

Abstract
We report on separations of ion isotopologues and isotopomers using ultrahigh-resolution traveling wavebased Structures for Lossless Ion Manipulations with serpentine ultralong path and extended routing ion mobility spectrometry coupled to mass spectrometry (SLIM SUPER IMS-MS). Mobility separations of ions from the naturally occurring ion isotopic envelopes (e.g., [M], [M+1], [M+2], … ions) showed the first and second isotopic peaks (i.e., [M+1] and [M+2]) for various tetraalkylammonium ions could be resolved from their respective monoisotopic ion peak ([M]) after SLIM SUPER IMS with resolving powers of ~400–600. Similar separations were obtained for other compounds (e.g., tetrapeptide ions). Greater separation was obtained using argon versus helium drift gas, as expected from the greater reduced mass contribution to ion mobility described by the Mason–Schamp relationship. To more directly explore the role of isotopic substitutions, we studied a mixture of specific isotopically substituted (15N, 13C, and 2H) protonated arginine isotopologues. While the separations in nitrogen were primarily due to their reduced mass differences, similar to the naturally occurring isotopologues, their separations in helium, where higher resolving powers could also be achieved, revealed distinct additional relative mobility shifts. These shifts appeared correlated, after correction for the reduced mass contribution, with changes in the ion center of mass due to the different locations of heavy atom substitutions. The origin of these apparent mass distribution-induced mobility shifts was then further explored using a mixture of Iodoacetyl Tandem Mass Tag (iodoTMT) isotopomers (i.e., each having the same exact mass, but with different isotopic substitution sites). Again, the observed mobility shifts appeared correlated with changes in the ion center of mass leading to multiple monoisotopic mobilities being observed for some isotopomers (up to a ~0.04% difference in mobility). These mobility shifts thus appear to reflect details of the ion structure, derived from the changes due to ion rotation impacting collision frequency or momentum transfer, and highlight the potential for new approaches for ion structural characterization.
Graphical Abstract
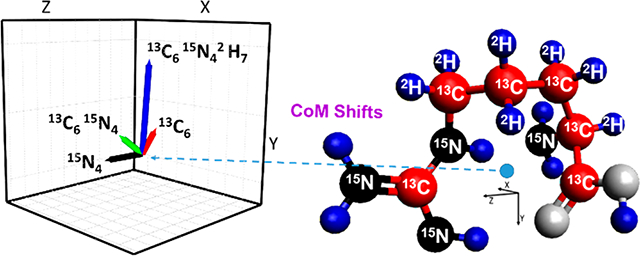
Isotopologues are molecules that differ only due to substitutions of isotopes for one or more of their atoms, while isotopomers are isotopic stereoisomers having the same number of each isotope but differing in their locations.1 Isotopic labeling with often “heavier” atoms (e.g., 15N, 13C, 2H) is broadly used with mass spectrometry (MS), especially for enabling more accurate quantification, for multiplexing with isobaric labeling (e.g., isobaric tags for relative and absolute quantitation, iTRAQ; tandem mass tag, TMT), and for the reduction of isobaric interferences.2,3 MS readily separates isotopologues due to their mass differences, and the development of high resolution (most notably Fourier Transform-based) MS over the last few decades has made distinguishing different isotopic substitutions (e.g., the 0.006 Da mass difference for 15N vs 13C substitution) routine.4 Conversely, isotopomers cannot be distinguished by MS alone since they have the same exact mass, unless fragment ions (e.g., obtained using MS/MS) reveal the sites of isotopic exchanges.
Ion mobility spectrometry (IMS) allows distinguishing ions in the gas-phase based on their size/shape in addition to m/z.5 IMS, most notably coupled to MS (IMS-MS), has shown broad utility for the resolution of isomeric species in proteomics, lipidomics, glycomics, and other areas.6-18 The mobility of an ion (K) in the low field regime can be predicted using the Mason–Schamp relationship (eq 1), where μ is the reduced mass of the ion–neutral molecule pair, k is the Boltzmann constant, z is the charge of the ion, T is the absolute temperature, N is the gas number density, and Ω is the ion–neutral momentum transfer (or collision) cross section.5,19
| (1) |
IMS resolution of isotopologues that, for example, constitute the naturally occurring isotopic envelopes of a compound’s mass spectrum (i.e., the [M], [M+1], [M+2], etc. ion “isotopic peaks”) would provide the basis for fundamental new understandings and applications, as emphasized over a decade ago by Valentine and Clemmer.20 Drift tube IMS (DTIMS) isotopologue separations of sufficient resolving power (Rp) are largely impractical due to the large voltage drops and/or extremely long linear drift paths needed.21-23 The most notable exception to date has been the separation of natural and deuterium (2H) substituted isotopologues for smaller compounds such as toluene, acetone, and benzene, using atmospheric pressure DTIMS with ~250 Rp.24 In the case of acetone versus its perdeuterated counterpart, a mobility difference of ~1.5% in nitrogen is predicted based solely upon the reduced mass contribution (μ; Mason–Schamp relationship, eq 1).24 However, the reported Rp would be insufficient for much larger ions, and even greater challenges would arise with the use of lighter drift gases (e.g., helium) due to the smaller reduced mass differences. The only other isotopologue or isotopomer ion separations of which we are aware have used Field Asymmetric Ion Mobility Spectrometry (FAIMS), where Rp values up to ~500 have been demonstrated based upon differences in mobilities at very high vs low electric fields in mixtures of gases.1,25-28 While these studies suggest that observed shifts for individual substitutions may be relatable to the ion’s structure, FAIMS incurs some significant, but not well characterized, extent of ion heating and potential structural perturbations due to the high fields used,29 as well as other possible contributions such as partial ion alignment in the field, making an understanding of the effects due to isotopic substitutions elusive.26, 28
In this work we have employed new ultrahigh precision and resolution IMS measurement capabilities using traveling wave (TW)-based Structures for Lossless Ion Manipulations (SLIM)6,8-11,30-36 in conjunction with MS for isotopologue and isotopomer separations. TW-based SLIM IMS benefits from the long and compact serpentine path lengths, where resolution has been shown to increase in proportion to the square root of path length.37 Additionally, directing the ions for additional passes through the SLIM, using a Serpentine Ultralong Path with Extended Routing (SUPER) implementation,30 further extends the path lengths achievable to >100 m, providing Rp up to ~1800,30 far greater than previously achievable with IMS. These measurements also benefit from the ability to inject very large ion populations via in-SLIM ion accumulation to provide significant ion signals after >1 km of SLIM SUPER IMS separations and improving the precision of mobility measurements achievable for higher Rp IMS measurements.7,31
Here we report relative ion mobility shifts for isotopologues and isotopomers that confirm the reduced mass contribution to mobility expected from eq 1. In addition, we report the contribution of additional mobility shifts, as well as our observation that these shifts appear to be correlated with changes to the ion center of mass (and by inference, ion rotation), due to the isotopic exchanges or substitutions, and lead to changes in Ω. These results show not only the utility of higher Rp IMS measurements, providing a greatly improved basis for understanding other factors contributing to an ion’s Ω, but also a potential basis for new insights into gas phase ion interactions and new approaches for the structural characterization of gas phase ions.
EXPERIMENTAL SECTION
Instrumentation.
The TW SLIM SUPER IMS-MS platform has been described previously.6-10,30,31 Briefly, it has a 13.5 m SLIM total ion path for each pass, using 0.5 mm width and used 1 mm length SLIM surface TW electrodes in a “6, 5” electrode array configuration (6 radio frequency electrodes interspaced with 5 TW electrode arrays). The SLIM used 1 to 1.8 mHz RF frequency and 300 V peak-to-peak (Vpp) RF amplitude for ion confinement between the SLIM surfaces, unless otherwise noted. Modifications to the previously described platform included SLIM fabrication from Rogers 4000 hydrocarbon ceramic laminates. This alternative SLIM material used here for some of the studies served to lower the SLIM capacitance, and thus lowered the RF power needed, minimizing possible warming of the SLIM components (and indirectly the SLIM chamber and the gas) compared to previous designs.
Ions exiting the SLIM were focused by an ion funnel through a conductance limit and transferred via a segmented quadrupole to an Agilent TOF MS incorporating a 1.5-m flight tube (Agilent Technologies, Santa Clara, CA). Data were recorded with a U1084A 8-bit ADC digitizer (Keysight Technologies, Santa Rosa, CA) and processed using in-house developed control software.
Experimental Conditions and Reagents: Naturally Occurring Isotopologues.
The tetraalkylammonium (TAA) salts and l-Met-l-Arg-l-Phe-l-Ala (MRFA) peptide were purchased from Sigma-Aldrich (Milwaukee, WI USA) and used without any further purification. The TAA salts (tetrahexyl, TAA-6; tetraheptyl, TAA-7, and tetraoctyl, TAA-8) were prepared to a final concentration of 1 μM in 50/50 water/ methanol with 0.5% acetic acid (v/v), while the MRFA peptide was prepared to 5 μM in 50/50 water/methanol with 0.5% acetic acid (v/v). All samples were infused at 0.5 μL/min for nanoelectrospray at 3000 V and 110 °C for the inlet capillary. IMS in the SLIM used 1.5 Torr for argon and 3.1 Torr for helium. TW parameters were optimized for separation for each analyte. In helium, a TW speed of 320 m/s was used for all analytes, with square TW amplitudes of: 23 V for MRFA, 21 V for TAA-6, 23 V for TAA-7, and 25 V for TAA-8. In argon, a TW speed of 300 m/s was used for all analytes, with square TW amplitudes of 30 V for MRFA, 28 V for TAA-6, 30 V for TAA-7, and 32 V for TAA-8. Each IMS spectrum represents a sum of 50 separations, with the exception of 350 separations used for the 2029.5 m separation of the MRFA peptide isotopologues.
The in-SLIM ion accumulation used in this study has been described in detail elsewhere.7-10,31 Briefly, ions can be accumulated in the first traveling wave region by halting the traveling wave in the second region. Ions were accumulated in-SLIM for 1–2 s for all SLIM SUPER IMS separations. An ion switch at the end of the second TW region permits ions to be routed either to the TOF-MS for detection or back to the beginning of the serpentine path for another “pass” through the SLIM module.
Experimental Conditions and Reagents: Arginine Isotopologues.
All reagents were purchased from Sigma-Aldrich, St Louis, MO, unless otherwise noted. The l-arginine isotopologues (Supporting Information), involving 15N4, 13C6, 13C615N4, and 13C615N42H7 isotopic substitutions in addition to “light” (i.e., unsubstituted) arginine, were purchased from Cambridge Isotope Laboratories Inc., Tewksbury, MA. One to 5 μM solutions of mixtures of light arginine along with its heavylabeled isotopologues in 50% methanol, 0.1% acetic acid were used. Ions were introduced via a multicapillary inlet with 500 μm i.d. stainless steel capillaries maintained at 130 °C. Ions were then focused by a high pressure ion funnel (maintained at 10 Torr) into a lower pressure ion funnel and accumulated in its ion funnel trapping region35,37 prior to injection into the SLIM chamber. The SLIM chamber was maintained at 50–100 mTorr higher pressure than the ion funnel trapping region to prevent/minimize gas introduction to the SLIM chamber from the interface. 2.5 Torr pressure was used for both nitrogen and helium drift gases. TW conditions were 9 V amplitude sine wave and 54 m/s speed for separations in nitrogen, and 12 V sine wave at 355 m/s for separations in helium.
Experimental Conditions and Reagents: Iodo-TMT Isotopomers.
The iodo-tandem mass tag (126, 127, 128, 129, 130, and 131 iodo-TMT) isotopomers were purchased from Thermo Fisher Scientific, Waltham, MA, USA. A mixture of all six isotopomers was prepared, with each component at a concentration of 10 μM, in 50/50 water/methanol with 0.5% acetic acid (v/v). SLIM SUPER IMS separations of this isotopomer mixture were performed in nitrogen drift gas (1.6 Torr) at a TW speed of 400 m/s and 35 V amplitude. 50 summations were performed to produce the arrival time distributions. Each experiment was conducted three times to produce the average normalized shift and standard deviation in Table 3. In order to plot the separation (arrival time distribution) of this isotopomer mixture, the protonated parent ion (m/z 458) was fragmented post-SLIM IMS separation to yield their six respective nonisobaric fragment ions (i.e., m/z 126–131). Each of these fragment ions was then extracted to plot the arrival distribution of the mixture (Figure 7).
Table 3.
Normalized IMS Shifts for the Set of iodo-TMT Isotopomers Studied as a Mixture Based on Averaged Triplicate Measurements in N2 Drift Gasa
| Isotopomer | Normalized shift |
|---|---|
| iodo-126 | 0 (reference) |
| iodo-127 | 0.06 ± 0.02 |
| iodo-128 | 0.53 ± 0.04 |
| iodo-129 | 0.66 ± 0.07 |
| iodo-130 | 0.97 ± 0.03 |
| iodo-131 | 1 (reference) |
Errors are based on 1 standard deviation.
Figure 7.

SLIM SUPER IMS separation of a mixture of six iodo-TMT isotopomers in nitrogen drift gas after 1354.5 m. The [M + H]+ parent ion (458 m/z) was fragmented post-SLIM IMS separation to yield nonisobaric reporter ions (126, 127, 128, 129, 130, and 131 m/z) used to construct the arrival time distribution (bottom left), revealing the separation of the six isotopomers. IMS-MS illustration of the fragment ions is also shown (bottom left). CoM vectors (bottom right) relative to the iodo-126 (red dot). 3D structures of the iodo-TMT are displayed (top). Hydrogen atoms were removed for ease of visualization.
Data Processing.
CoM calculations were performed using Python, and structures were visualized using the Avogadro Software. The lowest energy structure used for protonated iodo-TMT was optimized and calculated via a modified ISiCLE pipeline38 and visualized via the Avogadro software (for the x, y, z coordinates; Supporting Information). MarvinSketch (v.15.8.10, ChemAxon Ltd.; Budapest, Hungary) was used to generate an initial set of 100 conformers, optimized using MMFF9439 with a 1 kcal/mol diversity limit.
By running mixtures of isotopomers and isotopologues, we remove much of the uncertainty associated with small changes in conditions between separations (e.g., pressure fluctuations), as any such fluctuations should impact each ion in the same fashion. In this work we also corrected for the minor pressure fluctuations that can occur (more so in helium drift gas) between each SLIM SUPER IMS separation. These fluctuations are typically on the order of < ~5 mTorr in magnitude. These minor pressure changes may cause arrival times to shift between individual separations, leading to poorer S/N and resolution when signal averaging (e.g., summing 50) multiple separations to yield a final, output, arrival time distribution. In order to correct for this, we performed a linear shift alignment based on a Pearson correlation algorithm. This alignment uses a single, most intense, feature in a single separation (or acquisition) as a reference point and linearly shifts all other separations/ acquisitions to that reference point. (For an example, see Supporting Information.)
IMS separations were smoothed with a 3 to 25 point moving average filter and for the arginine isotopologues were fit to a Gaussian function to determine the peak centroid using Origin Pro 2016 software. The Gaussian fit centroids were used for subsequent data analysis. For other isotopologues and isotopomer measurements, a 5 to 20 moving average smooth was performed, respectively, and the peak apex was used for data analysis.
Calculation of Isotopologue IMS Shifts.
Contributions to ion mobility in the low field limit are broadly accepted as given by eq 1.5,19 Thus, if isotopomers have the same Ω, the relative mobilities of isotopologues with respect to their light analogues (KH/KL) will be inversely proportional to the ratio of the square root of their reduced masses (√μH/√μL). We refer to these values as theoretical relative isotopologue mobilities. In an analogous way to IMS calibration using different chemical species, we use selected nonisobaric isotopologues as calibrants for other isotopologues. To calibrate arrival times, we used the relative experimental arrival times of the arginine isotopologues using mixtures of “light” (i.e., unlabeled) and 13C615N4-labeled [arginine+H]+ versions to provide a mobility scale for the relative experimental mobilities of the other three isotopologues. We emphasize that the theoretical relative mobilities calculated for He and N2 drift gases assume only the reduced mass difference contribution and no structural contribution (i.e., no difference in Ω). By selecting a uniformly labeled isotopologue (i.e., 13C615N4, increasing arginine mass by 10 Da) we benefit from a significant reduced mass difference in establishing the mobility scale, as well as a likely minimal effect on the ion CoM relative to its unlabeled counterpart (see later discussion and Table 2).
Table 2.
Shifts in Relative Mobilities of the Mixture of Protonated Arginine Ion Isotopologues in Both Helium and Nitrogena
| Species [M + H]+ | m/z | Theoretical KL/KH(Helium) | Experimental KL/KH(Helium) | Theoretical KL/KH (Nitrogen) | Experimental KL/KH(Nitrogen) |
|---|---|---|---|---|---|
| unsubstituted | 175 | 1 (reference) | 1 (calibrant) | 1 (reference) | 1 (calibrant) |
| 15N4 | 179 | 1.00025 | 1.00073 ± (6 × 10−05) | 1.00156 | 1.00165 ± (1.7 × 10−04) |
| 13C6 | 181 | 1.00037 | 0.99983 ± (6 × 10−05) | 1.00232 | 1.00238 ± (2.3 × 10−04) |
| 13C615N4 | 185 | 1.00061 | 1.00061 ± (calibrant) | 1.00378 | 1.00378 ± (calibrant) |
| 13C615N42H7 | 192 | 1.00100 | 1.00022 ± (4 × 10−05) | 1.00623 | 1.00579 ± (2.2 × 10−04) |
Error represents the standard deviation for 6 averaged replicates.
Calculation of Electric Fields Experienced by Ions and Estimation of the Field Regime for TW SLIM IMS Separations.
Theoretical mobilities based upon eq 1 assume that ion mobility separations are performed within the low field limit above which the field can cause activating collisions and nonlinearly impact ion mobilities. The low field limit can be estimated by eq 2, where E is the electric field, N is the gas number density, m is the mass of the ion, M is the mass of the buffer gas, z is the ion charge, and Ω is the ion’s momentum transfer (or collision) cross section in the selected gas.40
| (2) |
In estimating the electric field in TW SLIM IMS separations one must consider that ions experience nonuniform electric fields which depend on the details of the SLIM design (e.g., electrode arrangement) and the TW parameters (the traveling waveform shape, amplitude, and speed). TW separations occur when the applied TW speed exceeds the ion mobility, K, where ions can no longer travel with the wave (i.e., surf). Under these conditions, with the application of symmetrical waveform, ions experience a dynamic potential, which oscillates in the direction between positive and negative values as they “roll over” the waves. The average magnitude of the field is highest when the TW speed is close to the ion’s mobility and decreases with higher applied TW speeds.
Of interest is not only the average electric field experienced by the ions but particularly the maximum value of the field, where significant ion heating is most likely to occur. To determine the maximum electric field experienced by the ions, the TW SLIM electrode array arrangement was modeled using SIMION 8.1, and MATLAB was used to visualize the electric fields. The electric field calculations did not take into account the contribution of the RF applied, as it is expected (and also previously shown) to have minimal contribution on ion heating in both an RF confining drift tube and SLIM IM instrumentation operating in a comparable pressure regime.41 The maximum value of the electric field generated by a 12 Vpp sine wave is 12.3 V/cm. This corresponds to E/N values of ~15 Td at 2.5 Torr pressure. The magnitude of the product expression of [m/(m + M)]1/2Ω/πz (eq 2) for protonated arginine in nitrogen is 39 and 22 Td in helium, based on the Ω values of ~70 Å2 (in helium) and 132 Å2 (in nitrogen).42 These values are higher than the present experimental E/N magnitude; therefore, the present SLIM TW IM studies are consistent with the low field limit regime. Moreover, we emphasize that the average electric field experienced by the ions in TW IMS separations is a TW speed dependent fraction of the maximum electric field value mentioned above.
Thus, while we cannot completely exclude the possibility of some degree of residual ion heating in the present studies, the TW conditions used and a range of observations are consistent with it being minimal or insignificant. Finally, we note that any amount of heating due to the TW will be essentially identical for all the isotopologue and isotopomer species in any study, and thus any impacts to relative mobility measurements would be expected to be minimal effect.
RESULTS AND DISCUSSION
In this work we have investigated the relative ion mobility shifts for mixtures of isotopologues and isotopomers using ultrahigh resolution IMS. These measurements involved naturally occurring isotopologues (i.e., the nonisobaric “isotopic peaks” for several compounds, and which include peaks that are due to sets of isotopomers), a mixture of distinct (arginine) isotopologues, and a mixture of distinct (iodoTMT) isotopomers.
Valentine and Clemmer first posited20 that the peaks constituting the isotopic peaks of mass spectra could also be resolved based on mobility differences due to their reduced mass differences given sufficiently high IMS Rp. This prediction implicitly assumes that natural isotopologues have essentially identical rotationally averaged ion-neutral momentum transfer (or collision) cross sections (Ω). Similarly, it has been assumed that differences in mobility (or Ω) for isotopomers due to the subtle effects of different isotope locations would be far too small to be detectable by low field (e.g., conventional drift tube) IMS.27
We note that resolution of some isotopomers has been achieved using FAIMS,1,25-28 where separations take place based on the mobility of ions in both high and low electric fields.1,25-28 However, the basis for these separations has not been established due to the fact that the FAIMS separation occurs in conjunction with significant ion heating (and likely induced structural changes) as well as other possible contributions such as partial ion dipole alignment due to the high fields involved.29,43
In TWIMS separations, as opposed to conventional DTIMS separations, ions experience nonuniform electric fields and detector arrival times vary depending on TW parameters.19 In consequence, calibrant ions are used to obtain mobility or Ω values from TWIMS measurements.5 The calibration procedure ideally aims to ensure that the effective temperature of calibrant ions is comparable to that of the species of interest,5 particularly if measurements are made under conditions that can lead to significant ion heating. Thus, the calibrant ions used in TW IMS measurements and the ions of interest ideally experience the same TW parameters, occur over a comparable mobility range, and are of the same compound class (i.e., they share structural similarity). Such selection of similar calibration compounds has been shown to significantly increase the accuracy of Ω determination using the commercially available Waters Synapt IMS-MS platform.44 We note that the Waters Synapt has been shown to cause ion heating due to a combination of factors, including most significantly in the ion injection step into the TW cell region, as well as to a lesser extent due to heating during the TW separation.45,46
The present studies aimed to minimize ion heating by using electric fields in the low field regime, where eq 1 is considered applicable. The use of TW sine waveforms rather than the stepped DC “square waves” (and generally larger average amplitude) served to reduce the maximum electric field experienced by ions for a given average field due to the TW and minimize possible ion heating due to the TW (Supporting Information). In this regard, previous studies have found generally good correlations between Ω determined using DTIMS in the low field regime and the TW-based Waters Synapt platform.47,45,48 Ion heating due to the TW is expected to be less significant in the present work due to the relatively low TW amplitudes used (e.g., 9 to 12 Vpp for arginine isotopologues), limiting the maximum field experienced by the ions during SLIM IMS separations ~12 V/cm, and within what is commonly considered to be the lo field regime. We also note that any residual ion heating should be very similar for a given set of isotopologues in any given measurement due to their essentially identical structures and similar m/z. This is consistent with our observation that relative mobilities were not impacted by changes in TW amplitude. Finally, we note that TW SLIM IMS measurements have provided relative mobilities (and Ω) after calibration that are comparable to those from drift tube measurements34,49 and that the separations of labile compounds and noncovalent complexes in reported studies7,9 have mirrored drift tube separations; observations supportive of minimal ion activation.
In this work we also exploited the ability to make high Rp comeasurements of the isotopologues (i.e., KL/KH) in the same separation (i.e., as mixtures) due to their different m/z, as employed for relative mobilities in electrophoretic separations,50,51 to further enhance measurement precision. For the arginine studies we used mixtures of “light” and 13C615N4 substituted [Arg+H]+ (due to its more significant mass difference as well as its relatively uniform distribution of added mass) as internal calibrants to establish the KL/KH scale. This corresponds to assuming that the mobility difference for this calibrant pair is only due to their reduced mass differences with the collision gas, an assumption that would not impact the relative shifts or direction of shifts found but only the absolute magnitude of the scale. For the isotopomer studies (where no reduced mass difference applies) we established a scale based upon the isotopomers having the smallest and largest mobilities. We expect the present relative mobilities to be both highly precise and robust. The studies here overall both confirm the predicted role of reduced mass on isotopologue ion mobility and reveal a new contribution to the ion Ω.
Mobility Differences for Naturally Occurring Isotopologues in Helium and Argon Drift Gases.
We examined the naturally occurring isotopologue mixtures present in the molecular ion isotopic envelopes and the resolution of the singly charged [M] (the monoisotopic molecular ion), [M+1], and [m+2] species. We note that the [M+1] and [M+2] peaks of the isotopic envelope are actually ensembles of isotopic permutations, (largely a mixture of isotopomers), a factor that may obscure any potential effects of isotopic substitutions at specific positions. Equation 1 predicts that a higher molecular weight drift gas would result in greater mobility differences for the isotopologues;20 mobility differences among isotopologues in argon would be expected to be nearly 10-fold greater than in helium assuming identical Ω. Table 1 shows the predicted differences in mobility for the observed isotopic peaks for the various TAA ions in both argon and helium drift gases.
Table 1.
Calculated Relative Mobilities (KL/KH) Relative to [M] Based on Respective Reduced Masses for the Naturally Occurring Isotopologues of Three Tetraalkylammonium Ions in Argon and Helium
| Ion | Isotopologues | Helium | Argon |
|---|---|---|---|
| TAA-6 [M] = 354.4 m/z | [M] vs [M+1] | 1.000016 | 1.000143 |
| [M] vs [M+2] | 1.000031 | 1.000284 | |
| TAA-7 [M] = 410.5 m/z | [M] vs [M+1] | 1.000012 | 1.000109 |
| [M] vs [M+2] | 1.000023 | 1.000215 | |
| TAA-8 [M] = 466.5 m/z | [M] vs [M+1] | 1.000009 | 1.000084 |
| [M] vs [M+2] | 1.000018 | 1.000168 |
Each TAA ion isotopologue mixture was assessed after 50 passes (679.5 m separations) in helium (Figure 1A). It can be seen that even after this SLIM SUPER IMS separation only very minor separation is achieved in helium, a 1–2 ms difference in arrival times between the [M] and [M+2] species for the lowest MW species (TAA-6) with our observed of Rp of ~400 to 500. As expected, the 679.5 m separations in argon (Figure 1B) provided significantly better resolution for the TAA ion isotopologues; for example, the resolution for [M+2] from [M] for TAA-6 was ~0.3.
Figure 1.

SLIM SUPER IMS separations of the natural isotopologues of various tetraalkyl ammonium ions: tetraalkylammonium 6 (354.4 m/z), tetraalkylammonium 7 (410.5 m/z), and tetraalkylammonium 8 (466.5 m/z). (A) helium (3.1 Torr) and (B) argon (1.5 Torr) for a 679.5m total path length with each isotopologue normalized to a 0 to 1 scale due to the significantly lower intensity of the [M+1] and [M+2] species relative to [M].
A 2029.5 m (150 pass) SLIM SUPER IMS separation for a singly charged tetrapeptide MRFA (where isotopic contributions due to 13C,15N, 18O, and 34S occur) in argon (Figure 2) shows similar shifts for the [M+1] and [M+2] and [M+3] species, and an apparent Rp of ~600. These results were entirely consistent with our observations for the TAA ions, and we believe that such observations will be general for other compounds.
Figure 2.

SLIM SUPER IMS separations of the natural isotopologues of protonated MRFA peptide in argon drift gas (1.5 Torr) after 2029.5 m of total path length (left). Table of the calculated shifts in relative mobility among its natural isotopologues, [M+1], [M+2], and [M+3], from [M], where [M] = [MRFA+H]+ (right). Note: some detector saturation for the [M] peak results in a somewhat distorted peak shape for this ion.
It is important to note that the [M+1] and [M+2] species for these naturally occurring isotopologues are largely mixtures of isotopomers: for the TAA ions, one and two 13C isotopic substitutions, respectively. Thus, the observed mobilities for the isotopic peaks each represent an unresolved set of isotopomers. Therefore, use of these naturally occurring isotopologues likely precludes the observation of more subtle effects arising from isotopic substitutions. However, we further note that any significant variation among the mobilities of the various isotopomers constituting the isotopic peaks may be manifested as a broader IMS peak; that is, the [M+1] and [M+2] would be broader than [M], which is only a single species. Indeed, Figure 3 shows that the TAA-6 ion [M+1] peak width is significantly larger (~25%) than the [M] peak width in argon drift gas (~16.75 ms in width at half-maximum vs ~13 ms in width at half-maximum for [M]), clearly suggesting the possibility of variations in the mobilities of the [M+1] component isotopomers.
Figure 3.

SLIM SUPER IMS separation of the TAA-6 [M] and [M+1] ions in argon drift gas with the same TW conditions as those used in Figure 2 after carefully controlling ion populations to avoid detector saturation for [M] (as evident in Figure 2). The ~25% broader [M+1] peak relative to the [M] peak suggests the possibility of variations in the mobilities of its component isotopomers.
These observations beg the question as to whether other more subtle ion mobility shifts may occur upon isotopic substitutions and whether these may be effectively hidden by the fact that the naturally occurring isotopologues (with the exception of the monoisotopic molecular ion; [M]) are primarily isotopomer mixtures. For this reason, we next studied a set of discrete arginine ion isotopologues.
Mass Distribution Induced Ion Mobility Shifts for Protonated Arginine Isotopologues.
Based on the results demonstrating that the naturally occurring isotopologues could be mobility separated due to their reduced mass differences, and the observation of increased peak widths for [M+1] vs [M] ions, we studied mixtures of various protonated arginine isotopologues in nitrogen and helium. Assuming no differences in the isotopologue Ω, relative theoretical isotopologue mobilities can be predicted using eq 1 based on the different reduced mass contribution, as done for the naturally occurring isotopologues. To establish a mobility scale, we proceeded in a manner analogous to IMS calibration using different chemical species. For the arginine isotopologues we used “light” (i.e., unsubstituted) and 13C615N4 substituted [arginine+H]+ as calibrants to determine the relative experimental mobilities for the other three isotopologues. While the choice of the calibrants is in principle arbitrary, our selection was based on the desire to (1) largely bracket the mobility range studied, (2) avoid the use of the 2H substituted isotopologue (that might be more likely to display an anomalous isotope effect, e.g., a primary kinetic isotope effect), and (3) have the added mass more uniformly distributed (see later discussion).
Figure 4 shows the SLIM SUPER IMS [Arg+H]+ isotopologue separations in N2 (a) and He (b), and Figure 5 plots their KL/KH in both gases. The relative mobilities and associated standard deviations for the protonated arginine ion isotopologues in the two gases are summarized in Table 2. The theoretical relative mobilities in the He and N2 drift gases given in Table 2 assume that the shifts in relative mobilities are based on their differences in reduced mass.
Figure 4.

Top: SLIM SUPER IMS arrival times for a mixture of [Arg +H]+ isotopologues (Gaussian fits) with the dashed line corresponding to the peak centroid for light arginine in nitrogen (a) after 68 m and helium (b) after 500 m. Bottom: structure of arginine along with locations of the heavy-atom substitution sites.
Figure 5.

Relative mobilities of the mixture of arginine isotopologues separated in helium (blue circles) and nitrogen (red squares) using light (i.e., unsubstituted) and 13C615N4 substituted [Arg+H]+ as internal calibrants, with theoretical relative mobilities based upon only a reduced mass contribution. Error bars represent standard deviations of 6 sets of IMS separations with different path lengths.
The separations in N2 over a 68 m path length can be largely attributed to the reduced mass contributions, which are larger than in He, with the most evident deviation being a small negative mobility shift for the 13C615N42H7 isotopologue. We note that the separations in N2 provided lower Rp than those in He due to greater m/z 175 ion losses from being at the upper end of the mobility range efficiently transmitted by our current platform.
In contrast, in He, where the reduced mass differences are nearly 6-fold less and higher Rp could also be achieved, significant deviations are observed from the trend predicted on the basis of the reduced mass differences. Particularly striking are the positive “shift” for the 15N4 isotopologue and the negative shift for the 13C6 isotopologue (i.e., the displacement from the line in Figure 5 we attribute to a Ω change). We note that the 13C6 isotopologue displays higher mobility than the 15N4 isotopologue, opposite to expectation from the reduced mass contribution. A large negative shift (i.e., higher mobility) is also observed for the 13C615N42H7 isotopologue.
The factors potentially contributing to changes in Ω for isotopologues (after consideration of their reduced mass differences) or isotopomers can include subtle structural variations due to changes in bond lengths and angles,52 charge location,53,54 polarizability, as well as vibrational motion arising from the isotopic substitutions upon ion–molecule collisions. However, spectroscopic information, as well as examples of computationally determined changes in individual bond lengths caused by H/D substitutions, where the most significant impact would be anticipated,52 indicate that such changes to Ω will be extremely small. Previous gas phase IR spectroscopy and computational studies of [Arg+H]+ tautomers indicate very small differences in free energy55,56 and provide no support for significant effects of isotopic substitutions upon ion structure. As the changes due to 14N/15N or 12C/13C substitutions are expected to be significantly smaller, structural changes due to isotope substitutions are thus conventionally considered to result in extremely small and undetectable impacts on Ω.57 Similarly, we consider it very unlikely that the isotopic substitutions have any significant impact on ion charge localization.
Our analysis of these data lead us to hypothesize that the observed relative mobility shifts are not due to the very small structural differences expected for the isotopologues, but arise primarily from a combination of the expected contribution due to their reduced mass differences and a second contribution previously unrecognized due to the changes in the ion center-of-mass (CoM). A CoM contribution has been speculated as a possible cause for differential mobility compensation voltage shifts for isotopomers in high field regime FAIMS measurements,28 but where the combined low and high field contributions and an uncertain degree of ion heating in FAIMS also challenge interpretation.1,25-28
A role of the CoM suggests a structural origin related to ion rotation and, more specifically, differences in collision frequency and momentum transfer due to the ion rotation and moment of inertia. While present computational methods do not explicitly include ion rotation in modeling of collisions for Ω calculations, the potential role of thermal excitation of ion rotation in ion mobility was considered two decades ago using simulations and suggested a significant contribution to Ω compared to static ions.58 One physical explanation for such a contribution is that an ion that is asymmetrical in any dimension (i.e., other than a perfect sphere with the CoM located at its exact center) will sweep out a larger volume as it rotates, providing a higher collision probability and differences in momentum transfer compared to the ion without rotation. Thus, a change in the ion CoM due to isotopic substitutions, and therefore the ion’s center of rotation, could be manifested by shifts in Ω that may be measurable given the increased Rp utilized in this work.
The [Arg+H]+ isotopologue CoM shift vectors have magnitudes in the range of 0.03 to 0.08 Å and are shown in Figure 6 for a widely accepted ground state structure.55,56,59 Since we can calculate the magnitude of the reduced mass contribution, the magnitude of the mobility shift due to a putative change in the CoM can be estimated by the deviation from this contribution (Figure 5). We note that the magnitude of the mobility scale given in Figure 5 has uncertainty due to our assumption that the mobility difference for the Arg+H+ light and 13C615N4 isotopologue “calibrants” is only due to their reduced mass differences, as discussed earlier. We note that while the absolute magnitudes of the mobility shifts will also depend on the ion rotational excitation, gas temperature and pressure, collision gas, etc., the relative magnitudes of based upon use of [Arg+H]+ light and 13C615N4 isotopologue calibrants are expected to be robust in the absence of sufficient heating that might induce ion structural changes. On this basis, a comparison of Figures 5 and 6 shows that the shift for the 15N4 isotopologue is largely opposite in direction to those for 13C6 and 13C615N42H7. Thus, we infer that the CoM changes for the 13C6 and 13C615N42H7 isotopologues move the CoM closer to a location where Ω would be minimized. Consistent with this, the [Arg+H]+ 15N4 isotopologue CoM change is in a largely opposite direction and leads to a larger Ω (Figure 5 and Supporting Information). Also consistent with this hypothesis is the observation that the Ω shift for the 13C615N42H7 isotopologue in N2 (0.0006 ± 0.0002) is in the same direction as in He and that the relative magnitude of this shift in He (0.0008 ± 0.0001) is comparable (within measurement error), even though the relative shift in N2 is smaller, due to the nearly 6-fold greater reduced mass contribution (Figure 5). We also note that the magnitudes as well as the directions of the experimental Ω shifts inferred from Figure 5 (after correction of mobilities for the reduced mass contributions) for arginine 15N4 and 13C6 isotopologues in helium are consistent with the calculated CoM changes, within measurement uncertainty. In particular, the directions of the observed helium relative mobility shifts (Ω) for the 13C6 as well 13C615N42H7 isotopologues are consistent with the directions of the vectors representing the CoM shifts (Supporting Information).
Figure 6.

Ground state structure of protonated arginine with labeled heavy mass substituted atoms (right), with CoM indicated by the blue dot. Inset (left) shows vectors representing the isotopologue CoM changes relative to the light isotopologue.
The CoM deviations are evident in the 2-dimensional projections of the protonated arginine structure, as well as in the angles of CoM shift vectors (Supporting Information). The directions of these vectors are consistent with the larger Ω of arginine 15N4 with respect to arginine 13C615N4 (one of the calibrants). Their angles are similar with respect to the X and Z axes but greatly differ with respect to the Y axis in threedimensional coordinates (Supporting Information). We emphasize that the CoM shifts calculated here are based on a single assumed structure, and experimental measurements may reflect contributions from additional unresolved ion conformers. However, the ensemble of lowest energy structures calculated and measured by IRMPD action spectroscopy60 involves the gas phase formation of a stable heterocycle for arginine. Thus, the NH2 group of the arginine side chain is the only peripheral moiety that is affected by isotopic substitution. The other peripheral moiety, the OH group, is not affected by the isotopic substitutions. Therefore, we expect that a change in the CoM to the periphery of the structure (i.e., substitution of 15N for 14N) would increase Ω and reduce K. Additionally, a change of the CoM due to the ring (i.e., from 12C to 13C and 1H to 2H) would similarly be expected to result in a Ω decrease. These expectations are consistent with the observed shifts in K and the approximate direction and magnitude of CoM changes.
Thus, the ion structure and the mass asymmetry impact of CoM changes appear to be correlated with the direction of the ion mobility shifts, as well as their magnitudes. Although our experimental shifts appear to be correlated with the CoM changes, our calculations assume that the previously calculated ground state structure of protonated arginine accurately reflects the protonated arginine studied under our experimental conditions. While we have not studied arginine isotopomers here, we emphasize that the use of isotopologues allows for the precise measurement of very small mobility shifts and for the role of the mass distribution shifts upon Ω to be determined after correction for the reduced mass contribution.
Isotopomer Separations Based on Mobility Shifts Arising from Ion CoM Changes.
In order to further explore the contributions of CoM vs those from the reduced mass contribution, as observed for the naturally occurring isotopologues as well as the arginine isotopologue mixture, we studied a mixture of six isotopomers: the tandem mass tag (iodo-TMT) −126 to −131 reagents.61 These compounds only differ in the locations of their heavy atom substitutions (Figure 7) and offer the benefit of facile dissociation after separation to provide discrete “reporter ions” for their identification. As noted earlier, while isotopomer separations have been previously reported with FAIMS based on their differences in mobility at both high and low electric fields,27,28 at low fields it has been believed that such isotopic substitutions would not result in measurable changes to the ion’s Ω.57 Figure 7 shows the overlaid peaks (based upon their respective reporter ions produced from fragmentation of the protonated parent ions) for the mixture of six iodo-TMT isotopomers after a 1354.5 m SLIM SUPER IMS separation in nitrogen drift gas. Also shown in Figure 7 is the 2-D IMS-MS plot for the reporter ions after separation and activation of this isotopomer mixture, illustrating how the reporter ion signal was extracted to construct the arrival time distribution. Also shown are the isotopomer CoM shifts for the calculated 3D structure used for the present CoM change calculations (a single low energy conformation, akin to our assumption for arginine). It is observed that as the labeled atoms are moved further away from the ion CoM (toward the ring), the isotopomers shift to lower mobility (i.e., later arriving, and larger Ω), consistent with our hypothesis; see the CoM vectors (Figure 7) having magnitudes ranging from 0.01 to 0.04 Å. We note that the evident pairing of isotopomers (i.e., 126/127, 128/129, and 130/131) can be attributed to the smaller CoM change 15N from the amide to the piperidine for each pair (~0.007 Å), and the larger shifts correspond to the larger CoM changes for pairs of carbon atoms (~0.02 and 0.04 Å, respectively).
These results provide the first observation of isotopomer separations by IMS in the low field regime. Since there is no present basis to establish an absolute mobility scale, we used a normalized separation scale (Table 3) with uncertainties based on averaged triplicate measurements within one standard deviation. It is important to note that in all our measurements the arrival time order was never observed to change, even for the most closely spaced isotopomers (e.g., iodo-TMT-128 and −129), supporting the robustness of the measurements. The greatest resolution achieved among isotopomers was ~0.2 between iodo-TMT 126 and 131. We plan to explore additional sets of isotopomers from other molecule classes (e.g., peptides or carbohydrates) as well as developing new computational approaches to help calculate CoM shifts and thus help guide analyte selection.
CONCLUSIONS
Herein, we have experimentally confirmed the expectation20 that the increased Rp provided by SLIM SUPER IMS enables the separation of the naturally occurring isotopic peaks due to changes in reduced mass of the ion–molecule collision partners, as described by the Mason–Schamp relationship (eq 1). Since [M+1] and [M+2] species provide mixtures of (e.g., TAA ion) isotopomers, while the parent monoisotopic ion [M] is a single species, our observation of increased peak width for [M+1] as compared to [M] (Figure 3) suggests the possibility of variations for the [M+1] isotopomer ion mobilities.
To explore this further, we studied the separation of various heavy-atom substituted protonated arginine isotopologues, where deviations were observed from their theoretical relative mobilities (and thus manifested as shifts in Ω), particularly in helium, where reduced mass contributions are minimized. The impacts of such isotopic changes upon Ω have not been previously observed and are now open to study using our ultrahigh resolution SLIM SUPER IMS platform. Upon correction for the reduced mass contributions and consideration of the likely ion structures, we found that these mass distribution-induced mobility shifts appeared to be correlated with changes to CoM of these arginine isotopologues.
To further test this hypothesis, we investigated the SLIM SUPER IMS separation of a mixture of six iodo-TMT isotopomers (i.e., where there is no change in reduced mass) and observed clear mobility shifts. To reiterate, fragmentation of the protonated parent ions was performed after IMS separation to yield nonisobaric fragment ions that were used to construct the arrival time distributions in Figure 7. The data also indicated that a change in the ion CoM away from the center of the ion structure decreased its mobility, consistent with our hypothesis.
Thus, these observed mobility shifts appear to reflect details of the ion structure and likely arise due to ion rotation about the changed CoM impacting collision frequency and overall momentum transfer. While further confirmation is needed, these observations suggest the potential for the use of such relative mobility shift information to evaluate potential computationally generated structures. The possibility that such studies could be conducted without the (problematic) requirement of greatly improved accuracy for Ω from, e.g., trajectory calculations, highlights the potential for new approaches for ion structural characterization.
While we cannot exclude other potentially significant contributions, the observed isotopologue mobility shifts in the present work appear largely attributable to the combination of changes in collision partner reduced mass and changes to the ion CoM. To further explore these shifts, we are evaluating approaches for further improving their precision, as well as computational approaches that also provide better precision to benefit their interpretation.
Finally, we speculate that increasingly high precision of relative mobility shift measurements for isotopologues and/or isotopomers can potentially provide important ion structural constraints when used in conjunction with equally high-quality computation of relative ion mobilities. In this case, each isotopologue’s or isotopomers shift could provide an additional and unique structural constraint and contribute to the refinement of the ion’s structure. We anticipate that such an approach may help improve the accuracy of collision cross section calculations, thus leading to better insight into how CoM shift can affect the resolution of various isomers or analytes with very similar mobilities. We also envision potential applicability to large biomolecular assemblies in the gas phase, such as those studied by Robinson and co-workers by IMS-MS,62 using an array of labeling of complex subelements (e.g., individual heavy labeled proteins) of the structures. There is also the potential for utilizing any compound’s naturally occurring isotopologues and isotopomers for such purposes with sufficient Rp and measurement dynamic range, which are significant attributes of SLIM IMS.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Frank Turecek (University of Washington), Prof. David Clemmer (Indiana University), Prof. Matt Bush (University of Washington), Dr. Niranjan Govind (Pacific Northwest National Laboratory), and Dr. Karol Kowalski (Pacific Northwest National Laboratory) for useful discussions. This research was supported by the National Institute of General Medical Sciences (P41 GM103493), the National Cancer Institute (R33 CA217699), and funding from the Pacific Northwest National Laboratory Quickstarter program. Additional support was provided by National Institute of Environmental Health Sciences grant U2CES030170. Experiments were conducted at the W. R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a DOE national scientific user facility at the Pacific Northwest National Laboratory (PNNL). PNNL is operated by Battelle under contract DE-AC05-76RL0 1830 for the DOE.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b02808.
Results for the MRFA peptide ions in helium. Comparison of ion injection methods. Calculations of the electric fields experienced by the ions, ion mobility shifts for the arginine isotopologues, and center of mass for the arginine isotopologues and iodo-TMT isotopomers. (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Pathak P; Baird MA; Shvartsburg AA Anal. Chem 2018, 90, 9410–9417. [DOI] [PubMed] [Google Scholar]
- (2).Chahrour O; Cobice D; Malone JJ Pharm. Biomed.Anal 2015, 113, 2–20. [DOI] [PubMed] [Google Scholar]
- (3).Lehmann WD Mass Spectrom. Rev 2017, 36, 58–85. [DOI] [PubMed] [Google Scholar]
- (4).Eliuk S; Makarov A Annu. Rev. Anal. Chem 2015, 8, 61–80. [DOI] [PubMed] [Google Scholar]
- (5).Gabelica V; Marklund E Curr. Opin. Chem. Biol 2018, 42, 51–59. [DOI] [PubMed] [Google Scholar]
- (6).Nagy G; Veliikovic D; Chu RK; Carrell AA; Weston DJ; Ibrahim YM; Anderton CR; Smith RD Chem. Commun 2019, 55, 306–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chouinard CD; Nagy G; Webb IK; Garimella SVB; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2018, 90, 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Nagy G; Attah IK; Garimella SVB; Tang K; Ibrahim YM; Baker ES; Smith RD Chem. Commun 2018, 54, 11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nagy G; Chouinard CD; Attah IK; Webb IK; Garimella SVB; Ibrahim YM; Baker ES; Smith RD Electrophoresis 2018, 39, 3148–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chouinard CD; Nagy G; Webb IK; Shi T; Baker ES; Prost SA; Liu T; Ibrahim YM; Smith RD Anal. Chem 2018, 90, 10889–10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wojcik R; Webb IK; Deng L; Garimella SV; Prost SA; Ibrahim YM; Baker ES; Smith RD Int. J. Mol. Sci 2017, 18, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mairinger T; Causon TJ; Hann S Curr. Opin. Chem. Biol 2018, 42, 9–15. [DOI] [PubMed] [Google Scholar]
- (13).Chen Z; Glover MS; Li L Curr. Opin. Chem. Biol 2018, 42, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ben-Nissan G; Sharon M Curr. Opin. Chem. Biol 2018, 42,25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hinz C; Liggi S; Griffin JL Curr. Opin. Chem. Biol 2018, 42, 42–50. [DOI] [PubMed] [Google Scholar]
- (16).Zhang X; Quinn K; Cruickshank-Quinn C; Reisdorph R; Reisdorph N Curr. Opin. Chem. Biol 2018, 42, 60–66. [DOI] [PubMed] [Google Scholar]
- (17).Zheng X; Smith RD; Baker ES Curr. Opin. Chem. Biol 2018, 42, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stuchfield D; Barran P Curr. Opin. Chem. Biol 2018, 42, 177–185. [DOI] [PubMed] [Google Scholar]
- (19).Shvartsburg AA; Smith RD Anal. Chem 2008, 80, 9689–9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Valentine SJ; Clemmer DE Anal. Chem 2009, 81, 5876–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kanu AB; Gribb MM; Hill HH Anal. Chem 2008, 80, 6610–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Siems WF; Wu C; Tarver EE; Hill HH Jr.; Larsen PR; McMinn DG Anal. Chem 1994, 66, 4195–4201. [Google Scholar]
- (23).Kirk AT; Bohnhorst A; Raddatz C-R; Allers M; Zimmermann S Anal. Bioanal. Chem 2019, DOI: 10.1007/s00216-019-01807-0 [DOI] [PubMed] [Google Scholar]
- (24).Kirk AT; Raddatz C-R; Zimmermann S Anal. Chem 2017, 89, 1509–1515. [DOI] [PubMed] [Google Scholar]
- (25).Baird MA; Pathak P; Shvartsburg AA Anal. Chem 2019, 91, 3687. [DOI] [PubMed] [Google Scholar]
- (26).Kaszycki JL; Baird MA; Shvartsburg AA Anal. Chem 2018, 90, 669–673. [DOI] [PubMed] [Google Scholar]
- (27).Kaszycki JL; Bowman AP; Shvartsburg AA J. Am. Soc. Mass Spectrom 2016, 27, 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shvartsburg AA; Clemmer DE; Smith RD Anal. Chem 2010, 82, 8047–8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Robinson EW; Shvartsburg AA; Tang K; Smith RD Anal. Chem 2008, 80, 7508–7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Deng L; Webb IK; Garimella SVB; Hamid AM; Zheng X; Norheim RV; Prost SA; Anderson GA; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2017, 89, 4628–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Deng L; Garimella SVB; Hamid AM; Webb IK; Attah IK; Norheim RV; Prost SA; Zheng X; Sandoval JA; Baker ES; Ibrahim YM; Smith RD Anal. Chem 2017, 89, 6432–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ibrahim YM; Hamid AM; Deng L; Garimella SV; Webb IK; Baker ES; Smith RD Analyst 2017, 142, 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Garimella SVB; Webb IK; Prabhakaran A; Attah IK; Ibrahim YM; Smith RD J.Am. Soc. Mass Spectrom 2017, 28, 1442–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Deng L; Ibrahim YM; Baker ES; Aly NA; Hamid AM; Xhang X; Zheng X; Garimella SVB; Webb IK; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Smith RD ChemistrySelect 2016, 1, 2396–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Deng L; Ibrahim YM; Hamid AM; Garimella SV; Webb IK; Zheng X; Prost SA; Sandoval JA; Norheim RV; Anderson GA; Tolmachev AV; Baker ES; Smith RD Anal. Chem 2016, 88, 8957–8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Garimella SV; Hamid AM; Deng L; Ibrahim YM; Webb IK; Baker ES; Prost SA; Norheim RV; Anderson GA; Smith RD Anal. Chem 2016, 88, 11877–11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hamid AM; Garimella SVB; Ibrahim YM; Deng L; Zheng X; Webb IK; Anderson GA; Prost SA; Norheim RV; Tolmachev AV; Baker ES; Smith RD Anal. Chem 2016, 88, 8949–8956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Colby SM; Thomas DG; Nunez JR; Baxter DJ; Glaesemann KR; Brown JM; Pirrung MA; Govind N; Teeguarden JG; Metz TO; Renslow RS Anal. Chem 2019, 91, 4346–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Halgren TA J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- (40).Mason EA; McDaniel EW Transport Properties of Ions in Gases; Wiley & Sons: New York, 2005. [Google Scholar]
- (41).Allen SJ; Bush MF J. Am. Soc. Mass Spectrom 2016, 27, 2054–2063. [DOI] [PubMed] [Google Scholar]
- (42).Davidson KL; Bush MF Anal. Chem 2017, 89, 2017–2023. [DOI] [PubMed] [Google Scholar]
- (43).Shvartsburg AA Anal. Chem 2014, 86, 10608–10615. [DOI] [PubMed] [Google Scholar]
- (44).Knapman TW; Berryman JT; Campuzano I; Harris SA; Ashcroft AE Int. J. Mass Spectrom 2010, 298, 17–23. [Google Scholar]
- (45).Morsa D; Gabelica V. r.; De Pauw E Anal. Chem 2011, 83, 5775–5782. [DOI] [PubMed] [Google Scholar]
- (46).Merenbloom SI; Flick TG; Williams ER J. Am. Soc. Mass Spectrom 2012, 23, 553–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hinnenkamp V; Klein J; Meckelmann SW; Balsaa P; Schmidt TC; Schmitz OJ Anal. Chem 2018, 90, 12042–12050. [DOI] [PubMed] [Google Scholar]
- (48).Morsa D; Gabelica V. r.; De Pauw E J. Am. Soc. Mass Spectrom 2014, 25, 1384–1393. [DOI] [PubMed] [Google Scholar]
- (49).Stow SM; Causon TJ; Zheng X; Kurulugama RT; Mairinger T; May JC; Rennie EE; Baker ES; Smith RD; McLean JA; Hann S; Fjeldsted JC Anal. Chem 2017, 89, 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lee TT; Yeung ES Anal. Chem 1991, 63, 2842–2848. [Google Scholar]
- (51).Nowak PM; Wozniakiewicz M; Gladysz M; Janus M; Koscielniak P Anal. Bioanal. Chem 2017, 409, 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Biernacki SW; Clerjaud B Phys. Rev. B: Condens. Matter Mater. Phys 2001, 63, No. 075201. [Google Scholar]
- (53).Perrin CL; Burke KD J. Phys. Chem. A 2015, 119, 5009–5018. [DOI] [PubMed] [Google Scholar]
- (54).Perrin CL; Dong YJ Am. Chem. Soc 2007, 129, 4490–4497. [DOI] [PubMed] [Google Scholar]
- (55).Bush MF; O’Brien JT; Prell JS; Saykally RJ; Williams ER J. Am. Chem. Soc 2007, 129, 1612–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rak J; Skurski P; Simons J; Gutowski MJ Am. Chem. Soc 2001, 123, 11695–11707. [DOI] [PubMed] [Google Scholar]
- (57).Kindy JM; Taraszka JA; Regnier FE; Clemmer DE Anal. Chem 2002, 74, 950–958. [DOI] [PubMed] [Google Scholar]
- (58).Shvartsburg AA; Mashkevich SV; Siu KWM J. Phys. Chem. A 2000, 104, 9448–9453. [Google Scholar]
- (59).Ling S; Yu W; Huang Z; Lin Z; Haranczyk M; Gutowski MJ Phys. Chem. A 2006, 110, 12282–12291. [DOI] [PubMed] [Google Scholar]
- (60).Forbes MW; Bush MF; Polfer NC; Oomens J; Dunbar RC; Williams ER; Jockusch RA J. Phys. Chem. A 2007, 111, 11759–11770. [DOI] [PubMed] [Google Scholar]
- (61).Thompson A; Schafer J. r.; Kuhn K; Kienle S; Schwarz J; Schmidt G. n.; Neumann T; Hamon C Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- (62).Sharon M; Robinson CV Annu. Rev. Biochem 2007, 76, 167–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.