host disease: A randomized double-blind study in patients undergoing allogeneic marrow transplantation. Clin Transplant 1987;1:21–8. [Google Scholar]
- 8.Emadi A, Jones RJ, Brodsky RA. Cyclophosphamide and cancer: golden anniversary. Nature reviews Clinical oncology 2009;6:638–47. [DOI] [PubMed] [Google Scholar]
- 9.DeZern AE, Petri M, Drachman DB, et al. High-dose cyclophosphamide without stem cell rescue in 207 patients with aplastic anemia and other autoimmune diseases. Medicine 2011;90:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood 2001;98:3456–64. [DOI] [PubMed] [Google Scholar]
- 11.Luznik L, O’Donnell PV, Symons HJ, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2008;14:641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasamon YL, Luznik L, Leffell MS, et al. Nonmyeloablative HLA-haploidentical bone marrow transplantation with high-dose posttransplantation cyclophosphamide: effect of HLA disparity on outcome. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2010;16:482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kasamon YL, Bolanos-Meade J, Prince GT, et al. Outcomes of Nonmyeloablative HLA-Haploidentical Blood or Marrow Transplantation With High-Dose Post-Transplantation Cyclophosphamide in Older Adults. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:3152–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunstein CG, Fuchs EJ, Carter SL, et al. Alternative donor transplantation after reduced intensity conditioning: results of parallel phase 2 trials using partially HLA-mismatched related bone marrow or unrelated double umbilical cord blood grafts. Blood 2011;118:282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCurdy SR, Kanakry JA, Showel MM, et al. Risk-stratified outcomes of nonmyeloablative HLA-haploidentical BMT with high-dose posttransplantation cyclophosphamide. Blood 2015;125:3024–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciurea SO, Zhang MJ, Bacigalupo AA, et al. Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood 2015;126:1033–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanate AS, Mussetti A, Kharfan-Dabaja MA, et al. Reduced-intensity transplantation for lymphomas using haploidentical related donors vs HLA-matched unrelated donors. Blood 2016;127:938–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghosh N, Karmali R, Rocha V, et al. Reduced-Intensity Transplantation for Lymphomas Using Haploidentical Related Donors Versus HLA-Matched Sibling Donors: A Center for International Blood and Marrow Transplant Research Analysis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34:3141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanakry CG, Ganguly S, Zahurak M, et al. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Science translational medicine 2013;5:211ra157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leung W, Campana D, Yang J, et al. High success rate of hematopoietic cell transplantation regardless of donor source in children with very high-risk leukemia. Blood 2011;118:223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ciceri F, Labopin M, Aversa F, et al. A survey of fully haploidentical hematopoietic stem cell transplantation in adults with high-risk acute leukemia: a risk factor analysis of outcomes for patients in remission at transplantation. Blood 2008;112:3574–81. [DOI] [PubMed] [Google Scholar]
- 22.Ciurea SO, Mulanovich V, Saliba RM, et al. Improved early outcomes using a T cell replete graft compared with T cell depleted haploidentical hematopoietic stem cell transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2012;18:1835–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seggewiss R, Einsele H. Immune reconstitution after allogeneic transplantation and expanding options for immunomodulation: an update. Blood 2010;115:3861–8. [DOI] [PubMed] [Google Scholar]
- 24.Smyth MJ, Swann J, Kelly JM, et al. NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. The Journal of experimental medicine 2004;200:1325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smyth MJ, Cretney E, Takeda K, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. The Journal of experimental medicine 2001;193:661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annual review of immunology 2011;29:235–71. [DOI] [PubMed] [Google Scholar]
- 27.Savani BN, Mielke S, Adams S, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia 2007;21:2145–52. [DOI] [PubMed] [Google Scholar]
- 28.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005;105:3051–7. [DOI] [PubMed] [Google Scholar]
- 29.Bachanova V, Cooley S, Defor TE, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood 2014;123:3855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee DA, Denman CJ, Rondon G, et al. Haploidentical Natural Killer Cells Infused before Allogeneic Stem Cell Transplantation for Myeloid Malignancies: A Phase I Trial. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2016;22:1290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denman CJ, Senyukov VV, Somanchi SS, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PloS one 2012;7:e30264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fei F, Lim M, George AA, et al. Cytotoxicity of CD56-positive lymphocytes against autologous B-cell precursor acute lymphoblastic leukemia cells. Leukemia 2015;29:788–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Wu HW, Sheard MA, et al. Growth and activation of natural killer cells ex vivo from children with neuroblastoma for adoptive cell therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19:2132–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shah N, Martin-Antonio B, Yang H, et al. Antigen presenting cell-mediated expansion of human umbilical cord blood yields log-scale expansion of natural killer cells with anti-myeloma activity. PloS one 2013;8:e76781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knorr DA, Ni Z, Hermanson D, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem cells translational medicine 2013;2:274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee DA. Regulatory Considerations for NK Cells Used in Human Immunotherapy Applications. Methods Mol Biol 2016;1441:347–61. [DOI] [PubMed] [Google Scholar]
- 37.Gaballa S, Ge I, El Fakih R, et al. Results of a 2-arm, phase 2 clinical trial using post-transplantation cyclophosphamide for the prevention of graft-versus-host disease in haploidentical donor and mismatched unrelated donor hematopoietic stem cell transplantation. Cancer 2016;122:3316–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciurea SO, Schafer JR, Bassett R, et al. Phase 1 clinical trial using mbIL21 ex-vivo expanded donor-derived NK cells after haploidentical transplantation. Blood 2017. [DOI] [PMC free article] [PubMed]
- 39.Oyer JL, Pandey V, Igarashi RY, et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: Clinical implications for cancer treatment. Cytotherapy 2016;18:653–63. [DOI] [PubMed] [Google Scholar]
- 40.Cichocki F, Cooley S, Davis Z, et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 2016;30:456–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Science translational medicine 2016;8:357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ophir E, Or-Geva N, Gurevich I, et al. Murine anti-third-party central-memory CD8(+) T cells promote hematopoietic chimerism under mild conditioning: lymph-node sequestration and deletion of anti-donor T cells. Blood 2013;121:1220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachar Lustig E, Or Geva N, Lask A, et al. Next Generation Veto Cells for Non-Myeloablative Haploidentical HSCT: Combining Anti-Viral and Graft Facilitating Activity. Blood 2016;128:3345–. [Google Scholar]
- 44.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nature reviews Immunology 2006;6:595–601. [DOI] [PubMed] [Google Scholar]
- 45.Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015;33:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han KP, Zhu X, Liu B, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine 2011;56:804–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu W, Jones M, Liu B, et al. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor alphaSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res 2013;73:3075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Foley B, Cooley S, Verneris MR, et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012;119:2665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanier LL, Ruitenberg JJ, Phillips JH. Functional and biochemical analysis of CD16 antigen on natural killer cells and granulocytes. Journal of immunology (Baltimore, Md : 1950) 1988;141:3478–85. [PubMed] [Google Scholar]
- 50.Gleason MK, Ross JA, Warlick ED, et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014;123:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wiernik A, Foley B, Zhang B, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clinical cancer research : an official journal of the American Association for Cancer Research 2013;19:3844–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gleason MK, Verneris MR, Todhunter DA, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Molecular cancer therapeutics 2012;11:2674–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vallera DA, Felices M, McElmurry R, et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22:3440–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lang P, Feuchtinger T, Teltschik HM, et al. Improved immune recovery after transplantation of TCRalphabeta/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone marrow transplantation 2015;50 Suppl 2:S6–10. [DOI] [PubMed] [Google Scholar]
- 55.Seidel UJ, Schlegel P, Grosse-Hovest L, et al. Reduction of Minimal Residual Disease in Pediatric B-lineage Acute Lymphoblastic Leukemia by an Fc-optimized CD19 Antibody. Molecular therapy : the journal of the American Society of Gene Therapy 2016;24:1634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlegel P, Lang P, Zugmaier G, et al. Pediatric posttransplant relapsed/refractory B-precursor acute lymphoblastic leukemia shows durable remission by therapy with the T-cell engaging bispecific antibody blinatumomab. Haematologica 2014;99:1212–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gokbuget N, Stanze D, Beck J, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood 2012;120:2032–41. [DOI] [PubMed] [Google Scholar]
- 58.Oriol A, Vives S, Hernandez-Rivas JM, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA Study Group. Haematologica 2010;95:589–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Messori A, Fadda V, Maratea D, Trippoli S. Acute lymphoblastic leukemia in first complete remission: temporal trend of outcomes in studies comparing allogeneic transplant with autologous transplant or chemotherapy. Annals of hematology 2013;92:1221–8. [DOI] [PubMed] [Google Scholar]
- 60.Kako S, Morita S, Sakamaki H, et al. A decision analysis of allogeneic hematopoietic stem cell transplantation in adult patients with Philadelphia chromosome-negative acute lymphoblastic leukemia in first remission who have an HLA-matched sibling donor. Leukemia 2011;25:259–65. [DOI] [PubMed] [Google Scholar]
- 61.Gupta V, Richards S, Rowe J, Acute Leukemia Stem Cell Transplantation Trialists’ Collaborative G. Allogeneic, but not autologous, hematopoietic cell transplantation improves survival only among younger adults with acute lymphoblastic leukemia in first remission: an individual patient data meta-analysis. Blood 2013;121:339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ravandi F, Othus M, O’Brien S, et al. Multi-Center US Intergroup Study of Intensive Chemotherapy Plus Dasatinib Followed By Allogeneic Stem Cell Transplant in Patients with Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia Younger Than 60. Blood 2015;126:796–. [Google Scholar]
- 63.Wang Y, Liu QF, Xu LP, et al. Haploidentical versus Matched-Sibling Transplant in Adults with Philadelphia-Negative High-Risk Acute Lymphoblastic Leukemia: A Biologically Phase III Randomized Study. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22:3467–76. [DOI] [PubMed] [Google Scholar]
- 64.Srour SA, Milton DR, Bashey A, et al. Haploidentical Transplantation with Post-Transplantation Cyclophosphamide for High-Risk Acute Lymphoblastic Leukemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2017;23:318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santoro N, Ruggeri A, Labopin M, et al. Unmanipulated haploidentical stem cell transplantation in adults with acute lymphoblastic leukemia: a study on behalf of the Acute Leukemia Working Party of the EBMT. Journal of hematology & oncology 2017;10:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anurathapan U, Pakakasama S, Rujkijyanont P, et al. Pretransplant immunosuppression followed by reduced-toxicity conditioning and stem cell transplantation in high-risk thalassemia: a safe approach to disease control. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2013;19:1259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gahrton G, Tura S, Ljungman P, et al. Allogeneic bone marrow transplantation in multiple myeloma. European Group for Bone Marrow Transplantation. N Engl J Med 1991;325:1267–73. [DOI] [PubMed] [Google Scholar]
- 68.Tricot G Graft-versus-myeloma effect: proof of principle. Blood 1996;3:3. [PubMed] [Google Scholar]
- 69.Krishnan A, Pasquini MC, Logan B, et al. Autologous haemopoietic stem-cell transplantation followed by allogeneic or autologous haemopoietic stem-cell transplantation in patients with multiple myeloma (BMT CTN 0102): a phase 3 biological assignment trial. The LancetOncology 2011;12:1195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lokhorst HM, Wu K, Verdonck LF, et al. The occurrence of graft-versus-host disease is the major predictive factor for response to donor lymphocyte infusions in multiple myeloma. Blood 2004;103:4362–4. [DOI] [PubMed] [Google Scholar]
- 71.Giralt S, Aleman A, Anagnostopoulos A, et al. Fludarabine/melphalan conditioning for allogeneic transplantation in patients with multiple myeloma. Bone marrow transplantation 2002;30:367–73. [DOI] [PubMed] [Google Scholar]
- 72.Kroger N, Sayer HG, Schwerdtfeger R, et al. Unrelated stem cell transplantation in multiple myeloma after a reduced-intensity conditioning with pretransplantation antithymocyte globulin is highly effective with low transplantation-related mortality. Blood 2002;100:3919–24. [DOI] [PubMed] [Google Scholar]
- 73.Kumar S, Zhang MJ, Li P, et al. Trends in allogeneic stem cell transplantation for multiple myeloma: a CIBMTR analysis. Blood 2011;118:1979–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castagna L, Mussetti A, Devillier R, et al. Unmanipulated Haploidentical Allogeneic Hematopoietic Cell Transplantation for Multiple Myeloma Using Post Transplant Cyclophosphamide Anti-Gvhd Prophylaxis. Blood 2016;128:3475–. [DOI] [PubMed] [Google Scholar]
- 75.Chakraverty R, Cote D, Buchli J, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. The Journal of experimental medicine 2006;203:2021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Childs R, Chernoff A, Contentin N, et al. Regression of metastatic renal-cell carcinoma after nonmyeloablative allogeneic peripheral-blood stem-cell transplantation [see comments]. N Engl J Med 2000;343:750–8. [DOI] [PubMed] [Google Scholar]
- 77.Omazic B, Remberger M, Barkholt L, et al. Long-Term Follow-Up of Allogeneic Hematopoietic Stem Cell Transplantation for Solid Cancer. Biology of Blood and Marrow Transplantation 2016;22:676–81. [DOI] [PubMed] [Google Scholar]
- 78.Alexander P, Delorme EJ, Hall JG. The effect of lymphoid cells from the lymph of specifically immunized sheep on the growth of primary sarcomata in rats. Lancet 1966:1186–9.
- 79.Ellman L, Katz DH, Green I, Paul WE, Benacerraf B. Mechanisms involved in the antileukemic effect of immunocompetent allogeneic lymphoid cell transfer. Cancer Res 1972;32:141–8. [PubMed] [Google Scholar]
- 80.SUMNER WC, FORAKER AG. Spontaneous regression of human melanoma: clinical and experimental studies. Cancer 1960;13:79–81. [DOI] [PubMed] [Google Scholar]
- 81.Kondo M, McCarty MF. Rationale for a novel immunotherapy of cancer with allogeneic lymphocyte infusion. Med Hypotheses 1984;15:241–77. [DOI] [PubMed] [Google Scholar]
- 82.O’Donnell PV, Luznik L, Jones RJ, et al. Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biology of Blood and Marrow Transplantation 2002;8:377–86. [DOI] [PubMed] [Google Scholar]
- 83.Dey BR, McAfee S, Colby C, et al. Anti-tumour response despite loss of donor chimaerism in patients treated with non-myeloablative conditioning and allogeneic stem cell transplantation. British Journal of Haematology 2005;128:351–9. [DOI] [PubMed] [Google Scholar]
- 84.Symons HJ, Levy MY, Wang J, et al. The allogeneic effect revisited: exogenous help for endogenous, tumor-specific T cells. Biology of Blood and Marrow Transplantation 2008. [DOI] [PMC free article] [PubMed]
- 85.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T- helper and a T-killer cell [see comments]. Nature 1998;393:474–8. [DOI] [PubMed] [Google Scholar]
- 86.Feau S, Garcia Z, Arens R, Yagita H, Borst J, Schoenberger SP. The CD4+ T-cell help signal is transmitted from APC to CD8+ T-cells via CD27GÇôCD70 interactions. Nat Commun 2012;3:948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ding ZC, Blazar BR, Mellor AL, Munn DH, Zhou G. Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood 2010;115:2397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stevanović S, Draper LM, Langhan MM, et al. Complete Regression of Metastatic Cervical Cancer After Treatment With Human Papillomavirus–Targeted Tumor-Infiltrating T Cells. Journal of Clinical Oncology 2015;33:1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14:2775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tai YT, Dillon M, Song W, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008;112:1329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chu J, Deng Y, Benson DM, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014;28:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amir AL, van der Steen DM, van Loenen MM, et al. PRAME-specific Allo-HLA-restricted T cells with potent antitumor reactivity useful for therapeutic T-cell receptor gene transfer. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17:5615–25. [DOI] [PubMed] [Google Scholar]
- 93.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124:188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology American Society of Hematology Education Program 2016;2016:567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schmid C, Labopin M, Nagler A, et al. Donor lymphocyte infusion in the treatment of first hematological relapse after allogeneic stem-cell transplantation in adults with acute myeloid leukemia: a retrospective risk factors analysis and comparison with other strategies by the EBMT Acute Leukemia Working Party. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2007;25:4938–45. [DOI] [PubMed] [Google Scholar]
- 96.Verzeletti S, Bonini C, Marktel S, et al. Herpes simplex virus thymidine kinase gene transfer for controlled graft-versus-host disease and graft-versus-leukemia: clinical follow-up and improved new vectors. Human gene therapy 1998;9:2243–51. [DOI] [PubMed] [Google Scholar]
- 97.Bonini C, Ferrari G, Verzeletti S, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 1997;276:1719–24. [DOI] [PubMed] [Google Scholar]
- 98.Oliveira G, Ruggiero E, Stanghellini MT, et al. Tracking genetically engineered lymphocytes long-term reveals the dynamics of T cell immunological memory. Science translational medicine 2015;7:317ra198. [DOI] [PubMed] [Google Scholar]
- 99.Philip B, Kokalaki E, Mekkaoui L, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014;124:1277–87. [DOI] [PubMed] [Google Scholar]
- 100.Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature medicine 2016;22:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011;365:1673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou X, Di Stasi A, Tey SK, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood 2014;123:3895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Imamura M, Tsutsumi Y, Miura Y, Toubai T, Tanaka J. Immune reconstitution and tolerance after allogeneic hematopoietic stem cell transplantation. Hematology 2003;8:19–26. [DOI] [PubMed] [Google Scholar]
- 104.Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science 2011;331:44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moretta L, Locatelli F, Pende D, Marcenaro E, Mingari MC, Moretta A. Killer Ig-like receptor-mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood 2011;117:764–71. [DOI] [PubMed] [Google Scholar]
- 106.Castagna L, Mavilio D. Re-discovering NK cell allo-reactivity in the therapy of solid tumors. J Immunother Cancer 2016;4:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Roberto A, Castagna L, Gandolfi S, et al. B-cell reconstitution recapitulates B-cell lymphopoiesis following haploidentical BM transplantation and post-transplant CY. Bone marrow transplantation 2015;50:317–9. [DOI] [PubMed] [Google Scholar]
- 108.Roberto A, Castagna L, Zanon V, et al. Role of naive-derived T memory stem cells in T cell reconstitution following allogeneic transplantation. Blood 2015. [DOI] [PMC free article] [PubMed]
- 109.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 2001;22:633–40. [DOI] [PubMed] [Google Scholar]
- 110.Stabile H, Nisti P, Morrone S, et al. Multifunctional human CD56 low CD16 low natural killer cells are the prominent subset in bone marrow of both healthy pediatric donors and leukemic patients. Haematologica 2015;100:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Romee R, Foley B, Lenvik T, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013;121:3599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guimond M, Balassy A, Barrette M, Brochu S, Perreault C, Roy DC. P-glycoprotein targeting: a unique strategy to selectively eliminate immunoreactive T cells. Blood 2002;100:375–82. [DOI] [PubMed] [Google Scholar]
- 113.Bastien JP, Krosl G, Therien C, et al. Photodepletion differentially affects CD4+ Tregs versus CD4+ effector T cells from patients with chronic graft-versus-host disease. Blood 2010;116:4859–69. [DOI] [PubMed] [Google Scholar]
- 114.Bastien JP, Roy J, Roy DC. Selective T-cell depletion for haplotype-mismatched allogeneic stem cell transplantation. Seminars in oncology 2012;39:674–82. [DOI] [PubMed] [Google Scholar]
- 115.Boumedine RS, Roy DC. Elimination of alloreactive T cells using photodynamic therapy. Cytotherapy 2005;7:134–43. [DOI] [PubMed] [Google Scholar]
- 116.Roy D-C, Lachance S, Roy J, et al. Donor Lymphocytes Depleted of Alloreactive T-Cells (ATIR101) Improve Event-Free Survival (GRFS) and Overall Survival in a T-Cell Depleted Haploidentical HSCT: Phase 2 Trial in Patients with AML and ALL. Blood 2016;128:1226–.27458004 [Google Scholar]
- 117.Roy D-C, Guerin M, Boumedine RS, et al. Reduction in Incidence of Severe Infections by Transplantation of High Doses of Haploidentical T Cells Selectively Depleted of Alloreactive Units. Blood 2011;118:3020–. [Google Scholar]
- 118.Mrozek K, Marcucci G, Nicolet D, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2012;30:4515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Armand P, Kim HT, Logan BR, et al. Validation and refinement of the Disease Risk Index for allogeneic stem cell transplantation. Blood 2014;123:3664–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cieri N, Oliveira G, Greco R, et al. Generation of human memory stem T cells after haploidentical T-replete hematopoietic stem cell transplantation. Blood 2015;125:2865–74. [DOI] [PubMed] [Google Scholar]
- 121.Ciceri F, Bonini C, Stanghellini MT, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. The Lancet Oncology 2009;10:489–500. [DOI] [PubMed] [Google Scholar]