

1. Introduction
Catecholamines, including dopamine and norepinephrine, are biological amines that display important roles in the homeostasis of several biological functions. Within the central nervous system (CNS), dopamine and norepinephrine play major roles as neurotransmitters, taking part in the proper functioning of critical neuronal pathways linked to motor function, cognition, emotion, memory and endocrine modulation. Impairments in catecholaminergic neurotransmission are implicated in neurologic and neuropsychiatric disorders. Moreover, catecholaminergic neurons represent important targets that mediate the deleterious effects of neurotoxic agents, such as metals and metalloids, pesticides, among others. Methylmercury (MeHg) is an organic mercury compound ubiquitously present in nature due to both natural and anthropogenic sources. MeHg is produced mainly as a consequence of the methylation of inorganic mercury in the aquatic environment (in a reaction catalyzed by reducing bacteria) and is biomagnified within the aquatic food chain, reaching high levels in predatory fish. Human populations, exposed to MeHg, mainly due to the ingestion of predatory fish, may develop neurological symptoms. Even though the glutamatergic neurotransmitter system has been noted as a major target mediating MeHg-induced neurotoxicity, several studies also affirm that the catecholaminergic system is involved in several toxic effects of MeHg. This chapter will focus on the toxic effects of MeHg toward the catecholaminergic system, with particular emphasis on the potential neurotoxic effects and neurological consequences resulting from MeHg exposure, as well as related mechanisms.
2. The catecholaminergic neurotransmitter system: general aspects
Catecholamines, including dopamine and norepinephrine (also called noradrenaline), are biological amines derived from the amino acid tyrosine. In order to synthesize dopamine, tyrosine is first converted to dihydroxyphenylalanine (DOPA) in a reaction catalyzed by tyrosine-hydroxylase (TH). DOPA is then decarboxylated by DOPA-decarboxylase to yield dopamine. The generation of norepinephrine from dopamine involves the hydroxylation catalyzed by dopamine β-hydroxylase. Figure 1 depicts the synthesis of dopamine and norepinephrine from the non-essential amino acid tyrosine.
Figure 1: Dopamine and norepinephrine biosynthesis.
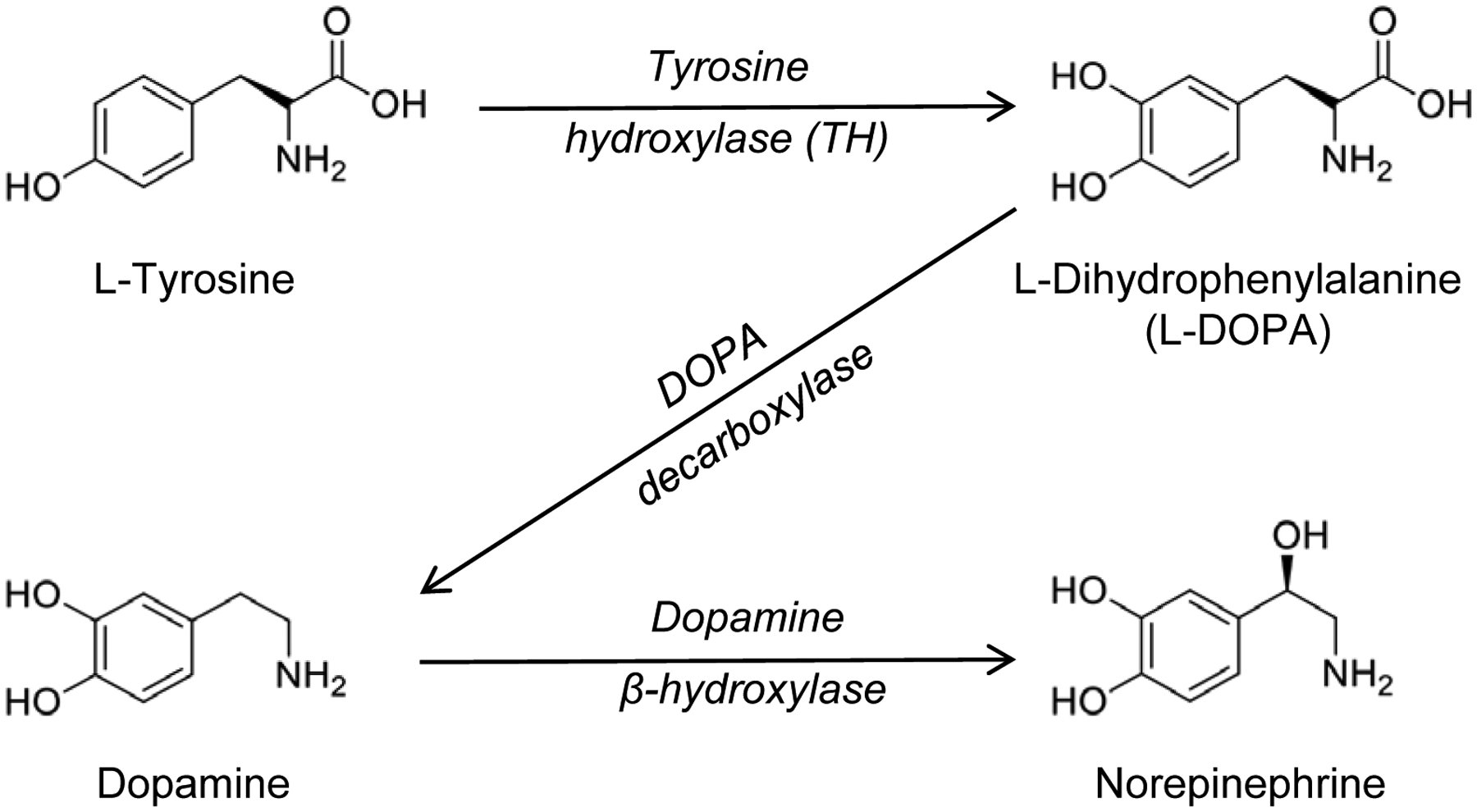
Tyrosine, a non-essential amino acid, is first converted to dihydroxyphenylalanine (DOPA) in a reaction catalyzed by tyrosine-hydroxylase (TH). DOPA is then decarboxylated by DOPA-decarboxylase to yield dopamine. The generation of norepinephrine from dopamine involves the hydroxylation catalyzed by dopamine β-hydroxylase, which is commonly used as a marker for noradrenergic neurons.
Within the central nervous system (CNS), dopamine and norepinephrine act as neurotransmitters; the cell groups producing catecholamines are localized in discrete brain regions and project their axon terminals to a wide range of target areas that play important roles in CNS functions (Kobayashi, 2001). Dopaminergic cells are mainly localized in the substantia nigra (A9 cell group) and ventral tegmental area (A10 cell group) (Lindvall et al., 1983). Neurons from the A9 group innervate the caudate-putamen to form the nigrostriatal pathway, which has a pivotal role in motor control (Gerfen,1992). Neurons from the A10 group project their fibers to the nucleus accumbens, amygdala, and prefrontal cortex, forming the mesocorticolimbic pathway, which is involved in emotion, motivation, and memory formation (Le Moal and Simon, 1991). Dopaminergic neurons from the A11-A14 groups are present in the mediobasal region of the hypothalamus, projecting their fibers to the median eminence and pituitary gland (Moore and Lookingland, 1995), modulating pituitary gland function.
The norepinephrine system originates from the locus coeruleus (A6 cell group) and the lateral tegmental area in the brain stem (A1, A2, A5, and A7 cell groups) (Moore and Card, 1984). Neurons from the A6 group innervate the cerebral cortex, amygdala, hippocampus and thalamus, forming the dorsal norepinephrinergic pathway. The ventral norepinephrinergic pathway represents an additional way that innervates mainly the hypothalamus and septum. These neurons are implicated in both cognitive (attention, memory) and vegetative functions, such as neuroendocrine and autonomic regulation (Robbins and Everitt, 1995).
Several behavioral and pharmacological approaches have been used to understand brain functions mediated by catecholamines. Using a mutant phenotype of mice genetically impaired in dopamine or norepinephrine biosynthesis, Kobayashi and coworkers reported behavioral and physiologic roles of these two catecholamines in the CNS (Kobayashi, 2001). Using mutant mice defective in dopamine biosynthesis, in which the expression of TH in norepinephrinergic neurons was rescued with the dopamine β-hydroxylase gene promoter (functional in norepinephrinergic cells), the authors observed a significant reduction of dopamine in various encephalic structures (forebrain, midbrain, hindbrain and pituitary gland) compared with wild-type animals, whereas norepinephrine levels were normal. The dopamine-deficient mice displayed a significant reduction in spontaneous locomotor activity, exhibited cataleptic behavior and were insensitive to the methamphetamine treatment, which normally induces the hyperactivity of locomotion (Kobayashi, 2001). These observations, derived from studies with genetically modified mice, are in line with the crucial role of dopamine in motor function in humans (Ko and Strafella, 2012).
In addition to motor dysfunction, dopamine-deficient mice displayed defects in certain emotional learning paradigms, such as the active avoidance; these defects seem to be related to dopamine depletion in nucleus accumbens (McCullough et al, 1993). In agreement with these data from experimental studies with genetically modified mice, pharmacological studies have shown the crucial role of dopamine in memory and cognition. In an in vivo study using mice treated with the D2 dopamine receptor agonist quinpirole (administered into the ventral pallidum), Lenard et al. (2017) showed that the activation of the D2 dopamine receptors in the ventral pallidum facilitates memory consolidation as well as memory-retention in inhibitory avoidance paradigm. In humans, striatal dopamine D2 receptors play a critical role in enabling the flexible updating and manipulation of information in working memory (Dodds et al., 2009). However, the role of dopamine in memory seems to depend on the type of receptor involved, given that D₃ receptor blockade may enhance cognitive performance in healthy individuals and treat cognitive dysfunction in individuals with a neuropsychiatric disorder (Nakajima et al., 2013).
With respects to norepinephrine, genetic manipulation procedures were able to generate heterozygous mutant mice with reduced norepinephrine metabolism in the brain; the release level through repetitive high K+ stimulation was reduced in the mutant mice (56% of the wild-type level). These heterozygous mutant mice displayed deficits in three kinds of associative learning paradigms, including active avoidance, cued fear conditioning, and conditioned taste aversion (Kobayashi, 2001). Notably, treatment with desipramine, an inhibitor of norepinephrine uptake, recovered the memory deficits in the mutant mice, confirming that the central norepinephrine system plays a key role in long-term memory formation of conditioned learning. With respect to brain regions involved in long-term memory of conditioned learning, several behavioral studies have indicated that the associative learning require the amygdala and its linking pathways, including the cerebral cortex (Everitt et al, 1991; Yamamoto et al., 1995). In line with the aforementioned evidence, Veyrac et al. (2009) showed that labetalol, a mixed β- and α1-adrenoceptor antagonist, blocked the improve in short-term olfactory memory and neurogenesis in the olfactory bulb of mice subjected to the environmental enrichment. In addition, desipramine, an inhibitor of norepinephrine uptake, has shown to improve cognition/memory in both animals (Feltmann et al., 2015) and humans (Mokhber et al., 2014).
The aforementioned evidence indicates that both catecholamines play important roles in motor control, emotional learning and memory formation. In line with this, disruption of these neurotransmitter systems lead to motor and cognitive impairments, as discussed below.
3. Catecholamines in neurological and neuropsychiatric disorders
As already mentioned, catecholaminergic neurotransmission is involved in the regulation of variety of neurophysiological and behavioral processes, from relatively simple endocrine events (for instance, peripheral regulation of blood pressure) to complex mental activities (i.e., motivation, attention, learning, cognition, and thinking, etc.) (Nieoullon, 2002; Bromberg-Martin et al., 2010; Cools and D’Esposito, 2011; Johansen et al., 2011; Huys et al., 2014; Montes et al., 2015). Disruption in the normal neurophysiology of catecholaminergic system has been implicated in the etiology of important neuropsychiatric disorders (van Os & Kapur, 2009; Del Campo et al., 2011; Tritsch and Sabatini, 2012; Sigitova et al., 2017). In this context, a significant progress in our knowledge concerning the role of dopaminergic transmission in neuropsychiatry diseases was achieved by showing that the first generation of therapeutic antipsychotic agents (i.e., chlorpromazine, haloperidol), used to treat schizophrenia, had the ability of blocking dopaminergic receptors (Creese et al. 1976). Likewise, the reported relationship between dopamine depletion in the caudate nucleus of rodents and the appearance of Parkinson’s diseases (PD)-related motor symptoms was instrumental in linking dopaminergic transmission with the neuropathology of this neurodegenerative condition. The major symptoms of PD, such as tremor, rigidity, difficulty to initiate movement and postural hypotension are associated with dopamine depletion in the substantia nigra and other interconnected brain regions. Although the degeneration of dopaminergic cells is more pronounced in PD, noradrenergic neurotransmission plays also an important role in the modulation of PD symptoms (Barbeau, 1970; Teychenne et al., 1985).
Nowadays, the importance of catecholaminergic neurotransmission in different types of motor, cognitive and affective disorders has been validated (Nieoullon, 2002). The pivotal role of dopamine and norepinephrine in the regulation of normal behavior in experimental animals and humans and the crucial causal role of disruption of catecholaminergic neurotransmission in the etiology of neuropsychiatric disorders is now well-established. For instance, in addition to schizophrenia and PD, disturbances of catecholaminergic neurotransmission have been implicated in attention deficit hyperactivity disorders (ADHD) (Van Enkhuizen et al., 2015), bipolar disorders (Van Enkhuizen et al., 2015; Sigitova et al., 2017), depression (Hamon and Bilier, 2013), substance-use or addiction disorders (Ewing and Myers, 1985; Beaulieu and Gainetdinov, 2011; Fitzgerald, 2013; Huys et al., 2014), eating disorders (Kaye et al., 2013; Volkow et al., 2011; 2013), Alzheimer’s disease (Trillo et al. 2013), obsessive compulsive disorder (OCD) (Tritsch and Sabatini, 2012; Pauls et al., 2014), autism spectrum disorders (ASD) (Quaak et al., 2013; Nguyen et al., 2014), among others.
In the case of schizophrenia, the dopaminergic hyperactivity in the prefrontal cortex is thought to play a central role in the psychotic symptoms of this condition. Although both pre- and post-synaptic dopamine receptors located in different brain areas seem to be involved in schizophrenia (Nikolaus et al., 2014; Howes et al., 2015), particular attention has been given to the increase in the D2 dopamine receptors in the high-affinity state in schizophrenic patients (Beaulieu and Gainetdinov, 2011).
Alterations in dopaminergic neurotransmission can explain satisfactorily the symptomatology found in about 2/3 of schizophrenic patients, but the remaining cases cannot be attributed exclusively to dopaminergic hyperactivity. In fact, other neurotransmitter systems interact with the dopaminergic transmission to produce the typical psychotic (hallucination, disorganized speech, delusions, etc.) or negative symptoms (social withdraw, apathy, cognitive deficits, etc.) found in this neurological condition. For instance, changes in the glutamatergic neurotransmission have been implicated in the etiology of schizophrenia, particularly the N-methyl-D-aspartate (NMDA) receptor hypofunctioning in specific brain areas (Howes et al., 2015). Noradrenergic neurotransmission has also been suggested to contribute in the etiology of some cases of schizophrenia (Fitzgerald, 2014). Thus, as in the case of other neurological or psychiatry disorders, the disruption in the delicate interaction between different neurotransmitters seems to be more important than isolated changes in a single neurotransmitter system.
Factors that can be important to the emergence of neuropathological disorders are the exposure to stressful situations or neurotoxic agents during critical periods of brain development (Rice and Barone Jr., 2000; Andersen, 2003; Rakers et al., 2017). Indeed, the delicate ontogeny of the nervous system can be disrupted by different stressors or neurotoxic chemicals (Rice and Barone Jr., 2000; Andersen, 2003; Grandjean and Landrigan, 2014; Rakers et al., 2017) and the normal pre-natal and post-natal development of catecholaminergic neurotransmission systems can be permanently altered by environmental stressors or neurotoxicants (Spear, 2000; Cory-Slechta et al., 2008). Accordingly, the association between exposures to environmental or industrial neurotoxicants with neurological disorders has been advocated in the literature. For instance, the increase in the incidence of autism or autism spectrum disorder (ASD) has been attributed to mercury exposure during brain development (Mutter et al., 2005; Austin & Shandley, 2008; Kern et al., 2011; 2016; Sealey et al., 2016). However, the points of evidence supporting the link between mercury and ASD are highly questionable (Stellfeld et al., 2003; Taylor et al., 2014; Rossignol et al., 2014).
Exposure of experimental animals and humans to high levels of neurotoxic xenobiotics during critical periods of brain development have been consistently linked with permanent behavioral abnormalities later in life (Rice and Barone Jr.; Kari et al., 2016; Heyer and Meredith, 2017). In contrast, epidemiological data showing a potential causal relationship between exposures to low levels of neurotoxicants (for instance, MeHg, Hg, Pb, polychlorinated biphenyls (PCBs), organophosphates, pyrethroids, etc.) and the occurrence of behavioral abnormalities are scarce (Petersen et al. 2008). Despite of this, the available studies indicate that exposure to MeHg, lead and PCBs have to be avoided (Rice and Barone Jr. 2000; Kraft et al., 2017; Heyer and Meredith, 2017).
4. The catecholaminergic neurotransmitter system as a potential target of neurotoxicants and related consequences
Several effects resulting from exposures to neurotoxicants are linked to misbalances in the proper functioning of specific neurotransmitter systems. Both dopaminergic and noradrenergic neurotransmission have been implicated. Of note, some neurochemical, histological and behavioral effects resulting from exposures to neurotoxicants have been useful to understand mechanisms concerning the role of such toxicants toward a specific neurotransmitter system. In this section, we will briefly introduce two noxious agents, paraquat (PQ) and N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride (DSP-4), which are known to cause neurotoxicity in the dopaminergic and noradrenergic neurotransmitter systems, respectively. Classical dopaminergic toxins, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), are not discussed here because of its nonspecific toxic roles; they act on both dopaminergic and noradrenergic neurotransmitter systems (Miyoshi et al., 1988; Archer and Fredriksson, 2006; Szot et al., 2012), making difficult to discriminate between specific outcomes.
Paraquat
(PQ; N,N′-dimethyl-4–4′-bipiridinium) is an herbicide widely used in several countries and its toxicity has been reported since approximately 50 years ago (Clark et al., 1966). Even though initial studies with PQ have reported its effects on lung, liver, and kidney, significant damage to the brain was seen in individuals who died from PQ intoxication (Grant et al., 1980). The molecular mechanisms mediating PQ-induced dopaminergic neurotoxicity seem to involve a redox cycling that generates superoxide anion (Day et al., 1999), leading to oxidative stress and neurodegeneration.
Experimental evidence has reported that PQ possesses marked neurotoxicity and induces degeneration of the rat nigrostriatal dopaminergic system (Liou et al., 1996). In addition, elevated levels of α-synuclein have been reported in the frontal cortex and ventral midbrain, as well as α-synuclein-positive inclusions in substantia nigra neurons of mice treated with PQ (Manning-Bog et al., 2002). The occurrence of dopaminergic neurotoxicity and α-synuclein up-regulation and aggregation suggests that the experimental PQ model may serve a useful tool to study Parkinson’s disease-like disorders (i.e., nigral cell loss and synuclein pathology). Notably, PQ exposure has been significantly associated with Parkinson’s disease (2.2-fold increase in risk for ever having used the chemical) (Pezzoli & Cereda., 2013).
It is noteworthy that experimental animals exposed to PQ have shown both motor (Park et al., 2005; Kang et al., 2010) and (Ait-Bali et al., 2016; Li et al., 2016) cognitive impairments, consistent with the aforementioned roles of dopamine in motor control and emotional learning (presented in item 2). Even though PQ exposure has been associated with increased risk for Parkinson’s disease (Pezzoli and Cereda., 2013), epidemiological studies on the effects of PQ toward motor and cognitive performance in humans are limited.
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine hydrochloride
(DSP-4) is a neurotoxin that selectively damages the locus coeruleus noradrenergic system. The first studies with DSP-4 date from approximately 4 decades ago, when Ross & Renyl (1976) found that DSP-4 inhibited the active uptake of norepinephrine in mouse cortical cerebral slices. In the same year, Ross (1976) observed that exposure of rats to DSP-4 decreased dopamine β-hydroxylase (DBH) activity in the brain, suggesting the loss of noradrenergic neurons. By observing that the inhibition of adrenaline uptake did not per se cause the fall in DBH activity, this author concluded that the binding of DSP-4 to the neuronal membrane was responsible for a degenerative process. This hypothesis is reinforced by the observation that desipramine, an inhibitor of norepinephrine uptake, prevented the decrease in brain DBH activity.
Because of its relative specific effects toward the noradrenergic system (serotoninergic and dopaminergic nerves are only slightly or not at all affected), DSP-4 has been an important toxic agent to understand the roles of the norepinephrine in the central nervous system (for a review, see Ross and Stenfors, 2015). DSP-4 readily passes the blood-brain barrier and cyclizes to a reactive aziridinium derivative that is accumulated into the noradrenergic nerve terminals via the noradrenaline transporter. DSP-4 exposure (at the dose 50 mg/kg i.p. in rodents) causes a rapid and long-lasting loss of noradrenaline and a slower decrease in the dopamine-β-hydroxylase enzyme activity and immunoreactivity in the regions innervated from locus coeruleus (A6 cell group). The neurotoxic effect is counteracted by pretreatment with noradrenaline uptake inhibitors (i.e. desipramine). MAO-B inhibitors of the N-propargylamine type, such as selegiline, also counteract the DSP4-induced neurotoxicity.
Taking advantage of DSP-4 as a specific toxin for the noradrenergic neurotransmitter system, it was possible to establish critical roles of norepinephrine for several physiological functions, such as learning, memory, pain and emotion, among others. By using a protocol of DSP-4 exposure in rats, Ogren and coworkers (1980) showed that DSP-4-induced degeneration of the locus coeruleus noradrenergic system was paralleled by a marked impaired avoidance acquisition, indicating that the locus coeruleus noradrenergic system may play a role in aversive learning (Ogren et al., 1980). Consistent with this observation, Archer et al., (1984), studying the effects of DSP-4 on active avoidance acquisition in rats, observed that DSP-4 significantly impaired avoidance learning and that pretreatment with desipramine antagonized the active avoidance impairment. In a study with rats, Liang (1998) observed that intra-amygdala infusion of DSP-4 impaired retention in the inhibitory avoidance task, suggesting the involvement of norepinephrine but not serotonin in memory storage processing in the applied experimental model.
The role of the noradrenergic system in modulating nociception was also proposed from studies using DSP-4. Zhong et al., (1985) showed that intrathecal injections of DSP-4 selectively depleted spinal noradrenaline and attenuated morphine analgesia in rats. In addition, by lesioning locus coeruleus noradrenergic neurons with DSP-4 in rats, Kudo et al (2010) proposed that central neuropathic pain may be facilitated by DSP-4 depleting locus coeruleus noradrenergic neurons.
Based on the aforementioned studies with the toxic agent DSP-4, the noradrenergic neurotransmitter system was invoked to play a critical role in modulating learning and pain. In addition, it is noteworthy that experimental studies with DSP-4 have also pointed to important roles of the noradrenergic neurotransmitter system in modulating mesolimbic dopamine transmission (Lategan et al., 1992) and emotionality (Harro et al., 1995).
5. Methylmercury: general aspects and major mechanisms of neurotoxicity
The mercury atom covalently bound to a carbon from the methyl group forms a reactive and soft electrophile center in the MeHg molecule. In fact, MeHg is a soft electrophile that has high affinity for soft nucleophiles (Pearson, 1963). In the biological scenario, we can found two types of physiologically relevant soft nucleophile centers, i.e., the sulfhydryl (-SH or thiol) or the selenohydryl (-SeH or selenol) groups. Thiol groups are found in a few endogenous low molecular mass molecules, such as cysteine and reduced and oxidized glutathione (GSH and GSSG), as well as in thousands of high molecular mass macromolecules (thiol-containing-proteins) (Table 1; Miseta et al. 2000; Go & Jones, 2013). For instance, the cysteinyl (Cys) proteome revealed that approximately 200,000 Cys residues are encoded in the human genome (Go & Jones, 2013). Evolutionary studies have indicated that cysteine was first selected to be part of the specific motif –C-(X)2-C-, which is found in metal binding proteins and in oxidoreductases.
Table 1 –
Relative quantity of molecules of biological significance containing the soft nucleophile groups thiol (-SH) or selenol (-SeH). These functional groups are the main targets of methylmercury (MeHg) and other soft electrophiles.
| High molecular mass thiol molecules (thiol-containing proteins) | |
| Occurrence | |
| –Cysteinyl residues (Cys) | in thousands of proteins |
| High molecular mass selenol molecules (selenoproteins) | |
| Occurrence | |
| –Selenocysteinyl residues (Sec) | in few number of proteins |
| Low molecular mass thiol molecules (-SH) | |
| Occurrence | |
| Endogenous | Cysteine glutathione (GSH) |
| exogenous (synthetic) | 2,3 dimercaptopropanol (BAL) 2,3 dimercaptosuccinic acid (DMSA) 2,3 dimercapto sulfonic acid (DMPS) |
| Low molecular mass selenol molecules (-SeH ) | |
| occurrence | |
| Endogenous | None |
| exogenous (synthetic) | Unstable |
According to Miseta et al. (2000), in archea, the ancient motif containing the two vicinal cysteine residues was evolutionary selected to binding metals. Here, possibly those metals with physiological function (i.e., Zn2+, Cu2+, etc).
Glutathione (GSH and GSSG) and thioredoxin [Trx(SH)2] systems have crucial roles in maintain the cellular homeostasis of redox sensitive cysteinyl- or thiol-containing proteins (Go and Jones, 2013). In fact, Trx and GSH systems have different role as regulators of disulfide-thiol redox equilibrium, because they regulate the redox state of distinct classes of thio-disulfide-containing proteins (Go et al. 2015; Jones 2015). Of particular toxicological significance, MeHg can target both antioxidant systems and their disruption can have profound impact in the neurotoxicity of MeHg (Carvalho et al. 2011; Farina et al. 2011b, 2013; Branco et al. 2017).
Low molecular mass molecules containing thiol groups have been synthesized for therapeutic purposes (Longcope et al., 1946; Vilensky and Redman, 2003; Blanusa et al. 2005). Table 1 shows some synthetic thiol molecules used in the treatment of intoxication with mercurials, lead and other toxic elements. In contrast, the existence of low mass selenol-containing biomolecules has not yet been clearly demonstrated and the occurrence of stable low mass selenol molecules under physiological conditions are improbable.
The occurrence of the selenol group is much more restricted than the thiol center and the –SeH group is found only a few number of selenoproteins (Hatfield et al. 2014; Rocha et al. 2017; Table 1). For instance, the mammalian genome codifies 20–30 selenoproteins, including important oxidoreductases involved in the metabolism of reactive species (e.g., glutathione peroxidase and thioredoxin reductase isoforms, Hatfield et al., 2014).
The affinity of MeHg for the –SH is high and the formation constant of RSH + MeHg →RS-HgMe (i.e. the formation of –Hg-S- bond ) is very high (~15–20; Rabenstein, 1978a). This implies that, under physiological conditions, MeHg will be not found in “a free form” or bound to other abundant ligands, such as Cl− (chloride, e.g., in stomach) or OH−. As a corollary, we may infer that the chemistry, biochemistry and toxicology of MeHg will be dictated by the chemistry of thiol ←→MeHg-sulfide exchange (Figure 2 and 3, Rabenstein 1978a,b; Rabenstein and Fairhurst, 1975; Rabenstein et al., 1974; 1982; Rabenstein and Reid, 1984; Farina et al. 2011; Dórea et al. 2013). On the other hand, MeHg can also coordinate with the selenol group of selenoproteins (Arnold et al., 1986; Rabenstein et al. 1986; and for review, see Farina et al. 2011). Of particular toxicological significance, the affinity of MeHg for the selenohydryl (selenol) groups is much higher than that of for the sulfhydryl (thiol) group (Sugiura et al., 1976; 1978). However, the formation constant of RSeH + MeHg →RSeHgMe has rarely been calculated chemically (Arnold et al., 1986). Essentially, the affinity of the –SeH for MeHg is too high, which makes the determination of the constant difficult. Recent in silico studies have confirmed the superior affinity of the selenol group for electrophilic mercury forms (e.g., Hg2+ and MeHg) over the analog thiol molecules (Asaduzzaman and Schreckenbach, 2011; Asaduzzaman et al., 2009).
Figure 2: Schematic representation of MeHg-thiol exchange in the body.
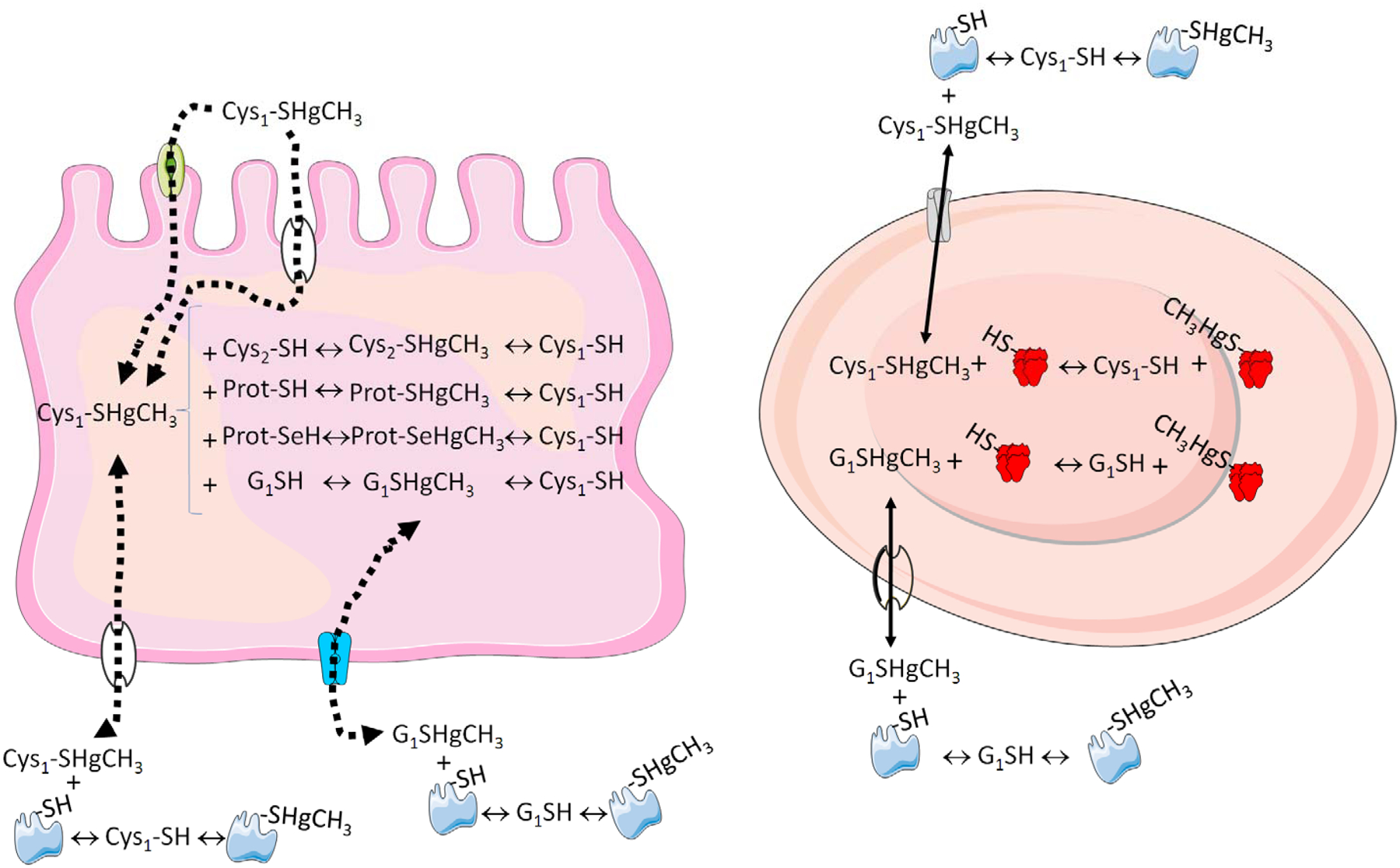
The absortion of MeHg-Cys complex (MeHg bound to a cysteine) in the intestine can be mediated by transporters or by exchange reactions. Inside the enterocyte (left part), the MeHg-Cys complex can exchange either with low molecular mass thiols (cysteine or glutathione, GSH) or with thiol-containing proteins. The complex can also exchange with selenol-containining proteins, i.e., selenoproteins (Prot-SeH). The low molecular mass sulfide-methylmercury complexes (GS-HgCH3 or Cys-S-HgCH3) can be transported to other cells or body fluids (e.g., interstitial fluid, capillary cells, which were omitted for the sake of clarity) and then can reach the plasma and blood cells (righ part). In the plasma, they can exchange with abundant proteins containing thiol (e.g. albumin) and can also be transported into the erythrocytes. Inside the erythrocytes, the low molecular mass thiols will delivery (exchange) the MeHg to hemoglobin and other thiol proteins, for instance, porphobilinogen synthase or aminolevulinate dehydratase (ALA-D, Rocha et al. 2012). The distribution of MeHg from plasma and erythrocytes to the tissues will involve the same kind of exchange reactions in the opposite direction (blood to tissue). In all the situations, the mobility of the low molecular mass complexes of MeHg-sulfides (MeHg-SG and MeHg-Cys) is expected to have a high facility to be redistributed than high molecular mass (thiol- or selenol-containing proteins).
Figure 3: Exchange reaction between low molecular mass methylmercury-glutathione sulfide (MeHg-SG) with different thiol-containing proteins.
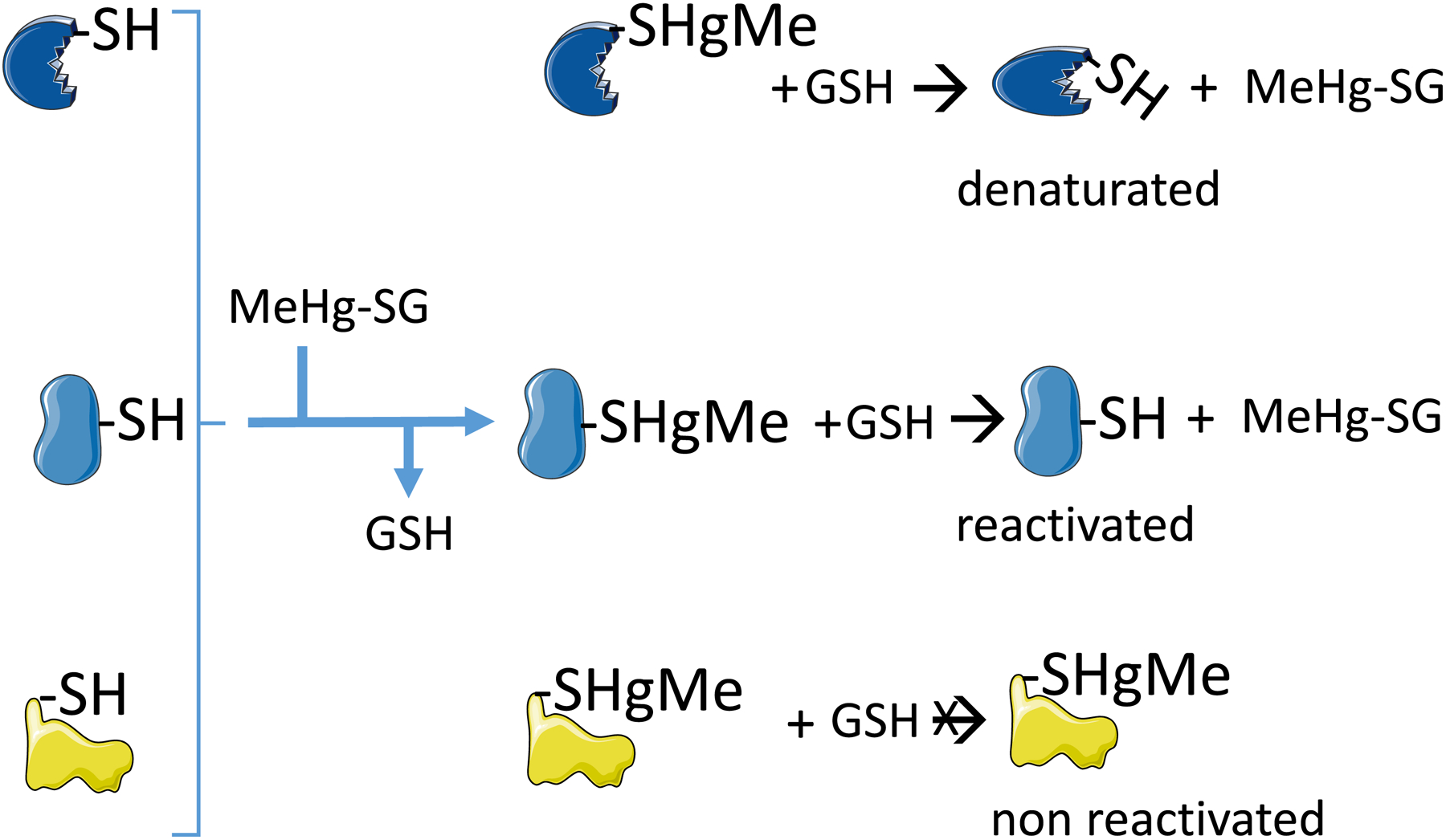
The first exchange (indicated by the reaction of the three proteins before the brace with GSH) results in the formation of three different protein-sulfide-methylmercury complexes. The protein-MeHg complexes are commonly inactive. The second set of exchange reactions can hypothetically reactivate the protein (represented by the second protein in the middle of the figure), or can release the denatured protein (i.e., the withdraw of the MeHg from the protein does not reestablish the activity, because the protein was denatured during the temporary interaction and is represented by the protein in the top of the figure). The reaction of MeHg with the protein thiol can cause a dramatic re-folding of the protein in such a way that MeHg-S-moiety will not be accessible to the medium and will not suffer the exchange reaction (represented by the third protein in the bottom of the figure).
Indeed, many studies have corroborated the ability of MeHg to inhibit selenoenzymes, both in vitro and after in vivo exposure (Carvalho et al., 2008; 2011, Farina et al., 2009; Franco et al., 2009; Wagner et al., 2010; Branco et al., 2011; 2012; 2014, 2017; Dalla Corte et al., 2013; Meinerz et al., 2017).
The higher affinity of MeHg for the –SeH than –SH groups may indicate that selenoproteins should be the primary targets of MeHg. However, the much lower concentration of –SeH groups when compared with –SH groups makes the scenario more complex and we have to weigh the superior affinity of –SeH for MeHg against the much greater concentrations of –SH groups in the biological milieu.
In short, any type of molecule containing thiol or selenol groups can be a potential target of MeHg. But our knowledge about the primary targets of MeHg is still elusive. In fact, the toxicity of MeHg is rather complex and involves the hierarchical interaction with multiple targets at different levels of cellular organization (Castoldi et al., 2000; Deny and Atchison 1996; Minnema et al., 1989; Sirois and Atchison, 2000; Atchison, 2005; Aschner et al., 2007). For instance, after being absorbed as a MeHg-cysteine complex, the MeHg derived from fish muscle proteins, can interact with thiol- and selenol-containing molecules (Figure 2 and 3; Farina et al., 2011a,b; Dórea et al., 2013). Since thiols are much more abundant than selenol groups, the probability that MeHg will target thiol-containing molecules is higher than targeting the selenol groups. Thus, the distribution of MeHg from non-target tissues to target tissues (particularly the brain) will be dictated by the exchange of MeHg from one thiol to another thiol- or selenol-containing molecule (Figure 2). However, in view of the high concentration of low mass thiol-containing molecules (i.e. GSH and cysteine), they are expected to have a central role MeHg distribution through the body and brain (Naganuma et al., 1980). In the blood, plasma and erythrocyte thiol-containing proteins (e.g. albumin and hemoglobin, etc.; Figure 2) possibly also play an important role in the distribution of MeHg. Accordingly, hemoglobin has the ability of binding MeHg and erythrocytes can retain a great proportion of MeHg in vertebrates (Naganuma et al., 1980; Doi and Tagawa, 1983; Clausing et al., 1984; Doi, 1991). The exchange of MeHg from the thiol group of hemoglobin to low molecular mass thiol (e.g. GSH and cysteine) will have a central role in the distribution of MeHg (Figure 2).
Selenoprotein P has an important role in the distribution of selenium to the brain and it is also one of the most abundant selenoproteins in the plasma (Burk and Hill, 2005). Although elevated levels of plasma selenoprotein P levels have been recently associated with high whole blood mercury (derived from fish and whale consumption) (Ser et al., 2017), its role in the distribution of MeHg to the brain has not been investigated. It is noteworthy that selenoprotein P can bind Hg+2-selenide complexes and it has been suggested that this type of interaction can facilitate the detoxification of Hg (Yoneda and Suzuki, 1997; Suzuki et al., 1998). However, the importance of selenoprotein P in MeHg distribution have been little investigated and, in contrast to humans exposed to mercury from seafood (Ser et al., 2017), the levels of selenoprotein P were decreased in rats intoxicated with high doses of MeHg (Usuki and Fujimura, 2016).
The complex biochemistry of MeHg movement from one thiol- or selenol-molecule to other molecules has not been studied in detail. Professor Dallas L. Rabenstein and collaborators were the pioneers investigating this type of MeHg-thiol exchange (Rabenstein & Fairhurst 1975; Rabenstein 1978b; Rabenstein et al. 1974; 1982; 1986; Rabenstein & Reid, 1984; Arnold et al. 1986). However, after them, little has been made and this subject is an important bottleneck in the molecular toxicology of MeHg. Although the exchange of MeHg from one thiol or selenol to another is well demonstrated, we cannot predict whether or not the temporary interaction of a given protein with MeHg will result in the inactivation of this protein even after MeHg leaving the thiol-containing protein (Figure 3). Consequently, we can hypothesize that the exchange of MeHg from one class of protein to another type of thiol- or selenol-containing protein can result or not in the inactivation of the proteins that have been temporarily bound with MeHg (Figure 3).
After reaching the brain, MeHg-thiol complex(es) are distributed to different cells types, including neural and glial cells (Atchison and Hare 1994; Aschner et al., 2007; Ni et al., 2012; Colón-Rodrigue et al., 2017). Astrocytes play a critical role in MeHg neurotoxicity and MeHg-induced impairment in glutamate up-take by astrocytes is tentatively one of the primary targets of MeHg (for review, see Aschner et al., 2007; Farina et al., 2011b). The inhibition of glutamate transport by MeHg causes an increase in the extracellular glutamate concentration, which can trigger excitotoxicity via activation of NMDA receptors. The activation of the glutamatergic receptors stimulates the entrance of Ca2+ into the neuronal cells and the activation of neurotoxic pathways. The intracellular overload of Ca2+ promotes the up-take of Ca2+ by the mitochondria, triggering reactive oxygen species overproduction, energy failure, mitochondrial permeability transition pore (mPTP) and cell demise (Atchison and Hare, 1994; Atchison, 2005; Limke et al., 2003; Roos et al., 2012; Farina et al., 2011b; 2013). The source of Ca2+ can be derived from the extracellular space and from intracellular stores (Atchison & Hare, 1994; Limke et al. 2003, Roos et al., 2012). Accordingly, in vitro and in vivo studies have demonstrated the protective role of Ca2+ channel blockers against the neurotoxicity of MeHg (Hare and Atchison 1995; Sakamoto et al., 1996; Castoldi et al., 2001; Gassó et al., 2001; Ramanathan and Atchison, 2011; Bailey et al., 2013).
Of particular neurotoxicological significance, MeHg also stimulates the ROS in astrocytes that can overwhelm the antioxidant cellular capacity (Aschner et al., 2007). Since astrocytes have substantial reserve of antioxidants, particularly the GSH system (Peuchen et al., 1997), the over-production of ROS/RNS can exhaust the astrocytes defenses, rendering the surroundings neurons more vulnerable to the neurotoxicity of glutamate and oxidative stress (Farina et al., 2011a,b).
MeHg can also be transported to the microglial cells and can trigger some biochemical changes similar to those provoked in astrocytes (Ni et al., 2010; 2011;2012). However, in vitro, microglial cells responded earlier to MeHg than astrocytes, indicating a potential role of this type of glial cells in the neurotoxicity of MeHg.
Although excitotoxicity has been shown to play a crucial role in the cytotoxicity of MeHg in vitro, the points of evidence indicating the participation of glutamatergic system under in vivo conditions are still limited (Juarez et al., 2002; 2005; Feng et al., 2014). The intracerebral infusion of high concentrations of MeHg caused a substantial increase in the extracellular levels of glutamate and oxidative damage (mediated by NMDA receptor activation) in the brain of rats (Juarez et al., 2002; 2005). Indirect points of evidence support the involvement of disruption in glutamatergic neurotransmitter system in the neurotoxicity of MeHg in developing and adult rodents (Farina et al., 2003; Manfroi et al., 2004; Carratu et al., 2006, Feng et al., 2006; Deng et al., 2014).
In addition to glutamatergic system, MeHg can also disrupt the functionality of cholinergic, dopaminergic, adrenergic and GABAergic neurotransmission in vertebrates (Hrdina et al., 1976; Sharma et al., 1982; Bartolome et al., 1987; Pereira et al., 1999; Zhou et al., 1999; Gimenez-Lort et al., 2001; Limke et al., 2004b; Aschner et al., 2007; Bradford et al., 2017). Indeed, MeHg can interact with thiol-containing proteins involved in the regulation of neurotransmission at different subcellular levels. For instance, MeHg can modify the activity of ionic channels, neurotransmitter receptors, neurotransmitters storage (uptake and release) and metabolism, etc. (Slotkin and Bartolome, 1987; Atchison and Hare 1994; Castoldi et al., 2001; Limke et al., 2004a,b; Atchison, 2005). In the next sections, we specifically discuss the effects of MeHg on the catecholaminergic system.
6. Methylmercury-mediated catecholaminergic toxicity
6.1. Dopamine
The effects of MeHg toward the dopaminergic neurotransmitter system have been extensively reported. In 1982, Bartolome and coworkers (1982) reported the effects of neonatal MeHg exposure on the biochemical development of CNS catecholamine synapses by evaluating synaptosomal and synaptic vesicular uptakes of [3H]catecholamines, TH activity, and levels and turnover rates of catecholamines. Most of the evaluated parameters were measured during the early postnatal (PN) period (from birth to PN day 40). The authors observed that neonatal MeHg exposure produced initial inhibition of [3H] dopamine synaptosomal uptake, followed by marked elevations of uptake from PN day 20 day onward. The observed changes in synaptosomal uptake were preceded by increases in the turnover rate of dopamine. However, the authors observed normal developmental patterns for other presynaptic terminal parameters (vesicular uptake, TH activity, and dopamine content). At that time, it was concluded that the alterations in uptake and turnover for dopamine indicate that the synaptic dynamics of developing central dopaminergic neurons are indeed affected by MeHg exposure.
Several years later, two groups reported increased dopamine release after MeHg exposure. Kalisch and Racz (1996) investigated the effects of in vitro MeHg exposure on endogenous dopamine efflux from mouse striatal slices. MeHg produced a concentration-dependent increase in the efflux of dopamine from mouse striatal slices. Notably, potassium-stimulated efflux of dopamine was enhanced by MeHg in both the presence and absence of Ca2+ in the medium, suggesting that under depolarizing conditions, dopamine efflux induced by MeHg has a Ca2+-independent component. The authors suggested that alterations in dopamine neurotransmission in the striatum might contribute to the symptoms of MeHg toxicity. In agreement with Kalisch and Racz (1996), Faro and colleagues (2000) investigating the effects of intrastriatal administration of MeHg on the dopaminergic system of rat striatum in conscious and freely-moving animals (using microdialysis coupled to liquid chromatography), observed that intrastriatal administration of MeHg (40μM to 4 mM) led to concentration-dependent increases in dopamine release from rat striatal tissue associated with significant decreases in extracellular levels of its main metabolites dihydroxyphenylacetic acid (DOPAC) and homovallinic acid (HVA). The authors explained these effects as a result of stimulated DA release and/or decreased DA intraneuronal degradation. The results from these two research groups reinforce the idea that increased dopamine release is an important effect of MeHg toward the dopaminergic neurotransmitter system. Further studies from Faro and coworkers (2002a) provided additional mechanistic information concerning MeHg-induced dopamine release in a microdialysis study. Evaluating the effects of striatal MeHg administration in dopamine release, the results demonstrated that MeHg increases the spontaneous DA release from rat striatum in a transporter-dependent manner, which is independent on vesicular stores and external Ca2+. MeHg also decreased KCl-evoked DA release.
Notably, the possibility of indirect effects in MeHg-induced dopamine release was proposed in 2002. Faro and coworkers (2002b) observed that intrastriatal infusion of MeHg in rats increased the extracellular dopamine levels with respect to basal levels. In contrast, this effect was significantly decreased in 400 μM MK-801 pretreated animals. Moreover, MeHg-induced increase in the extracellular dopamine levels was significantly decreased in 100 μM L-NAME or 7-NI (nitric oxide synthase inhibitors) pretreated animals, suggesting that MeHg acts, at least in part, through an overstimulation of NMDA receptors with possible NO production to induce DA release, and that administration of NMDA receptor antagonists and NOS inhibitors protects against MeHg-induced DA release from rat striatum. This study by Faro et al., (2002b) raised the possibility of indirect effects in MeHg-induced dopamine release through activation of other neurotransmitter systems, such as the glutamatergic one.
In agreement with these studies (Kalisch and Racz, 1996; Faro et al., 2000, 2002a, 2002b), an in vivo study by O’Kusky and coworkers (1988) showed that the treatment of rats with subcutaneous injections of MeHg during early postnatal development caused a significant increase in the concentrations of dopamine (28–29%) with a significant decrease in the concentration of 3,4-dihydroxyphenylacetic acid (DOPAC, 20–27%) at PN days 22–24. The authors pointed to altered metabolism of dopamine in the developing CNS during the pathogenesis of MeHg-induced movement and postural disorder.
The effects of MeHg on dopamine transporters have also been reported. By investigating whether MeHg exposure leads to changes in dopamine levels and dopamine transporter (DAT) function in synaptosomes from early postnatal rats, Dreiem and coworkers (2009) reported that MeHg exposure led to DAT inhibition and increased levels of released DA compared to control animals. Notably, the effects were much greater in synaptosomes prepared from PN day 7 rats than in synaptosomes from PND 14 or PND 21 animals. To our knowledge, this is the only study showing the effects of MeHg toward DAT function.
The direct deleterious effects of MeHg toward dopaminergic cells under in vitro conditions have been also reposted. Götz et al. (2002) used an in vitro approach to investigate the alterations induced by MeHg in primary dopaminergic cells isolated from the ventral mesencephalon of CD-1 embryonic mice. The morphometric analysis of DA neurons exposed to 1μM MeHg demonstrated a striking decrease in the number of neurites, indicative of cytoskeletal alteration. In addition, dopaminergic neurons displayed cell shrinkage and a significant increase in the number of nuclei with chromatin condensation. Based on these results, the authors concluded that MeHg is highly toxic to primary dopaminergic neurons. Götz and colleagues, Shao et al. (2015) investigated the effects of MeHg exposure on gene and protein profiles in a dopaminergic MN9D cell line. By performing proteomic analysis and evaluating differential protein expression, the authors suggested that MeHg and MPP+ (a classical dopaminergic toxin) share many similar signaling pathways leading to the pathogenesis of Parkinson’s disease.
MeHg also seems to affect the activity of monoamine oxidase (MAO), an enzyme involved in the metabolism of dopamine to DOPAC and/or HVA. Beyrouty et al. (2006) studied if oral exposure of adult female rats to MeHg before and during pregnancy would affect MAO activity in various brain regions of the offspring. The authors demonstrated that exposure to MeHg in rats before and/or during gestation resulted in a reduction of MAO activity in the developing embryo and brainstem of the female offspring with accompanying changes in auditory startle response. Based on these results, it is reasonable to posit a decreased metabolism of dopamine to DOPAC and HVA after MeHg exposure.
Dopamine receptors have also been proposed as potential targets involved in MeHg-toxicity. Coccini et al. (2011) treated rat dams with oral administrations of MeHg during the gestational days (GD) 7 to PN day 21. After treatments, the density (Bmax) and affinity (Kd) of dopamine D1-like (D1-Rs) and D2-like receptors (D2-Rs) were evaluated by saturation binding studies. Dopamine (DA) D1- and D2-like receptor Bmax and Kd were assessed in brain cortical and striatal membranes by saturation binding experiments using increasing concentrations of the specific ligands. The authors observed that the cerebral dopaminergic D1-like and D2-like receptors are differently impaired by developmental exposure to MeHg according to the brain area considered and/or to animal gender and time of growth, with some early changes persisting in time (D2-like receptors in males) or disappearing with time (D1-like receptors in males). To the best of our knowledge, this is the only study showing the effects of MeHg toward dopamine receptors function.
Studies in Caenorhabditis elegans (C. elegans) have also contributed to understand molecular mechanisms mediating MeHg effects toward the dopaminergic system. Martinez-Finley et al. (2013), using C. elegans as experimental model, tested the hypothesis that early-life exposure to MeHg and knockout (KO) of pdr-1 (mammalian: parkin/PARK2, mutations in this gene are a risk factor for PD) exacerbates MeHg toxicity and damage to the dopaminergic system. The authors observed that pdr-1 KO worms were more sensitive to MeHg than wild type worms, but MeHg did not exacerbate behavioral changes related to the absence of pdr-1. They concluded that the combination of early-life exposure to MeHg and pdr-1KO had significant effects on oxidative stress and aging, also suggesting that early-life MeHg exposure is a risk factor for loss of dopaminergic function later in life in wild type worms, but the combination of pdr-1 KO and early-life MeHg does not further exacerbate the already reduced dopaminergic function produced by pdr-1 KO alone. In another study with C. elegans, VanDuyn and Nass (2014) showed MRP-7 (multidrug resistance protein 7) loss-of-function mutations increase susceptibility of dopaminergic neurons to MeHg, attesting to gene x environment interactions.
Based to the studies mentioned in herein (6.1), it is reasonable to propose that the dopaminergic neurotransmitter system is indeed a target involved in MeHg-induced neurotoxicity, including developmental neurotoxicity. But what do the previously mentioned molecular effects represent in terms of behavioral outcomes? Reed and Newland (2009) studied the effects gestational exposure to MeHg on rat behavior during adulthood, as well as the involvement of dopaminergic system. The authors performed a protocol in which female rats were exposed in utero to MeHg, via maternal drinking water. As adults, the MeHg-exposed offspring were trained to lever press under a fixed interval schedule of reinforcement. Experiments with acute dose-effect curves were performed with dopamine (cocaine) and norepinephrine (desipramine) reuptake inhibitors, as well as a direct D1 or D2 agonist and antagonists. For high-rate behavior, MeHg-exposed rats were 2–3 times more sensitive to the rate-reducing effects of high doses of cocaine, but no differential effects of MeHg were seen with desipramine, suggesting MeHg’s effect is specific to dopaminergic receptors. In addition, no differential effects were seen with the specific D1 and D2 agonists. Notably, it seems that the observed effects were formed during gestational exposure and persisted into adulthood, suggesting that gestational MeHg exposure produces irreversible sensitivity to dopamine in rats.
6.2. Epinephrine
The effects of MeHg towards the adrenergic system have been less investigated than the dopaminergic system. Early in vitro studies reported that MeHg caused inhibition of norepinephrine uptake and stimulated its release from pre-loaded synaptosomes (Komulainen and Tuomisto, 1981; Rajanna and Hobson, 1985) and hippocampal slices (Gassó et al., 2000). The effects of perinatal in vivo exposure to MeHg have consistently indicated changes in brain norepinephrine levels in rats. However, the magnitude of brain norepinephrine decrease or increase varied depending on the schedule of exposure (period, dose, brain area evaluated, etc.)(Hrdina et al., 1976; Taylor and Distefanelo, 1976; Bartolome et al., 1982; 1984a; Tsuzuki, 1982; Lindstrom et al., 1991). For instance, MeHg exposure of suckling rats from birth to weaning caused dose and time dependent increase in brain norepinephrine levels and turnover. Of neurotoxicological significance, the levels of norepinephrine remained elevated even 20 days after the end of MeHg exposure (Bartolome et al., 1982). In contrast, the gestational exposure to MeHg did not cause changes in norepinephrine levels or turnover, but caused an increase in norepinephrine uptake by synaptosomes of suckling rats (Bartolome et al., 1984a).
The ontogeny of alfa- and beta-adrenergic receptors (α1, α2 and β) were modified by pre- and post-natal exposure to MeHg. The regions more affected were cerebellum>cerebral cortex>mid-brain and brain stem and, according to authors, the effects depended on the maturational profile of each brain region, and the regions that developed early were less affected than the ones that matured latter (Bartolome et al., 1987; Slotkin and Bartolome, 1986). In another study, the exposure of rats to a low dose of MeHg from conception-gestation to 50 days of life was associated with an increase in norepinephrine levels in the cerebellum, but not in frontal cortex, striatum, hypothalamus and hippocampus (Lindstrom et al., 1991).
In a comprehensive study, O’Kusky and co-workers examined the neurochemical alterations in rat brain noradrenergic system at three different stages of post-natal MeHg exposure (O’Kusky et al., 1988). MeHg exposure was started at the 5th post-natal day until day 15 (stage I, where MeHg treated pups did not show any overt sign of behavioral intoxication and gained weight but at lower rate than controls), day 18–21 (stage II, where significant changes in body weight were observed) and day 22–24 (stage III, which corresponded to the period of the onset of neurobehavioral impairments). The authors reported that norepinephrine levels were increased in spinal cord and caudate putamen, when compared with controls and body weight gain-matched controls (which were separated from their dams to growth at the same rate as did MeHg exposed rats). The increase in norepinephrine levels started before the installation of neurobehavioral impairments (started in the stage II and persisted until stage III), indicating that changes in norepinephrine levels occurred before the onset of neurobehavioral manifestations (O’kusky et al. 1988). In cerebral cortex, the variations in norepinephrine levels were very complex and body weight gain-matched controls had higher levels of norepinephrine than controls, whereas MeHg treated rats had the lowest levels of norepinephrine in the stage I. At stage II, the levels of norepinephrine were lower in controls and MeHg, when compared with body weight-matched controls. During the onset of neurological symptoms, there were no differences between the groups. Authors concluded that movement and postural disorders in suckling rats were associated with selective alterations in central catecholaminergic (dopaminergic and noradrenergic) and serotoninergic neurotransmitter systems.
The exposure of catfish (Clarias batrachus) to MeHg for long periods (90 and 180 days) caused a significant increase in brain norepinephrine levels. To some extent, this is agreement with results obtained with rats, where the levels of catecholamines (dopamine and norepinephrine) can be increased after different schedules of exposure to MeHg (Kirubagaran et al., 1990). Moreover, this indicates a conserved neurotoxic effect of MeHg in different vertebrates possibly reflecting the targeting of a similar group of conserved proteins involved in catecholaminergic neurotransmission.
One important aspect of MeHg neurotoxicity that has been little explored is its deleterious effect toward the sympathetic noradrenergic neurotransmission in heart and kidney. Bartolome and collaborators have demonstrated that MeHg exposure during the early post-natal period accelerated the maturation of cardiac sympathetic transmission, which was associated with increased norepinephrine levels and turnover. The authors have also demonstrated that MeHg caused heart and kidney overgrowth, which could be linked to sympathetic innervations over-activity (Bartolome et al., 1984). This topic should be further analyzed particularly in view of the points of evidence that MeHg exposure can cause hypertension in experimental animals (Groto et al., 2009) or increased diastolic and systolic blood pressure in humans exposed to MeHg during prenatal life (Sorensen et al., 1999; Thurston et al., 2007). The results from human exposed during the prenatal life indicate that MeHg can modify development of cardiac homeostasis (Sorensen et al., 1999) as observed in developing rats (Bartolome et al., 1984).
In fact, the disruption of normal cardiac function by inorganic mercury and the participation of sympathetic catecholaminergic system are well-documented (Henningsson et al., 1993; Torres et al., 2000; Beck et al., 2004; Michaeli-Yossef et al., 2007; Houston, 2011). However, the potential role of MeHg via fish consumption and catecholamines (particularly changes in noradrenergic sympathetic neurotransmission) as a risk for the development of cardiovascular pathologies is still debatable (Salonen et al., 1995; Guallar et al., 2002; Yoshizawa et al., 2002; Roman et al., 2011; Wennberg et al., 2012).
7. Concluding remarks
MeHg is a toxicant that targets a great variety of proteins involved in different cellular processes. Of particular neurotoxicological significance, MeHg targets several components of different neurotransmitter systems. MeHg exposure during perinatal phases of brain development can modify the fine dynamics of brain synapses (synaptogenesis, establishment of cell-cell contact, cell migration, etc.) by interacting with neurotransmitter receptors, ion channels, transport proteins, enzymes, etc. In short, MeHg can interfere with the synthesis, degradation, storage, release, re-uptake of different neurotransmitters, including dopamine and norepinephrine. The ultimate outcomes of disrupting such complex biochemical processes will be the behavioral teratology. More recently, the literature data have also indicated the dangers of interfering with normal brain development during the adolescence period. Indeed, the exposures to MeHg during adolescence have been shown to disrupt the dopaminergic neuropharmacology in rodents (Boomhower and Newland, 2016; 2017). If the experimental data derived from animal models were to be extrapolated to human, it might explain the growing incidence of behavioral and cognitive abnormalities in the human population. However, since now we can be exposed to thousands of potentially neurotoxic agents (Grandjean and Landingran, 2014), the understanding on how much a single potent neurotoxic agent, such as MeHg, effectively contributes to the disruption of behavior in developing organism will be a difficult task. In fact, our knowledge about the primary targets of MeHg, as well as of other neurotoxicants, is still incipient. The development of in silico methodologies to search for proteins containing moieties of the type –X-C-X-, X-U-X, -C-(X)2-C- or –C-(X)2-U- (where C=cysteinyl residues, U=selenocysteinyl residues and X=other residues than cys or sec) will be important to define potential targets of MeHg. Furthermore, the in vitro and in vivo identification of which of these proteins are actually targeted by MeHg will help in defining primary molecular targets of MeHg. As reported here, MeHg changes several neurochemical processes related to catecholaminergic neurotransmission, but it remains to be determined whether these are direct (primary) or indirect (secondary or tertiary) effects of MeHg neurotoxicity.
Acknowledgements:
The author would like to thank the colleagues/co-authors who have contributed to several studies referenced in this chapter. These studies were funded in part by grants from the National Institute of Environmental Health Sciences (grant numbers NIEHS R01ES07331, NIEHS R01ES10563 and NIEHS R01ES020852), as well as the Brazilian Agencies CNPq, CAPES, FAPERGS and FINEP.
REFERENCES
- Ait-Bali Y, Ba-M’hamed S, Bennis M (2016) Prenatal Paraquat exposure induces neurobehavioral and cognitive changes in mice offspring. Environ. Toxicol Pharmacol 48: 53–62. [DOI] [PubMed] [Google Scholar]
- Andersen SL (2003) Trajectories of brain development: Point of vulnerability or window of opportunity? Neurosc. Biobehav. Rev 27: 3–18. [DOI] [PubMed] [Google Scholar]
- Archer T, Fredriksson A (2006) Influence of noradrenaline denervation on MPTP-induced deficits in mice. J Neural Transm (Vienna). 113:1119–1129. [DOI] [PubMed] [Google Scholar]
- Archer T, Jonsson G, Ross SB (1984) A parametric study of the effects of the noradrenaline neurotoxin DSP4 on avoidance acquisition and noradrenaline neurones in the CNS of the rat.Br J Pharmacol. 82: 249–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold AP, Tan K-S, Rabenstein DL (1986) Nuclear magnetic resonance studies of the solution chemistry of metal complexes. 23. complexation of methylmercury by selenohydryl-containing amino acids and related molecules Inorg. Chem 25: 2433–2437. [Google Scholar]
- Asaduzzaman AM, Schreckenbach G (2011) Degradation mechanism of methyl mercury selenoamino acid complexes: a computational study. Inorg. Chem 50: 2366–2372. [DOI] [PubMed] [Google Scholar]
- Asaduzzaman AM, Khan MA, Schreckenbach G, Wang F (2009) Computational studies of structural, electronic, spectroscopic, and thermodynamic properties of methylmercury-amino acid complexes and their Se analogues. Inorganic chemistry. 49: 870–878. [DOI] [PubMed] [Google Scholar]
- Aschner M, Syversen T, Souza DO, Rocha JB, Farina M (2007) Involvement of glutamate and reactive oxygen species in methylmercury neurotoxicity. Braz J Med Biol Res 40: 285–291. [DOI] [PubMed] [Google Scholar]
- Atchison WD (2005) Is chemical neurotransmission altered specifically during methylmercury-induced cerebellar dysfunction? TIPS 26: 549–557. [DOI] [PubMed] [Google Scholar]
- Atchison WD, Hare MF (1994) Mechanisms of methylmercury-induced neurotoxicity (1994) FASEB J. 8: 622–629 [DOI] [PubMed] [Google Scholar]
- Austin DW, Shandley K (2008) An investigation of porphyrinuria in Australian children with autism. J. Toxicol. Environ. Health, A 71: 1349–1351. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Hutsell BA, Newland MC (2013). Dietary nimodipine delays the onset of methylmercury neurotoxicity in mice. Neurotoxicology, 37: 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeau A (1970) Dopamine and disease. Can. Med. Assoc. J 103: 824–832. [PMC free article] [PubMed] [Google Scholar]
- Bartolome J, Trepanier P, Chait EA, Seidler FJ, Deskin R, Slotkin TA (1982) Neonatal methylmercury poisoning in the rat: effects on development of central catecholamine neurotransmitter systems. Toxicol. Appl. Pharmacol 65: 92–99. [DOI] [PubMed] [Google Scholar]
- Bartolome J, Whitmore WL, Seidler FJ, Slotkin TA (1984a) Exposure to methylmercury in utero: Effects on biochemical development of catecholamine neurotransmitter systems. Life Sci. 35: 657–670. [DOI] [PubMed] [Google Scholar]
- Bartolome J, Trepanier PA, Chait EA, Barnes GA, Lerea L, Whitmore WL, Weigel SJ, Slotkin TA (1984b) Neonatal methylmercury poisoning in the rat: Effects on development of peripheral sympathetic nervous system. Neuronal participation in methylmercury-induced cardiac and renal overgrowth NeuroToxicol. 5: 45–54. [PubMed] [Google Scholar]
- Bartolome JV, Kavlock RJ, Cowdery T, Orband-Miller L, Slotkin TA (1987) Development of adrenergic receptor binding sites in brain regions of the neonatal rat: Effects of prenatal or postnatal exposure to methylmercury. NeuroToxicology, 8 : 1–14. [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 63: 182–217. [DOI] [PubMed] [Google Scholar]
- Beck C, Krafchik B, Traubici J, Jacobson S (2004) Mercury intoxication: it still exists. Pediatric. Derm 21:254–259. [DOI] [PubMed] [Google Scholar]
- Beyrouty P, Stamler CJ, Liu JN, Loua KM, Kubow S, Chan HM (2006) Effects of prenatal methylmercury exposure on brain monoamine oxidase activity and neurobehaviour of rats. Neurotoxicol Teratol. 28:251–259. [DOI] [PubMed] [Google Scholar]
- Blanusa M, Varnai VM, Piasek M, Kostial K (2005) Chelators as antidotes of metal toxicity: therapeutic and experimental aspects. Cur. Med. Chem 12: 2771–2794. [DOI] [PubMed] [Google Scholar]
- Boomhower SR, Newland MC (2017) Effects of adolescent exposure to methylmercury and d-amphetamine on reversal learning and an extra dimensional shift in male mice. Exp. Clin. Psychopharm 25: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomhower SR, Newland MC (2016) Adolescent methylmercury exposure affects choice and delay discounting in mice. Neurotoxicology. 57: 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford AB, Mancini JD, Atchison WD (2016) Methylmercury-dependent increases in fluo4 fluorescence in neonatal rat cerebellar slices depend on granule cell migrational stage and GABAA receptor modulation. J. Pharm. Exp. Ther 356: 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco V, Canário J, Holmgren A, Carvalho C (2011) Inhibition of the thioredoxin system in the brain and liver of zebra-seabreams exposed to waterborne methylmercury. Toxicol. App. Pharmacol 251: 95–103. [DOI] [PubMed] [Google Scholar]
- Branco V, Ramos P, Canário J, Lu J, Holmgren A, Carvalho C (2012) Biomarkers of adverse response to mercury: histopathology versus thioredoxin reductase activity. BioMed Res Int. 19: 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco V, Godinho-Santos A, Gonçalves J, Lu J, Holmgren A, Carvalho C (2014) Mitochondrial thioredoxin reductase inhibition, selenium status, and Nrf-2 activation are determinant factors modulating the toxicity of mercury compounds. Free Rad. Biol. Med 73: 95–105. [DOI] [PubMed] [Google Scholar]
- Branco V, Caito S, Farina M, Rocha JB, Aschner M, Carvalho C (2017) Biomarkers of mercury toxicity: Past, present, and future trends. J. Toxicol. Environ. Health, B 20: 119–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco V, Coppo L, Solá S, Lu J, Rodrigues CM, Holmgren A, Carvalho C (2017) Impaired cross-talk between the thioredoxin and glutathione systems is related to ASK-1 mediated apoptosis in neuronal cells exposed to mercury, Red. Biol Available online 1 June 2017, ISSN 2213–2317, 10.1016/j.redox.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Matsumoto M, Hikosaka O (2010) Dopamine in motivational control: Rewarding, aversive, and alerting. Neuron, 68: 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk RF, Hill KE (2005) Selenoprotein P: An extracellular protein with unique physical characteristics and a role in selenium homeostasis. Ann. Rev. Nut 25: 215–235. [DOI] [PubMed] [Google Scholar]
- Carratù MR, Borracci P, Coluccia A, Giustino A, Renna G, Tomasini MC, Raisi E, Antonelli T, Cuomo V, Mazzoni E, Ferraro L (2006) Acute exposure to methylmercury at two developmental windows: Focus on neurobehavioral and neurochemical effects in rat offspring. Neuroscience 141: 1619–1629. [DOI] [PubMed] [Google Scholar]
- Carvalho CML, Chew E-H, Hashemy SI, Lu J, Holmgren A (2008) Inhibition of the human thioredoxin system: A molecular mechanism of mercury toxicity J. Biol. Chem 283: 11913–11923. [DOI] [PubMed] [Google Scholar]
- Carvalho CML, Lu J, Zhang X, Arnér ESJ, Holmgren A (2011) Effects of selenite and chelating agents on mammalian thioredoxin reductase inhibited by mercury: Implications for treatment of mercury poisoning. FASEB J. 25: 370–381. [DOI] [PubMed] [Google Scholar]
- Castoldi A, Barni S, Turin I, Gandini C, Manzo L (2000) Early acute necrosis, delayed apoptosis and cytoskeletal breakdown in cultured cerebellar granule neurons exposed to methylmercury. J. Neurosci. Res 59: 775–787. [DOI] [PubMed] [Google Scholar]
- Castoldi AF, Coccini T, Ceccatelli S, Manzo L (2001) Neurotoxicity and molecular effects of methylmercury. Brain Res. Bul 55: 197–203. [DOI] [PubMed] [Google Scholar]
- Clark DG, McElligott TF, Hurst EW (1966) The toxicity of paraquat. Br J Ind Med. 23:126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausing P, Riedel B, Gericke S, Grün G, Müller L (1984) Differences in the distribution of methyl mercury in erythrocytes, plasma, and brain of Japanese quails and rats after a single oral dose. Arch. Tox 56: 132–135. [DOI] [PubMed] [Google Scholar]
- Coccini T, Roda E, Castoldi AF, Poli D, Goldoni M, Vettori MV, Mutti A, Manzo L (2011) Developmental exposure to methylmercury and 2,2’, 4,4’, 5,5’–hexachloro-biphenyl (PCB153) affects cerebral dopamine D1-like and D2-like receptors of weanling and pubertal rats. Arch. Toxicol 85:1281–1294. [DOI] [PubMed] [Google Scholar]
- Colón-Rodrígue A, Hannon H, Atchison WD (2017) Effects of methylmercury on spinal cord afferents and efferents—A review, NeuroToxicol. 60: 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, D’Esposito M (2011) Inverted-U-shaped dopamine actions on human working memory and cognitive control. Biol. Psych 69: e113–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory-Slechta DA, Virgolini MB, Rossi-George A, Thiruchelvam M, Lisek R, Weston D (2008) Lifetime consequences of combined maternal lead and stress. Basic Clin. Pharmacol. Toxicol 102: 218–227. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH (1976) Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 192: 481–483. [DOI] [PubMed] [Google Scholar]
- Dalla Corte CL, Wagner C, Sudati JH, Comparsi B, Leite GO, Busanello A, Soares FAA, Aschner M, Rocha JBT (2013) Effects of diphenyl diselenide on methylmercury toxicity in rats. BioMed Res. Int, art. no. 983821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS (1999) A mechanism of paraquat toxicity involving nitric oxide synthase. Proc Natl Acad Sci U S A. 96:12760–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo N, Chamberlain SR, Sahakian BJ, Robbins TW (2011) The roles of dopamine and noradrenaline in the pathophysiology and treatment of attention-deficit/hyperactivity disorder. Biol. Psych 69: e145–e157. [DOI] [PubMed] [Google Scholar]
- Deng Y, Xu Z, Xu B, Liu W, Wei Y, Li Y, Feng S, Yang T (2014) Exploring cross-talk between oxidative damage and excitotoxicity and the effects of riluzole in the rat cortex after exposure to methylmercury. Neurotox. Res 26: 40–51. [DOI] [PubMed] [Google Scholar]
- Denny MF, Atchison WD (1996) Mercurial-induced alterations in neuronal divalent cation homeostasis.Neurotoxicology 17: 47–61. [PubMed] [Google Scholar]
- Dodds CM, Clark L, Dove A, Regenthal R, Baumann F, Bullmore E, Robbins TW, Müller U (2009) The dopamine D2 receptor antagonist sulpiride modulates striatal BOLD signal during the manipulation of information in working memory. Psychopharmacology (Berl). 207: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi R (1991) Individual difference of methylmercury metabolism in animals and its significance in methylmercury toxicity In: Advances in mercury toxicology (section 2) 77–98. Springer US. [Google Scholar]
- Doi R, Tagawa M (1983) A study on the biochemical and biological behavior of methylmercury. Toxicol Appl Pharmacol 69 : 407–416. [DOI] [PubMed] [Google Scholar]
- Dórea JG, Farina M, Rocha JB. Toxicity of ethylmercury (and Thimerosal): a comparison with methylmercury (2013) J. App. Toxicol 33: 700–711. [DOI] [PubMed] [Google Scholar]
- Dreiem A, Shan M, Okoniewski RJ, Sanchez-Morrissey S, Seegal RF (2009) Methylmercury inhibits dopaminergic function in rat pup synaptosomes in an age-dependent manner. Neurotoxicol Teratol. 31: 312–317. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Morris KA, O’Brien A, Robbins TW (1991) The basolateral amygdala-ventral striatal system and conditioned place preference: further evidence of limbic-striatal interactions underlying reward-related processes. Neuroscience. 42: 1–18. [DOI] [PubMed] [Google Scholar]
- Ewing JA, Myers RD (1985) Norepinephrine, alcohol, and alcoholism, In The Catecholamines in Psychiatric and Neurologic Disorders, edited by Lake C. Raymond and Ziegler Michael G., Butterworth-Heinemann, 137–152. [Google Scholar]
- Farina M, Aschner M, Rocha JB (2011a) Oxidative stress in MeHg-induced neurotoxicity. Toxicol. Appl. Pharmacol 256:405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Rocha JB, Aschner M (2011b) Mechanisms of methylmercury-induced neurotoxicity: evidence from experimental studies. Life Sci. 89: 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Avila DS, Da Rocha JBT, Aschner M (2013) Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury Neurochem. Int 62: 575–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina M, Dahm KCS, Schwalm FD, Brusque AM, Frizzo MES, Zeni G, Souza DO, Rocha JBT (2003) Methylmercury increases glutamate release from brain synaptosomes and glutamate uptake by cortical slices from suckling rat pups: Modulatory effect of ebselen. Toxicol. Sci 73: 135–140. [DOI] [PubMed] [Google Scholar]
- Farina M, Campos F, Vendrell I, Berenguer J, Barzi M, Pons S, Suñol C (2009) Probucol increases glutathione peroxidase-1 activity and displays long-lasting protection against methylmercury toxicity in cerebellar granule cells. Toxicol Sci. 112: 416–426. [DOI] [PubMed] [Google Scholar]
- Faro LR, do Nascimento JL, Alfonso M, Durán R (2002a) Mechanism of action of methylmercury on in vivo striatal dopamine release. Possible involvement of dopamine transporter. Neurochem Int. 40:455–465. [DOI] [PubMed] [Google Scholar]
- Faro LR, do Nascimento JL, Alfonso M, Durán R (2002b) Protection of methylmercury effects on the in vivo dopamine release by NMDA receptor antagonists and nitric oxide synthase inhibitors. Neuropharmacology. 42: 612–618. [DOI] [PubMed] [Google Scholar]
- Faro LR, do Nascimento JL, San José JM, Alfonso M, Durán R (2000) Intrastriatal administration of methylmercury increases in vivo dopamine release. Neurochem Res. 25: 225–229. [DOI] [PubMed] [Google Scholar]
- Feltmann K, Konradsson-Geuken Å, De Bundel D, Lindskog M, Schilström B (2015) Antidepressant drugs specifically inhibiting noradrenaline reuptake enhance recognition memory in rats. Behav Neurosci. 129: 701–708. [DOI] [PubMed] [Google Scholar]
- Feng S, Xu Z, Liu W, Li Y, Deng Y, Xu B (2014) Preventive effects of dextromethorphan on methylmercury-induced glutamate dyshomeostasis and oxidative damage in rat cerebral cortex. Biol. Trace Elem. Res 159: 332–345. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PJ (2013) Elevated norepinephrine may be a unifying etiological factor in the abuse of a broad range of substances: Alcohol, nicotine, marijuana, heroin, cocaine, and caffein. Substance Abuse: Research and Treatment, 7: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ (2014) Is elevated norepinephrine an etiological factor in some cases of schizophrenia?. Psych. Res 215: 497–504. [DOI] [PubMed] [Google Scholar]
- Franco JL, Posser T, Dunkley PR, Dickson PW, Mattos JJ, Martins R, Bainy AC, Marques MR, Dafre AL, Farina M (2009) Methylmercury neurotoxicity is associated with inhibition of the antioxidant enzyme glutathione peroxidase. Free Rad. Biol. Med 47: 449–457. [DOI] [PubMed] [Google Scholar]
- Gassó S, Suñol C, Sanfeliu C, Rodrıguez-Farré E, Cristòfol RM (2000) Pharmacological characterization of the effects of methylmercury and mercuric chloride on spontaneous noradrenaline release from rat hippocampal slices. Life Sci. 67: 1219–1231. [DOI] [PubMed] [Google Scholar]
- Gassó S, Cristofol RM, Selema G, Rosa R, Rodríguez‐Farré E, Sanfeliu C (2001). Antioxidant compounds and Ca2+ pathway blockers differentially protect against methylmercury and mercuric chloride neurotoxicity. J. Neurosci. Res 66: 135–145. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1992) The neostriatal mosaic: multiple levels of compartmental organization. J Neural Transm Suppl. 36: 43–59. [DOI] [PubMed] [Google Scholar]
- Gimenez-Llort L, Ahlbom E, Dare E, Vahter M, Ögren SO, Ceccatelli S (2001) Prenatal exposure to methylmercury changes dopamine-modulated motor activity during early ontogeny: age and gender-dependent effects. Environmental toxicology and pharmacology. 9: 61–70. [DOI] [PubMed] [Google Scholar]
- Go YM, Jones DP (2013) The redox proteome. J. Biol. Chem 288: 26512–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go YM, Chandler JD, Jones DP (2015) The cysteine proteome. Free. Rad. Biol. Med 84: 227–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz ME, Koutsilieri E, Riederer P, Ceccatelli S, Daré E (2002) Methylmercury induces neurite degeneration in primary culture of mouse dopaminergic mesencephalic cells. J Neural Transm (Vienna). 109: 597–605. [DOI] [PubMed] [Google Scholar]
- Grandjean P, Landrigan PJ (2014) Neurobehavioural effects of developmental toxicity. Lancet Neurol. 13: 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant H, Lantos PL, Parkinson C (1980) Cerebral damage in paraquat poisoning. Histopathology. 4: 185–95. [DOI] [PubMed] [Google Scholar]
- Grotto D, de Castro MM, Barcelos GR, Garcia SC, Barbosa F Jr (2009) Low level and sub-chronic exposure to methylmercury induces hypertension in rats: nitric oxide depletion and oxidative damage as possible mechanisms. Archives of toxicology.83: 653–662. [DOI] [PubMed] [Google Scholar]
- Guallar E, Sanz-Gallardo MI, van’t Veer P, Bode P, Aro A, Gómez-Aracena J, Kark JD, Riemersma RA, Martín-Moreno JM, Kok FJ (2002) Heavy Metals and Myocardial Infarction Study Group.. Mercury, fish oils, and the risk of myocardial infarction. N. Engl. J. Med 347: 1747–1754. [DOI] [PubMed] [Google Scholar]
- Hamon M, Blier P (2013) Monoamine neurocircuitry in depression and strategies for new treatments. Prog. Neuro-Psychopharmacol. Biol. Psych 45: 54–63. [DOI] [PubMed] [Google Scholar]
- Hamon M, Blier P Monoamine neurocircuitry in depression and strategies for new treatments (2013) Progress in Neuro-Psychopharmacology and Biological Psychiatry, 45: 54–63. [DOI] [PubMed] [Google Scholar]
- Harro J, Oreland L, Vasar E, Bradwejn J (1995) Impaired exploratory behaviour after DSP-4 treatment in rats: implications for the increased anxiety after noradrenergic denervation. Eur Neuropsychopharmacol. 5: 447–55. [DOI] [PubMed] [Google Scholar]
- Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN (2014) Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem. Sci 39:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningsson C, Hoffmann S, McGonigle L, Winter JS (1993) Acute mercury poisoning (acrodynia) mimicking pheochromocytoma in an adolescent. The Journal of pediatrics.122:252–253. [DOI] [PubMed] [Google Scholar]
- Heyer DB, Meredith RM (2017) Environmental toxicology: Sensitive periods of development and neurodevelopmental disorders. NeuroToxicol. 58: 23–41. [DOI] [PubMed] [Google Scholar]
- Howes O, McCutcheon R, Stone J (2015) Glutamate and dopamine in schizophrenia: an update for the 21st century. J. Psychopharmacol 29: 97–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdina PD, Peters DA, Singhal RL (1976) Effects of chronic exposure to cadmium, lead and mercury of brain biogenic amines in the rat. Res. Com. Chem. Pathol. Pharmacol 15: 483–493. [PubMed] [Google Scholar]
- Huys QJ, Tobler PN, Hasler G, Flagel SB (2014) The role of learning-related dopamine signals in addiction vulnerability. Prog Brain Res. 211: 31–77. [DOI] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, Ledoux JE (2011) Molecular mechanisms of fear learning and memory. Cell, 147: 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP (2015) Redox theory of aging. Redox Biol. 5: 71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez BI, Martınez ML, Montante M, Dufour L, Garcıa E, Jimenez-Capdeville ME (2002) Methylmercury glutamate extracellular levels in frontal cortex of awake rats. Neurotoxicol. Teratol 24: 767–771. [DOI] [PubMed] [Google Scholar]
- Juárez BI, Portillo-Salazar H, González-Amaro R, Mandeville P, Aguirre JR, Jiménez ME (2005) Participation of N-methyl-D-aspartate receptors on methylmercury-induced DNA damage in rat frontal cortex. Toxicology 207: 223–229. [DOI] [PubMed] [Google Scholar]
- Kalisch BE, Racz WJ (1996) The effects of methylmercury on endogenous dopamine efflux from mouse striatal slices. Toxicol Lett. 89: 43–49. [DOI] [PubMed] [Google Scholar]
- Kang MJ, Gil SJ, Lee JE, Koh HC (2010) Selective vulnerability of the striatal subregions of C57BL/6 mice to paraquat. Toxicol Lett. 195: 127–134. [DOI] [PubMed] [Google Scholar]
- Karri V, Schuhmacher M, Kumar V (2016) Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: A general review of metal mixture mechanism in brain. Env. Tox. Pharm 48: 203–213. [DOI] [PubMed] [Google Scholar]
- Kaye WH, Wierenga CE, Bailer UF, Simmons AN, Bischoff-Grethe A (2013). Nothing tastes as good as skinny feels: The neurobiology of anorexia nervosa TINS 36: 110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern JK, Geier DA, Adams JB, Mehta JA, Grannemann BD, Geier MR. (2011) Toxicity biomarkers in autism spectrum disorder: a blinded study of urinary porphyrins. Pediatrics International. 53:147–153. [DOI] [PubMed] [Google Scholar]
- Kern JK, Geier DA, Sykes LK, Haley BE, Geier MR (2016) The relationship between mercury and autism: A comprehensive review and discussion. J. Trace Elem. Med. Biol 37:8–24. [DOI] [PubMed] [Google Scholar]
- Kirubagaran R, Joy KP (1990) Changes in brain monoamine levels and monoamine oxidase activity in the catfish, Clarias batrachus, during chronic treatments with mercurial. Bul. Environ. Cont. Toxicol 45: 88–93. [DOI] [PubMed] [Google Scholar]
- Kobayashi K (2001) Role of catecholamine signaling in brain and nervous system functions: new insights from mouse molecular genetic study. J Investig Dermatol Symp Proc. 6: 115–21. [DOI] [PubMed] [Google Scholar]
- Komulainen H, Tuomisto J (1981) Interference of methyl mercury with monoamine uptake and release in rat brain synaptosomes. Bas. Clin. Pharm. Tox 48: 214–22. [Google Scholar]
- Kraft AD, Aschner M, Cory-Slechta DA, Bilbo SD, Caudle WM, Makris SL (2016) Unmasking silent neurotoxicity following developmental exposure to environmental toxicants. Neurotoxicol. Teratol 55:38–44. [DOI] [PubMed] [Google Scholar]
- Kudo T, Kushikata T, Kudo M, Kudo T, Hirota K (2010) A central neuropathic pain model by DSP-4 induced lesion of noradrenergic neurons: preliminary report. Neurosci Lett. 481:102–104. [DOI] [PubMed] [Google Scholar]
- Lategan AJ, Marien MR, Colpaert FC (1992) Suppression of nigrostriatal and mesolimbic dopamine release in vivo following noradrenaline depletion by DSP-4: a microdialysis study. Life Sci. 50:995–999. [DOI] [PubMed] [Google Scholar]
- Le Moal M, Simon H (1991) Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev. 71: 155–234. [DOI] [PubMed] [Google Scholar]
- Lénárd L, Ollmann T, László K, Kovács A, Gálosi R, Kállai V, Attila T, Kertes E, Zagoracz O, Karádi Z, Péczely L (2017) Role of D2 dopamine receptors of the ventral pallidum in inhibitory avoidance learning. Behav Brain Res. 321: 99–105. [DOI] [PubMed] [Google Scholar]
- Liang KC (1998) Pretraining infusion of DSP-4 into the amygdala impaired retention in the inhibitory avoidance task: involvement of norepinephrine but not serotonin in memory facilitation. Chin J Physiol. 41: 223–233. [PubMed] [Google Scholar]
- Limke TL, Heidemann SR, Atchison WD (2004) Disruption of intraneuronal divalent cation regulation by methylmercury: Are specific targets involved in altered neuronal development and cytotoxicity in methylmercury poisoning? NeuroToxicology, 25: 741–760. [DOI] [PubMed] [Google Scholar]
- Limke TL, Otero-Montañez JK, Atchison WD (2003). Evidence for interactions between intracellular calcium stores during methylmercury-induced intracellular calcium dysregulation in rat cerebellar granule neurons. J Pharm. Exp.Ther 304: 949–958. [DOI] [PubMed] [Google Scholar]
- Limke TL, Bearss JJ, Atchison WD (2004b) Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Tox. Sci 80: 60–68. [DOI] [PubMed] [Google Scholar]
- Lindström H, Luthman J, Oskarsson A, Sundberg J, Olson L (1991) Effects of long-term treatment with methyl mercury on the developing rat brain. Environ. Res 56: 158–169. [DOI] [PubMed] [Google Scholar]
- Liou HH, Chen RC, Tsai YF, Chen WP, Chang YC, Tsai MC (1996) Effects of paraquat on the substantia nigra of the wistar rats: neurochemical, histological, and behavioral studies. Toxicol Appl Pharmacol. 137: 34–41. [DOI] [PubMed] [Google Scholar]
- Liu W, Xu Z, Yang T, Xu B, Deng Y, Feng S (2016) Memantine, a low-affinity NMDA receptor antagonist, protects against methylmercury-induced cytotoxicity of rat primary cultured cortical neurons, involvement of Ca2+ D dyshomeostasis antagonism, and indirect antioxidation effects. Mol. Neurobiol 18: 1–7. [DOI] [PubMed] [Google Scholar]
- Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA (2002) The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: paraquat and alpha-synuclein. J Biol Chem. 277: 1641–1644. [DOI] [PubMed] [Google Scholar]
- Martinez-Finley EJ, Chakraborty S, Slaughter JC, Aschner M (2013) Early-life exposure to methylmercury in wildtype and pdr-1/parkin knockout C. elegans. Neurochem Res. 38:1543–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Sokolowski JD, Salamone JD (1993) A neurochemical and behavioral investigation of the involvement of nucleus accumbens dopamine in instrumental avoidance. Neuroscience. 52(4):919–925. [DOI] [PubMed] [Google Scholar]
- Meinerz DF, Branco V, Aschner M, Carvalho C, Rocha JBT (2017) Diphenyl diselenide protects against methylmercury-induced inhibition of thioredoxin reductase and glutathione peroxidase in human neuroblastoma cells: A comparison with ebselen (2017) J. App. Toxicol First published: 6 April 2017, DOI: 10.1002/jat.3458 [DOI] [PubMed] [Google Scholar]
- Michaeli-Yossef Y, Berkovitch M, Goldman M (2007) Mercury intoxication in a 2-year-old girl: a diagnostic challenge for the physician. Pediatric Nephr. 22: 903–906. [DOI] [PubMed] [Google Scholar]
- Minnema DJ, Cooper GP, Greenland RD (1989) Effects of methylmercury on neurotransmitter release from rat brain synaptosomes. Toxicol. Appl. Pharmacol 99: 510–521. [DOI] [PubMed] [Google Scholar]
- Miseta A, Csutora P (2000) Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol. Biol. Evol 17: 1232–1239. [DOI] [PubMed] [Google Scholar]
- Miyoshi R, Kito S, Ishida H, Katayama S (1988) Alterations of the central noradrenergic system in MPTP-induced monkey parkinsonism. Res Commun Chem Pathol Pharmacol. 62: 93–102. [PubMed] [Google Scholar]
- Mokhber N, Abdollahian E, Soltanifar A, Samadi R, Saghebi A, Haghighi MB, Azarpazhooh A (2014) Comparison of sertraline, venlafaxine and desipramine effects on depression, cognition and the daily living activities in Alzheimer patients. Pharmacopsychiatry. 47: 131–140. [DOI] [PubMed] [Google Scholar]
- Montes DR, Stopper CM, Floresco SB (2015) Noradrenergic modulation of risk/reward decision making. Psychopharmacol. 232:2681–2696. [DOI] [PubMed] [Google Scholar]
- Moore KE, Lookingland KJ (1995) Dopaminergic neuronal systems in the hypothalamus In: Psychopharmacology (Bloom FE, Kupfer DJ, eds). New York: Raven Press, 1995, 245–256. [Google Scholar]
- Moore RY, Card JP (1984) Noradrenaline-containing neuron systems In: Handbook of Chemical Neuroanatomy (BjoÈrklund A, HoÈ kfelt T, eds), Vol. 2 Amsterdam: Elsevier, 1984, 123–156. [Google Scholar]
- Mutter J, Naumann J, Schneider R, Walach H, Haley B (2005) Mercury and autism: accelerating evidence. Neuroendocrinol. Lett 26:439–446. [PubMed] [Google Scholar]
- Mutter J, Curth A, Naumann J, Deth R, Walach H (2010) Does inorganic mercury play a role in Alzheimer’s disease? A systematic review and an integrated molecular mechanism (2010) J. Alz. Dis, 22: 357–374. [DOI] [PubMed] [Google Scholar]
- Naganuma A, Koyama Y, Imura N (1980) Behavior of methylmercury in mammalian erythrocytes. Tox. Appl. Pharmacol 54: 405–410. [DOI] [PubMed] [Google Scholar]
- Nakajima S, Gerretsen P, Takeuchi H, Caravaggio F, Chow T, Le Foll B, Mulsant B, Pollock B, Graff-Guerrero A (2013) The potential role of dopamine D₃ receptor neurotransmission in cognition. Eur. Neuropsychopharmacol 23: 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Roth A, Kyzar EJ, Poudel MK, Wong K, Stewart AM, Kalueff AV (2014) Decoding the contribution of dopaminergic genes and pathways to autism spectrum disorder (ASD). Neurochem Int. 66: 15–26. [DOI] [PubMed] [Google Scholar]
- Ni M, Li X, Yin Z, Jiang H, Sidoryk-Węgrzynowicz M, Milatovic D, Aschner M (2010). Methylmercury induces acute oxidative stress, altering Nrf2 protein level in primary microglial cells. Tox. Sci 116: 590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Li X, Yin Z, Sidoryk-Wegrzynowicz M, Jiang H, Farina M, Rocha JB, Syversen T, Aschner M (2011) Comparative study on the response of rat primary astrocytes and microglia to methylmercury toxicity. Glia 59: 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Li X, Rocha JB, Farina M, Aschner M (2012). Glia and methylmercury neurotoxicity. J. Tox. Env. Health, A, 75: 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieoullon A (2002) Dopamine and the regulation of cognition and attention Prog. Neurobiol 67: 53–83. [DOI] [PubMed] [Google Scholar]
- Nikolaus S, Hautzel H, Müller HW (2014) Neurochemical dysfunction in treated and nontreated schizophrenia–a retrospective analysis of in vivo imaging studies. Rev. Neurosci 25: 25–96. [DOI] [PubMed] [Google Scholar]
- O’Kusky JR, Boyes BE, McGeer EG (1988) Methylmercury-induced movement and postural disorders in developing rat: regional analysis of brain catecholamines and indoleamines. Brain Res. 439:138–146. [DOI] [PubMed] [Google Scholar]
- Ogren SO, Archer T, Ross SB (1980) Evidence for a role of the locus coeruleus noradrenaline system in learning. Neurosci Lett. 20: 351–356. [DOI] [PubMed] [Google Scholar]
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J (2005) Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 361:133–139. [DOI] [PubMed] [Google Scholar]
- Pauls DL, Abramovitch A, Rauch SL, Geller DA (2014) Obsessive-compulsive disorder: an integrative genetic and neurobiological perspective. Nat Rev Neurosci. 15: 410–424. [DOI] [PubMed] [Google Scholar]
- Pearson RG (1963) Hard and soft acids and bases. J Am. Chem. Soc 85:3533–3539. [Google Scholar]
- Pereira ME, Morsch VM, Christofari RS, Rocha JB (1999) Methyl mercury exposure during post-natal brain growth alters behavioral response to SCH 23390 in young rats. Bull Environ Contam Toxicol. 63: 256–262. [DOI] [PubMed] [Google Scholar]
- Peuchen S, Bolaños JP, Heales SJ, Almeida A, Duchen MR, Clark JB (1997) Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system. Prog. Neurobiol 52: 261–281. [DOI] [PubMed] [Google Scholar]
- Pezzoli G, Cereda E (2013) Exposure to pesticides or solvents and risk of Parkinson disease. Neurology. 80:2035–2041. [DOI] [PubMed] [Google Scholar]
- Quaak I, Brouns MR, Van de Bor M (2013) The dynamics of autism spectrum disorders: how neurotoxic compounds and neurotransmitters interact. Int. J. Environ Res Public Health 10: 3384–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabenstein DL (1978a) The chemistry of methylmercury toxicology (1978) J. Chem. Educ 54: 292–296. [Google Scholar]
- Rabenstein DL (1978b) The aqueous solution chemistry of methylmercury and its complexes. Acc. Chem. Res 11: 100–107. [Google Scholar]
- Rabenstein DL, Fairhurst MT (1975) Nuclear magnetic resonance studies of the solution chemistry of metal complexes. XI. The binding of methylmercury by sulfhydryl-containing amino acids and by glutathione. J. Ame Chem. Soc 97: 2086–2092. [DOI] [PubMed] [Google Scholar]
- Rabenstein DL, Isab AA, Reid RS (1982) A proton nuclear magnetic resonance study of the binding of methylmercury in human erythrocytes (1982) BBA – Mol. Cell Res 720: 53–64. [DOI] [PubMed] [Google Scholar]
- Rabenstein DL, Ozubko R, Libich S, Evans CA, Fairhurst MT, Suvanprakorn C (1974) Nuclear magnetic resonance studies of the solution chemistry of metal complexes. x. determination of the formation constants of the methylmercury complexes of selected amines and amino carboxylic acids. J. Coor. Chem 3: 263–271. [Google Scholar]
- Rabenstein DL, Reid RS (1984) Nuclear Magnetic Resonance Studies of the Solution Chemistry of Metal Complexes. 20. Ligand-Exchange Kinetics of Methylmercury(II)-Thiol Complexes. Inor. Chem 23: 1246–1250. [Google Scholar]
- Rabenstein DL, Reid RS, Yamashita G, Tan KS, Arnold AP (1986) Determination of thiols and selenols by titration with methylmercury with end point detection by nuclear magnetic resonance spectrometry. Anal. Chem 58:1266–1269. [Google Scholar]
- Rocha JB, Saraiva RA, Garcia SC, Gravina FS, Nogueira CW (2012) Aminolevulinate dehydratase (δ-ALA-D) as marker protein of intoxication with metals and other pro-oxidant situations. Toxicol. Res 1: 85–102. [Google Scholar]
- Rajanna B, Hobson M (1985) Influence of mercury on uptake of [3H]dopamine and [3H]norepinephrine by rat brain synaptosomes. Toxicol. Lett 27: 7–14. [DOI] [PubMed] [Google Scholar]
- Rakers F, Rupprecht S, Dreiling M, Bergmeier C, Witte OW, Schwab M (2017) Transfer of maternal psychosocial stress to the fetus. Neuroscience Biobehav. Rev Available online 22 February 2017, ISSN 0149–7634, 10.1016/j.neubiorev.2017.02.019. [DOI] [PubMed] [Google Scholar]
- Ramanathan G, Atchison WD (2011). Ca 2+ entry pathways in mouse spinal motor neurons in culture following in vitro exposure to methylmercury. Neurotoxicol. 32: 742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MN, Newland MC (2009) Gestational methylmercury exposure selectively increases the sensitivity of operant behavior to cocaine. Behav Neurosci. 123: 408–417. [DOI] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ (1995) Central norepinephrine neurons and behavior In: Psychopharmacology: the Fourth Generation of Progress (Bloom FE, Kupfer DJ, eds). New York: Raven Press, 1995, pp 363–372. [Google Scholar]
- Roman HA, Walsh TL, Coull BA, Dewailly É, Guallar E, Hattis D, Mariën K, Schwartz J, Stern AH, Virtanen JK, Rice G (2011) Evaluation of the cardiovascular effects of methylmercury exposures: current evidence supports development of a dose-response function for regulatory benefits analysis. Environ Health Perspect. 119: 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos D, Seeger R, Puntel R, Vargas Barbosa N (2012) Role of calcium and mitochondria in MeHg-mediated cytotoxicity. J. Biomed. Biotechnol 2012: 248764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SB, Renyl AL (1976) On the long-lasting inhibitory effect of N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP 4) on the active uptake of noradrenaline. J Pharm Pharmacol. 28: 458–459. [DOI] [PubMed] [Google Scholar]
- Ross SB, Stenfors C (2015) DSP4, a selective neurotoxin for the locus coeruleus noradrenergic system. A review of its mode of action. Neurotox Res. 2015 27: 15–30. [DOI] [PubMed] [Google Scholar]
- Ross SB (2015) Long-term effects of N-2-chlorethyl-N-ethyl-2-bromobenzylamine hydrochloride on noradrenergic neurones in the rat brain and heart. Br J Pharmacol. 58: 521–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto M, Ikegami N, Nakano A (1996). Protective effects of Ca2+ channel blockers against methyl mercury toxicity. Bas. Clin. Pharm. Toxicol 78: 193–199. [DOI] [PubMed] [Google Scholar]
- Salonen JT, Seppanen K, Nyyssonen K, Korpela H, Kauhanen J, Kantola M, Tuomilehto J, Esterbauer H, Tatzber F, Salonen R (1995) Intake of mercury from fish, lipid peroxidation, and the risk of myocardial infarction and coronary, cardiovascular, and any death in Eastern Finnish men. Circulation 91: 645–655 [DOI] [PubMed] [Google Scholar]
- Sealey LA, Hughes BW, Sriskanda AN, Guest JR, Gibson AD, Johnson-Williams L, Pace DG, Bagasra O (2016) Environmental factors in the development of autism spectrum disorders. Env. Int 88: 288–298. [DOI] [PubMed] [Google Scholar]
- Ser PH, Omi S, Shimizu-Furusawa H, Yasutake A, Sakamoto M, Hachiya N, Konishi S, Nakamura M, Watanabe C (2017) Differences in the responses of three plasma selenium-containing proteins in relation to methylmercury-exposure through consumption of fish/whales. Tox. Let 267: 53–58. [DOI] [PubMed] [Google Scholar]
- Shao Y, Figeys D, Ning Z, Mailloux R, Chan HM (2015) Methylmercury can induce Parkinson’s-like neurotoxicity similar to 1-methyl-4- phenylpyridinium: a genomic and proteomic analysis on MN9D dopaminergic neuron cells. J Toxicol Sci 40: 817–828. [DOI] [PubMed] [Google Scholar]
- Sharma RP, Aldous CN, Farr CH (1982) Methylmercury induced alterations in brain amine syntheses in rats. Tox.Let 13: 195–201 [DOI] [PubMed] [Google Scholar]
- Sigitova E, Fišar Z, Hroudová J, Cikánková T, Raboch J (2016) Biological hypotheses and biomarkers of bipolar disorder. Psyc. Clin. Neurosci [DOI] [PubMed] [Google Scholar]
- Sirois JE, Atchison WD (2000) Methylmercury affects multiple subtypes of calcium channels in rat cerebellar granule cells. Toxicol. Appl. Pharmacol, 167: 1–11. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Bartolome J (1987) Biochemical mechanisms of developmental neurotoxicity of methylmercury NeuroToxicology 8: 65–84. [PubMed] [Google Scholar]
- Sørensen N, Murata K, Budtz-Jørgensen E, Weihe P, Grandjean P (1999) Prenatal methylmercury exposure as a cardiovascular risk factor at seven years of age. Epidemiology. 1:370–375. [PubMed] [Google Scholar]
- Spear LP (2000) The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev 24: 417–463. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Hojo Y, Tamai Y, Tanaka H (1976) Selenium protection against mercury toxicity. Binding of methylmercury by the selenohydryl-containing ligand. J. Am. Chem. Soc 98: 2339–2341. [DOI] [PubMed] [Google Scholar]
- Sugiura Y, Tamai Y, Tanaka H (1978) Selenium protection against mercury toxicity: high binding affinity of methylmercury by selenium-containing ligands in comparison with sulfur-containing ligands. Bioinorg. Chem 9:167–180. [DOI] [PubMed] [Google Scholar]
- Suzuki KT, Sasakura C, Yoneda S (1998) Binding sites for the (Hg-Se) complex on selenoprotein P. Biochim. Biophys. Acta - Prot Struc Mol Enz 1429: 102–112. [DOI] [PubMed] [Google Scholar]
- Szot P, Knight L, Franklin A, Sikkema C, Foster S, Wilkinson CW, White SS, Raskind MA (2012) Lesioning noradrenergic neurons of the locus coeruleus in C57Bl/6 mice with unilateral 6-hydroxydopamine injection, to assess molecular, electrophysiological and biochemical changes in noradrenergic signaling. Neuroscience. 216: 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor LL, DiStefano V (1976) Effects of methylmercury on brain biogenic amines in the developing rat pup. Toxicol Appl Pharmacol 38: 489–497. [DOI] [PubMed] [Google Scholar]
- Teychenne PF, Feuerstein G, Lake CR, Ziegler MG (1985) Central catecholamine systems: Interaction with neurotransmitters in normal subjects and in patients with selected neurologic diseases. In The Catecholamines in Psychiatric and Neurologic Disorders, Butterworth-Heinemann, 1985, Pages 91–119. [Google Scholar]
- Torres AD, Rai AN, Hardiek ML (2000) Mercury intoxication and arterial hypertension: report of two patients and review of the literature. Pediatrics. 105:e34-. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Sabatini BL (2012) Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 76:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston SW, Bovet P, Myers GJ, Davidson PW, Georger LA, Shamlaye C, Clarkson TW (2007) Does prenatal methylmercury exposure from fish consumption affect blood pressure in childhood? Neurotoxicol. 28: 924–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trillo L, Das D, Hsieh W, Medina B, Moghadam S, Lin B, Dang V, Sanchez MM, De Miguel Z, Ashford JW, Salehi A (2013) Ascending monoaminergic systems alterations in Alzheimer’s disease. Translating basic science into clinical care. Neurosci. Biobehav.Rev 37: 1363–1379. [DOI] [PubMed] [Google Scholar]
- Tsuzuki Y (1982) Effect of methylmercury exposure on different neurotransmitter systems in rat brain. Toxicol Lett 13: 159—162. [DOI] [PubMed] [Google Scholar]
- Usuki F, Fujimura M (2016) Decreased plasma thiol antioxidant barrier and selenoproteins as potential biomarkers for ongoing methylmercury intoxication and an individual protective capacity. Arch. Tox 90: 917–926. [DOI] [PubMed] [Google Scholar]
- VanDuyn N, Nass R (2014) The putative multidrug resistance protein MRP-7 inhibits methylmercury-associated animal toxicity and dopaminergic neurodegeneration in Caenorhabditis elegans. J Neurochem. 128:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Os J, Kapur S. Schizophrenia (2009) The Lancet 374: 635–645 [DOI] [PubMed] [Google Scholar]
- Van Enkhuizen J, Janowsky DS, Olivier B, Minassian A, Perry W, Young JW, Geyer MA (2015) The catecholaminergic-cholinergic balance hypothesis of bipolar disorder revisited. Eur. J. Pharmacol 753: 114–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veyrac A, Sacquet J, Nguyen V, Marien M, Jourdan F, Didier A (2009) Novelty determines the effects of olfactory enrichment on memory and neurogenesis through noradrenergic mechanisms. Neuropsychopharmacology. 34:786–795. [DOI] [PubMed] [Google Scholar]
- Vilensky JA, Redman K (2003) British anti-Lewisite (dimercaprol): an amazing history. Ann. Emerg. Med 41: 378–383. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Baler RD (2011) Reward, dopamine and the control of food intake: Implications for obesity. Trends Cog. Sci 15: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang G-J, Tomasi D, Baler RD (2013) Obesity and addiction: Neurobiological overlaps. Obes. Rev 14: 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C, Sudati JH, Nogueira CW, Rocha JBT (2010) In vivo and in vitro inhibition of mice thioredoxin reductase by methylmercury. BioMetals, 23: 1171–1177. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Fujimoto Y, Shimura T, Sakai N (1995) Conditioned taste aversion in rats with excitotoxic brain lesions. Neurosci Res. 22: 31–49. [DOI] [PubMed] [Google Scholar]
- Yoneda S, Suzuki KT (1997) Equimolar Hg-Se complex binds to selenoprotein P. Biochem. Biophys. Res. Com 231: 7–11. [DOI] [PubMed] [Google Scholar]
- Yoshizawa K, Rimm EB, Morris JS, Spate VL, Hsieh C-C, Spiegelman D, Stampfer MJ, Willett WC (2002) Mercury and the risk of coronary heart disease in men (2002) New Eng. J. Med 347: 1755–1760. [DOI] [PubMed] [Google Scholar]
- Zhong FX, Ji XQ, Tsou K (1985) Intrathecal DSP4 selectively depletes spinal noradrenaline and attenuates morphine analgesia. Eur J Pharmacol. 116: 327–330. [DOI] [PubMed] [Google Scholar]
- Zhou T, Rademacher DJ, Steinpreis RE, Weis JS (1999). Neurotransmitter levels in two populations of larval Fundulus heteroclitus after methylmercury exposure. Comp. Biochem. Phys. C 124: 287–294. [DOI] [PubMed] [Google Scholar]