

Abstract
Brome mosaic virus (BMV) is a representative member of positive-strand RNA viruses. The 1a replicase from BMV is a membrane protein of unknown structure with a methyltransferase N-terminal domain and a putative helicase activity in the C-terminal domain. In order to make a functional prediction of the helicase activity of the BMV 1a C-terminal domain, we have built a model of its structure. The use of fold recognition servers hinted at two different superfamilies of helicases [superfamily 1 (SF1) and superfamily 2 (SF2)] as putative templates for the C-terminal fragment of BMV 1a. A structural model of BMV 1a in SF2 was obtained by means of a fold recognition server (3D-PSSM). On the other hand, we used the helicase motifs described in the literature to construct a model of the structure of the BMV 1a C-terminal domain as a member of the SF1. The biological functionality and statistic potentials were used to discriminate between the two models. The results illustrate that the use of sequence profiles and patterns helps modeling. Accordingly, the C-terminal domain of BMV 1a is a potential member of the SF1 of helicases, and it can be modeled with the structure of a member of the UvrD family of helicases. The helicase mechanism was corroborated by the model and this supports the hypothesis that BMV 1a should have helicase activity.
Keywords: Helicase, Threading, Modeling, Brome mosaic virus, 1a
Introduction
Helicases are proteins that catalyze the separation of RNA or DNA duplexes into single strands using the energy generated by the NTP hydrolysis [1]. Although helicases form a wide and diverse group of proteins, they all share common sequence patterns, the so-called “helicase motifs”. These motifs consist of short conserved amino acids that play a key role in diverse steps of the helicase activity. Based on these motifs, helicases have been classified into five main groups [2] (see Fig. 1). The majority of helicases belong to superfamily 1 (SF1) or superfamily 2 (SF2). Concerning their biological roles, the DNA helicases are involved in replication, recombination and repair processes [3]. RNA helicases function in RNA transcription, editing, splicing, ribosome biogenesis, RNA export, translation, RNA turnover and organelle gene expression [4]. In addition, it has been suggested that helicases act as RNA chaperones modulating the RNA structure in diverse metabolic processes [5].
Fig. 1.
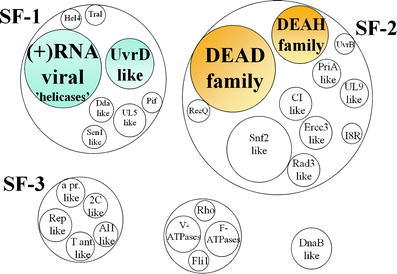
Scheme of the main groups of helicases (adapted from Gorbalenya and Koonin [2]). The distinct groups of helicases within the large families are represented by the small circles inside the larger circles. This classification is based on the extend of amino acid similarity and the organization of the helicase motifs. The helicase groups used in the modeling are hinted with a shaded background.
Helicases also play an important role in viral replication. They assure that the viral templates are repeatedly available for every new round of replication [5]. This is particularly relevant for positive-stranded RNA viruses, where the terminal regions of the genomes are highly structured. Furthermore, the recent discovery that some helicases can influence RNA-protein interactions [6] suggests that they may function in other processes of virus multiplication like viral RNA translation.
Positive strand RNA viruses cause important human diseases such as hepatitis, encephalitis, hemorrhagic fevers and SARS. To study their replication, the Brome mosaic virus (BMV), a member of the alphavirus-like superfamily of plant, animal and human viruses, has been proven to be a useful model system [7]. Two viral proteins are sufficient for BMV replication, the polymerase 2a and the replicase 1a. The latter protein has an N-terminal m7G methyltransferase and a covalent GTP-binding activity necessary for capping of the three viral RNAs. The C-terminal half contains a helicase-like domain that was shown to be essential for BMV RNA synthesis. However, its putative helicase activity has not yet been tested because the protein could not be purified sufficiently.
To make functional predictions about the putative 1a helicase activity, here we develop a 3D model of the 1a C-terminal domain structure. It was unclear if 1a helicase is a member of the SF1 or the SF2 of helicases. Therefore, we report a model of BMV 1a protein for each superfamily. These two models will help us to recognize the superfamily type of the helicase domain of BMV 1a by a further evaluation of the structures using statistic potentials and an analysis of its functionality.
Materials and methods
The sequence of the target protein BMV was retrieved from the Swissprot database [8] under code V1A_BBMV. The databases used for searching sequence and structural homologies were downloaded respectively from Swissprot [8] and the Protein data bank (PDB) [9]. The protein sequences of these databases were formatted into BLAST format [10] and Psi-Blast [10, 11] was used to search homologous sequences of the target protein. Also, the Pfam-A database [12] of protein profiles was downloaded for searching homologous sequences by means of Hidden Markov Models with the program HMMER [13]. The databases of motifs PROSITE [14] and PRINTS [15] were used on the web for searching local motifs. The alignments between the target protein and the templates were obtained by means of fold recognition using 3D-PSSM [16] and FUGUE [17]. Several partial models of the structure of the target sequence were obtained by distance restraints (extracted from the alignment) with the program MODELLER [18, 19] and energy minimized with GROMOS [20]. Statistical energies per residue were calculated with ProsaII [21] for all models and were used to validate their conformations. Finally, the protein models complexed with DNA were obtained by superimposition with STAMP [22] on the structure of the DNA-helicase complex of PcrA with DNA (PDB code 3pjr). All runs were performed on a Pentium Xeon and protein figures were obtained with RasMol [23] and PREPI in a Silicon Graphics Octane workstation.
Results
Search and sequence alignment
The sequence of BMV was compared with the profiles of the PFAM database by means of Hidden Markov Models. The result splits the sequence in two domains: the Nt domain matches the PFAM domain of “viral methyltransferase” (e-value 3.5e-162) (see Fig. 2a), and the Ct domain matched the “viral_helicase1” domain of PFAM (e-value 6.6e-75) (see Fig. 2b). We focused on the structure and function of the Ct domain in order to prove the putative helicase activity.
Fig. 2.


PFAM identified domains of Ct and Nt domains of BMV 1a. a The alignment of BMV 1a sequence obtained by HMM in PFAM for 400 residues of the N-terminal domain matched the viral methyltransferase domain. b The alignment of BMV 1a sequence obtained by HMM in PFAM for the last 350 residues at the C-terminal domain matched the viral_helicase1 domain.
Homologous sequences of the Ct helicase domain of BMV were searched with Psi-Blast [10] among the sequences of the PDB without success. Also, searching by Hidden Markov was unsuccessful because of the lack of structures for the viral_helicase PFAM domain. Therefore, the search was done on the sequences of the Swissprot database until convergence was achieved, and the PSSM matrix was kept and reused to search on the sequences of PDB. This procedure succeeded in matching a P-loop pattern on the sequence of some crystallized helicases (PDB codes 3pjr, 2pjr, 1pjr, 1qhh, 1qhg and 1uaa, which show the common feature of the UvrD helicase domain) and the viral_helicase 1 domain (the sequence-pattern included in the local alignment was GVAGCGKT). Indeed, these helicases are members of the same SCOP fold identified as “P-loop containing nucleotide triphosphate hydrolase”. The sequence-pattern GVAGCGKT was also found using Prosite [14] (pattern PS00017, ATP_GTP_A ATP/GTP-binding site motif A). Nevertheless, the local alignments were too short for modeling the structure of the Ct domain.
According to this result, we searched the template with the servers FUGUE [17] and 3D-PSSM [16] on the web. Both servers scored helicases from the PDB, although only 3D-PSSM scored on the first ranks (codes: 1hv8, 1pjr, 1uaa, 1a1v, see Table 1) with an acceptable alignment for model building a fragment of about 150–250 residues, being the alignment obtained for 1hv8, the largest one that included the ATP binding site motif in a correct position (see Fig. 3a). The results with FUGUE were discarded because the Z-score was less than marginal; the best score was 4.68 and it was given to gluconate kinase, while UvrD-helicase ranked on the 8th position with a Z-score of 2.33 and a wrong alignment at the ATP binding site motif. According to 3D-PSSM results, then, the model for the Ct domain of BMV can be obtained either as a member of the DEAD and DEAH box helicase family (SF2 helicases) or as a member of the UvrD-helicases family (SF1 helicases). For a model of the Ct domain as a member of the DEAD/H box helicase family we should use the known structure of MjDEAD, with PDB code 1hv8, or the structure of the Hepatitis C virus NS3 (HCV-NS3), with PDB code 1a1v. On the other hand, the modeling as a member of the UvrD-helicases family should be done with the structure of PcrA, with PDB codes 1pjr and 3pjr, or the structure of RepA, with PDB code 1uaa.
Table 1.
Fold recognition from 3D-PSSM and FUGUE servers for the C-terminal region of BMV 1a
| PBD code | Name | SF | Description | 3D-PSSM | FUGUE | ||
|---|---|---|---|---|---|---|---|
| e-Value | Percentage of identity | Z-score | Percentage of identity | ||||
| 1pjr and 3pjr | PcrA | 1 | DNA helicase from Bacillus stearothermophilus | – | – | 2.33 | 6% (of 632 residues) |
| 1uaa | RepA | 1 | DNA helicase from E.Coli | 1.01 | 12% (of 306 residues) | 2.33 | 5% (of 623 residues) |
| 1hv8 | MjDEAD | 2 | RNA helicase from Methanococcus jannaschii | 1.53 | 12% (of 353 residues) | – | – |
| 1a1v | NS3 | 2 | RNA helicase from Hepatitis C virus | 0.176 | 11% (of 135 residues) | – | – |
First ranked e-values (and Z-scores) for the alignment of BMV 1a C-terminal region (residues 448–961) with the sequences of helicases with known structure hinted by means of the fold recognition server 3D-PSSM. Scores and alignments obtained by FUGUE are also given for the UvrD-helicases (sequences of PcrA and RepA). The percentage of identity and the total number of residues aligned with the query are shown. PDB codes are used to identify the helicase proteins: HCV helicase NS3 (1a1v), MjDEAD (1hv8), UvrD helicase RepA (1uaa), and PcrA (1pjr). The largest number of residues aligned is obtained with MjDEAD
Fig. 3.

Partial region of the Ct helicase domain of BMV 1a. a Best alignment obtained by 3D-PSSM with the sequence of ATP-dependent RNA helicase MjDEAD from M. jannaschii (helicase belonging to the SF2, PDB code 1hv8). b Alignment obtained by manual alignment of the helicase motifs found in BMV 1a sequence with the of ATP-dependent DNA helicase PcrA from Geobacillus stearothermophilus (helicase belonging to SF1, PDB code 3pjr). The sequences corresponding to motif I, motif Ia, motif II, motif III, and motif IV are colored and hinted in bold.
Model building
The model of a region of 254 residues of the Ct domain of BMV 1a as a member of the DEAD/DEAH box helicase family (model for SF2) was obtained when this sequence was aligned with MjDEAD, an ATP-dependent RNA helicase from the archeobacteria M. jannaschii, according to the alignment obtained from the 3D-PSSM fold- recognition program and using the program MODELLER [18, 24]. Although the 3D-PSSM program had also found a good alignment for HCV-NS3, a viral_helicase, the number of residues aligned was lower than for MjDEAD. Gaps within the modeling region were modeled as loops [25] and a set of 50 putative models were obtained. The sequence numbering of the model was restarted from 1 to 254 (corresponding to amino acids 643–896 of the 1a protein sequence).
In a similar manner, a model of the Ct domain of BMV 1a as a member of the UvrD-helicase family (model for SF1) was obtained with the alignment with PcrA (yielding the largest alignment by means of FUGUE), an ATP dependent DNA helicase from B. stearothermophilus. This alignment was obtained manually by forcing the first five helicase motifs on BMV 1a to coincide with those of PcrA (see Fig. 3b), using the alignment of the SF2 model as a basis. The steps followed in this process were: (1) we obtained a structural alignment between the structures of SF2 (MjDEAD) and SF1 (PcrA) with the program STAMP; (2) to obtain the alignment between PcrA and BMV 1a we combined the MjDEAD-PcrA alignment with the one previously used in the SF2 model (MjDEAD-BMV 1a) using the common aligned residues; and (3) we manually rearranged the sequence to align the SF1 helicase motifs described for PcrA with those expected in BMV 1a, [3] while maintaining the alignment from step 2 in the zones that did not include these motifs. We used this alignment and the program MODELLER [26] to model build the C-terminal fragment of BMV 1a as a member of the SF1.
An energy profile was calculated by statistical potentials of mean force with the program ProsaII for each modeled conformation. The conformation with the smallest pseudo-energy for each superfamily type model was selected and minimized. The optimization was performed with 10,000 steps of steepest-descent minimization with the program GROMOS using the in vacuo force field and the reaction field approach [27]. The purpose of this refinement was the optimization of the side-chain orientations to avoid physical bad contacts. This optimization was performed without solvent molecules in order to avoid large modifications of the conformation of the model. SHAKE [28] was applied on the last 5,000 steps of the optimization and the electrostatic energy was calculated using the twin-range method with cut-off radii of 9 and 15 Å, updating a list of long-range interactions every ten steps, and with an additional reaction field at 15 Å [29].
After energy minimization, the model conformation for SF2 (hereafter named “superfamily-2 preliminary model”, SF-2 preliminary) still shows a positive pseudo-energy. Therefore, the model was modified by manual intervention, introducing helical conformations in coiled regions of the model where a classical β–α–β type motif was expected according to the prediction of secondary structure obtained with PSIPRED [30]. The correct pairs of α-helix H-bonds, where the pair had been lost due to misalignments, were reinforced. The regions modified by forcing the helical conformation are indicated in Fig. 4. The resulting model was optimized with GROMOS using the previous procedure, and the new model was named “superfamily-2 model” (SF2 model). A combined ProsaII pseudo-energy, obtained by a linear combination of “surface” and residue “pair” energies, was used to hint the region of the protein core where the energy of the modified model was better than the original (see Fig. 5). Although this approach resulted in an overall improvement, some regions showed worst pseudo-energy after this modification, mainly for the “surface” pseudo-energy.
Fig. 4.

Secondary structures of the models of BMV 1a. The sequence of the C-terminal fragment of BMV 1a used for model building is aligned with its secondary structure prediction obtained with psi-pred [30] and with the secondary structures obtained by means of DSSP for the SF1 model, SF-2 preliminary and SF2 model (code E is used for β-strand, H for helix and C for coil). Regions forced into helical conformation in the SF-2 model are underlined and hinted in red.
Fig. 5.
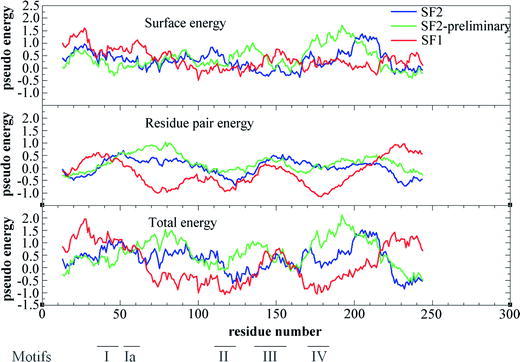
Pseudo-energies of residue-pair and surface by Prosa II. The pseudo-energy is calculated with the potentials of Prosa II (residue pair energy, surface energy and total energy) and the average of a window of 25 residues is plotted versus the middle point of the window along the sequence. The energy for the SF2 model is plotten in blue, the energy for the SF-2 preliminary in green and the energy for the SF1 model in red. The regions corresponding to SF1 helicase motifs of are indicated at the bottom of the figure.
However, the best pseudo-energies were obtained after energy minimization with GROMOS of the model of BMV 1a as a member of helicase SF1. This model was named “superfamily-1 model” (SF1 model). After energy optimization, the secondary structures of the three models were calculated with the program DSSP [31] and compared with the predicted secondary structure (see Fig. 4). The results were acceptable for the three models. Only the β-strandpredicted between residues 199 and 212 of the model is not found by DSSP, probably due to the lack of an additional β-strand formed by residues not considered in the model. The interval of residues identified by negative pseudo-energy indicates the correct alignment for the five helicase motifs comprised within the interval limits between the first 50 residues and 210. The model produces a single positive region of the pseudo-energy that falls in a region without helicase motifs. The surface energy according to ProsaII is very similar for the three models, being mostly positive. The energy between pairs of residues is mostly positive for the SF2 model and specially for the SF2 preliminary model, while it is mostly negative in the case of the SF1 model, indicating the best packing interaction for this last model (see Fig. 5).
In the SF1 model, the region with lowest pseudo-energy is found between residues 65 and 140 and from residues 165 to 205, containing some structural motifs functionally characterized in helicases or related to them. In the case of the SF2 model, the region with lowest pseudo-energy is restricted between residues 100 and 150, a region also containing some helicase motifs (see Fig. 5).
Sequence structural motifs and biological function
Helicases of SF1 and SF2 present seven characteristic helicase motifs: motif I (also named Walker A Box), Ia, II (also named Walker B box), III, IV, V and VI. Except for motifs III and IV, which differ in sequence, function and relative position, the rest of the motifs have strong similarities between the two superfamilies. It has been reported that in the SF1 of helicases, the motifs I, II, III and IV are responsible for ATP (or another NTP) binding and hydrolysis, while motifs Ia, III and V interact with the nucleic-acid substrate. Motif VI would connect these two groups, transferring the energy from the ATPase activity to the helicase activity. In the SF2 helicases, the ATPase activity depends on motifs I, II and VI, while interaction with the nucleic-acid substrate depends on motifs Ia, IV and V. Motif III would be the connector in these helicases [3].
We analyzed the presence of these motifs in the models obtained from the structures of helicases of SF1 and SF2 by checking their sequence, as well as their ability to perform a biological function (see Fig. 6). In order to emphasize the helicase function of the C-terminal domain of BMV 1a, we also modeled the interaction with DNA. The model should show an interaction with RNA. As, however, the homology with any of the available structures of helicases complexed with RNA was not good enough to allow the proper superimposition of protein structures, we sustain the model of docking with DNA as the most reliable approach. The partial model of BMV in complex with DNA was obtained by superimposition of the model and the structure of the member of the SF1, taken from the PDB with code 3pjr, which includes the binding with DNA. The protein part of 3pjr was removed from the superimposition, while DNA remained docked with the model of BMV 1a. The docking of DNA and BMV1a was adjusted manually with Turbo-Frodo [32] and the side chains were refined energetically with GROMOS. Moreover, using a similar procedure, ATP was also added to the SF1 model, while for the SF2 model this was not possible because of the disposition of the residues and their side chains in the putative ATP binding site.
Fig. 6.
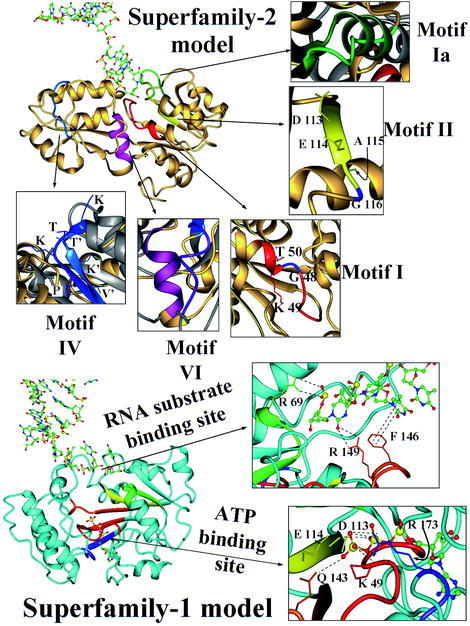
General view of the SF2 and SF1 models. Ribbon plots of the SF2 model (at the top) and SF1 model (at the bottom). Helicase motifs of helicase SF1s and SF2s are identified by colors, while the original crystallographic structure of MjDEAD (with PDB code 1hv8) is in grey and the models are in gold (for SF2 model) and cyan (for SF1 model). A detailed view of the motifs of SF2 model is shown at the top of the figure: motif I is in red (plus a conserved Gly in blue), motif Ia in green, motif II in yellow (showing a different DEAG, instead of DEAD/DEAH signature and indicating the Gly residue in blue), motif IV is in blue and motif VI in magenta. The motifs of SF1 model are indicated in red (motif I), green (motif Ia), yellow (motif II), orange (motif III) and blue (motif IV), and a detailed view of the ATP binding site implicating motif I (Lys 49), motif II (Asn 113, Glu 114), motif III (Gln 143), and motif IV (Arg173) is indicated at the margin (sequence numbering according to the model, see text). A detailed view of the DNA binding is also indicated, showing the interactions of motif Ia (Arg 69) and motif III (Arg 149 and Phe 146) with the nucleotide chain.
Superfamily-2 model
We scanned the modeled region of the BMV 1a sequence and searched for the conserved residues described by Hall and Matson [3] among the motifs of the SF2 helicases. Only motifs I, Ia, IV and VI were correctly assigned, while motifs III and V were not found in the complete BMV 1a sequence. Motif II was found with a Gly residue instead of the Asp or His residues in the last position of the DEAD/DEAH box signature (residue 116 of the model).
After the structural alignment between the SF2 model and the structure of MjDEAD, the locations and environment of the aligned residues were compared (see Fig. 6). Only the amino acids located around the positions of motifs I, II and IV in the SF2 model retained similar features of the SF2 of helicases. Moreover, motif Ia had an α-helix conformation in MjDEAD (and typically in the SF2 helicases) while this helix became unfolded in the SF2 model. Also motif IV did not form a β-strand conformation because of the lack of the partner strand with which it forms a network of main-chain hydrogen bonds because this strand was not included in the sequence of the fragment modeled.
Motif I is a P-loop motif that can be found in the SF-2 model between residues 40 and 50. This motif was already found by sequence search using PSI-BLAST and it is involved in the binding of ATP. The sequence showed two conserved Gly residues (G46 and G48) and one Lys (K49) that could interact with the phosphate group of ATP, and the side chains of the residues of the motif presented a spatial distribution in the model similar than in the structure of MjDEAD.
Motif II is involved in divalent cation binding (Mg+2) and in the catalysis of ATP. Two conserved residues (Asp and Glu) are necessary for kinase activity and are involved in the hydrolysis of ATP. Both residues were found in the SF2 model (D113 and E114). However, the last position of the conserved region that defines this motif in SF2 helicases, the so-called “DEAD/H box”, was in this case a Gly residue instead of His or Asp, and this might affect the ATPase mechanism. Furthermore, in this model the region around motif VI only presented two of the three conserved Arg residues that would bind the γ-phosphate of ATP after hydrolysis.
The structure of the SF2 helicases interacts with the nucleic acid through motifs Ia, IV and V. In the SF2 model, DNA binding would be sustained by two major interactions with the phosphate groups: (1) Arg 212 and Arg 213 (in motif VI); and (2) Arg 69 and Lys 70 (in motif Ia) plus some additional residues (Arg 207 and Lys 244). Nevertheless, we could not find the correspondence in this model for the characteristic interactions of motifs IV and V of the SF2 helicases.
Superfamily-1 model
We scanned the BMV 1a sequence to find the presence of the motifs of SF2 helicases described by Hall and Matson [3]. Five of them were found in the sequence of the C-terminal model-built fragment of BMV 1a: motif I, motif Ia, motif II, motif III and motif IV. Motifs V and VI were found in the complete sequence of BMV 1a sequence, but were neglected from model building in order to compare its conformation with the SF2 model.
These motifs were forced to be located in similar 3D positions as described for the structure of PcrA. Once the model was built and DNA and ATP molecules were added to the structure, we checked the spatial disposition of the side-chains of the residues in the helicase motifs, so we could asses their putative biological function by analyzing the possible interactions between the these motifs and the helicase substrates. In this sense, we studied the implication of motifs I, II, III and IV in the hydrolysis of the NTP molecule. We found that the side-chains of the conserved residues of these motifs are at bond distances from the phosphates of ATP in the SF1 model. Two conserved residues of motif I, Lys 49 and Gly 48, are 3.9 and 3.0 Å, respectively, from the oxygens of the β- and γ-phosphate of ATP. In motif II, the carboxylic oxygens of Asp 113 and Glu 114 are 5.4 and 4.9 Å, respectively, from the β- and γ-phosphate of ATP. And the carbonyl oxygen of the side-chain of Gln 143 in motif III, which is a conserved residue in SF1 helicases, is at 4.7 Å from the γ-phosphateof ATP.
The interaction between DNA and motifs Ia and III was used to analyze the putative interaction of the model with RNA. We neglected the interactions of motif V because it was not considered in the SF1 model. We found the side-chains of Arg 69 in motif Ia at 3.7 Å from a phosphate group of the polymer of nucleotides. On the other hand, some of the interactions found in the crystallographic structure of PcrA in complex with DNA were lost or weakened by the distance when the structure of the SF1 model was binding DNA. For example, the aromatic ring of Phe 146 of the model should have a π-interaction with one of the nitrogenated bases of DNA at 3.4 Å and the guanidinium nitrogen of Arg 149 a hydrogen bond with a ribose-phosphate. Instead, the closest nucleotide to Phe 146 was at 5.8 Å and the closest phosphate to Arg 149 at 5.5 Å.
Nevertheless, the topology of motif III retains the location near motif II (closest distance at 3.1,Å) as found in all SF1 helicases. Consequently, the positions of these residues and their known biological implication experimentally corroborated in other SF1 helicases, substantiate the SF1 model by evincing its plausible helicase activity.
Discussion
In the present work, the use of homology searches showed that the C-terminal domain of the BMV 1a has a P-loop sequence pattern plus several similarities with other helicases. Also, by means of the predicted sequence pattern-motifs, BMV 1a was expected to be a helicase [2] similar to the rest of the alphavirus-like helicases. Nevertheless, because of the lack of clear experimental evidence of its helicase activity and likely membership to one of the superfamilies of helicases, we needed further proof confirming the function of the C-terminal domain of BMV 1a as a helicase. Furthermore, it was unclear if this helicase belonged to the SF1 or SF2 main groups of helicases.
The methods of fold recognition were able to recognize the “P-loop containing nucleotide triphosphate hydrolase” structural superfamily and fold in SCOP [33] and the “tandem AAA-ATPase domain” family of a C-terminal fragment of BMV 1a. Two independent fold-recognition servers based on threading (3D-PSSM and FUGUE) identified a set of helicases of SF1 and SF2, as well as other types of non-helicase proteins that contained a P-loop. Also, the confidence degree of the 3D-PSSM server and the scores obtained by FUGUE for the C-terminal domain of BMV 1a, stated that this was a helicase. Nevertheless, the recognition as a member of the SF1 or SF2 remained unclear, and the alignment with a template sequence of known structure was either insufficient or uncertain. Consequently, we computed two putative models: (1) by means of threading with an automatic alignment from a fold recognition server; and (2) a model obtained by a manual alignment forcing the location of the sequence-motifs described for the SF1 helicases.
Both models of the structure of the fragment of the C-terminal domain of BMV 1a, as member of the SF1 (SF1 model) and as a member of the SF2 (SF2 model) of helicases, were proposed, refined and compared. The final assessment of the structure of the modeled fragment was obtained by statistical pseudo-potentials. The pseudo-energies calculated with ProsaII were used to obtain some clues for the identification of BMV 1a and its functional annotation as helicase. Furthermore, these energies were used for the validation of the models (both SF1 and SF2). The energy between pairs of residues resulted positive for the SF2 model while for the SF1 model it was mostly negative. However, this energy was not as negative as for the crystallographic structures of PcrA (as member of SF1 helicases) and the structure of MjDEAD (as member of SF2 helicases). Also, the putative interactions with ATP and DNA were better arranged for the SF1 model. The model building of a partial region of the C-terminal fragment of BMV 1a, containing five helicase motifs of the SF1, and the correct interaction of these residues with a polymer of nucleic and an ATP molecule, reinforced the assumption of BMV 1a being a member of the SF1 of helicases.
In conclusion, this work attempts to introduce the use of hypothetical local alignments, based on functional information of sequence patterns, to force or readjust a larger alignment and derive a conformational model that is more useful for interpreting the structure/function relationship of a protein than other approaches of fold prediction. The methods of threading or fold prediction can give clues about the structure of a protein sequence. However, this needs further arrangements based on scientific knowledge and the manual intervention of a curator. These final arrangements can be obtained by using sequence patterns or local profiles containing the functional and structural correlation. In this particular example, most coincidences concluded that the most likely structure of the C-terminal fragment of BMV 1a was the SF1 model. While threading methods concluded that this fragment was a member of the “P-loop containing nucleotide triphosphate hydrolase” fold and probably a member of SF2 helicases, we needed additional manual intervention to construct the model as a member of SF1 helicases. Therefore, although we can not claim a unique answer to the 3D conformation of the C-terminal domain of BMV 1a, the energetic and sequence trends proved the conclusion that the C-tail of BMV 1a was likely a member of the SF1 helicases with a conformation similar to the structure of PcrA. First, this example would answer the main question about the function and structure of the C-terminal fragment of BMV 1a. However, in addition, this example would also confirm the use of functional sequence patterns and scientific intuition to obtain a sensible answer on the deepest knowledge and more detailed description of the biological function and conformation of a protein. Answering the possibility of the helicase mechanism as a member of the SF1 helicases also supports the original hypothesis that BMV 1a should have helicase activity. Further experiments should be performed to corroborate the hypothesis. Nevertheless our results clearly hint at this direction.
Acknowledgements
This work has been supported by grants from Fundación Ramón Areces and the Spanish Ministerio de Ciencia y Tecnología (MCyT BIO2002-03609, BMC 2001-0834).
References
- 1.Caruthers JM, McKay DB. Curr Opin Struct Biol. 2002;12:123–133. doi: 10.1016/S0959-440X(02)00298-1. [DOI] [PubMed] [Google Scholar]
- 2.Gorbalenya AE, Koonin EV. Curr Opin Struct Biol. 1993;3:419–429. [Google Scholar]
- 3.Hall MC, Matson SW. Mol Microbiol. 1999;34:867–877. doi: 10.1046/j.1365-2958.1999.01659.x. [DOI] [PubMed] [Google Scholar]
- 4.Tanner NK, Linder P. Mol Cell. 2001;8:251–262. doi: 10.1016/S1097-2765(01)00329-X. [DOI] [PubMed] [Google Scholar]
- 5.Kadare G, Haenni AL. J Virol. 1997;71:2583–2590. doi: 10.1128/jvi.71.4.2583-2590.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jankowsky E, Gross CH, Shuman S, Pyle AM. Science. 2001;5:121–125. doi: 10.1126/science.291.5501.121. [DOI] [PubMed] [Google Scholar]
- 7.Noueiry AO, Ahlquist P. Annu Rev Phytopathol. 2003;41:77–98. doi: 10.1146/annurev.phyto.41.052002.095717. [DOI] [PubMed] [Google Scholar]
- 8.Boeckmann B, Bairoch A, Apweiler R, Blatter M-C, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westbrook J, Feng Z, Chen L, Yang H, Berman HM. Nucleic Acids Res. 2003;31:489–491. doi: 10.1093/nar/gkg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Bunschuh R, Olsen R, Hwa T. Nucleic Acids Res. 2001;29:351–361. doi: 10.1093/nar/29.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy S, Griffiths-Jones S, Howe K, Marshall M, Sonnhammer E. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddy SR. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 14.Hofmann K, Bucher P, Falquet L, Bairoch A. Nucleic Acids Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Attwood TK, Bradley P, Flower DR, Gaulton A, Maudling N, Mitchell AL, Moulton G, Nordle A, Paine K, Taylor P, Uddin A, Zygouri C. Nucleic Acids Res. 2003;31:400–402. doi: 10.1093/nar/gkg030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley LA, MacCallum RM, Sternberg MJE. J Mol Biol. 2000;299:501–522. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- 17.Shi J, Blundell TL, Mizuguchi K. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- 18.Eswar N, Bino J, Mirkovic N, Fiser A, Ilyin VA, Pieper U, Stuart AC, Marti-Renom MA, Madhusudhan MS, Yerkovich B, Sali A. Nucleic Acids Res. 2003;31:3375–3380. doi: 10.1093/nar/gkg543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez R, Pieper U, Melo F, Eswar N, Martí-Renom M, Madhusudhan M, Mirkovic N, Sali A. Nat Struct Biol. 2000:S986–S990. doi: 10.1038/80776. [DOI] [PubMed] [Google Scholar]
- 20.van Gunsteren W, Billeter S, Eising A, Hünenberger P, Früger P, Mark A, Scott W, Tironi I. Biomolecular simulation: the GROMOS96 manual and user guide. Zürich: Verlag der Fachvereine; 1996. [Google Scholar]
- 21.Sippl MJ. J Mol Biol. 1996;260:644–648. doi: 10.1006/jmbi.1996.0427. [DOI] [PubMed] [Google Scholar]
- 22.Russell R, Barton G. Proteins. 1992;14:309–323. doi: 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- 23.Sayle R, Millner-White E. Science. 1995;20:374–374. doi: 10.1016/S0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 24.Martí-Renom MA, Stuart A, Fisher A, Sánchez R, Melo F, Sali A. Ann Rev Biophys Biomolec Struc. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 25.Fiser A, Do RK, Sali A. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sánchez R, Sali A. Proteins. 1997;S1:50–58. doi: 10.1002/(SICI)1097-0134(1997)1+<50::AID-PROT8>3.3.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 27.Hünenberger PH, van Gunsteren WF. J Chem Phys. 1998;108:6117–6134. doi: 10.1063/1.476022. [DOI] [Google Scholar]
- 28.Kräutler V, van Gunsteren WF, Hünenberger PH. J Comput Chem. 2001;22:501–508. doi: 10.1002/1096-987X(20010415)22:5<501::AID-JCC1021>3.0.CO;2-V. [DOI] [Google Scholar]
- 29.Hünenberger PH, van Gunsteren WF. J Chem Phys. 1998;108:6117–6134. doi: 10.1063/1.476022. [DOI] [Google Scholar]
- 30.McGuffin L, Bryson K, Jones D. Bioinformatics. 2000;16:404–405. doi: 10.1093/bioinformatics/16.4.404. [DOI] [PubMed] [Google Scholar]
- 31.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2630. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 32.Roussel A, Fontecilla-Camps JC, Cambillau C. J Mol Graph. 1990;8:86–88. doi: 10.1016/0263-7855(90)80087-V. [DOI] [PubMed] [Google Scholar]
- 33.LoConte L, Brenner S, Hubbard T, Chothia C, Murzin A. Nucleic Acids Res. 2002;30:264–267. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]