

Abstract
Hydrogen tunneling in enzymatic C-H activation requires a dynamical sampling among ground-state enzyme-substrate conformations, which transiently generates a tunneling-ready state (TRS). The TRS is characterized by an H-donor-acceptor distance (DAD) of 2.7 Å, ca. 0.5Å shorter than the dominant DAD of optimized ground states. Recently, a high-resolution, 13C electron-nuclear double-resonance (ENDOR) approach was developed to characterize the ground state structure of the complex of linoleic acid (LA) substrate with soybean lipoxygenase (SLO). The resulting enzyme-substrate (E-S) model revealed two ground-state conformers with different distances between the target C11 of LA and the catalytically active cofactor (Fe(III)-OH): the active conformer “a”, with a van der Waals donor-acceptor distance (DAD, 3.1 Å) between C11 and metal-bound hydroxide; an inactive conformer “b”, distance almost 1 Å-longer. Herein, the structure of the E-S complex is examined for a series of six variants in which subtle structural modifications of SLO have been introduced either at a hydrophobic sidechain near bound substrate or at a remote residue within a protein network whose flexibility influences H-transfer. A remarkable correlation is found between the ENDOR-derived population of the active ground-state conformer “a” and the kinetically-derived differential enthalpic barrier for D vs. H transfer, ΔEa, with the latter increasing as the fraction of conformer “a” decreases. We propose that ΔEa provides a ‘ruler’ for the DAD within the TRS. ENDOR measurements further corroborate the previous identification of a dynamical network coupling the buried active site of SLO to the surface. This study shows that subtle imperfections within the initial ground state structures of E-S complexes are accompanied by compromised geometries at the TRS.
Keywords: ENDOR, lipoxygenase, temperature-dependent KIEs, donor-acceptor distance, hydrogen tunneling, tunneling ready states
Graphical Abstract
INTRODUCTION
Cleavage of C-H bonds is one of the most pervasive and fundamental reactions catalyzed by enzymes. Accumulated evidence supports an expanded, Marcus-like quantum mechanical tunneling mechanism for H-transfer that incorporates an additional donor-acceptor distance-sampling coordinate.1–6 The ability to stably label C-H bonds with D and T has further enabled the characterization of bond-selective kinetic isotope effects (KIEs). Large, weakly temperature-dependent KIEs, with a small difference between the enthalpic barriers for homolytic C-D and C-H bond cleavage, ΔEa = Ea(D) – Ea(H) ≅ 0, are hallmarks of nonadiabatic quantum H-tunneling behavior in native enzymes,7–13 and represent true deviations from the classical “over the barrier” view of catalysis.14, 15 Because of their sensitivity to barrier width, enzyme-catalyzed hydrogen atom transfer reactions provide a unique window into reaction barrier shape, illuminating the importance of protein conformational sampling that leads to reduced donor-acceptance distances (DADs) conducive to tunneling16–21
According to the prevailing nonadiabatic proton-coupled electron transfer (PCET) model, which reproduces the kinetic phenomena seen in many natively evolved enzyme catalyzed C-H activation reactions (Figure 1), the dominant DAD undergoes a progressive reduction from the typically observed van der Waals distances in the ground state of 3.2–3.4 Å (Figure 1A; a conformer) through thermodynamically averaged conformational sampling to a more compacted a* conformer (DAD ~ 2.7–2.9 Å) that undergoes further thermally activated environmental reorganization (Marcus λ term) to reach a tunneling-ready state (TRS) configuration (Figure 1B). At the TRS, the donor and acceptor hydrogenic wave functions (Figure 1C) are degenerate and exhibit strong probability overlap in the native enzyme. Accumulating evidence further indicates a role for specific and dynamical protein networks in achieving the TRS; in general, such networks couple reactivity within buried active sites to remote regions of an enzyme in contact with solvent.22–27
Figure 1.
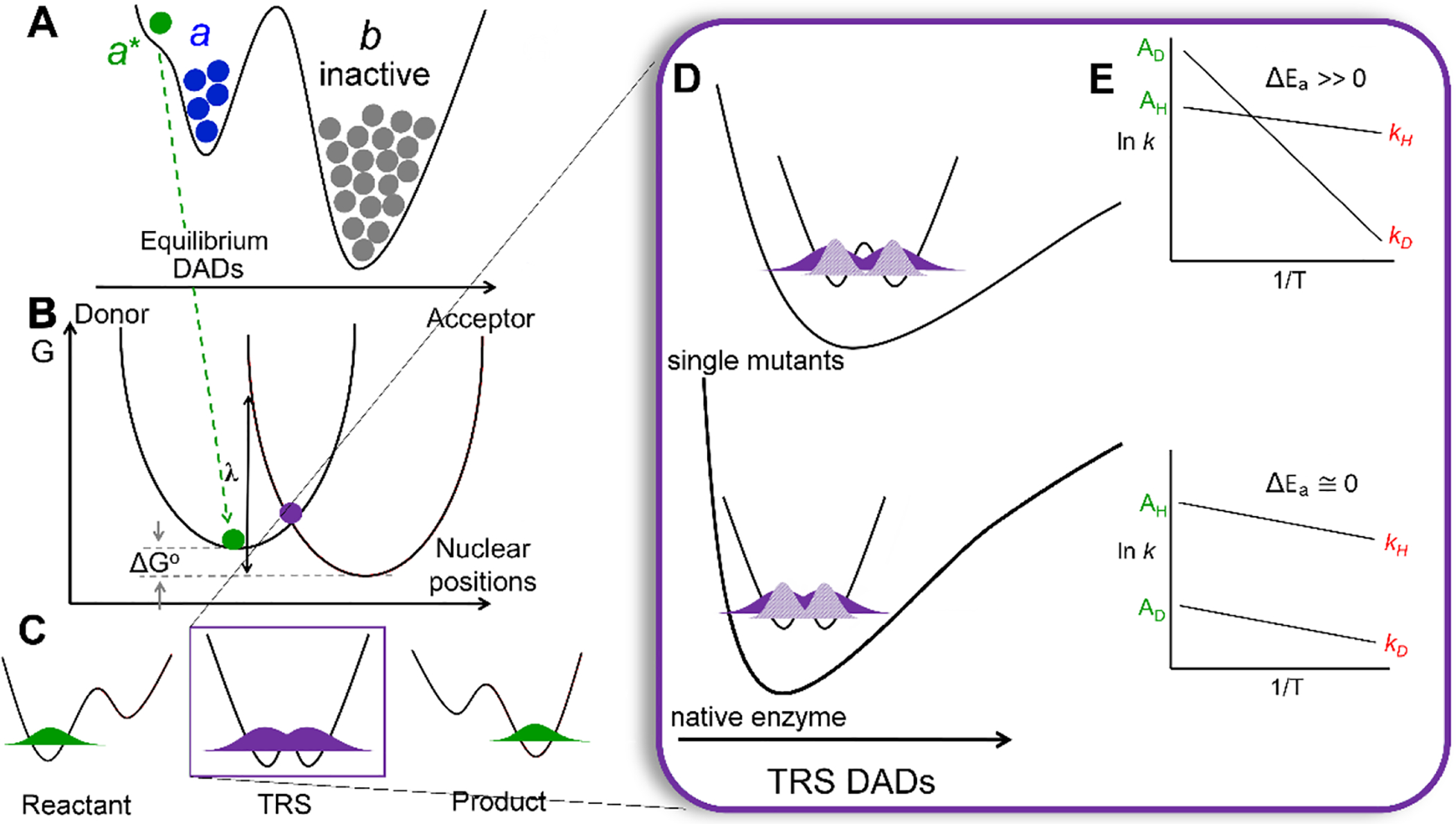
Representation of the nonadiabatic PCET theoretical model for enzymes catalyzing C-H activation (A-D) and kinetic phenomena that characterize them (E). (A) Potential-energy surface (PES) showing the distribution of enzyme into active and inactive ground state populations, a (blue) and b (gray), respectively. Subsequent and transient fluctuations lead to short-lived substates, represented as conformer a* with DADs reduced from ground state configurations (green dot). (B) Slice along Marcus reaction environmental coordinate within the tunneling-active a* conformation involving the nuclear ‘heavy atom’ positions and defined by the Marcus reorganization energy, λ, and driving force, ΔGo. (C) The positioning of the hydrogenic wave function, in the reactant (donor, left) and product (acceptor, right) states, and in the TRS (center, purple dot in B) where it is degenerate across the donor and acceptor wells. (D) The PES of the DAD sampling coordinate. The small ΔEa values characteristic of native enzymes (bottom) indicate a short DAD that enables efficient overlap for both protium and deuterium with rigid DAD sampling frequencies. When the average DAD is elongated, as occurs in single site mutations, the PES of the DAD sampling coordinate is softened (top). In such as case, the tunneling efficiency of the broader wave function for H can be maintained (dark purple). However, the less diffuse wave function for D (light purple) results in a greater dependence on local sampling of the donor and acceptor for effective wave function overlap. This is the origin of the disproportionate increase in the enthalpy barrier for D (ΔEa >> 0). (E) Cartoon representations of Arrhenius plots for the cleavage of C-H or C-D bonds, to show the distinction between native (WT) enzymes (ΔEa ~ 0) and impaired, single mutant enzymes (ΔEa >> 0). For illustration purposes, the slope of the line for H-transfer is represented as unchanged.
An important feature that distinguishes nonadiabatic tunneling models, from classical models of catalysis is their ability to rationalize the commonly observed increase in the magnitude of ΔEa upon insertion of strategically placed site-specific mutations (local and/or remote), ascribed to a disruption of the optimized donor and acceptor alignment at the TRS of native enzyme. SLO has been a particularly important model system in this regard, due to its large kinetic isotope effect (WT KIE ~ 80 at 30°C) during the initial, and rate limiting, C-H abstraction step, which is weakly temperature dependent (ΔEa ~ 0.9 kcal/mol) (Scheme 1). Relevant to the work described herein, a series of well-characterized SLO mutations has been generated in which mutations with progressively reduced sidechain volume exhibit a regular increase in ΔEa from close to zero in WT enzyme, with little to no impact on the magnitude of the KIE.28 The physical origin of these selective increases in ΔEa can be understood in the context of a perturbation-induced elongation of the DAD that is accompanied by a decreased force constant for the DAD sampling mode (Figure 1D top). The more confined DAD and sampling coordinate characteristic of WT enables wave function overlap for both isotopes with similar energies (Figure 1D, bottom). However, increases in the DAD, as occurs in the mutants, exacerbate the differential reactivity of D transfer, a consequence of its less diffuse wave function and greater dependence on DAD sampling. This is the origin the increase in the enthalpic barrier for D relative to H transfer (i.e. ΔEa >> 0). Both linear and quadratic treatments of an anharmonic DAD sampling coordinate, and its exponential coupling to the tunneling rate, have been described.29–31 While the quadratic analysis is the more mathematically complete, the series of SLO mutants characterized herein can be well fit within the linear region of the function.5, 32
Scheme 1.
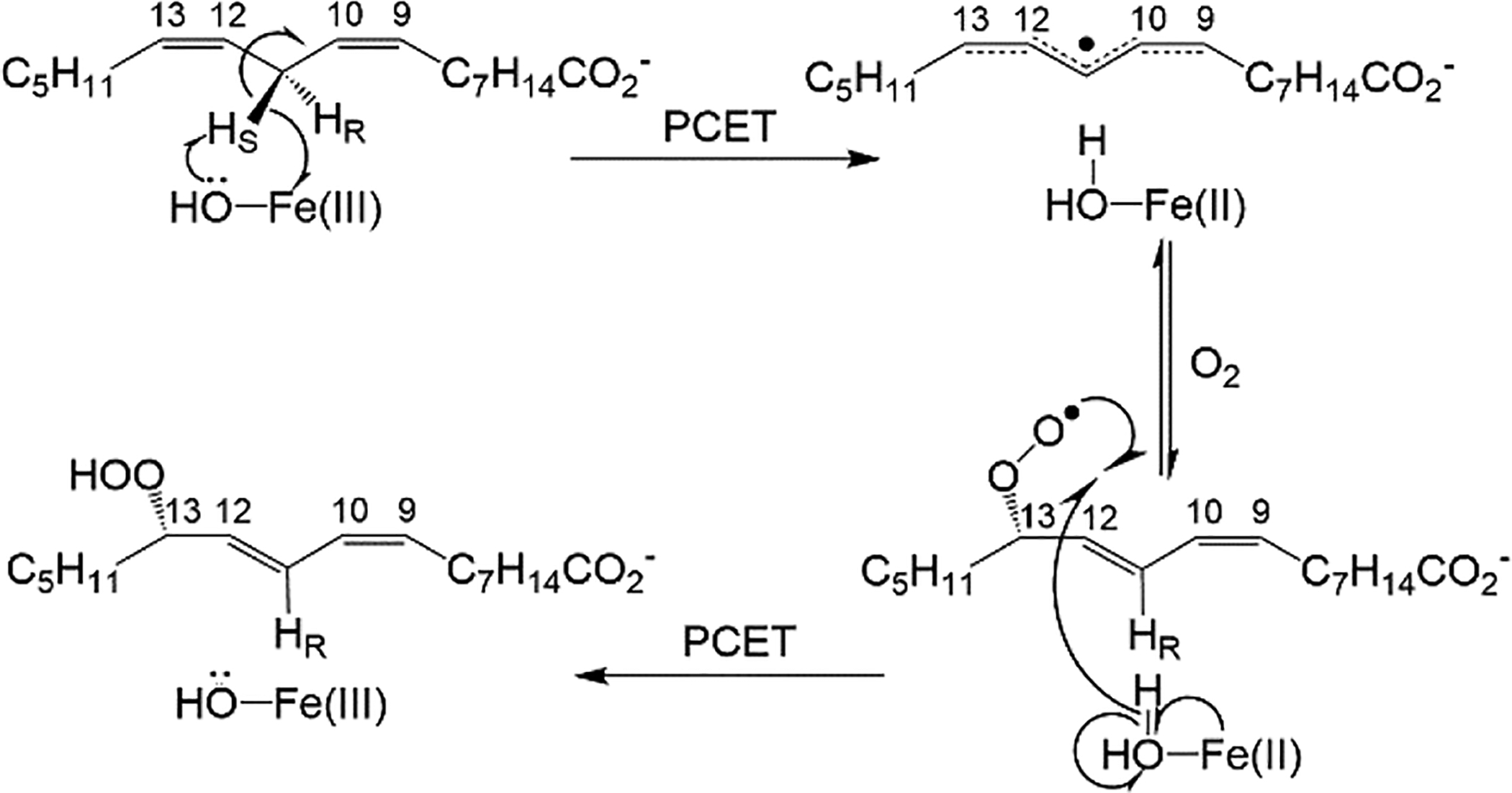
Mechanism of linoleic acid (per)oxidation by SLO is initiated by a rate-determining hydrogen abstraction of pro-S hydrogen at carbon-11 (C11). Both the C-H abstraction and subsequent O2 insertion are associated with PCET processes. Select carbons are numbered for reference. In WT SLO, O2 insertion is site-selective at carbon-13. The DAD relevant to this study is between C11 of LA and oxygen from the metal-bound hydroxide.
While the mounting kinetic evidence from studies on the temperature dependence of the kinetic isotope effect, ΔEa, sheds light on the compactness of the TRS, it has remained unclear how the structure of the ground state enzyme-substrate (E-S) complex is modulated by size reducing mutations and, more importantly, how ground-state perturbations impact and correlate with the transient compaction of the DAD. Herein, we apply the high-resolution capabilities of electron-nuclear double resonance (ENDOR) spectroscopy to compare the conformational distributions of ground-state E-S complexes for the native (wild-type, WT) SLO to those of a series of selected six site-directed mutations .33, 34 In conjunction with averaged X-ray structures for SLO in the absence of substrate,22, 28, 35 ENDOR has the power to quantify distributions of active (conformer a) and inactive (conformer b) ground-state E-S complexes, allowing correlation of these distributions with catalytic function.31
We recently applied 13C ENDOR spectroscopy to SLO using a high-spin (S=5/2) Mn2+ spin-reporter substituted for the native Fe2+ cofactor. This approach provides an exceptional spatial ‘horizon’ of up to 6 Å for measuring the through-space Mn-13C metal-to-substrate-carbon distances in WT and in a number of SLO variants.34 While the Mn-SLO form is inactive, Mn behaves as a faithful structural surrogate for Fe, with nearly superimposable X-ray models of the Mn-substituted and native SLO enzymes (cf. Figure 2A);34 this behavior is consistent with findings from other lipoxygenase species and Mn/Fe substitution in Mn and Fe mononuclear superoxide dismutases.36, 37 The lack of activity in Mn-substituted Fe lipoxygenases is likely attributed to the intrinsic electronic properties of the metal ions.34, 38 The structural equivalence of the Mn and Fe enzymes, In conjunction with the agreement of ENDOR derived and X-ray derived ground state DADs in LOXs,39 indicate that metal substitution does not significantly influence substrate positioning.
Figure 2.
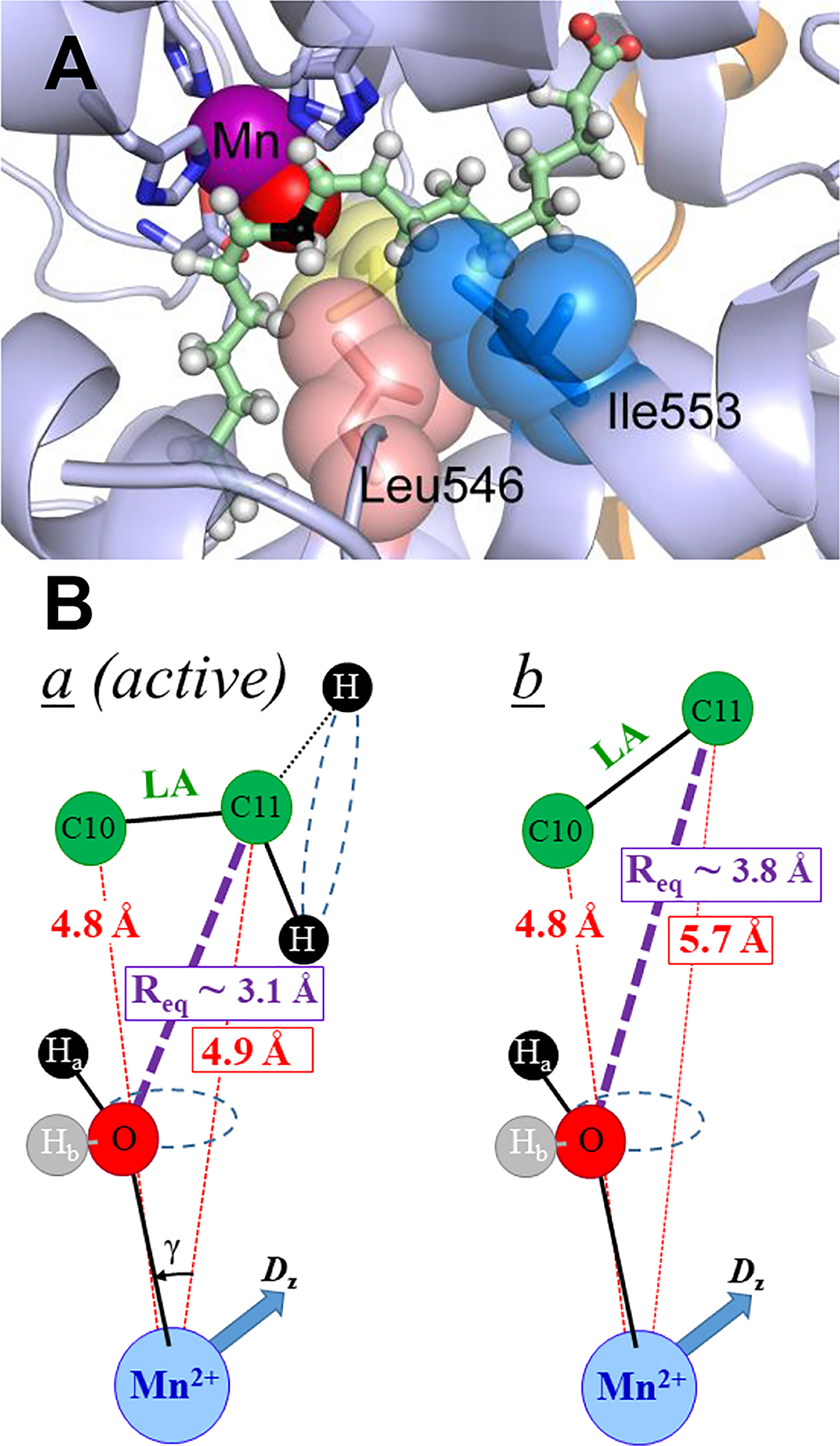
(A) 1.14Å structure of Mn-substituted SLO active site (PDB: 4WFO) resolved in the absence of substrate. Catalytically important residues are shown as spheres; yellow spheres represent Leu754. The substrate (green sticks; reactive carbon, C11 is in black) was modeled using previous ENDOR geometries of the dominant ground state conformer.31 The Mn2+(OH2) cofactor is shown as purple and red spheres, respectively. (B) ENDOR-derived model structures of the a and b conformers. Reproduced with permission from ref 34. Copyright 2017 J. Am. Chem. Soc.
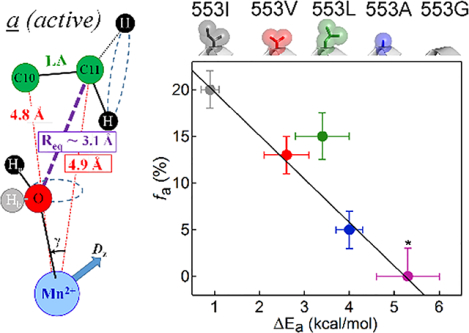
Using the developed ENDOR approach with specifically 13C enriched substrate, the reactive carbon (C11) of LA was shown to adopt two ground-state conformers, a and b in WT Mn-SLO, distinguished by different distances between the target C11 and the H-acceptor (metal-bound oxygen), whereas its neighbor, C10, had essentially the same position in both conformers (Figure 2B). The metal-to-carbon distances for C10 thus serves as an important reference in positioning LA with respect to the Mn-O vector. In the active, a conformer the 13C11 Mims pulsed ENDOR40 response yields a metal-C11 distance of rC11-Mn ≅ 4.9 Å, which corresponds to a van der Waals ‘C11↔O’ DAD of ~ 3.1Å (see Figure 2B and reference34 for details). The second, b, conformer has C11 positioned ~5.7 Å from Mn, nearly 1 Å farther than in a, and this has been assigned as an inactive ground state structure. Even within the active conformer, the observed van der Waals DAD distance (3.1Å) is significantly beyond that for efficient H-atom wave function overlap (2.7Å). This property highlights the importance of conformational sampling that transiently compacts the DAD from van der Waals ground state structures (conformer a) to the reduced DADs of a*, together with a thermally activated environmental reorganization that creates the degenerate TRSs from which facile tunneling can proceed.3, 6, 41
In the initial ENDOR study of SLO, an extreme active-site single mutation, Ile553Gly, showed impaired catalysis and correspondingly eliminated the occupancy (signal) of the a conformer,34 suggesting an important link between ground state (conformer a) substrate alignment and the enzyme’s ability to reach a finely tuned TRS. Here, (i) we expand the ENDOR investigation of enzyme control of substrate positioning in SLO to include the complete Ile553X series of aliphatic mutations that vary with side chain size and shape. The results reveal a remarkable correspondence between the ability of an enzyme to achieve a closely packed ground state E-S conformer a, and its ability to access an activated protein substate, a*, characterized by short DADs and high frequencies for DAD sampling. In addition, (ii) we examine the influence on substrate positioning of amino acid side chains identified within a dynamical protein network that links the buried active site to the remote solvent-protein interface.
METHODS
ENDOR Sample Preparation:
Wild-type and mutant SLOs were expressed in E. coli BL21(DE3) Codon Plus RIL cells in M9-based minimal medium, depleted of iron and supplemented with manganese chloride (99.99%) as previously described.34 SLO proteins were purified as described elsewhere. All proteins were 90% pure as assessed by SDS-PAGE analysis and contained 0.7–0.9 Mn per protein as determined by ICP-OES. ENDOR samples were prepared anaerobically. Each sample contained 0.5–0.7 mM Mn-substituted SLO and 1 equiv. of respective LA in 0.1 M sodium borate, pH 9.0, with 15% (v/v) ethylene glycol.
EPR/ENDOR Measurements:
Q-band echo-detected EPR spectra (two-pulse echo sequence, π/2-τ-π-τ-echo) and Davies/Mims pulsed ENDOR34 spectra were collected on a spectrometer that has been described,42, 43 whose microwave amplifier had been replaced, yielding higher powers. All measurements were done with a helium immersion dewar, at 2 K. The Mims pulsed ENDOR sequence (three-pulse echo sequence, π/2−τ−π/2−T−π/2−τ−echo, with rf pulse (Trf) inserted in the interval, T, between second and third pulses34) was used to probe the 13C hyperfine coupling of C10, C11 nuclei of labeled LA substrate.
As described in SI, we have refined the data acquisition and spectrum decomposition process in three ways. (i) With the availability of enhanced microwave power, we were able to maximize the Mims ENDOR S/N ratio of 13C ENDOR response by optimizing, (increasing) τ, the interval, in the Mims ENDOR sequence (Figure S3); this causes minimal changes in line shapes. (ii) The signals from the selectively 13C labelled substrate were isolated from those of natural-abundance 13C by subtraction of a corresponding spectrum with the natural-abundance substrate (Figures S4, S5). (iii) The simulation program previously used overestimated the contributions of the a conformer, so as described in SI, a purpose-written (ad hoc) program was written that corrects that problem.
RESULTS AND DISCUSSION
Mn2+ EPR:
The S = 5/2 Mn2+ ion of Mn-SLO complexed with LA experiences a small zero-field splitting interaction, and its resulting EPR spectrum is the sum of the ‘envelopes’ of the five orientation-dependent Δms = ± 1 transitions between adjacent pairs of the six electron-spin ms substates, −5/2 ≤ ms ≤ +5/2. Each envelope has a well-defined shape that is spread over a range of fields defined by its ms values and the magnitudes of the zero-field splitting (ZFS) parameters, and each envelope is weighted by the thermal population of the contributing ms levels.44 As reported previously,34 the EPR spectra collected from LA complexes of Mn-SLO are characteristic of a small ZFS, with the principal ZFS parameter, D, much less than the microwave energy at Q-band (35 GHz):45 These spectra show an intense central feature that is associated with transitions between the ms = +1/2 and −1/2 electron-spin substates, and that exhibits a sextet arising from 55Mn (I = 5/2) hyperfine interactions, A ~ 91 G. This feature “rides on” and is flanked by significantly broader signals from the four “satellite” envelopes involving the other electron-spin substates (ms ±5/2 ⇔ ±3/2; ±3/2 ⇔ ±1/2; Figure S1.
The EPR spectra of the LA complexes of WT SLO and the four mutants characterized herein (Ile553X) closely overlay (Figure S1), which indicates that the mutations cause negligible change to the Mn2+ ion and its coordination sphere, reflected in the ZFS parameters; as reported earlier, this is the case for the I553G variant, which has the most extreme change in the size of the hydrophobic side chain.34 Such behavior is expected, because the site of mutation Ile553, though near the substrate, is far from the metal ion. Nonetheless, as more precise and detailed confirmation that the mutations do not perturb the catalytic metal ion, we tested for changes in the positions/orientation of the Mn2+-coordination sphere ligands, including histidyl nitrogen’s and H2O, through comparison of the 14N, 1H Davies ENDOR spectra of WT-SLO and SLO-I553G.34 In all cases the data show that the ZFS tensor is unchanged.
13C ENDOR Protocol:
A 13C of labelled LA experiences an electro-nuclear dipolar interaction that arises from through-space dipolar interaction between the spin of the Mn2+ center and the 13C. Although this spin is partially delocalized over the ligands to Mn2+, for the interaction with LA 13C, the components of this interaction matrix can be written in its principal axis coordinate frame as a point dipole coupling, T(r), with the Mn2+ ion of SLO whose magnitude is determined by the 13C dipolar parameter, T(r), which is fixed by the length (r) of the Mn2+ → 13C vector (r):
| Eq 1 |
The ZFS interaction of the S = 5/2 Mn2+ ion causes the EPR spectrum at each field within the EPR envelope to arise from a well-defined set of orientations of the molecular frame, as defined by the ZFS tensor, relative to the external magnetic field. This in turn allows the collection of ENDOR spectra in which the external field has a well-defined orientation, or set of orientations, relative to the Mn-13C vector, r, for labelled LA. As a result, the orientation of r relative to the ZFS tensor axes, and the distance r (as derived from T(r)) can be determined for a frozen solution through collection/analysis of a 2D field-frequency Mims ENDOR pattern across the EPR envelope34, 46 Figure S2 shows such a 2D pattern, and its simulation, for 13C10 of LA bound to the WT SLO. The simulations incorporate the low temperature of our experiments, 2 K, which causes the EPR signal at the low-field edge of the Mn2+ EPR spectrum to be associated with the ms −5/2 ⇔ −3/2 envelope, and to arise from molecular orientations in which the field lies along the y-direction of the ZFS tensor. A key result of these simulations is that the (Mn2+ → 13C10/11) vectors lie essentially in the x–z plane of the ZFS tensor. As a consequence of the simplification provided by this orientation, in ENDOR spectra collected for both 13C10/11 at the low-field edge of the EPR spectrum, the magnetic field is perpendicular to the Mn-13C10/11 vectors, and one observes an essentially single-crystal like spectrum, a simple doublet whose offsets from the 13C Larmor frequency, δv(ms), directly yield the dipolar parameter, T(r) and thus the Mn-13C distance, r (eq 2),
| Eq 2 |
where the ms are the electron-spin quantum numbers associated with the ms −5/2 ⇔ −3/2 envelope.
When an observed spectrum is a superposition of signals from two (or more) conformers, it can be decomposed through simulation into sums of spectra for the individual conformers, giving the percentage contributions, fi and the distance, ri, for each conformer, i. As described in Materials and Methods and SI, in this study, we have obtained 13C ENDOR spectra with substantially higher signal/noise ratio (Figure S3) and have developed superior protocols for their decomposition into the fractional contributions from individual conformers than used previously (Figure S4, S5, S6, S7). As described in SI, the previous protocol gave the correct Mn-13C distances, but overestimated the a conformer contribution. As an example of the improved analysis, Figure 3A presents the 13C11 Mims ENDOR spectrum for native SLO and its decomposition into contributions from a and b conformers: ra = 4.99 Å, fa = 20%; rb = 5.65 Å, fb = 80% (Table S1, S2); this contrasts with a value for fa = 50% computed in the original study (the ra is unchanged). Similarly, the previous simulations suggested the presence of a majority conformer of 13C10 in WT enzyme at a Mn-13C10 distance of 4.9 Å, plus a small minority at a longer distance; the current protocol shows that within error, C10 occupies a single position at the same distance of 4.9 Å (Figure S6). The improved decomposition protocol applied to the enhanced 13C ENDOR spectra has enabled a robust comparison of the conformational occupancies across the suite of SLO variants interrogated. However, we emphasize that the trends in occupancy, as reported herein, are independent of the decomposition protocol employed.
Figure 3.
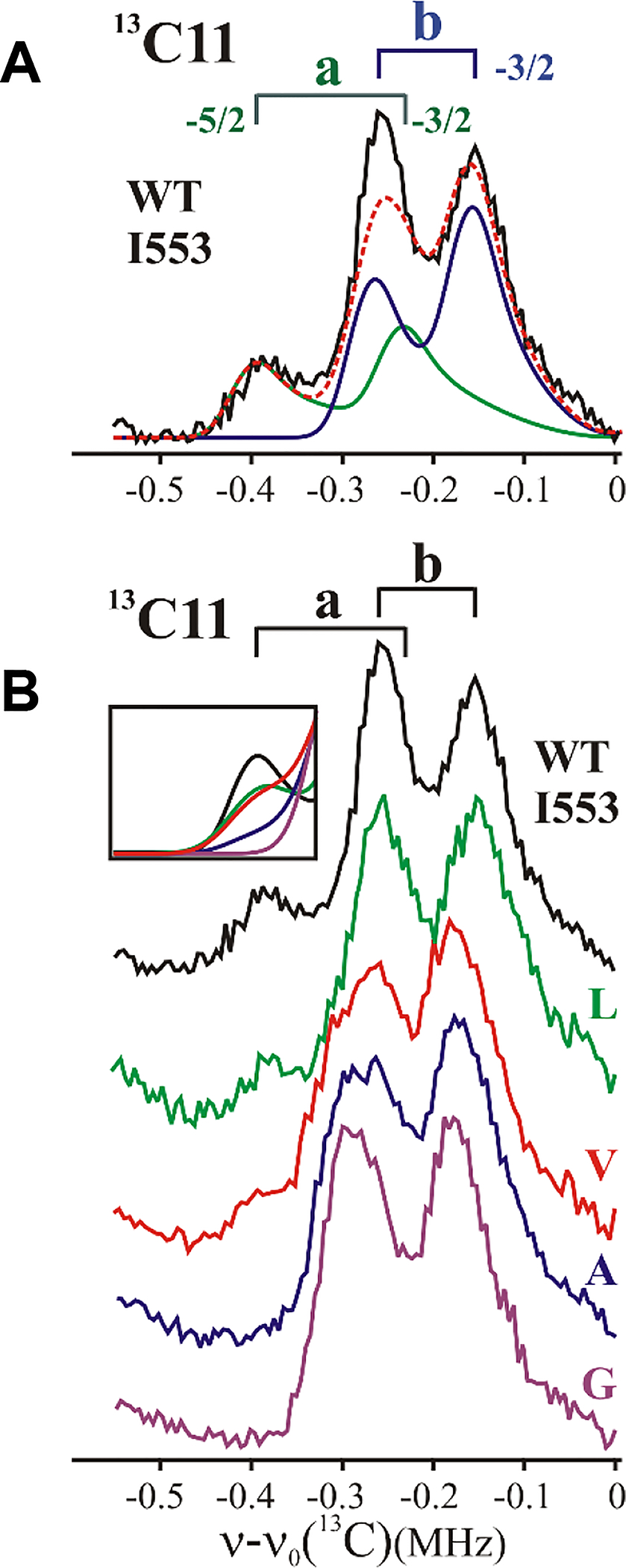
13C Mims ENDOR spectrum for (A) WT SLO and decomposition into a, b conformers (see Materials and Methods and SI); (B) Ile553X variants. Inset: overlay of simulations containing percentage of a conformer, fa (Figure S7). simulation parameters, Tables S1, S2. Mims ENDOR conditions, magnetic field = 11000 G, microwave frequency ~34.8 GHz, MW pulse length (π/2) = 50 ns, τ = 1500 ns, repetition rate = 100 Hz, and T = 2 K.
An additional parameter in the simulations is the ENDOR linewidth, which carries important information about the precise substrate positioning. As described previously,34 the linewidth of an ENDOR signal for the 13C, W(ms), is the sum of intrinsic contributions plus a contribution from a distribution in Mn2+−13C distances around the average distance, r, which leads to a distribution in ENDOR offsets from the 13C Larmor frequency, δv(ms) (eq. 2). These two contributions cannot be determined individually for WT enzyme, but we can assign a change in linewidth, δW, caused by mutation to a change (δr) in the distribution of distances through use of the following relationship for the features in the low-field, single-crystal-like Mn-SLO/LA(13C) ENDOR spectra:
| Eq. 3 |
Size/Shape of a Single Active Site Sidechain Strongly Impacts Ground-State Substrate Positioning:
Having previously established that replacing the large sec-butyl aliphatic sidechain of Ile553 with the H-atom of Gly in SLO leads to a complete loss of the 13C11 signal characteristic of the reactive ground-state conformer a, namely fa → 0,34 we posed the question whether a systematic variation in the 553 sidechain volume would lead to detectable trends in fa. Figure 3B presents 13C11 Mims ENDOR spectra for active-site Ile553X SLO variants using the complete series of naturally available aliphatic sidechains ranging from Ile (wild-type) to Gly. As is visually evident in these spectra, their decomposition into contributions from a and b conformers (Figure S7) reveals a monotonic decrease in occupancy of the active a conformer with decreasing sidechain volume (Figure 4A), starting with an occupancy of fa ~20% of the a conformer for Ile (WT) and ending with an undetectable amount of this conformer for Gly; the relative occupancy of the unreactive b conformer, whose DA D is almost 1 Å greater, increases correspondingly.
Figure 4.
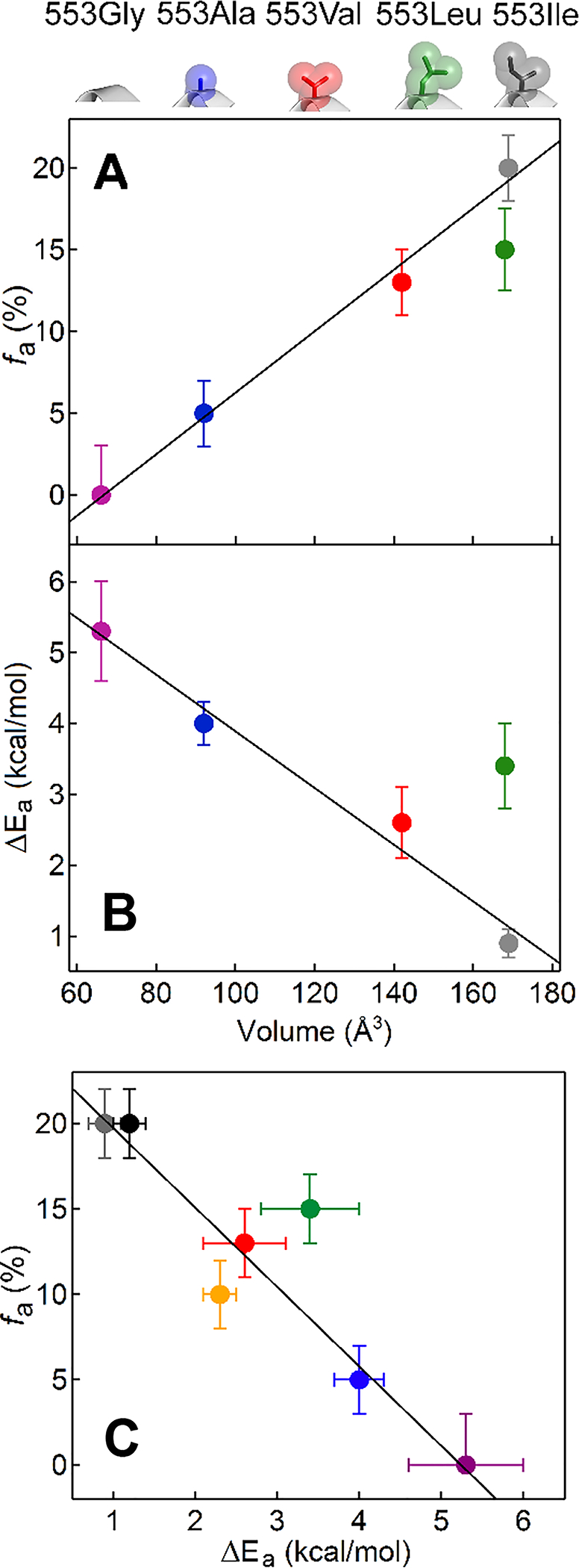
(A) ENDOR-detected fa versus 553 volume. For reference, the structures of the sidechains for the ENDOR data are shown at the top of the panel and color coded. (B) Kinetic-determined ΔEa versus 553 volume. (C) ENDOR-detected fa versus kinetic-determined ΔEa. In addition to the color coding for Ile553X variants, the black and gold points represent the Tyr317Ser and Tyr317Leu variants (cf. Figure 5 and S9), respectively. In all panels, linear fits (black lines) were determined in the absence of the outlier Ile553Leu.
Within the limits of resolution, the ENDOR offset, δv, (eq (2)), for the 13C11 a conformer, and thus its calculated Mn-13C11 distance, r, does not vary as residue 553 varies; the offset for the b conformer slightly increases for the smaller residues, indicating a slight shortening of the longer distance in this conformer. However, for both conformers the 13C11 ENDOR linewidths (W), and thus the distributions in Mn-13C11 distances, δr (eq 3), increase for every sidechain with volume smaller than 553Ile (Figure 3B, Table S1). Note that the absence of mutation-induced changes in the Mn2+ coordination sphere discussed above means that such changes cannot be the source of the broadening of the 13C Mims ENDOR lines. This broadening instead reveals a ‘softening’ of the minimum on the potential-energy surface for LA binding. This softening is functionally significant, since such behavior is postulated to play a key role in enabling enhanced DAD sampling among catalytically impairing I553X variants with reduced side chain volume (Figure 1D, top panel).
Considering 13C10, the ENDOR offset/Mn-13C10 distance of this carbon also remains largely unchanged in this series of variants. The 13C ENDOR linewidth and thus the distribution in Mn-13C10 distances is unchanged compared to WT for X = Val, but the linewidth increases modestly for X = Ala, and substantially for X = Gly (Figure S6, S8) indicating an increase in the distance distributions, δr (Eq 3, Table S3).
As an important corollary experiment, 13C10/11 ENDOR spectra were collected for LA bound to Ile553Leu, a mutation of identical sidechain volume but different shape than the native isoleucine residue. This mutation decreases the occupancy of conformer a compared to WT by an amount comparable to that caused by Ile553Val (Figure 4). Thus, unsurprisingly, the a conformation occupancy is not merely responsive to the side chain volume (Table S1 and Figure 4A). The result with Ile553Leu instead indicates that substrate positioning is finely tuned by subtle interactions that vary with the shape of substituents on the β-carbon of 553 of equal volume. Perhaps more importantly for function, even this iso-volume substitution ‘softens’ the potential-energy minimum on the substrate-binding surface for the active conformation, as evidenced by an increased ENDOR linewidth, that once again is correlated with the elevation of its ΔEa above that seen for WT enzyme.
Correlation Between fa and ΔEa:
The kinetic features that emerged from mutational studies of the 553 sidechain in SLO have already played an important role in the development of prevailing theoretical models for nonadiabatic C-H activation reactions.1, 2, 28, 47 These variants show very modest rate and kinetic isotope effect differences with random trends in Ea. The major difference is a systematic trend in ΔEa with sidechain volume (Figure 4B), from 0.9 kcal/mol in Ile553 to 5.3 kcal/mol in Ile553Gly (Table S2). As shown in Figure 4C, the decrease in the percentage of a conformer (fa) from Ile553 to Ile553Gly parallels the increase in the experimental ΔEa. These results support the conclusion that tight packing within the ground state a conformer of the WT enzyme is sufficient to promote effective wave function overlap upon formation of the TRS (Figure 1B, purple dot and 1C) with little-to-no contributions from the DAD sampling coordinate (Figures 1D, bottom). Moreover, as indicated by the decreased fa and accompanying increased ENDOR linewidth, LA in the single site mutants is less precisely positioned (softer potential energy surface) in the ground state conformer a structure. As a consequence of the correspondingly observed increases in the ΔEa for hydrogen transfer, this ground state effect is proposed to be propagated into a longer DAD at conformer a* with softer DAD sampling coordinate, which together lead to the increased contribution of DAD thermal sampling (Figure 1D, top).
The ENDOR measurements thus provide compelling evidence for a close correspondence between the ability of an enzyme active site to stabilize a closely packed ground state conformation a, with favorable van der Waals DADs, and the ability to access a compacted conformer a* that can achieve the TRS with short DADs that results in little contribution from the DAD sampling coordinate.
Mutation Within a SLO Protein Dynamical Network and Control of Substrate Positioning by Residues Far from the Active Site:
Reports on conformational networks that link sites remote from the active site to active-site structure have increasingly emerged.48–51 Recently, a hydrogen deuterium exchange (HDX) study on SLO identified a solvated loop, 317–334, lying at the surface of the protein, at least 15Å from the active site, with mutations in this loop influencing H-atom transfer.22
A model of the SLO structure shows a network of residues that extends from the active site to this loop at the protein surface (Figure 5A). This network originates with Ile553 and Leu546 at the active site (Figure 2A). Continuing from Ile553 to the solvent via the proposed dynamical network it is possible to identify a single hydrogen bond between the hydroxyl functional groups of Ser749 and Tyr317, with the Tyr317 residing closer to the protein surface. Substitution of Tyr317 by the hydrophobic side chain Leu breaks this network and generates kinetic properties that differ from WT enzyme and instead are similar to the kinetically impaired active-site Ile553Leu mutant.22, 28 Remarkably, the 13C11 ENDOR spectrum for the remote Tyr317Leu mutant also shows a corresponding decrease in a conformer occupancy (Figure 5B), with a magnitude of change similar to the decrease for Ile553Leu (Figure 4A).
Figure 5.
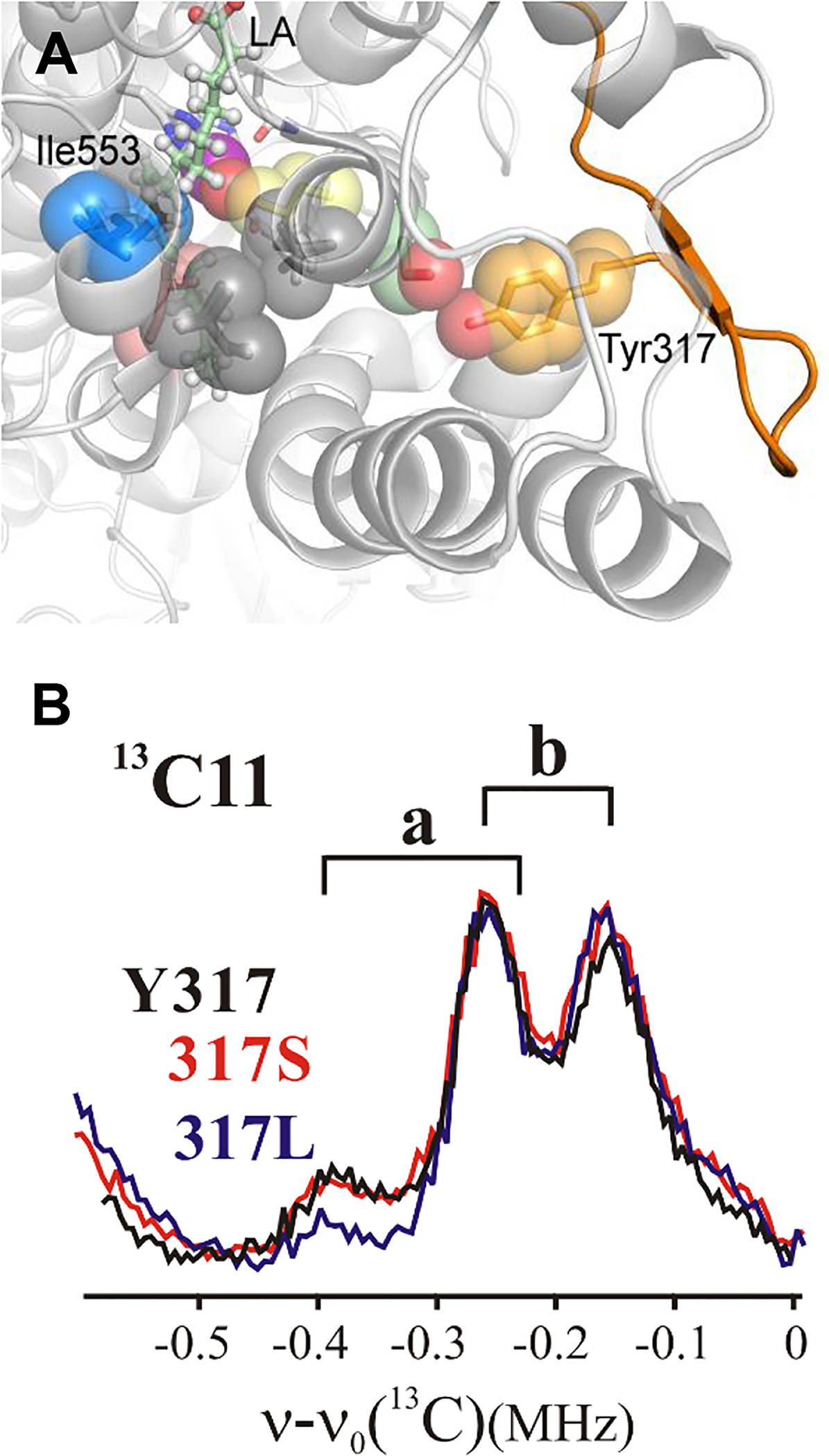
(A) Catalytically-linked protein network from solvated loop (orange cartoon) to the active site. The gray colored residues near the active site, Ile552 and Val750, have not been pursued in the present study. Kinetics for Tyr317X variants are presented in Table S4. (B) Background-subtracted 13C11 Mims ENDOR spectra for Tyr317 mutants compared to WT; Conditions as in Figure 3.
Given the remoteness of Tyr317 from the active site, it was, important to determine whether the impact of inserting Leu at this position might reflect a ‘generic’ effect of remote mutation, and thus be unrelated to a break in the H-bonding connectivity along the network. A more conservative variant, Tyr317Ser had been generated on the premise that this substitution would preserve the H-bonded network; this expectation was borne out by the observation of its kinetic properties similar to native enzyme.22 Notably for the present study, the 13C11/10 ENDOR spectra of the Tyr317Ser variant likewise shows both an a occupancy and ENDOR linewidths identical to those of the WT enzyme (Figure 5B). In short, a non-conservative mutation of hydrophilic Tyr317 to the hydrophobic, non-H-bonding Leu (Tyr317Leu) perturbs both enzyme kinetics and substrate conformation (Figure 4C), whereas mutation to a conservative hydrophilic, H-bonding serine (Tyr 317Ser) changes neither.
This correlation between kinetic and ENDOR behavior in fact buttresses the earlier HDX-MS finding, of a spatially resolved dynamical network in SLO that emerged from data in the absence of bound substrate (to circumvent an anticipated protective effect of LA on the H/D exchange process). The correlation of ΔEa with the ENDOR results for the E-S complex of Tyr317Leu thus indicates that the HDX-derived conformational landscape for apo-SLO is indeed relevant to catalytic complexes of enzyme. This conclusion in turn is compatible with findings from Kern and co-workers, who have shown that an overall protein conformational landscape can be quite similar for substrate free vs. substrate bound enzyme forms, although in detail, substrate is expected to shift the relative distribution of each conformer within the ensemble.52
Interrelationship of Kinetic and ENDOR Measurements:
The present study enables the determination of hydrogen transfer DADs in ground-state (conformer a) E-S complexes that could not be visualized by X-ray crystallography.39 The ENDOR-derived a conformer for WT SLO and its variants places the target C11 of LA within a van der Waals DAD in the ground state. This positioning does not correspond to a TRS, but the occupancy of such an ‘active’ conformer (conformer a) is considered a prerequisite for efficient tunneling (Figure 1).53 In the context of theoretical formalisms for nonadiabatic C-H activation reaction,3, 5, 6, 32 multiple tiers of distance and energetic optimization are necessary to produce the tunneling. The system first adopts the reactive, ground-state E-S configuration (designated a; Figure 1A), then reaches weakly populated compacted ground state configurations on the reaction coordinate (a*: Figure 1A–C) via conformational sampling. The transition from a* (Figure 1B green dot) to the TRS (Figure 1 Band C purple dot) is controlled by the Marcus reorganization energy (λ) associated with the tunneling coordinate. As C-H activating enzymes have evolved, they have attained highly tuned active sites generally characterized by reduced DADs for conformer a* that are close to the dominant protium tunneling distance of 2.7 – 2.9 Å. As such, native enzymes exhibit relatively little need of DAD sampling for either H- or D-transfer, producing the characteristic ΔEa ~ 0 (Figure 1D, E, bottom).
However, single site ‘impairing’ SLO mutants (e.g. Ile553X) are often associated with modest impacts on kcat and Dkcat, along with measurable increases in the ΔEa. The results presented in Figure 4C show a previously undetected correlation between increasing ΔEa and decreasing fa, with the near-zero ΔEa for WT SLO corresponding to the maximum fa seen for any of the variants studied here. Most significantly, the ENDOR data indicate an inverse correlation between the fractional population of active conformer a corresponding to van der Waals DADs and the magnitude of ΔEa. This both supports the view that the a conformation, with its van der Waals DAD, is the catalytically relevant ground-state structure, and shows that the ability to achieve a close positioning of the substrate target in the ground state (fa) translates to small values for ΔEa.
Pursuing this further, the available parameters, Dkcat and ΔEa, can be used in combination with the analytical expression derived from the nonadiabatic formalism that calculates the rate constant for PCET, kPCET,5 to yield two physical parameters: the equilibrium DAD associated with conformer a* and the frequency of the DAD sampling coordinate, ω. The corresponding values32 for the SLO Ile553X series (Table S2) are plotted in Figure 6, showing that decreases in fa, which are correlated with increases in ΔEa (Figure 4), likewise are associated with elongations of the DAD at conformer a* (Figure 6A) and a softening of the DAD sampling potential (i.e. reduced frequency for the DAD sampling mode, ω; Figure 6B).
Figure 6.
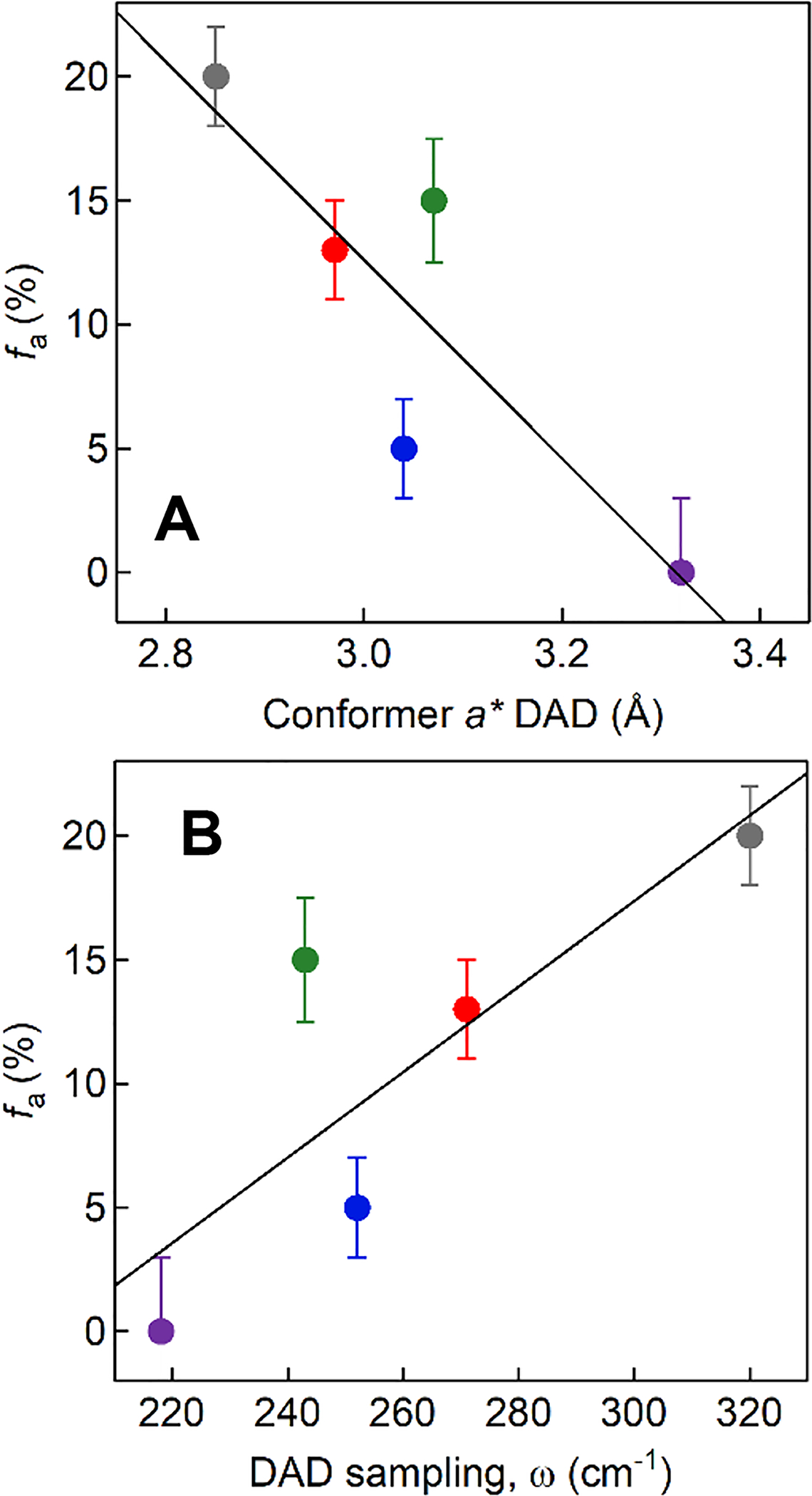
ENDOR-detected fa versus the computed values for the DADs in conformer a* (A) and the frequency of the DAD sampling mode, ω (B). The color coding for Ile553X variants is same as in Figure 4: WT, gray; Ile553Val, red; Ile553Leu, green; Ile553Ala, blue; Ile553Gly, purple. Values for a* and ω were determined using the analytical rate expression for the rate constant for PCET, kPCET, that includes quadratic terms;5 the values are listed in Table S2. The data are illustrative and were obtained using a harmonic potential for the DAD sampling mode. Analysis via a Morse potential is expected to lead to small changes in the absolute values for a* and ω,53 but not in the observed trends.
We propose that the magnitude of ΔEa offers a “ruler” for the degree of active-site compactness in a* with native enzymes and its subsequent impairment in activity-impacting mutants.15, 54 This is supported by the impact of active-site packing defects generated by mutation of single protein side chains both proximal to and remote from the reacting bonds. These subtle changes systematically suppress the optimized ground-state substrate configuration (conformer a) and this effect is propagated into elongated DADs at conformer a*. Some of the perturbation at a* can be recovered by the onset of DAD sampling coordinate that is amplified for deuterium relative to protium transfer (Figure 1-D, top).
CONCLUSIONS
The key finding in the present work is the strong inverse correlation between the occupancy of the active ground state structure, fa and the dependence of effective H- tunneling on enhanced distance sampling, as revealed in the difference between the enthalpic barriers for D and H abstraction, ΔEa, (Figure 4C). This linkage shows the critical role of ground state positioning in achieving correspondingly optimized TRSs that enable efficient quantum H-transfer. The active-conformation occupancy, fa, is not only highly sensitive to packing defects generated by mutation of a single hydrophobic side chain (Ile553) in contact with bound substrate (Figure 3), but also is modulated by a remote residue through the previously identified,22 long-range, catalytically-linked protein network (Figure 5).
The data show that any perturbation, even a quite small one, can alter both the fraction of reactive conformers in the ground state (conformer a) and the disposition of the donor and acceptor as the enzyme-substrate complex moves from conformer a to conformer a* and on to the TRS along the Marcus reaction coordinate. It appears that the mutant enzymes are simply never able to fully recover a WT TRS state from their impaired conformer a* geometries. In the case of protium transfer, the impact on rate can be fairly minimal, but the shorter wavelength spread for D within the impaired conformer a* state dictates a greater dependence on DAD sampling.
The present results, underscored by mounting recent experimental evidence,6, 55 illustrate how the study of tunneling in C–H activation reactions affords a unique and incisive window into the geometric and dynamic contributions that control enzymatic fitness. This study further illustrates how ENDOR provides an exquisitely sensitive probe of enzyme control of substrate conformation, revealing functionally significant changes in the ground state E-S structure generated by subtle ‘point’ (single-residue) perturbations.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank an anonymous reviewer whose remarks helped to markedly improve the manuscript.
Funding Sources
Financial support was provided by the National Institutes of Health (NIH): GM111097 to BMH; and GM025765 to JPK. ARO was supported by NIH GM113432.
ABBREVIATIONS
- SLO
soybean lipoxygenase
- ENDOR
electron nuclear double resonance
- TRS
tunneling-ready state
- DAD
donor-acceptor distance
Footnotes
Supporting Information. ENDOR spectral fits, tabulated kinetic parameters and ENDOR-derived fa for WT and SLO variants. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession codes
SLO, UniProt P08170
REFERENCES
- [1].Kuznetsov AM, and Ulstrup J (1999) Proton and hydrogen atom tunneling in hydrolytic and redox enzyme catalysis, Can. J. Chem 77, 1085–1096. [Google Scholar]
- [2].Knapp MJ, Rickert KW, and Klinman JK (2002) Temperature-dependent isotope effects in soybean lipoxygenase-1: correlating hydrogen tunneling with protein dynamics, J. Am. Chem. Soc 124, 3865–3874. [DOI] [PubMed] [Google Scholar]
- [3].Hammes-Schiffer S (2006) Hydrogen tuneling and protein motion in enzyme reactions, Acc. Chem. Res 39, 93–100. [DOI] [PubMed] [Google Scholar]
- [4].Klinman JK (2006) Linking protein structure and dynamics to catalysis: the role fo hydrogen tunneling, Phil. Trans. R. Soc. B 361, 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Soudackov AV, and Hammes-Schiffer S (2016) Proton-coupled electron transfer reactions: analytical rate constants and case study of kinetic isotope effects in lipoxygenase, Faraday Discuss. 195, 171–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klinman JP, Offenbacher AR, and Hu S (2017) Origins of enzyme catalysis: experimental findings for C-H activation, new models, and their relevance to prevailing theoretical constructs, J. Am. Chem. Soc 139, 18409–18427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Basran J, Patel S, Sutcliffe MJ, and Scrutton NS (2001) Importance of barrier shape in enzyme-catalyzed reactions. Vibrationally assisted hydrogen tunneling in tryptophan tryptophylquinone-dependent amine dehydrogenases, J. Biol. Chem 276, 6234–6242. [DOI] [PubMed] [Google Scholar]
- [8].Francisco WA, Knapp MJ, Blackburn NJ, and Klinman JP (2002) Hydrogen tunneling in peptidylglycine a-hydroxylating monooxygenase, J. Am. Chem. Soc 124, 8194–8195. [DOI] [PubMed] [Google Scholar]
- [9].Maglia G, and Allemann RK (2003) Evidence for environmentally coupled hydrogen tunneling during dihydrofolate reductase catalysis, J. Am. Chem. Soc 125, 13372–13373. [DOI] [PubMed] [Google Scholar]
- [10].Agrawal N, Hong B, Mihai C, and Kohen A (2004) Vibrationally enhanced hydrogen tunneling in the Escherichia coli thymidylate synthase catalyzed reaction, Biochemistry 43, 1998–2006. [DOI] [PubMed] [Google Scholar]
- [11].Sikorski RS, Wang L, Markham KA, Rajagopalan PT, Benkovic SJ, and Kohen A (2004) Tunneling and coupled motion in the Escherichia coli dihydrofolate reductase catalysis, J. Am. Chem. Soc 126, 4778–4779. [DOI] [PubMed] [Google Scholar]
- [12].Klinman JK (2009) An integrated model for enzyme catalysis emerges from studies of hydrogen tunneling, Chem. Phys. Lett 471, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stojkovic V, Perissinotti LL, Willmer D, Benkovic SJ, and Kohen A (2012) Effects of the donor-acceptor distance and dynamics on hydride tunneling in the dihydrofolate reductase catalyzed reaction, J. Am. Chem. Soc 134, 1738–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sühnel J, and Schowen RL (1991) Theoretical basis for isotope effects, In Enzyme Mechasnism from Isotope Effects (Cook PF, Ed.), 3–35, CRC Press, Boca Raton, Fl. [Google Scholar]
- [15].Klinman JP, and Offenbacher AR (2018) Understanding biological hydrogen transfer through the lens of temperature dependent kinetic isotope effects, Acc. Chem. Res 51, 1966–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cha Y, Murray CJ, and Klinman JK (1989) Hydrogen tunneling in enzyme reactions, Science 243, 1325–1330. [DOI] [PubMed] [Google Scholar]
- [17].Kohen A, Cannio R, Bartolucci S, and Klinman JP (1999) Enzyme dynamics and hydrogen tunneling in a thermophilic alcohol dehydrogenase, Nature 399, 496–499. [DOI] [PubMed] [Google Scholar]
- [18].Kohen A (2003) Kinetic isotope effects as probes for hydrogen tuenneling, coupled motion and dynamics contributions to enzyme catalysis, Prog. React. Kinet. Mech 28, 119–156. [Google Scholar]
- [19].Pudney CR, Johannissen LO, Sutcliffe MM, Hay S, and Scrutton NS (2010) Direct analysis of the donor-acceptor distance and relationship to isotope effects and the force constant for barrier compression in enzyme H-tunneling reactions, J. Am. Chem. Soc 132, 11329–11335. [DOI] [PubMed] [Google Scholar]
- [20].Hu S, Sharma SC, Scouras AD, Soudackov AV, Carr CAM, Hammes-Schiffer S, Alber T, and Klinman JK (2014) Extremely elevated room-temperature kinetic isotope effects quantify the critical role of barrier width in enzymatic C-H activation, J. Am. Chem. Soc 136, 8157–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sharma SC, and Klinman JK (2015) Kinetic detection of orthogonal protein and chemical coordinates in enzyme catalysis: double mutants of soybean lipoxygenase, Biochemistry 54, 5447–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Offenbacher AR, Hu S, Poss EM, Carr CA, Scouras AD, Prigozhin DM, Iavarone AT, Palla A, Alber T, Fraser JS, and Klinman JP (2017) Hydrogen-deuterium exchange of lipoxygenase uncovers a relationship between the distal, solvent exposed protein motions and the thermal activation barrier for catalytic proton-coupled electron tunneling, ACS Cent. Sci 3, 570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nibali VC, and Havenith M (2014) New insights into the role of water in biological function: studying solvated biomolecules using terahertz absorption spectroscopy in conjunction with molecular dynamics simulations, J. Am. Chem. Soc 136, 12800–12807. [DOI] [PubMed] [Google Scholar]
- [24].Jimenez-Oses G, Osuna S, Gao X, Sawaya MR, Gilson L, Collier SJ, Yeates TO, Tang Y, and Houk KN (2014) The role of distal mutations and allosteric regulation on LovD active site dynamics, Nat. Chem. Biol 10, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Saavedra HG, Wrabl JO, Anderson JA, Li J, and Hilser VJ (2018) Dynamic allostery can drive cold adaptation in enzymes, Nature 558, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nguyen V, Wilson C, Hoemberger M, Stiller JB, Agafonov RV, Kutter S, English J, TTheobald DL, and Kern D (2017) Evolutionary drivers of thermoadaptation in enzyme catalysis, Science 355, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zaragoza JP, Nguy A, Minnetian N, Deng Z, Iavarone AT, Offenbacher AR, and Klinman JP (2019) Detecting and characterizing the kinetic activation of thermal networks in proteins: Thermal transfer from a distal, solvent-exposed loop to the active site in soybean lipoxygenase, J. Phys. Chem. B [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meyer MP, Tomchick DR, and Klinman JK (2008) Enzyme structure and dynamics affect hydrogen tunneling: the impact of a remote side chain (I553) in soybean lipoxygenase-1, Proc. Natl. Acad. Sci. U. S. A 105, 1146–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benabbas A, Salna B, Sage JT, and Champion PM (2015) Deep proton tunneling in the electronically adiabatic and non-adiabatic limits: comparison of the quantum and classical treatment of donor-acceptor motion in a protein environment, J. Chem. Phys 142, 114101. [DOI] [PubMed] [Google Scholar]
- [30].Soudackov AV, and Hammes-Schiffer S (2015) Nonadiabatic rate constants for proton transfer and proton-coupled electron transfer reactions in solution: effects of quadratic term in the vibronic coupling expansion, J. Chem. Phys 143, 194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Salna B, Benabbas A, and Champion PM (2017) Proton-coupled electron transfer and the “linear approximation” for coupling to the donor-acceptor distance fluctuations, J. Phys. Chem. A 121, 2199–2207. [DOI] [PubMed] [Google Scholar]
- [32].Hu S, Soudachkov AV, Hammes-Schiffer S, and Klinman JP (2017) Enhanced rigidifcation within a double mutant of soybean lipoxygenase provides experimental support for vibronically nonadiabatic proton-coupled electron transfer models, ACS Catal 7, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang TC, Wolfe MD, Neibergall MB, Mekmouche Y, Lipscomb JD, and Hoffman BM (2003) Substrate binding to NO-ferro-naphthalene 1,2-dioxygenase studied by high-resolution Q-band pulsed 2H-ENDOR spectroscopy, J. Am. Chem. Soc 125, 7056–7066. [DOI] [PubMed] [Google Scholar]
- [34].Horitani M, Offenbacher AR, Carr CAM, Yu T, Hoeke V, Cutsail GEI, Hammes-Schiffer S, Klinman JK, and Hoffman BM (2017) 13C ENDOR spectroscopy of lipoxygenase-substrate complexes reveals the structural basis for C-H activation by tunneling, J. Am. Chem. Soc 139, 1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Minor W, Steczko J, Stec B, Otwinowski Z, Bolin JT, Walter R, and Axelrod B (1996) Crystal structure of soybean lipoxygenase L-1 at 1.4 A resolution, Biochemistry 35, 10687–10701. [DOI] [PubMed] [Google Scholar]
- [36].Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller A-F, Teixeria M, and Valentine JS (2014) Superoxide dismutases and superoxide reductases, Chem. Rev 114, 3854–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Newie J, Kasanmascheff M, Bennati M, and Feussner I (2016) Kinetics of a bis-allylic hydroperoxide synthesis in the iron-containing lipoxygenase 2 from Cyanothece and the effects of manganese substitution, Lipids 51, 335–347. [DOI] [PubMed] [Google Scholar]
- [38].Wennman A, Oliw EH, Karkehabadi S, and Chen Y (2016) Crystal structure of manganese lipoxygenase of the rice blast fungus Magnaporthe oryzae, J. Biol. Chem 291, 8130–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Neau DB, Bender G, Boeglin WE, Barlett SG, Brash AR, and Newcomer ME (2014) Crystal structure of a lipoxygenase in complex with substrate. The arachidonic acid-binding site of 8R-lipoxygenase., J. Biol. Chem 289, 31905–31913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schweiger A, and Jeschke G (2001) Principles of Pulse Electron Paramagnetic Resonance, Oxford University Press, Oxford, UK. [Google Scholar]
- [41].Li P, Soudachkov AV, and Hammes-Schiffer S (2018) Fundamental insights into proton-coupled electron transfer in soybean lipoxygenase from quantum mechanical/molecular mechanical free energy simulations, J. Am. Chem. Soc 140, 3068–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Davoust CE, Doan PE, and Hoffman BM (1996) Q-band pulsed electron spin-echo spectrometer and its application to ENDOR and ESEEM, J. Magn. Reson 119, 38–44. [Google Scholar]
- [43].Xu W, Lees N, Adedeji D, Wiesner J, Jomaa H, Hoffman BM, and Duin E (2010) Paramagnetic intermediates of (E)-4-hydroxy-3-methylbut-2–3nyl diphosphate synthase (GcpE/IspG) under steady-state and pre-steady-state conditions, J. Am. Chem. Soc 132, 14509–14520. [DOI] [PubMed] [Google Scholar]
- [44].Coffino AR, and Peisach J (1996) Simulation of Mn(II) EPR spectra using a full spin-Hamiltonian approach, J. Magn. Reson 111, 127–134. [DOI] [PubMed] [Google Scholar]
- [45].Magnusson OT, Reed GH, and Frey PA (1999) Spectroscopic evidence for the participation of an allylic analogue of the 5’-deoxyadenosyl radical in the reaction of lysine 2,3-aminomutase, J. Am. Chem. Soc 121, 9764–9765. [Google Scholar]
- [46].Hoffman BM, Martinsen J, and Venters RA (1984) General theory of polycrystalline ENDOR patterns. g and hyperfine tensors of arbitrary symmetry and relative orientation, J. Magn. Reson 59, 110–123. [Google Scholar]
- [47].Edwards SJ, Soudackov AV, and Hammes-Schiffer S (2010) Impact of distal mutation on hydrogen transfer interface and substrate conformation in soybean lipoxygenase, J. Phys. Chem. B 114, 6653–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang L, Goodey NM, Benkovic SJ, and Kohen A (2006) Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase, Proc. Natl. Acad. Sci. U.S.A 130, 15753–15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Fraser JS, Clarkson MW, Degnan SC, Erion R, Kern D, and Alber T (2009) Hidden alternative structures of proline isomerase essential for catalysis, Nature 462, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hekstra DR, White KI, Socolich MA, Henning RW, Srajer V, and Ranganathan R (2016) Electric-field-stimulated protein mechanics, Nature 540, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vaughn MB, Zhang J, Spiro TG, Dyer RB, and Klinman JP (2018) Activity-related microsecond dynamics revealed by temperature-jump Forster resonance energy transfer measurements on thermophilic alcohol dehydrogenase, J. Am. Chem. Soc 140, 900–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chakrabarti KS, Agafonov RV, Pontiggia F, Otten R, Higgins MK, Schertler GF, Oprian DD, and Kern D (2016) Conformational selection in a protein-protein interaction revealed by dynamic pathway analysis, Cell Reports 14, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Salna B, Benabbas A, Russo D, and Champion PM (2017) Tunneling kinetics and nonadiabatic proton-coupled electron transfer in proteins: the effect of electric fields and anharmonic donor-acceptor interactions, J. Phys. Chem. B 121, 6869–6881. [DOI] [PubMed] [Google Scholar]
- [54].Klinman JK, and Kohen A (2013) Hydrogen tunneling links protein dynamics to enzyme catalysis, Annu. Rev. Biochem 82, 471–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hu S, Offenbacher AR, Thompson EM, Gee CL, Wilcoxen J, Carr CA, Prigozhen DM, Yang V, Alber T, Britt RD, Fraser JS, and Klinman JP (2019) Biophysical characterization of a disabled double mutant of soybean lipoxygenase: the “undoing” of precise substrate positioning relative to metal cofactor and an identified dynamical network, J. Am. Chem. Soc 141, 1555–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.