

Abstract
The current need for novel antibiotics is especially acute for drug-resistant Gram-negative pathogens1,2. These microorganisms have a highly restrictive permeability barrier, which limits penetration of most compounds3,4. As a result, the last class of antibiotics acting against Gram-negative bacteria was developed in the 60s2. We reason that useful compounds can be found in bacteria that share similar requirements for antibiotics with humans, and focus on Photorhabdus symbionts of entomopathogenic nematode microbiomes. Here we report a new antibiotic that we name darobactin, from a screen of Photorhabdus isolates. Darobactin is coded by a silent operon with little production under laboratory conditions, and is ribosomally synthesized. Darobactin has an unusual structure with two fused rings that form post-translationally. The compound is active against important Gram-negative pathogens both in vitro and in animal models of infection. Mutants resistant to darobactin map to BamA, an essential chaperone and translocator that folds outer membrane proteins. Our study suggests that bacterial symbionts of animals harbor antibiotics that are particularly suitable for development into therapeutics.
It is difficult to find compounds acting against Gram-negative bacteria1,2. This problem is largely responsible for the antimicrobial resistance crisis we are currently experiencing. Pathogens such as Escherichia coli, Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii have acquired resistance to most, and in some cases to all, antibiotics currently available in the clinic. The WHO has recently classified these drug resistant pathogens as a critical priority for global human health5.
Gram-negative bacteria evolved an outer membrane to protect themselves from unwanted compounds3,4. The few molecules that penetrate across this barrier make up our shrinking arsenal of antibiotics acting against Gram-negative bacteria. Most of these compounds are natural products made by soil microorganisms, primarily Actinomycetes – aminoglycosides, tetracyclines and β-lactams. The last class of antibiotics to act against Gram-negative bacteria, the synthetic fluoroquinolones, were introduced half a century ago. Since then, discovery has been largely limited to narrow-spectrum compounds2,6.
We reasoned that useful compounds will be present in microorganisms whose requirements for antibiotics may be similar to our own. Nematode symbionts, Photorhabdus and Xenorhabdus, seem to represent such a group of microorganisms. These nematophilic bacteria are members of the gut microbiome of nematodes and are closely related to other Enterobacteriaceae, such as E. coli. Nematodes invade insect larvae and release their symbionts. Nematophilic bacteria first produce neurotoxins to immobilize the prey, and then release various antimicrobials to fend off invading environmental microorganisms7,8. However, the most immediate competitors probably come not from the environment, but from other members of the nematode gut. Interestingly, Gram-negative bacteria that are common opportunistic pathogens of humans are abundant in the microbiome of entomopathogenic nematodes9. The antimicrobial compounds of nematophilic bacteria must be non-toxic to the nematode, and be able to spread well through the larvae. This suggests antimicrobials with low toxicity and good pharmacokinetics active against Gram-negative pathogens.
Identification of darobactin
We screened a small library of Photorhabdus and Xenorhabdus strains, a total of 67 isolates belonging to 28 species (Extended Data Table 1), against E. coli. In the classical format, producing bacteria are spotted on a nutrient agar plate overlaid with a target microorganism. Most of the tested bacteria did not produce zones of inhibition, and we reasoned that this may be due to poor expression of “silent” biosynthetic gene clusters (BGCs) in vitro. We therefore prepared concentrated extracts from the bacterial cultures and spotted them on overlay plates. A concentrated extract from P. khanii HGB1456 produced a small zone of E. coli growth inhibition on a Petri dish, while spotting a colony had no effect (Fig. 1a). Bioassay-guided isolation of the extract by HPLC produced an active fraction (Extended Data Fig. 1a). High resolution ESI-MS analysis identified a compound with a molecular mass of 966.41047 which is consistent with a molecular formula C47H56O12N11+ ([M+H]+calc 966.41044). This mass did not have a match in Antibase, suggesting the presence of a novel compound. Mass spectrometric fragmentation and NMR studies (Extended Data Fig. 1b–h) led to the identification of the structure of the active compound which we named darobactin (Fig. 1b). Darobactin is a modified heptapeptide with an amino acid sequence W1-N2-W3-S4-K5-S6-F7. NMR studies revealed two unusual macrocycle cross linkages in darobactin: an unprecedented aromatic-aliphatic ether linkage between the C7 indole of W1 and the β-carbon of W3, and a carbon-carbon bond between the C6 indole of W3 and the β-carbon of K5. The tryptophan-lysine bond is made between two unactivated carbons, which is unique for an antibiotic. We then sequenced the genome of P. khanii HGB1456 (accession number WHZZ00000000) and searched for biosynthetic gene clusters (BGCs) encoding non-ribosomal peptide synthetases (NRPSs). There were 10 NRPSs in the genome, but none of them could be predicted to form the darobactin peptide. We then directly compared the sequence of this 7 amino acid peptide against the genome of P. khanii and found a perfect match near the C-terminus of an open reading frame coding for a 58 amino acid long peptide. The ribosomal synthesis of darobactin suggests that the amino acid backbone is in L-configuration. The macrocycle cross linkages generate two chiral centers at the β-carbons of W3 and K5, which have R and S configurations, respectively, based on NOE correlations and molecular modeling (Extended Data Fig. 2).
Figure 1. Darobactin produced by a silent operon of P. khanii is a bactericidal antibiotic.
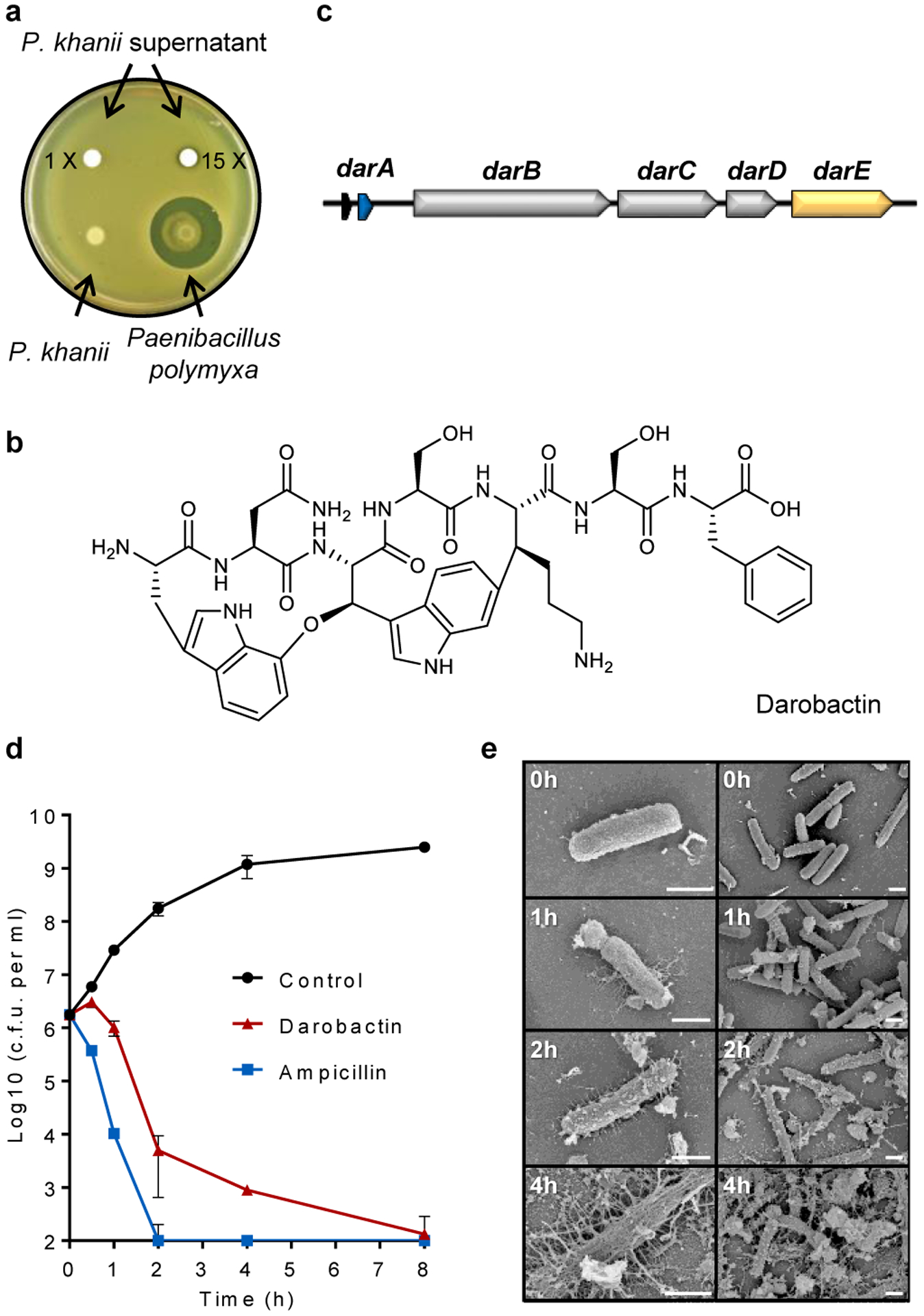
a, P. khanii was grown in liquid culture, then concentrated culture supernatants tested for inhibition of E. coli MG1655. P. khanii concentrated supernatant produced a zone of inhibition on an E. coli lawn, while unconcentrated supernatant or a colony overlay did not. Paenibacillus polymyxa produces polymyxin and serves as a positive control. b, Darobactin structure. c, The BGC consists of the structural gene darA (colored in blue), darBCD (transporter encoding genes, in grey) and darE (encoding a radical SAM enzyme, in orange). In addition, a relE-like gene (black) ORF can be co-located with the BGC at different positions. d, Time-dependent killing of E. coli MG1655 by darobactin. An exponential culture of E. coli MG1655 was challenged with 16xMIC antibiotics. n=3 biologically independent samples, symbols are mean, error bars are SD. e, SEM analysis of E. coli MG1655 treated with 16xMIC darobactin (Scale bar, 1 μm).
The putative operon coding for darobactin (Fig. 1c; Extended Data Fig. 3) is typical of RiPPs that code for a variety of ribosomally-produced natural products, including the antibiotics nisin, a food preservative and thiostrepton. This dar operon consists of the propeptide encoded by darA, a small relE-type ORF, darBCD coding for an ABC-type transenvelope exporter (darB,D make up the transporter proper, and darC codes for a membrane fusion protein), and darE for a radical SAM enzyme. The radical SAM class of enzymes catalyze free radical-based reactions that can link unactivated carbons10. This would explain the formation of the tryptophan-lysine C-C bond in darobactin. Such a Trp-Lys C-C bond was recently reported in a peptide pheromone, streptide, from Streptococcus thermophilus11. There is little overall homology between the two enzymes, but DarE contains the SAM and SPASM domains characteristic of this group. The operon does not contain a separate enzyme for making the ether bond in the first ring. RiPP operons often code for a protease that cleaves out the active peptide; this was not present in the dar operon. Hence, generic proteolysis may be involved in maturation of the propeptide. To link the putative BGC with darobactin production, we generated a markerless knockout mutant in which the complete BGC darABCDE was deleted from P. khanii DSM3369 by double crossover. Darobactin production was abolished in the resulting mutant strain (Extended Data Fig. 4a,c,d). Importantly, darobactin was produced heterologously from the dar operon cloned into E. coli (Extended Data Fig. 4b,d). This shows that the dar operon is sufficient for making darobactin. Surprisingly, it appears that the DarE radical SAM enzyme catalyzes the formation of both the Trp-Lys C-C bond, and the C-O-C Trp-Trp ether bond. The chemistries of these two reactions are quite different, and the mechanism of DarE catalysis clearly requires a separate investigation.
We find that the dar operon is common in Photorhabdus, and detected it in 15 different species for which the genome sequence is available (Extended Data Fig. 4e). The dar operon was only absent in P. bodei. Synteny of the genomes containing the dar locus with that of P. bodei helped determine the boundaries of the operon (Extended Data Fig. 3a,c). We also tested production of darobactin in several different Photorhabdus, and found that it is the highest in a strain of P. khanii DSM 3369. We switched to this strain for the isolation of darobactin, but even in this isolate, production is low, 3 mg l−1, only 2-fold higher than in P. khanii HGB1456, and requires unusually long fermentation, 10–14 days. This probably explains why darobactin has been overlooked.
We then expanded the search for dar-type operons in databases of bacterial genome sequences (NCBI), using the propeptide and the dar encoding peptide as queries. The two searches identified homologues of the dar operon that appear to code for four darobactin analogs. We therefore propose the name darobactin A for the first compound, and darobactin B-E for the predicted analogs of this class of antibiotics. In P. australis and P. asymbiotica, the sequence data suggest the presence of darobactin B, which contains two amino acid changes on the N-terminus (SKSF→TKRF). In multiple Yersinia species either the second amino acid (N→S) or the fifth amino acid (K→R), or both, are modified. We named these analogs darobactin C, D, and E (Extended Data Fig. 4e,f). Interestingly, darobactin C sequence is present in Yersinia pestis, the causative agent of plague, and in Y. frederiksenii from the human gut microbiome. Darobactin A is the most common, and a corresponding propeptide sequence is present in 6 sequenced Photorhabdus species, 7 Yersinia species, Vibrio crassostreae, and Pseudoalteromonas luteoviolacea, all of which are γ-proteobacteria. All species containing dar operons are associated with animals. Apparently, combinatorial reshuffling of the dar operon produced a family of genes, and the 5 analogs were selected in the course of evolution from a total of 1.28×109 (207). The GC content of the dar operon is 32%, significantly lower as compared to the rest of the genome of P. khanii and other γ-proteobacteria which is 45%. This suggests that the operon was horizontally acquired from a microorganism in which darobactin evolved. While the nature of this intriguing microorganism is unknown, it is not an actinomycete – their genomes have a characteristically high GC content, >55%12.
Identifying the target
Darobactin had reasonable activity against a range of Gram-negative bacteria, with an MIC of 2 μg ml−1 against important drug-resistant pathogens, E. coli and K. pneumoniae, including polymyxin resistant, ESBL (extended spectrum β-lactamase) and carbapenem resistant clinical isolates (Table 1; Supplementary Table 1). The compound is bactericidal (Fig. 1d), with an MBC of 8 μg ml−1 against E. coli. There was little activity against Gram-positive bacteria. Interestingly, the compound was also largely inactive against gut commensals, including Bacteroides, the main group of Gram-negative symbionts13. Disrupting the microbiome by antibiotics, especially early in life, is a major concern, given the important role of symbiotic bacteria in many aspects of human health, such as shaping the immune system during development14.
Table 1.
MIC and cytotoxicity of darobactin against pathogens, intestinal gut bacteria and human cell lines.
| Organism and genotype | μg ml−1 | |
|---|---|---|
| Dar | Amp | |
| Pathogenic bacteria (MIC) | ||
| Pseudomonas aeruginosa PAO1 | 2 | >128 |
| P. aeruginosa pmrB 523C>T | 2 | >128 |
| P. aeruginosa JMI 1045324 | 16 | N.D. |
| Shigella sonnei ATCC 25931* | 2 | 4 |
| Klebsiella pneumoniae ATCC 700603 | 2 | 128 |
| K. pneumoniae ESBL JMI 1052654 | 2 | >128 |
| K. pneumoniae ATCC700603 (SHV-18) | 4 | >128 |
| K. pneumoniae ATCC BAA-1705 (KPC) | 4 | >128 |
| Escherichia coli ATCC 25922 | 2 | 8 |
| E. coli AR350 (mcr-1) | 2 | >128 |
| E. coli ESBL JMI 1043856 | 2 | >128 |
| E. coli ATCC BAA-2340 (KPC) | 2 | >128 |
| E. coli MG1655 +10% serum | 2 | 4 |
| E. coli MG1655 | 4 | 4 |
| Salmonella Typhimurium LT2 ATCC 19585* | 4 | 2 |
| Moraxella catarrhalis ATCC 25238* | 8 | <0.25 |
| Acinetobacter baumannii ATCC 17978 | 8 | 64 |
| Enterobacter cloacae ATCC 13047* | 32 | >128 |
| Proteus mirabilis KLE 2600*1 | 64 | >128 |
| Staphylococcus aureus HG003 | >128 | 0.5 |
| Clostridium bifermentans KLE 2329*1 | >128 | 1 |
| Mycobacterium tuberculosis mc26020 | >128 | 16 |
| Symbiotic gut bacteria (MIC) | ||
| Bifidobacterium longum ATCC BAA-999* | >128 | 0.25 |
| Bacteroides fragilis ATCC 25285* | >128 | 128 |
| Bacteroides xylanisolvens KLE 2253*1 | >128 | 1 |
| Bacteroides dorei KLE 2422*1 | >128 | 1 |
| Bacteroides caccae KLE 2423*1 | >128 | 2 |
| Bacteroides vulgatus KLE 2303*1 | >128 | 2 |
| Bacteroides nordii KLE 2369*1 | >128 | 4 |
| Lactobacillus reuteri ATCC 23272* | >128 | 1 |
| Enterococcus faecalis KLE 2341*1 | >128 | 4 |
| Faecalibacterium prausnitzii KLE 2243*1 | >128 | 64 |
| Haemophilus parainfluenzae KLE 2367*1 | >128 | 128 |
| Stenotrophomonas maltophilia KLE 11416*1 | >128 | >128 |
| Human cell line (IC50) | ||
| HepG2 | >128 | >128 |
| FaDu | >128 | >128 |
| HEK293 | >128 | >128 |
Cultivated under anerobic conditions.
Human stool isolate, Kim Lewis laboratory collection.
Dar; darobactin, Amp; ampicillin. N.D.; No data.
Darobactin is a large, 965 Da molecule, while the cutoff for compounds to permeate the outer membrane is around 600 Da15. We therefore considered that darobactin, similarly to polymyxin, might be targeting LPS of the outer membrane. Adding purified LPS to a culture of E. coli protected cells from polymyxin, but had no effect on darobactin activity (Extended Data Fig. 5a). Addition of darobactin to E. coli caused blebbing of the membrane, and eventual swelling and lysis of cells (Fig. 1e; Extended Data Fig. 6, Supplementary Video 1). Transcriptome analysis revealed that darobactin rapidly (in 15–30 min) induced the sigma E and Rcs envelope stress responses, and more broadly activated genes from all five envelope stress pathways (Extended Data Fig. 7, Supplementary Discussion). In order to probe binding of darobactin to the target, we performed a ligand protection thermal proteome analysis. This however did not point to a particular protein whose denaturation was protected by darobactin. At the same time, the proteome showed that the abundance of periplasmic chaperones Spy and DegP was dramatically increased, and that of outer membrane proteins, especially NanC, LamB, and OmpF, were decreased (at least in part due to a decrease in the respective transcripts) in response to darobactin treatment (Extended Data Fig. 8, Supplementary Table 2, Supplementary Discussion). Microscopy, transcriptome, and proteome data point to a defect in the cell envelope. We then sought to obtain mutants resistant to darobactin in order to identify its target. Plating of E. coli on solid medium containing darobactin at 4xMIC produced resistant mutants with a frequency of 8×10−9. In order to obtain mutants resistant to higher levels of the compound, we performed an evolutionary experiment in liquid medium16 (Fig. 2a). Repeatedly reinoculating a culture into media with progressively increasing levels of the antibiotic produced mutants with high resistance to darobactin, with MICs greater than 128 μg ml−1 (Fig. 2a). Sequencing the mutants showed that in all three strains, there were 2–3 mutations in the same gene coding for BamA, an essential outer membrane protein17 (Fig. 2b). After transferring the 3 bamA mutations from the resistant Strain-3 into a clean E. coli background by allelic replacement, we confirmed that they are solely responsible for darobactin resistance (MIC of 128 μg ml−1). Ability to raise mutants resistant to high levels of the compound suggests lack of off-target activity. In order to sustain an infection in the presence of antibiotic, the pathogen should be both resistant and virulent. We therefore tested whether darobactin resistant mutants retain virulence. Injecting mice with 107 cells of E. coli ATCC 25922 caused 60% mortality within 24 hours. By contrast, there was no death at 24 hr when the animals were inoculated with E. coli carrying either single or triple mutations in bamA (Fig. 2c, Extended Data Fig. 5b). E. coli virulence is thus strongly compromised by bamA mutations conferring resistance to darobactin.
Figure 2. Multiple mutations in bamA confer darobactin resistance.
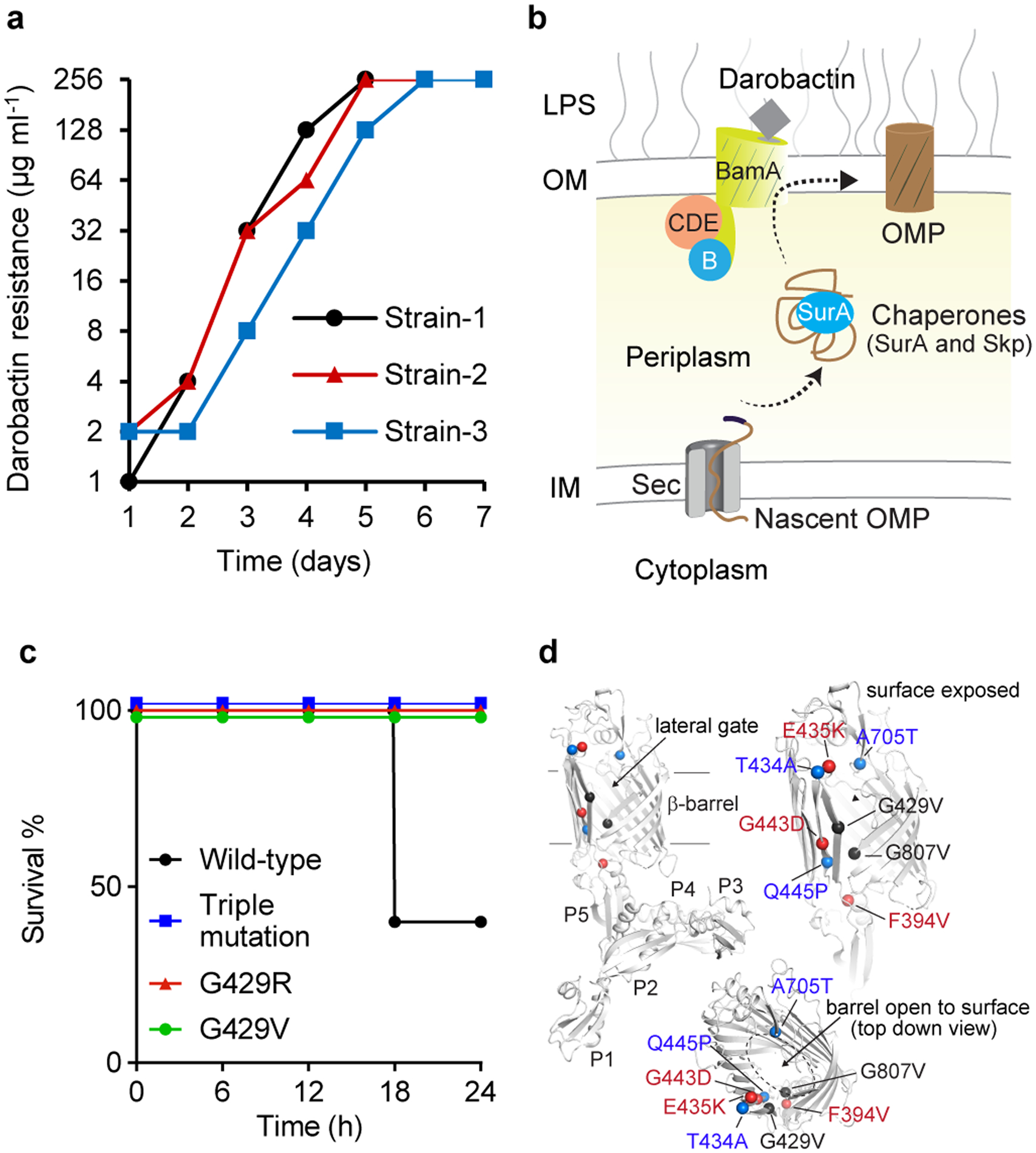
a, Darobactin resistant mutants were generated by serial passaging of E. coli MG1655 at sub-MIC concentrations of darobactin daily, leading to a steady shift in darobactin concentration permitting E. coli MG1655 growth. This experiment was performed in three biologically independent samples. The three mutants obtained harbored 2–3 mutations in bamA. b, Schematic of the Bam complex18. c, Mice were injected with 107 c.f.u. E. coli ATCC 25922, wild-type or containing mutations in bamA; the triple mutations evolved in Strain-3 (Fig. 2a), or single spontaneous resistant mutations of G429 to R or V, n=5 per group. Mice were monitored for survival. d, Darobactin resistance mutations (colored spheres) mapped on to the BamA protein structure (gray) shown as cartoon with the barrel domain and the individual POTRA domains indicated.
BamA is the central component of the BamABCDE complex17 (Fig. 2b). One proposed mechanism for BAM is that nascent porins are inserted from the periplasm into the outer membrane by the central component BamA, which serves to catalyze both folding and insertion. BamA is not an enzyme, and its β-barrel structure does not obviously lend itself to inhibition by small molecules. BamA is targeted by large Lectin-like bacteriocins, LlpA19, and a group from Genentech developed an antibody that inhibits the protein in E. coli20. In a recent study, a small molecule synthetic compound MRL-494 was reported to act against BamA, importantly, without the need to penetrate across the outer membrane21. MRL-494 is active against E. coli and K. pneumoniae with an MIC of 15 and 62 μg ml−1, correspondingly, but also acts against Gram-positive bacteria by disrupting their cytoplasmic membrane.
We observed direct inhibition of BAM by darobactin using an in vitro protein refolding assay. Isolated BAM complex is integrated into lipid nanodiscs, and its ability to fold the protease OmpT is measured (Extended Data Fig. 5c). Darobactin inhibited BAM-dependent folding of OmpT with an apparent IC50 of 0.68–1 μM (Fig. 3a; Extended Data Fig. 5d), consistent with the E. coli MIC of 1.9 μM. Darobactin had no effect on OmpT activity in the absence of BAM (Extended Data Fig. 5e), and a linear peptide of the same sequence as darobactin had no inhibitory activity on BAM (Extended Data Fig. 5f). We then tested darobactin resistant mutants in the same assay. The IC50 of mutant 1a was increased dramatically, to 120 μM. In mutants 2 and 3, the IC50 was unchanged, but the folding activity was strongly decreased (Fig. 3a). The mechanism by which mutants 2 and 3 confer resistance is unclear and will require additional study.
Figure 3. Darobactin inhibits BAM activity, binds to and induces selection of the closed-gate conformation of BamA-b.
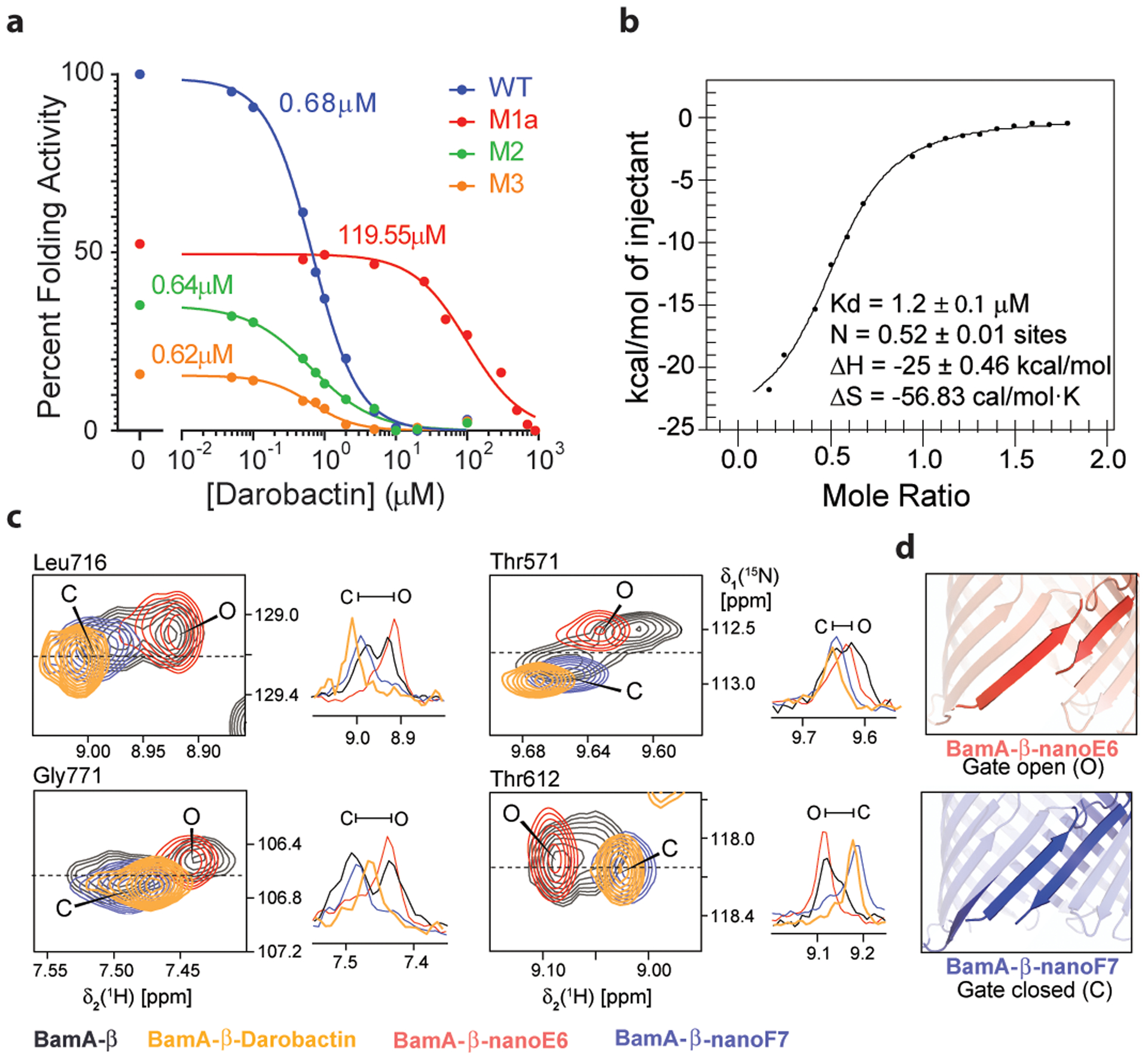
a, The assay in Extended Data Fig. 5c was used to measure BAM activity, wild type and resistant mutants, in the presence of increasing concentrations of darobactin. IC50 value are indicated in the figure for each mutant. Confidence intervals 95% for IC50 WT 0.61 to 0.75 μM, M1a (G429V, T434A and G807V; Methods) 68 to 148 μM, M2 (F394V, E435K and G443D) 0.50 to 0.83 μM, M3 (T434A, Q445P and A705T) 0.38 to 0.94 μM (Prism v8.2). The experiment was repeated independently at least three times with similar results. b, Specific binding of darobactin to BamA/BAM. Mole Ratio is the protein/ligand ratio. Plot of ITC experiments of WT BAM titrated with darobactin showing a Kd of 1.2 μM, N of 0.52, ΔH of −25 kcal/mol, and ΔS of =-56 cal/mol·K. The experiment was repeated independently two times with similar results. c, 2D-close up and 1D-cross sections from 2D [15N, 1H]-TROSY spectra of BamA-β in LDAO micelles for four selected amino acid residues, as indicated on top of each panel. Color code: Apo BamA-b (black), equimolar BamA-b:Darobactin (orange), BamA-b+nanoF7 (blue) and BamA-b+nanoE6 (red). Resonances corresponding to open and closed conformation have been indicated as O and C, respectively. The experiment was repeated independently two times with similar results. d, Conformation of the gate region in crystal structures of BamA-b+nanoE6 and BamA-b+nanoF7, respectively (PDB: 6QGW, 6QGX7).
Using ITC experiments, we also observed that darobactin directly and specifically interacts with BamA of BAM with a measured Kd of 1.2 μM, with no binding observed for the linear peptide (Fig. 3b; Extended Data Fig. 5g,h).
In order to characterize the interaction of BamA with darobactin at the atomic level, we performed a high-resolution NMR study. A stepwise titration of unlabeled darobactin to [U-15N,2H]-labeled BamA β-barrel (BamA-β) was carried out and monitored by solution NMR spectroscopy. Upon addition of 0.5 molar equivalent darobactin, significant changes were observed in the NMR spectrum of BamA-β, which became more prominent at 1 molar equivalent (Extended Data Fig. 5i; Supplementary Data 1). In contrast, a linear scrambled darobactin peptide had no effect on the NMR spectrum (Extended Data Fig. 5j; Supplementary Data 1). We had previously shown that BamA-β exists as an interchanging two-state ensemble of a gate-closed and a gate-opened conformation and that each of these two conformers can be stabilized by a conformation-specific nanobody, nanoF7 for the gate-closed and nanoE6 for the gate-opened structure22,23. Strikingly, we found that darobactin stabilized a single one of these two conformations (Fig. 3c,d). The darobactin-stabilized conformation resembles for most residues the closed-gate conformation, as evidenced by the high similarity of NMR spectral positions of BamA-β+nanoF7 and BamA-β+darobactin and the clear difference from BamA-β+nanoE6 (Fig. 3c,d). These findings strongly suggest that darobactin stabilizes a closed lateral gate upon binding to BamA, preventing exit of substrates into the outer membrane. Interestingly, most mutations conferring resistance to darobactin are located at the lateral gate of BamA (Fig. 2d).Taken together, these findings are consistent with darobactin inhibiting BamA and disrupting the formation of a functional outer membrane. Future studies will determine the mechanism by which darobactin kills bacterial cells by acting against this target.
Animal efficacy
Given the attractive mode of action and lack of cytotoxicity (Table 1), we next examined the efficacy of darobactin in mouse models of infection. Single-dose pharmacokinetic analysis shows that darobactin achieves good exposure, with an intraperitoneal (ip) injection of 50 mg kg−1 leading to a peak blood level of 94 μg ml−1, and a half-life of 1 hour (Extended Data Fig. 9a). Importantly, the blood levels of the compound were maintained above the MIC of E. coli for 8 hours, an excellent predictor for efficacy. We also did not notice any toxicity with this dose of darobactin. Next, efficacy of the compound was examined in a mouse septicemia model. For this, we examined wildtype and polymyxin-resistant P. aeruginosa (PAO1 and pmrB 523C>T), carbapenemase producing K. pneumoniae (KPC), and wildtype and polymyxin-resistant E. coli (ATCC 25922 and AR350 mcr-1) (Fig. 4a,b,c). Carbapenem resistant K. pneumoniae cause 30–40% mortality in the US and 40–50% in Europe24,25. Polymyxin-resistant E. coli mcr-1 is of particular concern, since the resistance locus is present on a plasmid and can rapidly spread26.
Figure 4. Darobactin is efficacious in mouse infection models.
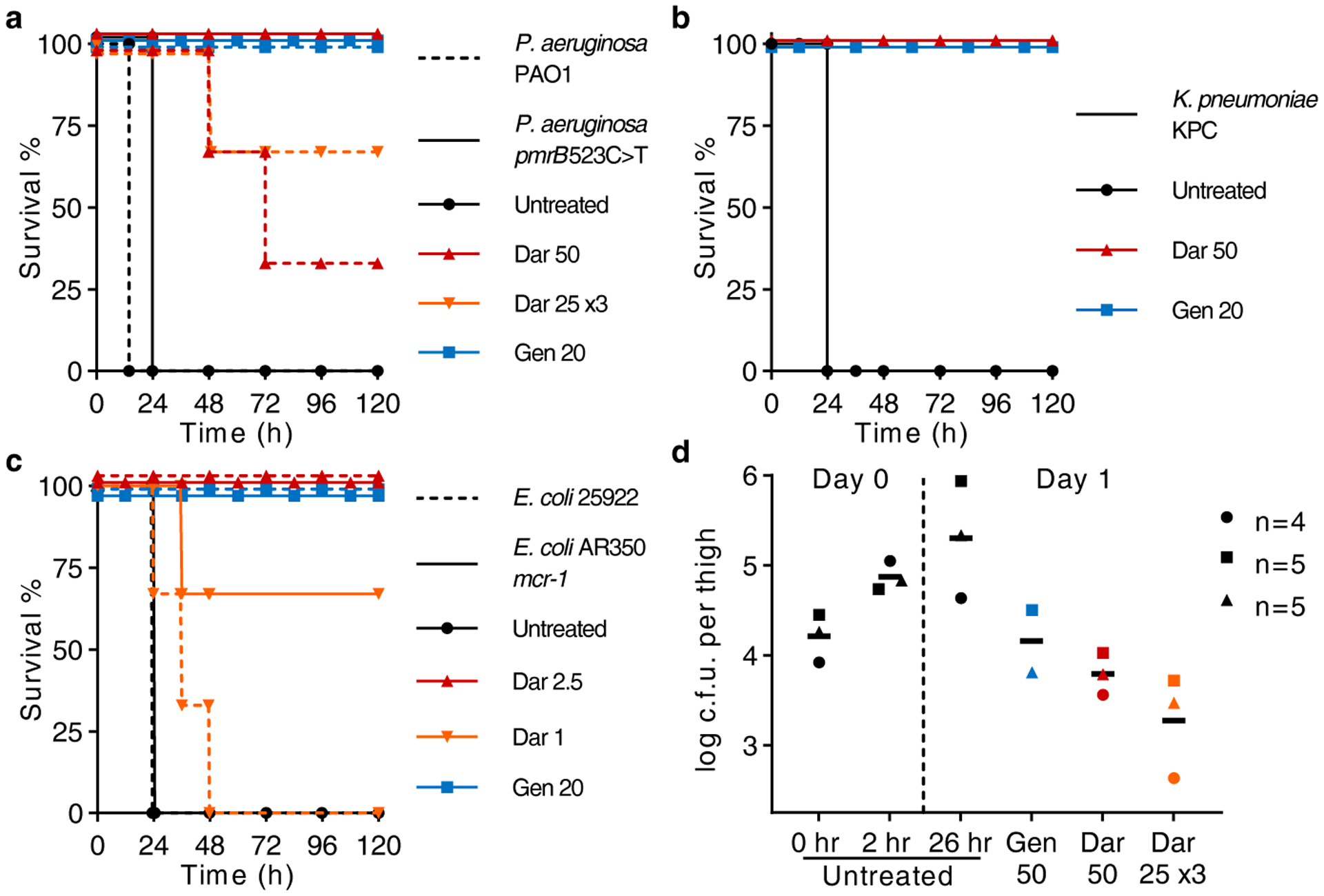
a, b, c, Mice were given a lethal inoculum of bacteria (ip), and antibiotics were administered 1 h later. a, Darobactin (Dar) was tested against P. aeruginosa, PAO1 wild type and pmrB 523C>T (resistant to polymyxin) septicemia, n=3 per group. ‘25 ×3’ refers to three doses given every 6 h. b, Darobactin was tested against K. pneumoniae, carbapenemase producing (KPC), n=3 per group. c, Determining the minimum curative dose of darobactin against E. coli wild type (ATCC 25922) and polymyxin-resistant clinical isolate (AR350), n=3 per group. d, In a neutropenic thigh model darobactin was given as a single dose (ip) at 2 h post infection, or administered three times; at 2, 8, and 14 h. Thighs were removed and plated for CFU at 26 h. Experiment was repeated three times, symbols represent average of group in each experiment (n=4 or 5), lines are mean of experiments. Gentamicin (Gen) was used as a positive control. All doses are mg kg−1.
To initiate septicemia, mice were infected intraperitoneally and one hour after introducing the pathogens, darobactin was administered. Untreated controls all died within 24 hours, but a single dose of darobactin completely protected the animals infected with E. coli, K. pneumoniae, and polymyxin resistant P. aeruginosa (Fig. 4a,b,c). Darobactin given in three doses of 25 mg kg−1 cured 2 out of 3 mice for wildtype P. aeruginosa PAO1 (Fig. 4a). Darobactin was then tested in a mouse thigh infection with E. coli mcr-1. In this model, animals are made neutropenic with cyclophosphamide treatment, and the ability of the antibiotic to kill the pathogen is tested in the absence of an immune response. Darobactin, given as either a single injection of 50 mg kg−1, or as three injections of 25 mg kg−1 every 6 hours, significantly decreased the pathogen burden at 26 hours, and was more efficacious than gentamicin (50 mg kg−1) (Fig. 4d; Extended Data Fig. 9b). These experiments suggest that darobactin is a promising lead compound for developing a therapeutic against Gram-negative pathogens.
Discussion
The number of novel compounds acting against Gram-negative bacteria is small, comprising mainly of β-lactamase inhibitors – avibactam27, vaborbactam28, and aspergillomarasmin29; arylomycin analogs that target the LepB signal peptidase30 are in development by Genentech31.
An intriguing new discovery platform is in development, based on emerging rules of permeation that determine properties required for compounds to breach the permeability barrier of Gram-negative bacteria32. However for now, perhaps the most practical approach is to mine untapped groups of microorganisms that may harbor new chemistry. These include uncultured bacteria, from which teixobactin was discovered16; and, surprisingly, several of the most abundant soil taxa – Acidobacteria, Verrucomicrobia, Rokubacteria and Gemmatimonadetes33. Several dozen compounds with antimicrobial properties have been isolated from nematophiles34, but only the odilorhabdins (produced by Xenorhabdus nematophila) hit a specific target and show efficacy in certain animal models of infection35,36. The compounds identified so far represent a small fraction of what is coded by the genomes of nematophilic bacteria and are expressed well under laboratory conditions; darobactin is coded by a silent operon. Nematophilic bacteria split from other Enterobacteriaceae around 370 m years ago37. Since then, they would have acquired, by horizontal transmission from the biosphere, antibiotics that may be of use to us.
Darobactin is indeed a typical example of a compound acquired by horizontal transmission of a BGC operon from an unknown microorganism. It acts against an attractive target on the surface of the cell. The BamA chaperone and translocator helps fold and insert β-barrel proteins such as porins into the outer membrane. BamA itself is an outer membrane β-barrel protein. Drugs in general, and natural products in particular, normally target enzymes with their well-defined catalytic centers, rather than chaperones. According to our data, darobactin stabilizes the closed lateral gate conformation of BamA, preventing it from opening and inserting its substrates into the membrane. Darobactin is a large molecule, which is probably necessary for its unusual mode of action. The location of the target on the surface resolves the intractable problem of penetration across the permeability barrier of Gram-negative bacteria. There are only two essential proteins exposed on the surface of the outer membrane – BamA; and LptD17. There is little doubt that nature produced more than one type of compounds acting against these targets.
Methods
Screening conditions
Photorhabdus and Xenorhabdus strains used for this study were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) and kindly provided by Dr. Heidi Goodrich-Blair (University of Madison-Wisconsin, currently at The University of Tennessee). Strains were inoculated in 10 ml Luria Bertani Broth (LBB) in 50 ml falcon tubes and incubated overnight, then diluted 1:100 in new falcon tubes with 10 ml LBB, Nutrient Broth (NB) or Tryptic Soy Broth (TSB) and incubated for 8 days, at 28˚C with shaking at 200 rpm. Culture aliquots (1 ml) were centrifuged at 12,000 g for 10 min, and supernatants (750 μl) collected and dried by centrifugal evaporation. Dried samples were resuspended in 50 μl water or 50% dimethyl sulfoxide to generate 15x concentrated extracts, then 3 μl spotted onto E. coli overlays. Overlays were prepared from an exponential culture of E. coli (grown for 2–5 h after dilution 1:100 from an overnight culture in cation-adjusted Mueller Hinton II Broth (MHIIB) and incubated at 37˚C with shaking at 220 rpm), diluted to OD600 0.03 in MHIIB, and used to cover cation-adjusted Mueller Hinton II Agar (MHIIA) plates; excess culture was removed, and overlays left to dry in a biosafety cabinet. Overlays spotted with culture extracts were incubated at 37˚C overnight, and activity evaluated by zones of inhibition.
Strain fermentation and purification of darobactin
P. khanii strains were inoculated in a 500 ml Erlenmeyer flask with 200 ml LBB and incubated at 28˚C with aeration at 200 rpm overnight, then diluted 1:100 into a 2 litre Erlenmeyer flask with 1 litre TSB and incubated for 10 to 14 days. Cells were removed by centrifugation at 8,000g for 10 min, and the culture supernatant was incubated overnight with XAD16N resin (20–60 mesh, Sigma-Aldrich), under agitation, to bind darobactin. Darobactin was eluted from the XAD16N resin by using 1 litre 50% methanol with 0.1% formic acid (FA). The eluate was concentrated using a rotary evaporator, and subjected to cation-exchange (SP Sepharose XL, GE Healthcare) chromatography. The concentrated eluate was loaded on to the activated cation-exchange resin and the resin washed with 0.1% (v/v) FA dd-water. The compound was eluted by step-gradients of 50 mM ammonium acetate pH 5, pH 7, and pH 8. The bioactive eluates were combined and freeze-dried, then resuspended in 0.1% (v/v) FA water. The solution was subjected to reverse-phase high performance chromatography (RP-HPLC) on a C18 column (Agilent, C18, 5 μm; 250×10 mm, Restek). HPLC conditions: Solvent A - water + 0.1 % (v/v) FA, Solvent B - acetonitrile + 0.1 % (v/v) FA; the initial concentration of 2% Solvent B was maintained for 2 min, followed by linear gradient to 26% Solvent B over 12 min; flow rate 5 ml min−1; UV detection by diode-array detector from 210 to 400 nm. Darobactin eluted at 12.5 min, with a purity of 97% by UV.
Structure elucidation
Mass spectrometric analysis:
The exact mass of darobactin was determined using a Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, Bremen, Germany) equipped with a heated electrospray ionization (HESI-II) source operated in positive ionization mode. Darobactin was prepared in water + 0.1% FA and introduced into the mass spectrometer by direct infusion at a constant flow rate of 5 μl min−1. The ion source conditions were set as follows: Ion spray voltage, 1.50 kV; capillary temperature, 125°C; spray current, 50 μA; sheath gas, 0 and aux gas, 2. The MS/MS spectrum for darobactin was acquired in HCD (Higher-Energy Collisional Dissociation) mode and collision energy of 55 eV was applied for the fragmentation. The mass analyzer was calibrated according to the manufacturer’s directions. The data acquisition and processing was performed using Xcalibur software (Thermo Fisher Scientific, Inc.).
NMR studies:
All NMR data were recorded on a Bruker AVANCE II 700 MHz NMR spectrometer with 5 mm TXI probehead and a Bruker AVANCE NEO 600 MHz NMR spectrometer equipped with 5 mm TCI cryoprobe. Complete assignments were obtained using 2D experiments including COSY(cosydfesgpphpp), TOCSY(dipsi2esfbgpph), 1H-15N_HSQC (hsqcetfpf3gpsi), 1H-13C_HSQC (hsqcedetgpsisp2.3), 1H-13C_HMBC (hmbcgplpndprqf), and ROESY (roesyesgpph). All NMR experiments were performed with 5 mg of darobactin solubilized in 500 μl of aqueous solvent containing 94% (v/v) water, 4% (v/v) deuterium oxide, and 2% (v/v) deuterated FA. Additional 1D 1H, and 2D HMBC and ROESY NMR experiments were performed with 5 mg of darobactin solubilized in 500 μl of 2:1 (v/v) mixture of water and deuterated acetonitrile including 2% (v/v) deuterated FA.
Modeling of isomers:
Modeling of the 4 possible darobactin isomers was performed in Schrodinger 2018–2. The four isomers first underwent conformational search in Macromodel module (Schrödinger) with MMFF forcefield. Mixed torsional/low-mode sampling method was used with a maximum of 10,000 steps. The lowest energy conformer for each isomer was then subjected to geometry optimization using Jaguar (Schrödinger) at B3LYP/6–31G (d, p) level with fine grid density and the ultrafine accuracy level of SCF. All the simulations were performed for gas phase.
Identification of biosynthetic gene cluster
The genome of P. khanii HGB1456 was sequenced by both Pacbio technology and Illumina Miseq, and assembled by SPAdes 3.1138. The resulting data were initially analyzed using antibiotic and secondary metabolite analysis shell (antiSMASH39). Each predicted BGC was then analyzed manually, taking into account the number and identity of predicted amino acids. Since this initial approach did not yield any putative darobactin BGCs, a direct screening for the core peptide sequence WNWSKSF was done on all Photorhabdus genomes available in public databases using Basic Local Alignment Search Tool (BLAST). In P. khanii the seven amino acid sequence of darobactin was located close to the C-terminus of an open reading frame coding for 58 amino acids, upstream of an ABC transporter and a radical SAM enzyme, suggesting a RiPP operon. This putative BGC was identified in the other darobactin producers P. luminescens DSM3368 and P. khanii DSM3369. The boundaries of the cluster were determined by comparison with the P. bodei genome which did not contain the operon. Furthermore, the GC content of the dar cluster was clearly lower than the rest of the average GC content in the genome (32% vs 45%).
In order to identify other bacterial species that potentially produce darobactin-like compounds, homologous enzymes were searched using the radical SAM protein sequence (DarE) as input in BLAST. The genomic context of each hit was analyzed manually to confirm the presence of a DarA-like propeptide in the vicinity of the radical SAM protein. In addition, a search using the propeptide DarA as input was done, delivering the same hits.
Generation of a darobactin deletion mutant and heterologous expression
To delete the dar BGC (darABCDE) from the genome of the producer strain P. khanii DSM3369, a plasmid was constructed by assembly of 5 fragments, which enables marker less genome modification. Therefore, chromosomal DNA was isolated using the innuprepBacteria DNA Kit (AnalytikJena, Jena, Germany). Fragments (i) up- and (ii) downstream of the BGC were amplified (size ~1kb) using the primer pairs 5’-TTTGACGTTGGAGTCCACGTGTTATGGACGTGGCAAACGCGGTTCTTGAC-3’, 5’-TTGAAATATCAGGATAGCATTGCGCTCGCTCACCCCGGTCACATAGTTCG-3’, as well as 5’-ATGCTATCCTGATATTTCAAATGCAAGTAAAATGTTTCATCATAATAACC-3’ and 5’-TTCTTGACGAGTTCTTCTGAGATGGGTTGATATCCACTGATATAAATCTC-3’. (iii) The R6K origin of replication (ori), the origin of transfer (oriT) and the levan sucrase gene sacB from Bacillus subtilis were amplified in one piece from the vector pNPTS13840 using the primers 5’-TCGAGCTCTAAGGAGGTTATAAAAAATGAACATCAAAAAGTTTGCAAAACAAGCA-3’ and 5’-ACGTGGACTCCAACGTCAAA-3’. (iv) The arabinose inducible expression system of pKD4641 with the adjacent beta-lactamase (bla) promoter was amplified using the primers 5’-ACTCTTCCTTTTTCAATATTATTGAAGCAT-3’ and 5’-TGCATTTTTTATAACCTCCTTAGAGCTCGAATTCC-3’, and (v) the aph gene from pCAP0342 conferring resistance to kanamycin, was amplified using the primers 5’-TCAGAAGAACTCGTCAAGAAGGCGA-3’ and 5’-TCAATAATATTGAAAAAGGAAGAGTATGATTGAACAAGATGGATTGCACG-3’. All fragments were amplified by Q5 DNA polymerase (New England Biolabs, Ipswich, USA), gel purified with 1% or 2% TAE agarose gels and DNA was retrieved with the Large Fragment DNA Recovery Kit (Zymo Research, Irvine, USA). Subsequently all fragments were fused by isothermal assembly, generating the plasmid pNB02.
After assembly, E. coli WM3064 cells were transformed with pNB02 by electroporation and correct assembly was corroborated by PCR and restriction analysis following standard procedures. Conjugation between E. coli WM3064 and P. khanii DSM3369 was performed by growing both strains to an OD600 of ~ 0.6. After washing twice with LB medium, cells were mixed in 1:3 ratio of E. coli and P. khanii, plated out on LB agar supplemented with diaminopimelinic acid (0.3 mM) and incubated at 37°C for 3 h, followed by overnight incubation at 30°C. The bacterial lawn was resuspended in LB medium and plated on LB agar with kanamycin (50 μg ml−1) in serial dilution. Kanamycin resistant single cross over transconjugants were grown in LB medium to an OD600 of ~0.6. Then, expression of SacB was induced by adding arabinose (0.2% w/v), followed by 2 h incubation. Subsequently, the culture was plated out on LB agar supplemented with 0.2% (w/v) arabinose and 10% sucrose and incubated at 30°C for 48 h. Single colonies were picked on LBKan and LBAra/Suc agar. Sensitivity to kanamycin indicated plasmid loss and therewith a successful double crossover event. Clones were picked and analyzed for BGC loss by PCR using the primers 5’-ATCTCCATCAAAGCGCTACC-3’ and 5’-CCGCGCTGCAACTCGAAATC-3’. The knock out strain is called P. khanii DSM3369 ΔdarABCDE.
For heterologous expression of the darobactin A BGC in E. coli and to complement P. khanii DSM3369 ΔdarABCDE, the expression plasmid pNB03 was used. To avoid issues with the regulation system between the propeptide and the modifying enzymes, all intergenic regions were removed and the genes darA-darE were expressed streamlined under the control of the arabinose inducible araB promoter.
pNB03 was created by amplification of (i) the p15A ori from pACYC177 (primers 5’-GGTCGACGGATCCCCGGAATAGCGGAAATGGCTTACGAAC-3’ and 5’-CTCTAAGGAGGTTATAAAAAGCGGCCGCATCCCTTAACGTGAGTTTTC-3’), (ii) the arabinose expression system and kanamycin resistance of pNB02 (primers 5’-AAGCAGCTCCAGCCTACATCAGAAGAACTCGTCAAGAAGGCGA-3’ and 5’-TTTTTATAACCTCCTTAGAGCTCGAATTCC-3’), as well as (iii) the oriT and the aac(3) gene conferring resistance to apramycin from pIJ77343 (primers 5’-ATTCCGGGGATCCGTCGACC-3’ and 5’-TGTAGGCTGGAGCTGCTT-3’). Subsequently, all fragments were gel purified and assembled as described previously. E. coli TOP10 cells were transformed with the vector and correct assembly was corroborated.
To introduce the dar BGC into P. khanii DSM3369 ΔdarABCDE, (i) pNB03 was linearized using the primers 5’-TCCCTTAACGTGAGTTTTCG-3’ and 5’-TTTTATAACCTCCTTAGAGCTCGAA-3’, (ii) darA was amplified using 5’-GCTCTAAGGAGGTTATAAAAATGCATAATACCTTAAATGAAACCGTTAAA-3’ and 5’-AATAGCATTCATTTATGGCTCTCCTTTTAAATTTCCTGGAAGCTTT-3’, (iii) darB-darE were amplified using 5’-AAAGCTTCCAGGAAATTTAAAAGGAGAGCCATAAATGAATGCTATT-3’ and 5’-CGAAAACTCACGTTAAGGGATTACGCCGCGATGGTTTGTTTTATT-3’. All fragments were gel purified and assembled as described above. The resulting vector pNB03-darABCDE was transferred to E. coli TOP10 cells and correct assembly was corroborated.
The empty pNB03 as well as pNB03-darABCDE were transferred to P. khanii DSM3369 ΔdarABCDE by triparental conjugation. In brief, conjugation between P. khanii DSM3369 ΔdarABCDE, E. coli TOP10 carrying the expression plasmid and E. coli ET pUB307, harboring the pUB307 conjugation helper plasmid was carried out as before (cell ratio 3:1:1). Since P. khanii DSM3369 is naturally resistant to carbenicillin and the kanamycin resistance of pUB307 lacks the bla promoter, final selection took place on LB agar supplemented with kanamycin and carbenicillin. Kanamycin resistant transconjugants were grown in LBKan, the plasmid was isolated and the identity verified by PCR. For heterologous expression, the vector pNB03-darABCDE was transferred in E. coli BW25113 (arabinose non-utilizer) by electroporation.
Subsequently, P. khanii DSM3369 WT, P. khanii DSM3369 ΔdarABCDE + pNB03, P. khanii DSM3369 ΔdarABCDE + pNB03-darABCDE, and E. coli + pNB03-darABCDE were grown in LB or LBKan supplemented with 0.2 % (w/v) arabinose for 5–7 days and analyzed by LCMS.
Then, centrifuged culture supernatant was desalted on self-packed C18 columns by washing with 5% acetonitrile, then eluting with 80% acetonitrile in water + 0.1% FA. A Dionex UltiMate 3000 HPLC system was coupled to a high-resolution electrospray ionization quadrupole time-of-flight mass spectrometer (QqTOF-ESI-HRMS) from Bruker Daltonics instrument (Bremen, Germany). Dionex Acclaim 120 C18 (5 μm 4.6x 100 mm) was used for the separation with solvent-A - water and solvent-B - 100% methanol. The initial concentration of 10% solvent-B was maintained for 10 min, followed by a linear gradient to 100% over 30 min. MS parameters were as follows: nebulizer gas 1.6 bar; gas temperature, 200°C; gas flow, 8 litre min−1; capillary voltage, 4500 V; endplate offset, 500 V; positive ion mode.
Minimum inhibitory concentration (MIC)
The MIC was determined by microbroth dilution. Under aerobic conditions E. coli strains, P. aeruginosa strains, A. baumannii ATCC17978, K. pneumoniae strains, and S. aureus HG003, overnight cultures were diluted 1:100 in MHIIB and incubated at 37˚C with aeration at 220 rpm. Exponential cultures (OD600 0.1 to 0.9) were diluted to OD600 0.001 (approximately 5 × 105 c.f.u. ml−1) in MHIIB and 98 μl aliquots were transferred into round bottom 96-well plates containing 2 μl of darobactin solutions diluted serially 2-fold. After overnight incubation at 37˚C, the darobactin MIC was determined as the minimum concentration where no growth of strains could be detected by eye. For the Mycobacterium tuberculosis susceptibility testing, an exponentially growing culture of strain mc26020 (ΔlysAΔpanCD) was diluted to an OD600 of 0.003 (approx. 5×105 cells ml−1) and seeded into 96-well plates containing darobactin dilutions. The plates were incubated for five days, then resazurin was added to each well to a final concentration of 2.5 μg ml−1. The plates were incubated for an additional two days, at which point the MIC was determined by eye. The MIC against intestinal pathobionts and symbionts (Shigella sonnei, Salmonella enterica Typhimurium LT2, Moraxella catarrhalis, Enterobacter cloacae, Bifidobacterium longum, Bacteroides fragilis and Lactobacillus reuteri (ATCC 25931, 19585, 25238, 13047, BAA-999, 25285 and 23272, respectively), KLE collection bacteria were isolated from stool under anaerobic conditions and identified by 16S sequencing] was determined under anaerobic conditions (Coy Vinyl Anaerobic chamber, 37°C, 5% H2, 10% CO2, 85% N2). Overnight cultures grown in Brain Heart Infusion (BHI) broth, supplemented with 0.5% Yeast Extract, 0.1% L-Cysteine hydrochloride, and 15 μg ml−1 Hemin (BHI-Ych), were diluted 1:100 in BHI-Ych. 96-well assay plates were prepared by 2-fold dilution of darobactin, and included a positive growth control. After 24 hours incubation, the MIC was determined. All MIC assays were performed at least in triplicate. The MIC against clinical isolates (Supplementary Table 1) of E. coli, K. pneumoniae and P. aeruginosa was evaluated by JMI laboratories (Iowa, USA).
Cytotoxicity
A microplate Alamar Blue assay (MABA/resazurin) was used to determine the cytotoxicity of darobactin. Exponentially growing FaDu pharynx squamous cell carcinoma (ATCC HTB-43), HepG2 liver hepatocellular carcinoma (ATCC HB-8065), and HEK293-RFP human embryonic kidney red fluorescent protein tagged (GenTarget SC007) cells, all cultured in Eagle’s Minimum Essential Medium supplemented with 10% fetal bovine serum were seeded into a 96-well, flat bottom, tissue culture treated plate (Corning) and incubated at 37°C with 5% CO2. After 24 h, the medium was aspirated and replaced with fresh medium containing test compounds (2 μl of a 2-fold serial dilution in water to 98 μl of media). After 72 h of incubation at 37°C with 5% CO2, resazurin (Acros Organics) was added to each well to a final concentration of 0.15 mM. After three hours, the absorbance at 544 nm and 590 nm were measured using a BioTek Synergy H1 microplate reader. Experiments were performed with biological replicates.
Time-dependent killing
An overnight culture of E. coli MG1655 was diluted 1:10,000 in MHIIB and incubated at 37˚C for 2 h with aeration at 220 rpm. E. coli was treated with 16xMIC antibiotic (darobactin 64 μg ml−1 and ampicillin 64 μg ml−1) and the time each antibiotic was added was defined as 0 h. At each time point, 100 μl aliquots were collected and centrifuged at 12,000g for 5 min, pellets washed with 100 μl PBS and resuspended in 100 μl PBS and 10-fold serially diluted suspensions were plated onto MHIIA. After overnight cultivation at 37˚C, colonies were counted and c.f.u. per ml was calculated. Experiments were performed in biological triplicate.
Resistance studies
E. coli MG1655 from an exponential culture were washed in PBS, then inoculated onto 30 MHIIA plates containing 4xMIC darobactin, at a density of 5×107 c.f.u. per plate. After 2 days of cultivation at 37˚C plates were examined for colonies, colonies counted, and restreaked to test for resistance stability then tested by 16S sequencing to ensure colonies were E. coli. To evolve resistance to darobactin in liquid culture, an overnight culture of E. coli MG1655 was diluted 1:100 in 1 ml MHIIB containing 0.5x, 1x, 2x and 4xMIC darobactin and incubated at 37˚C for 24 h with aeration at 220 rpm. The darobactin concentration that inhibited growth of E. coli below OD600 2.0 was defined as the MIC, and the culture at 0.5xMIC (OD600 > 2) was used to re-inoculate tubes with 0.5x, 1x, 2x, and 4x the new MIC at 1:100. This was repeated until cultures were able to grow in 256 μg ml−1 darobactin, and cultures were then maintained in 256 μg ml−1 darobactin until the end of the experiment. Experiments were performed with three independent cultures. For the mutation analysis, over 3 million paired end Illumina reads were sequenced for each resistant mutant and mapped to the E. coli MG1655 genome (GenBank Accession U00096.3) using Geneious 11.0.4. SNPs were called using the default parameters, and the generated variant call format (VCF) files were manually filtered to remove calls with a quality score of less than 1000.
Scanning microscopy
E. coli MG1655 samples were prepared as for the time-dependent killing experiments. After washing the cells with PBS, 10 μl cell suspensions were spotted onto Aclar film coated with 0.1% poly-L-lysine. E. coli cells were fixed by 2.5% glutaraldehyde in 0.1 M sodium cacodylate containing 0.15% Alcian blue and 0.15% safranin O, for 24 h at 4˚C. The samples were washed in 0.1 M sodium cacodylate for 5–10 min, infiltrated with 1% osmium tetroxide for 30 min, washed three times in 0.1 M sodium cacodylate, and then dehydrated by a graded series of ethanol concentrations (30%, 50%, 70%, 85%, 95% and 100%) for 5–10 min for each concentration. The dehydration step with 100% ethanol was repeated three times. Critical point drying was performed by SAMDRI®-PVT-3D (Tousimis) from liquid CO2. The samples were mounted onto an aluminium sample mount using double sided conductive carbon adhesive tape, and coated with 5 nm platinum by sputter coating (Cressington 208HR). The samples were imaged with Hitachi S-4800 (Hitachi) at 3.0 kV.
Fluorescence microscopy
E. coli MG1655 was cultured in MHIIB until stationary phase, inoculated into fresh MHIIB at 1:10,000, and grown for 2 h at 37°C. Cells were concentrated 50-fold in MHIIB, placed on top of a MHIIB/darobactin (64 μg ml−1) 1.5% low melting agarose pad containing FM4–64 (10 μg ml−1) and Sytox Green (0.5 μM) dyes from Molecular Probes (Eugene, OR), and observed under a ZEISS LSM 710 confocal microscope using a 63X oil immersion objective lens. The two signals from FM4–64 and Sytox Green were collected after excitation at 488 nm, alongside a DIC image. Acquisition recording DIC, FM4–64 and Sytox Green signals was performed every 30 minutes under a temperature of 37°C maintained through a thermostatic chamber. Images were acquired by Zen Software at a resolution of 1024 × 1024 and lane average of 8, and processed with Fiji software44. On the panels shown in Extended Data Fig. 6, Enhance Contrast process was performed, and HyperStackReg plugin was used to correct for the x-y drift in Supplementary Video 1.
LPS binding assay
LPS binding assay was performed based on the MIC assay. Aliquots (100 μl) of an OD600 0.001 E. coli MG1655 culture grown in MHIIB were transferred into 96-well plate containing purified LPS from E. coli O55:B5 (Sigma L4524, 0.5–100 μg ml−1) and darobactin or polymyxin B. The antibiotic concentrations that inhibited E. coli MG1655 growth were determined in the absence or presence of LPS.
Construction of bamA recombinant mutant in E. coli MG1655 and ATCC 25922
Linear DNA product comprising the mutated bamA gene (1300 A>G, 1334 A>C and 2113 G>A) was amplified by PCR, using the primers bamA-recF (5’-ACTATCTGGATCGCGGTTATGC-3’) and bamA-recR (5’-TTCACAGCAGTCTGGATACGAG-3’), and the genomic DNA from E. coli darobactin- resistant mutant (Strain-3) template. Approximately 500 ng of column-purified mutated bamA product was used to transform electrocompetent cells of E. coli MG1655-pKD46 to perform λ Red recombination41,45. The subsequent steps have been adapted from “Quick and Easy E. coli Gene Deletion Kit” (GeneBridges, Heidelberg). Briefly, 30 μl of an overnight culture of E. coli MG1655-pKD46 was used to inoculate a microtube containing 1.4 ml of LB complemented with ampicillin (100 μg ml−1). After 2 h of shaking at 30°C, 0.4% of L-arabinose was added, and the tube was transferred for shaking at 37°C for 1 h. Cells were washed and concentrated with ice cold 10% glycerol prior to electroporation. The recovery step was performed for 3 h at 37°C with shaking. First selection was performed using resistance to darobactin (32 μg ml−1). Several transformant clones were then restreaked with double selection for resistance to darobactin (32 μg ml−1) and sensitivity to ampicillin (100 μg ml−1, at 30°C). The bamA locus was amplified and the presence of the mutations (1300 A>G, 1334 A>C and 2113 G>A leading to Thr434Ala, Gln445Pro and Ala705Thr, respectively) was confirmed by sequencing.
For virulence testing, to transfer mutations into E. coli ATTC 25922 leading to the Strain-3 triple bamA mutant, the same strategy was used. During the manipulation of E. coli ATTC 25922 to construct the bamA recombinant mutant, two spontaneous bamA mutants with single SNPs were isolated on darobactin plates (16 μg ml−1): 1285 G>A, leading to Gly429Arg and 1286 G>T, leading to Gly429Val.
Transcriptome analysis
For the challenge experiments, 3 mls of E. coli BW25113 at an OD600 of 0.5, representing mid-log phase, was exposed to darobactin in biological triplicate at 4 μg ml−1 for 15, 30 mins and 1 hour. After exposure, cells were immediately pelleted at 4°C by centrifugation for 2 mins at 2000 rpm in 1 ml aliquots. The supernatants were removed and samples were immediately frozen in liquid nitrogen at −80°C until they were processed for total RNA isolation. Total RNA was extracted by automation using the NucleoMag RNA extraction kit on the EpMotion Robotic liquid handler. For the resulting total RNA, RIN values were obtained to check for RNA quality using the 2200 TapeStation instrument from Agilent Genomics. rRNAs were subtracted from the total RNA to yield only messenger RNA for library construction using NEB bacterial rRNA depletion kit at half reactions with a total RNA input maximum of 400 ng. The rRNA depleted samples were quality checked using an Agilent Bioanalyzer with the Agilent Pico chip for RNA detection for less than 0.5% of rRNA remaining in each sample. 2–5 ng of the rRNA depleted samples was used as the input material to construct each cDNA library for RNA sequencing using the NEBNext Ultra Directional RNA Library prep kit from Illumina. The resulting libraries were quality checked using Agilent High Sensitivity DNA chips to ensure proper library size distribution and the absence of small adapters. Libraries were quantified and normalized by qPCR and then sequenced using the NextSeq 500 High Output Kit at 150 cycles producing approximately 9 million, 75 base-pair, paired-end reads for each library. These reads were mapped to E. coli strain BW25113 using clc_assembler v4.4.2.133896 (CLC Genomics Workbench 11.0). Differential expression was computed using edgeR::exactTest46 in R v3.5.1 with unnormalized gene counts (n= 4626 genes) for each treatment at time t vs. matched control, where t ∈ {15, 30, 60}. The gene count matrix was restricted to genes at minimum present in all replicates from one treatment condition resulting in n= 4514 genes. Volcano plots were created using plot_volcano from soothsayer (https://github.com/jolespin/soothsayer) in Python v3.6.6. Directed networks (DiNetwork) were constructed and plotted using NetworkX and Matplotlib Python packages, respectively. Heatmaps were generated using seaborn and operon plots were created, once again, with Matplotlib.
Two-dimensional thermal proteome profiling (2D-TPP)
Thermal proteome profiling was performed as previously described47,48. Briefly, E. coli BW25113 cells were grown aerobically at 37°C with shaking until OD578 ~ 0.7. For living cell experiments, darobactin was then added at five different concentrations and incubated for 10 min. For experiments in which protein synthesis was inhibited, cells were treated with 0.2 mg ml−1 chloramphenicol for 10 min prior to addition of darobactin. For lysate experiments, cells were disrupted with five freeze-thaw cycles prior to darobactin treatment. Aliquots of treated cells or lysates were then heated for 3 min to ten different temperatures in a PCR machine (Agilent SureCycler 8800). After cell lysis, protein aggregates were removed and the remaining soluble proteins were digested according to a modified SP3 protocol49,50, as previously described51. Peptides were labeled with TMT10plex (ThermoFisher Scientific), fractionated onto six fractions under high pH conditions and analyzed with liquid chromatography coupled to tandem mass spectrometry, as previously described47. Protein identification and quantification was performed using IsobarQuant52 and Mascot 2.4 (Matrix Science) against the E. coli Uniprot FASTA (Proteome ID: UP000000625). Data was analyzed with the TPP package for R52 followed by an FDR-controlled method for functional analysis of dose-response curves51. Data is available in Supplementary Information as Supplementary Table 2.
Cloning, expression and purification of BAM and BAM mutants for nanodiscs
To prepare the BAM mutants, the pJH114 plasmid (a gift from Harris Bernstein at NIDDK)18 was used as a template using an Agilent QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA); oligonucleotide sequences are available upon request. The plasmids encoding wildtype BAM (pJH114), with BamE carrying a C-terminal His-tag, and the corresponding BamA mutant genes 1–3 (M1, G429V and G807V; M2, F394V, E435K and G443D; M3, T434A, Q445P and A705T) were cloned under an IPTG promoter and sequences were confirmed. The primers used for mutation are as below: BAM_mutation1_G429V (5’-TTCAACTTTGTTATTGGTTAC-3’), BAM_mutation1_G807V (5’-TTTAACATCGTTAAAACCTGG-3’), BAM_mutation2_ F394V (5’-CGTCTGGGCGTCTTTGAAAC-3’), BAM_mutation2_ E435K (5’-TACGGTACTAAAAGTGGCGTG-3’), BAM_mutation2_G443D (5’-TTCCAGGCTGATGTGCAGCAG-3’), BAM_mutation3_T434A (5’-GGTTACGGTGCTGAAAGTGGC-3’), BAM_mutation3_Q445P (5’-GCTGGTGTGCCGCAGGATAAC-3’) and BAM_mutation3_A705T (5’-TCGGATGATACTGTAGGCGG-3’). Plasmids were transformed into BL21(DE3) cells, plated onto an LB-carbenicillin agar plate and incubated overnight at 37°C. After transforming the plasmids into E. coli BL21 (DE3), they were isolated and resequenced. The sequence of M2 and M3 was unchanged, but an additional mutation, T434A, appeared in M1, which we refer to as M1a. This additional mutation matches the T434A mutation in M3, and may have been selected during cell growth, possibly stabilizing the protein. A 50 ml overnight culture was prepared from a single colony in 2xYT medium supplemented with 100 μg ml−1 of ampicillin. The cells were then centrifuged and resuspended into 5 ml of fresh 2xYT medium and then 1 ml added to five 2 litre baffled flasks containing 1 litre of 2xYT medium supplemented with 50 μg ml−1 of ampicillin. These cultures were grown at 37°C until an OD600 between 0.8 – 1.0, then induced with 0.5 mM IPTG and cells were harvested after 3 hours. Purification was performed as previously described18. Briefly, cells were resuspended in lysis buffer (1xPBS, 10 μg ml−1 DNaseI, 200 μM PMSF, 2 μM leupeptin, 1.5 nM pepstatin A) and lysed by three passages through an Emulsiflex C-3 homogenizer (Avestin) at 18,000 psi. The lysate was then centrifuged at 6,000 × g for 20 min and the supernatant was centrifuged at 200,000 × g for 90 min at 4°C. The membrane pellets were resuspended into solubilization buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.5% DDM, and 37 mM imidazole) using a dounce homogenizer, which were then stirred at 4°C for 4 hours. Solubilized membranes were then centrifuged at 200,000 × g for 40 min at 4°C to collect the supernatant, which was then used for purification using a 5 ml Ni-NTA column using an ÄKTA system (GE Healthcare) using Buffer A (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% DDM, and 37 mM imidazole) and Buffer B (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% DDM, and 1M imidazole). Fractions containing the protein were pooled, concentrated to 5 ml for size-exclusion chromatography using a 16/60 Sephacryl S-300 HR column at a flow rate of 1.0 ml min−1 in 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.6% C8E4. The peak fractions were pooled and concentrated as necessary.
Reconstitution of the BAM into nanodiscs
Membrane scaffold protein MSP1E3D1 was expressed and purified from E. coli as previously described53,54. BAM was purified by size-exclusion chromatography in 25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1.0% OG and concentrated to 100 μM. Nanodisc reconstitution was performed in a final volume of 300 μl by adding 20 μM of purified BAM, 100 μM of MSP1E3D1, and 2 mM of E. coli polar lipids (Avanti Polar Lipids) to a buffer containing 25 mM Tris-HCl, pH 7.5 and 150 mM NaCl. Bio-beads SM2 (Biorad) were added to the mixture and incubated at 4°C overnight. The Bio-beads were spun down and the supernatant incubated with 300 μl of HisPur™ Ni-NTA Resin (ThermoFisher Scientific) for 30 min at 4°C. The BAM-inserted nanodiscs were then eluted from the Ni-NTA resin with 25 mM Tris-HCl, pH 7.5, 150 mM NaCl and 400 mM imidazole. The elution was then loaded onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) in 25 mM Tris-HCl, pH 7.5 and 150 mM NaCl. The peak fractions were then pooled and concentrated to 40 μM.
BAM folding assay
OmpT and SurA (periplasmic chaperone) were expressed and purified from E. coli as previously reported55,56. Solution 1 contains 0.4 μM BAM-nanodiscs, 0.6 μM of the fluorogenic peptide (Abz-Ala-Arg-Arg-Ala-Tyr(NO2)-NH2), and 0.1 mg ml−1 LPS in 25 μl of 20 mM Tris-HCl, pH 6.5. Empty nanodiscs were used as a negative control. Solution 2 contains 20 μM urea-denatured OmpT with 140 μM SurA in 25 μl of 20 mM Tris-HCl, pH 6.5. To initiate the BAM folding reaction, Solution 2 was incubated at room temperature for 10 min and then mixed with Solution 1. Darobactin was added to Solution 1 and incubated for 10 min prior to being mixed with Solution 2. The fluorescence signal was monitored at 430 nm (excitation at 325 nm) using a SpectraMax M2e fluorescent plate reader (Molecular Devices) for 60 min with readings every 8 seconds. Data was then analyzed and plotted using the online IC50 Calculator tool (AAT Bioquest), and Graphpad Prism v8.2.
Isothermal Titration Calorimetry (ITC)
All ITC experiments were carried out at 25˚C with the NanoITC microcalorimeter (TA, Inc.) in duplicate. BAM (300 μl) at a concentration of 20 μM in 1xPBS supplemented with 0.05% DDM was placed in the sample cell, and the ligand (darobactin or the linear peptide) with a concentration of 200 μM in the syringe (50 μl) was injected in 20 successive injections with a spacing of 300 s with a stirring rate of 300 rpm. Control experiments in the absence of BAM were performed under identical conditions to determine the heat signal from injection of the ligand to the buffer only. The resulting data were analyzed and fit to the independent binding model using the NanoAnalyze software package (TA, Inc).
Sample preparation of BamA-β in LDAO micelles for NMR
The protein construct comprising the β-barrel of E. coli BamA (residues 426–810, C690S, C700S; termed BamA-β) was established previously and sample production followed published protocols22. In brief, protein expression was carried out in E. coli BL21(DE3) Lemo cells in M9 medium containing15NH4Cl and D2O. Once the OD600 reached 0.8, expression into inclusion bodies was induced by 1 mM IPTG at 37°C for 12 h. The harvested cells were resuspended in Buffer A (20 mM Tris pH 8.0 and 300 mM NaCl) and lysed by sonication. Inclusion bodies were harvested by centrifugation at 30,000xg for 1 h and solubilized into 20 mM Tris pH 8.0 and 6 M guanidinium hydrochloride for 2 h. The solubilized sample was loaded onto Ni-NTA beads preequilibrated with buffer A supplemented with 6 M guanidium hydrochloride. The protein was eluted with buffer A, containing also 6 M guanidium hydrochloride and 200 mM imidazole. Refolding was carried out in 20 mM Tris, 150 mM NaCl, pH 8.0 and 0.5% w/v LDAO at 4°C. The refolded sample was dialysed in 20 mM Tris, pH 8.0 overnight. Afterwards, folded BamA-β was purified by ion exchange in 20 mM Tris pH 8.0, 0.1% LDAO and the protein was eluted with a linear gradient of 0.5 M NaCl. Finally, BamA-β was loaded onto a size-exclusion chromatography column (HiLoad 16/600 Superdex 200 pg, GE) in 20 mM phosphate buffer pH 7.5, 150 mM NaCl and 0.1% LDAO yielding a monomeric sample.
Solution NMR Spectroscopy
A sample was concentrated to an initial protein concentration of 250 μM. Darobactin was added stepwise from a stock solution to 0.5:1, 1:1 and 2:1-fold stoichiometry darobactin:BamA-β. At each titration step, a 2D [15N,1H]-TROSY experiment with 64 transients was recorded on a 700 MHz Bruker spectrometer equipped with a cryogenic probe at 37°C. 1024 and 128 complex points were acquired in the direct and indirect dimension, respectively, and zero-filled to 2048 and 256 points during processing. As a control experiment, a linear scrambled peptide WNKWSFS was synthesized, and added at 230 μM to BamA-β. The NMR spectra of 0:1,1:1 and 2:1-fold stoichiometry with darobactin:BamA-β, and 0:1, and 1:1 with the peptide WNKWSFS are provided as raw data (NMR raw data file). From these, spectra shown in Figure 3c and Extended Data Figure 5i,j have been produced. The data format is readable by the standard NMR software TOPSPIN 3.6.2. An upper limit estimate for the dissociation constant KD was obtained from a quantification of the relative amounts of ligand-free and ligand-bound BamA from NMR signal intensities under consideration of the spectral noise (Fig. 3c).
Animal studies
All animal studies were performed at Northeastern University (Boston, MA), approved by Northeastern IACUC, and were performed up to institutional animal care and use policies. Experiments were not randomized nor blinded, as it was not deemed necessary. Female CD-1 mice (20–25 grams, experimentally naive, 6 weeks old) from Charles River Labs were used for all studies.
Virulence Model
E. coli ATCC 25922, both wildtype and with bamA mutations leading to darobactin resistance (see recombinant mutant methods), were tested in an acute infection model. An overnight culture (OD600 of 2.0) of E. coli was diluted 1:10 in MHIIB. Mice were infected with 0.1 ml of bacterial suspension, 2×107 c.f.u. for all strains (determined from plate counts), and monitored for survival. At 24 h post-infection, mice were euthanized via CO2 asphyxiation, unless already dead. The spleen and a piece of liver (lower lobe) were aseptically removed, weighed, homogenized, serially diluted and played on LBA and MacConkey agar for c.f.u. titres.
Pharmacokinetic analysis
Mice were injected intraperitoneally with a single dose of 50 mg kg−1 darobactin, in 10% PEG-200. Blood samples were collected from 3 mice at each time point (0.25, 0.5, 1, 2, 3, 5, 8 and 24 hours) via a tail snip, 10 μl of blood was diluted into 90 μl of chilled saline, then centrifuged at 1,000g for 5 min. The diluted plasma was decanted into a fresh tube and kept at −80°C. Blood was collected from an untreated mouse and diluted in saline, and a standard curve generated by addition of known concentrations (0.1, 1, 10, and 100 μg ml−1) of darobactin to decanted supernatant. All of the samples were run on LC/MS to determine the concentration of compound in the blood. An Agilent 1260 Infinity liquid chromatography system and 6460 triple quadrupole (QqQ) system (Agilent Technologies) were used to quantify darobactin. Thermo Scientific Accucore C18 column (50×2.1 mm, 2.6 μm) was used for the separation with a flow rate of 200 μl min−1 with solvent-A - 0.1% (v/v) FA in water and solvent-B - 0.1% (v/v) FA in acetonitrile. The initial concentration of 2% solvent-B was maintained for 2 min, followed by a linear gradient to 70% over 10 min. MS parameters were as follows: gas temperature, 300°C; gas flow, 7 l min−1; capillary voltage, 3500 V; fragmentor voltage, 100 V; scan type, MRM; transition parent ion 483.8 to product ions 211.3, 160.1, 120.1, and 103.1 with collision energy 42, 46, 50, and 94 V respectively. MassHunter qualitative and quantitative analysis B.05 (Agilent Technologies) was used to quantify the darobactin peaks.
Septicemia Model
Darobactin was tested in a septicemia protection model against E. coli, wild type (ATCC 25922) or MDR (AR350, CDC), Pseudomonas aeruginosa, wild type PAO1 and a spontaneous polymyxin-resistant mutant (pmrB 523C>T mutation), and KPC Klebsiella pneumoniae (ATCC BAA1705). Mice were infected with 0.5 ml of bacterial suspension in BHI with 5% mucin (1×106 cells for E. coli and K. pneumoniae, 8×106 and 4×106 cells for P. aeruginosa wt and pmrB mutant respectively) via intraperitoneal injection. This dose achieves >90% mortality within 24 h post infection. At 1 h post-infection, mice received treatments with darobactin from 50 mg kg−1 down to 1 mg kg−1 administered by intraperitoneal injection. Infection control mice were treated with 20 mg kg−1 gentamicin as positive controls and the vehicle alone as a negative control. Survival was monitored for 7 days.
Thigh Infection Model
Darobactin was tested in a neutropenic thigh infection model against MDR E. coli AR350 (CDC). Mice were rendered neutropenic via cyclophosphamide injections 4 days (150 mg kg−1) and 1 day (100 mg kg−1) prior to infection. An overnight culture (OD600 of 2.0) of E. coli was diluted 1:1000. Mice were infected with 100 μl of the prepared inoculum into the right thigh with the actual inoculum being 104-105 c.f.u. (determined from plate counts), and one group of mice was euthanized and thighs collected and processed for time 0 counts. At 2 h post-infection, mice began treatments with darobactin at 25 mg kg−1 (given for 3 doses, q6h) or 50 mg kg−1 (given once), or gentamicin at 20 (one experiment, n=4) or 50 mg kg−1 (two experiments, n=5 each) as a positive control, administered by intraperitoneal injection. At the time of treatment, one group of infected mice was euthanized and thighs were collected and processed for c.f.u. At 26 h post-infection, mice were euthanized via CO2 asphyxiation. The right quadricep muscles were aseptically removed, weighed, homogenized, serially diluted and plated on MHIIA for c.f.u. titres. This experiment was repeated on three separate occasions with one experiment containing 4 and two experiments containing 5 mice per group.
Statistics
Confidence intervals for IC50 values in the BAM folding assay were calculated by Prism v8.2 using non-linear regression [inhibitor] vs response, constraining bottom to 0. Significance in transcriptome data for a differentially expressed gene was determined by |log2FC| ≥ 2 and FDR < 0.001, differential expression was computed using edgeR::exactTest46 in R v3.5.1 with unnormalized gene counts (n=4626 genes) for each treatment at time t vs. matched control. For thermal proteome profiling, significant hits (false discovery rate <1%) were calculated as described in Sridharan et al. (2019)52.
Data Availability
All data supporting the findings of this study are available within the paper and its Supplementary Information or in databases. The genome of P. khanii HGB1456 has been deposited to Genbank with identifier WHZZ00000000.The transcriptomic dataset (Extended Data Figure 7) has been deposited to NCBI Sequence Read Archive with identifier PRJNA530781.The mass spectrometry proteomics (Extended Data Figure 8; Supplementary Table 2) data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013319. Source Data for Figures 2c, 4 and Extended Data Figures 5b, 9 are provided with the paper. All other data available from corresponding author.
Extended Data
Extended Data Figure 1. Structural determination of darobactin.
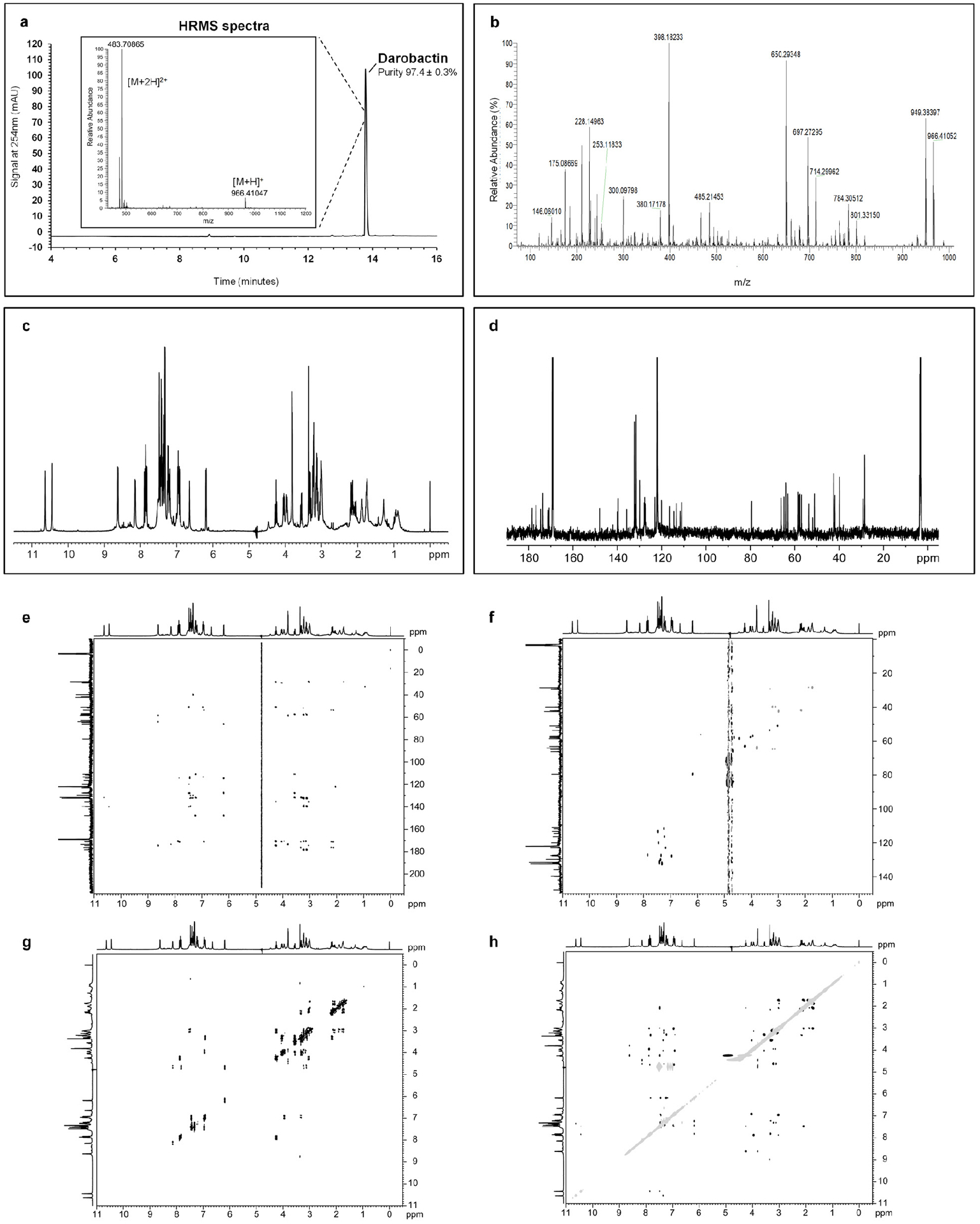
a, HPLC chromatogram of Darobactin, in inset, HRMS spectra of Darobactin showing a peak at m/z 966.41047 corresponding to [M+H]+ ion and another at m/z 483.70865 corresponding to [M+2H]2+ ion. b, High Energy Collisional Dissociation-MS/MS spectra (HCD-MS/MS) of Darobactin. c, 1H NMR spectrum of Darobactin. d, 13C NMR spectrum. e, HMBC NMR spectrum. f, HSQC NMR spectrum. g, COSY NMR spectrum. h, ROESY NMR spectrum.
Extended Data Figure 2. NMR assignments of darobactin.
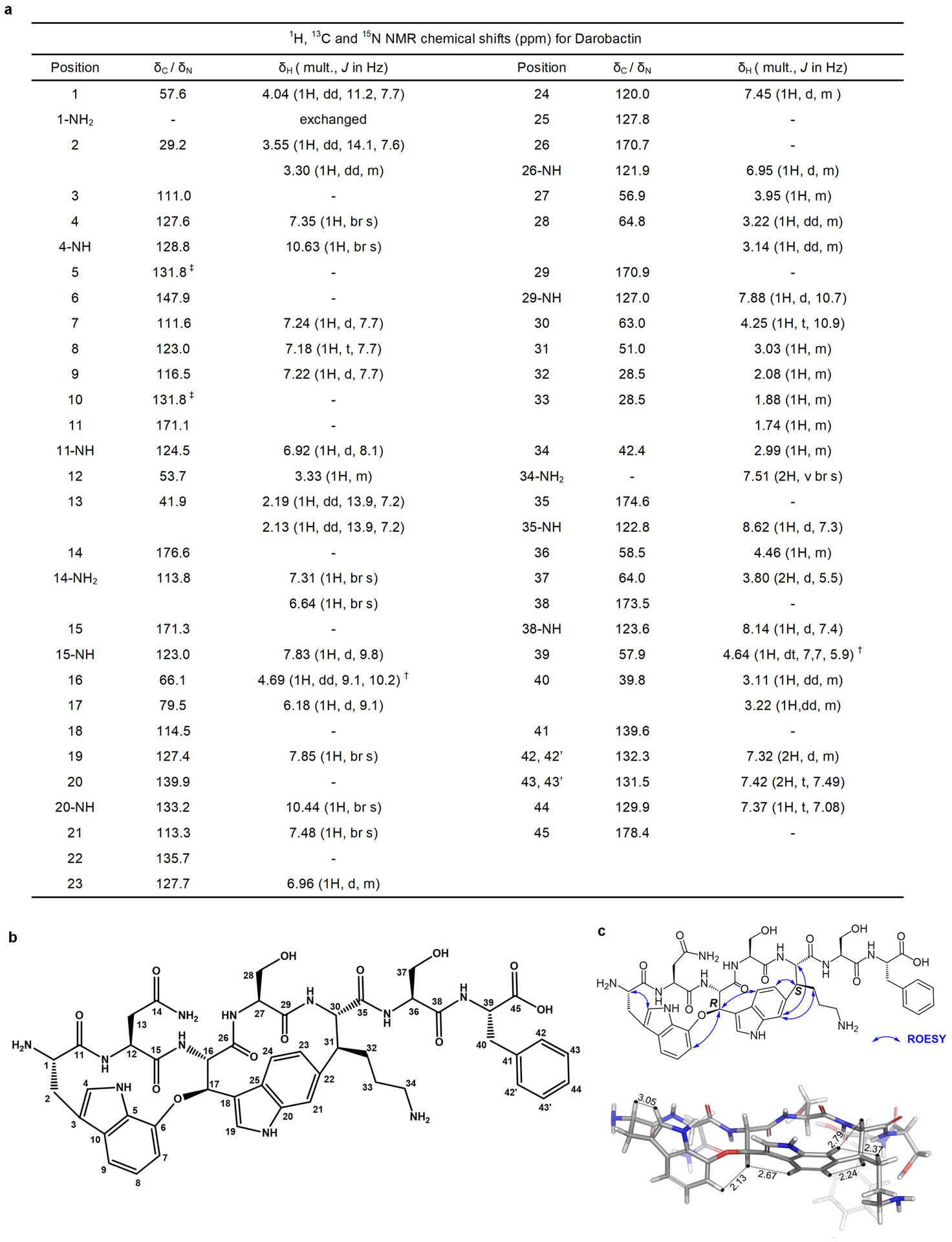
a, 1H, 13C and 15N NMR chemical shifts (ppm) for darobactin. † Due to overlap with residual water peak at 4.6 ppm, the multiplicity and J coupling values were from a different 1H- NMR spectrum of Darobactin in water: deuterated acetonitrile (2:1, v/v). ‡ Two partially overlapped peaks were observed at 131.79 ppm and 131.83 ppm. b, Structure of darobactin with numbering for NMR assignments. c, Key ROESY correlations (top) and 3D model of darobactin (bottom).
Extended Data Figure 3. Biosynthetic gene cluster (BGC) of darobactin in selected bacterial strains.
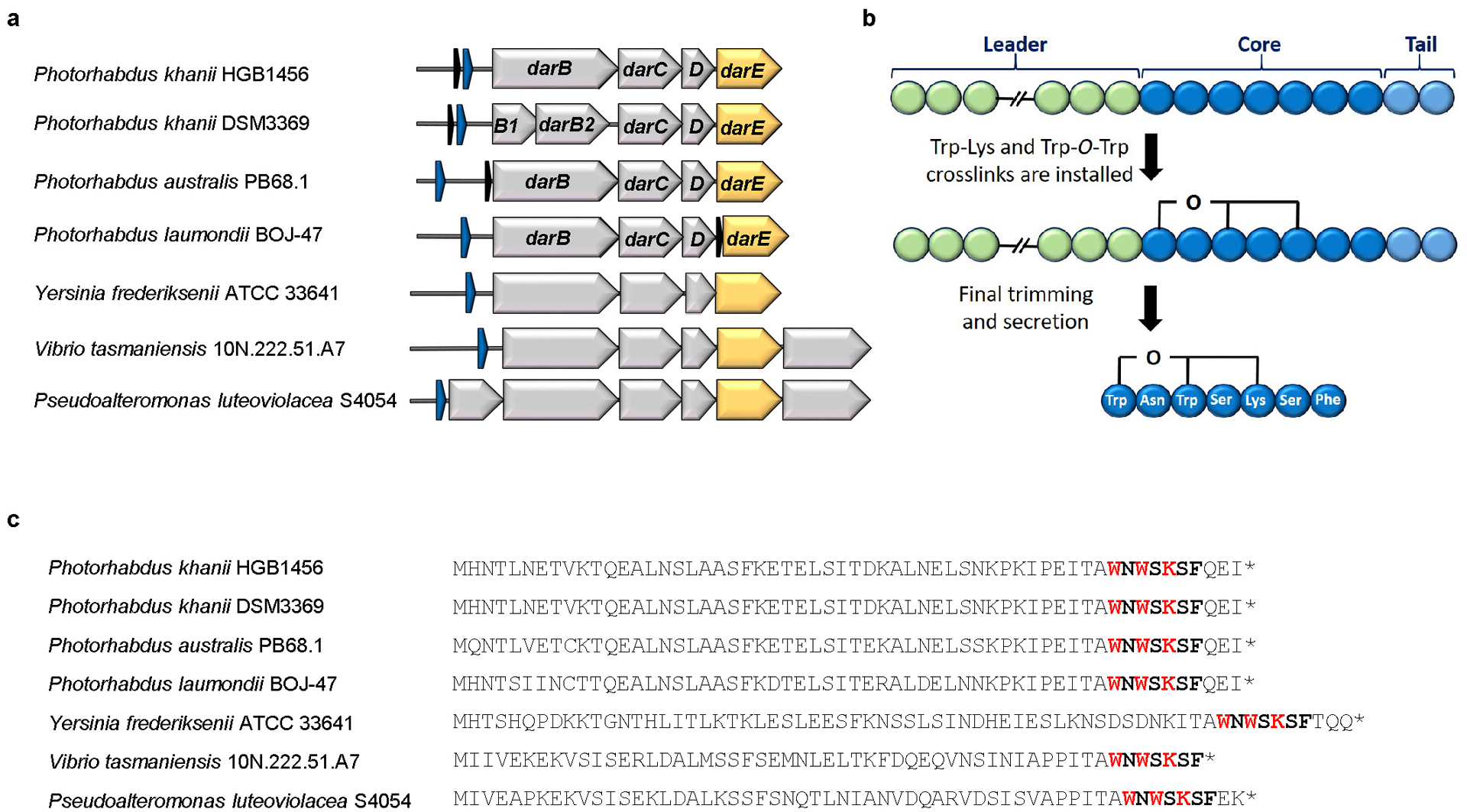
a, The BGC consists of the structural gene darA (colored in blue), darBCD (transporter encoding genes, in grey) and darE (encoding a radical SAM enzyme, in orange). In addition a relE-like gene (black) ORF can be co-located with the BGC at different positions. The BGC can be detected in most Photorhabdus strains in a conserved genetic region. In addition, homologous BGCs (related genes show the identical color code) are in Yersinia, Vibrio and Pseudoalteromonas strains. b, Biosynthetic hypothesis. The propeptide encoded by darA consists of 58 amino acids. The crosslinks are installed on the linear propeptide by DarE. In a next step the leader and tail regions are cleaved off and darobactin is secreted by the ABC transporter DarBCD. c, Amino acid sequence of the propeptide from selected bacterial strains. The darobactin core peptide is highlighted in bold and the amino acids involved in the crosslinking in bold red. The star indicates the stop codon.
Extended Data Figure 4. Darobactin knockout strain and heterologous expression, and putative structures and producers of darobactin A-E.
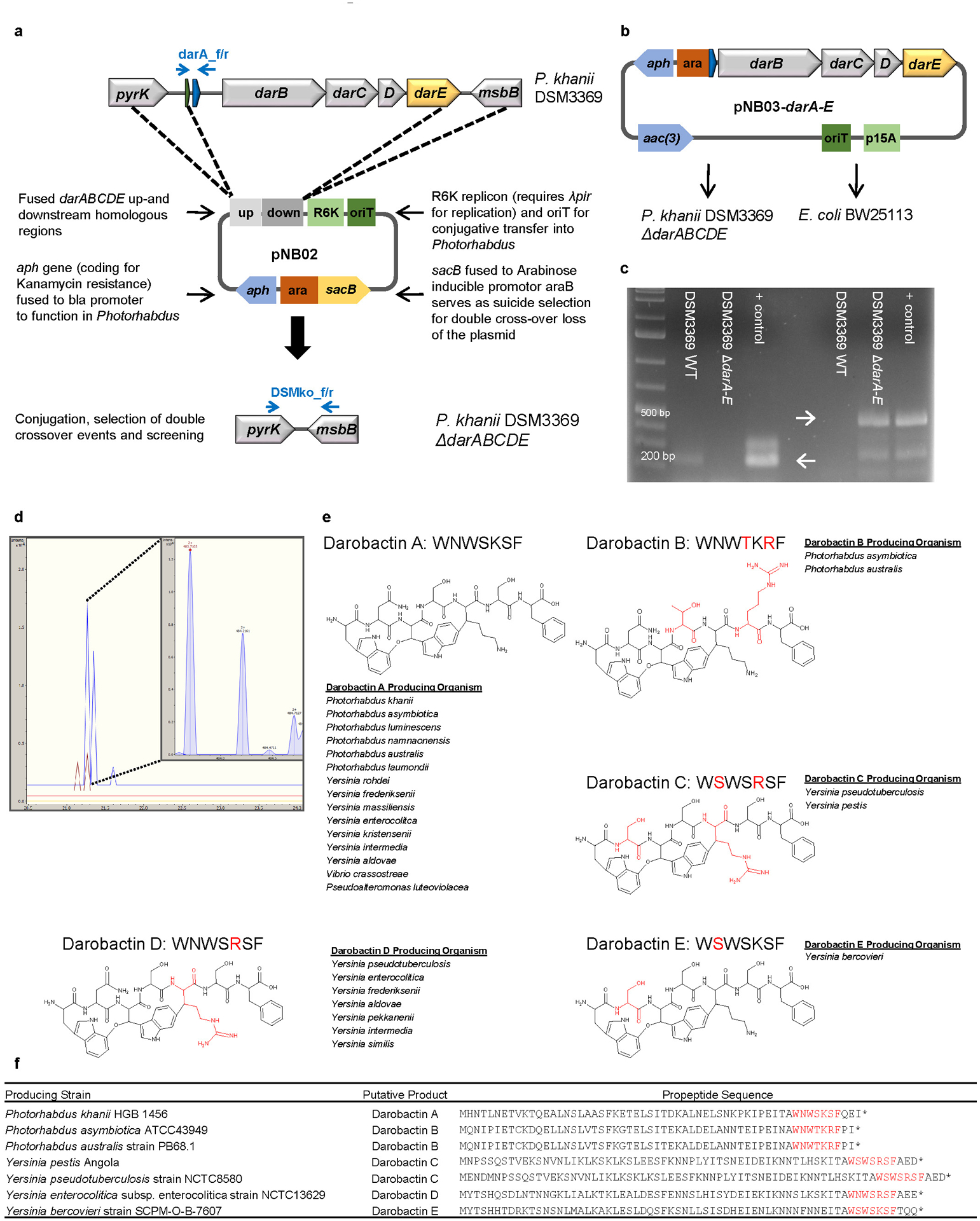
a, Scheme of the double cross-over knock out vector pNB02 and the targeted genomic region. b, Scheme of the darobactin BGC expression plasmid. c, Test PCRs on P. khanii DSM3369 ΔdarABCDE, proving the loss of the darobactin BGC; left: Amplification of darA (primers darA_f/r) resulting in a 177 bp fragment in the WT and in no fragment in the mutant; right: After loss of pNB02 (indicated by sensitivity to Kan) amplification of a 450 bp fragment if the BGC is deleted (primers DSMko_f/r); positive control: pNB03-darA-E and pNB02, respectively; primer positions indicated in blue in scheme a. Raw DNA gel is provided in Supplementary Figure 1. d, LC-MS extracted ion chromatogram (EIC) at m/z=483.7089 ± 0.001, yellow: P. khanii DSM3369 ΔdarABCDE + pNB03 red: P. khanii DSM3369 ΔdarABCDE + pNB03-darA-E, brown: E. coli BW25113 + pNB03-darA-E blue: P. khanii DSM3369 WT, inset: HRMS spectrum of the ion peak showing the double charged [M+2H]2+ ion corresponding to darobactin. Data (c and d) are representative of at least three independent biological replicates. e, Putative darobactin analogs B-E were drawn based on the amino acid sequence present in the darobactin BGC. The proposed producing organisms were identified by a BLASTP search of the 7 amino acid sequence of darobactin A, and confirming the presence of darBCDE downstream of the propeptide. Amino acid changes from darobactin A are highlighted in red. f, The table shows the propeptide sequence of the various darobactin analogs.
Extended Data Figure 5. Darobactin mechanism of action and resistance studies.
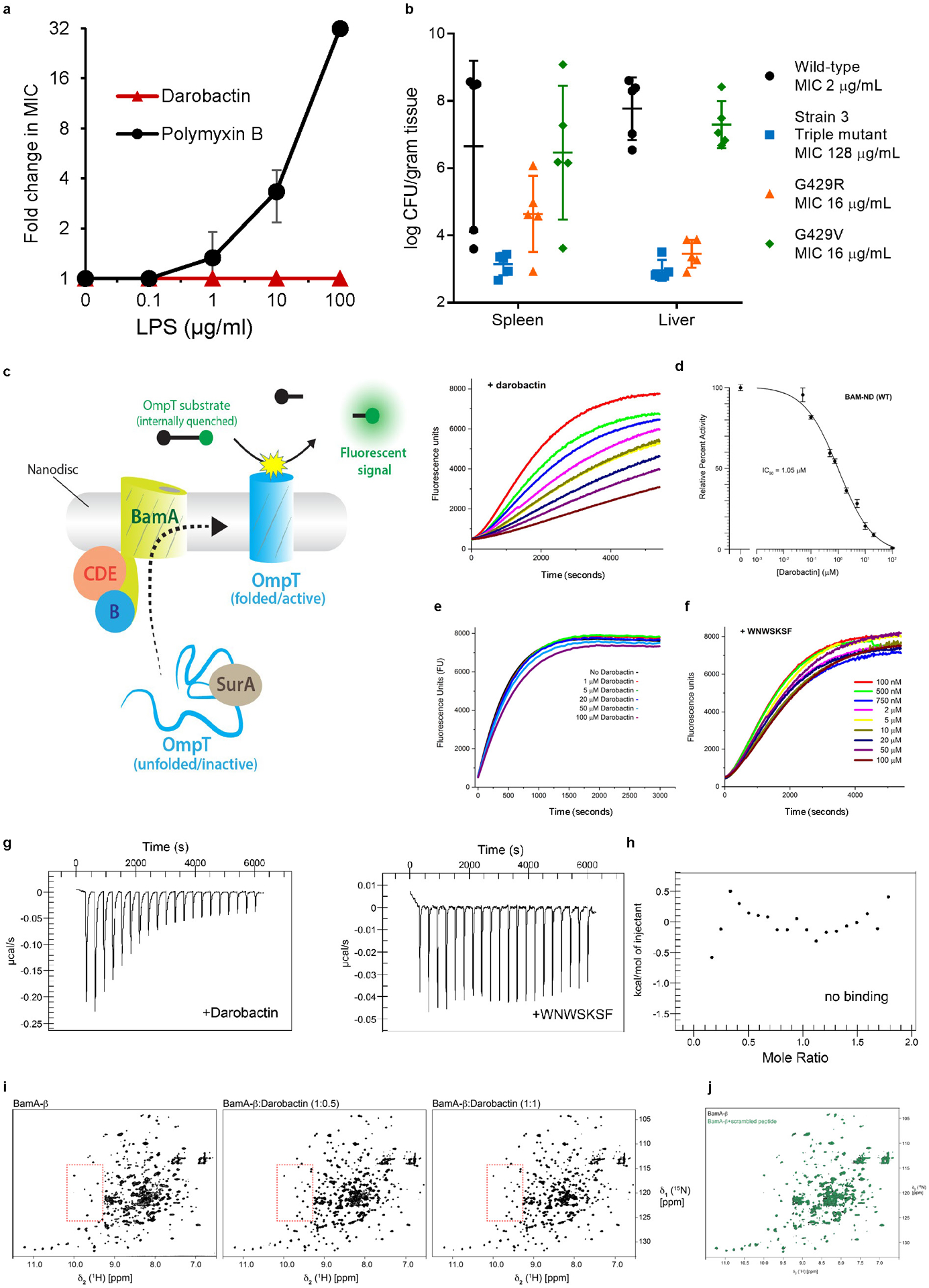
a, Darobactin and polymyxin B MIC against E. coli MG1655 were performed in the presence of LPS. Addition of LPS antagonized polymyxin activity, but not darobactin. Data are from triplicate experiments, symbols are mean, error bars SD. b, Groups of five mice were infected ip with 107 E. coli ATCC 25922, then at 24 h euthanized (if not already dead), livers and spleens harvested, homogenized, and plated for c.f.u. The wild-type E. coli caused 60% death and was at high c.f.u. burdens in liver and spleen. All three darobactin resistant bamA mutants had reduced virulence, with 100% survival in all groups at 24 h. The burden of bacteria of the Strain-3 (Fig. 2a) triple bamA mutant was close to limit of detection (LOD) in organs, G429R was at low but detectable levels, whereas G429V was at relatively high loads in organs. n=5, lines are mean, error bars are SD. c, Schematic of the BAM activity assay with BAM (BamA-E) first being inserted into lipid nanodiscs. Unfolded OmpT, along with the periplasmic chaperone SurA, is then mixed with the BAM-nanodiscs, where BAM folds OmpT into the nanodisc. OmpT, a protease, cleaves an internally-quenched peptide which produces a fluorescent signal. d, BAM-nanodisc (ND) assays performed in the presence of increasing concentrations of darobactin (left panel). The results show that darobactin is able to specifically inhibit BAM-ND activity in a dose-dependent manner. This data was then normalized against the ‘no darobactin’ sample and the highest concentration of darobactin, and plotted and an IC50 calculated using the online IC50 Calculator tool (AAT Bioquest) (right panel). n=3 biologically independent experiments. Symbols are mean, error bars are SD. e, As a control to the BAM-ND assays, we prepared OmpT-ND and assayed OmpT-ND activity in the presence of increasing concentrations of darobactin. To prepare the OmpT-ND, we first expressed OmpT as inclusion bodies and then refolded using previously reported methods. We then incorporated OmpT into nanodiscs using the same methods as described for BAM. The assays were performed using 0.4 μM of OmpT-ND. The results show that darobactin has virtually no effect on OmpT-ND activity, thereby confirming that darobactin is not affecting OmpT activity itself, or disrupting the nanodiscs themselves. A representative plot is shown from a triplicate experiment. f, The WNWSKSF peptide does not inhibit BAM-ND. As a control to darobactin, the BAM-ND assays were performed in the presence of increasing concentrations of a linear peptide WNWSKSF. The results show that the WNWSKSF peptide has only minimal effects on BAM-ND activity, even at the highest concentrations. A representative plot is shown from a triplicate experiment. g,h, Specific binding of darobactin to BamA/BAM. Mole Ratio is the protein/ligand ratio. g, Plot of ITC experiments of WT BAM titrated with darobactin showing a Kd of 1.2 μM, N of 0.52, ΔH of −25 kcal/mol, and ΔS of =-56 cal/mol·K. The experiment was repeated independently two times with similar results. h, Plot of ITC experiments of WT BAM titrated with the peptide WNWSKSF showing no binding within the same concentration range used for darobactin. The experiment was repeated independently two times with similar results. i, j, 2D [15N, 1H]-TROSY spectra of 250 μM BamA-β in 0.1% w/v LDAO. i, BamA-β in the absence (left) and in the presence of darobactin in the molar ratio 1:0.5 (middle) and 1:1 (right). The red dashed line outlines an exemplary spectral region experiencing substantial spectral changes during the titration. The experiment was repeated independently two times with similar results. j, An overlay of apo BamA-b (black) (250 μM) with BamA-b+scrambled linear peptide WNKWSFS (green) (230 μM). The experiment was performed once as is typical for NMR.
Extended Data Figure 6. Darobactin disrupts the outer membrane and causes lysis of E. coli.
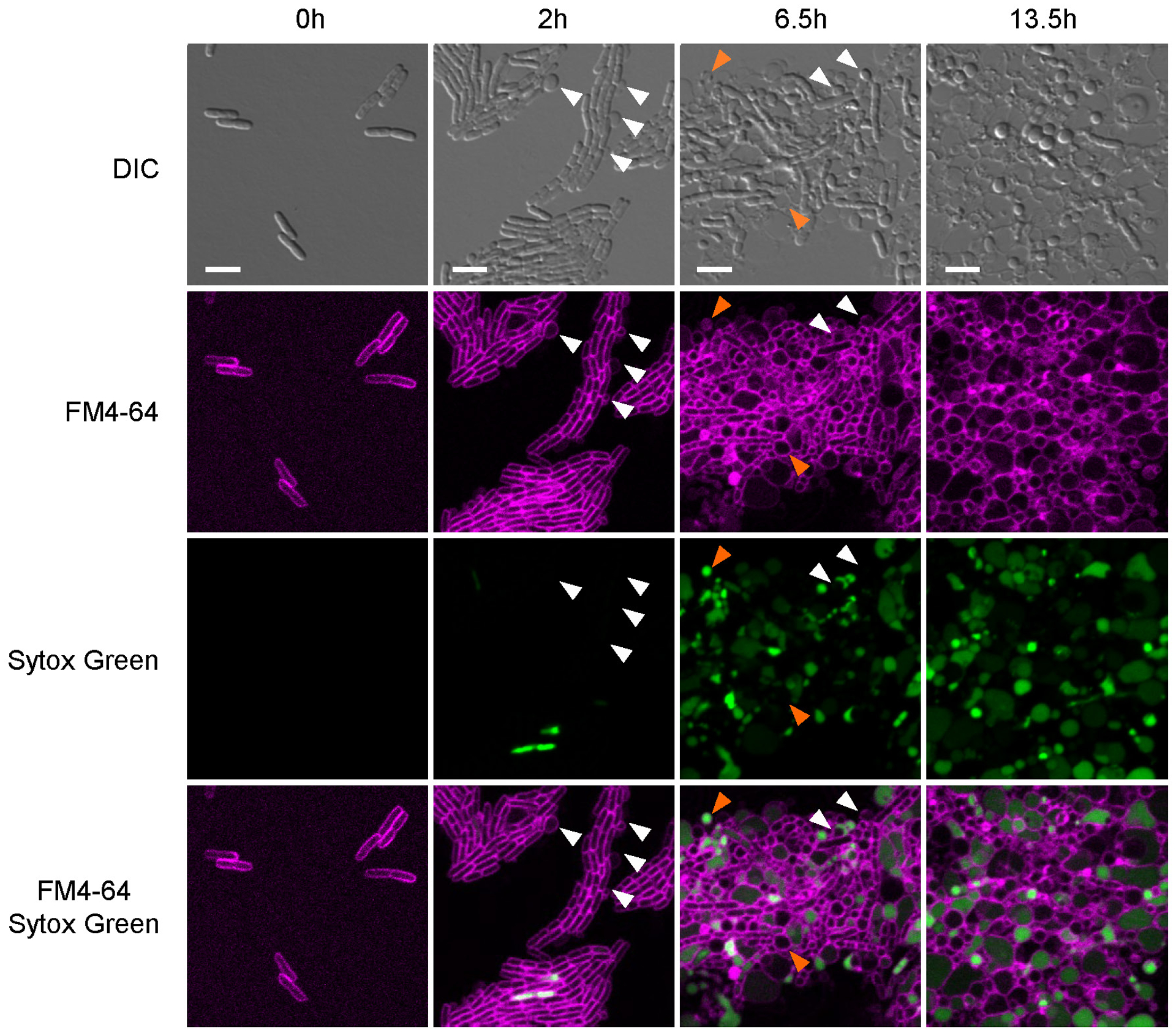
E. coli MG1655 cells were placed on top of an agarose pad containing darobactin and the fluorescent dyes FM4–64, to stain the membrane (false-colored here in magenta), and Sytox Green, to show membrane permeabilization (false-colored here in green), and observed over time at 37°C under the microscope. For each time indicated, representative panels show the killing progression of E. coli MG1655 with darobactin. White arrows highlight membrane blebbing, and orange arrows highlight swelling and lysis. Scale bar, 5 μm. This figure is representative of three biologically independent experiments performed with similar results.
Extended Data Figure 7. Transcriptome analysis of darobactin treatment shows activation of envelope stress pathways.
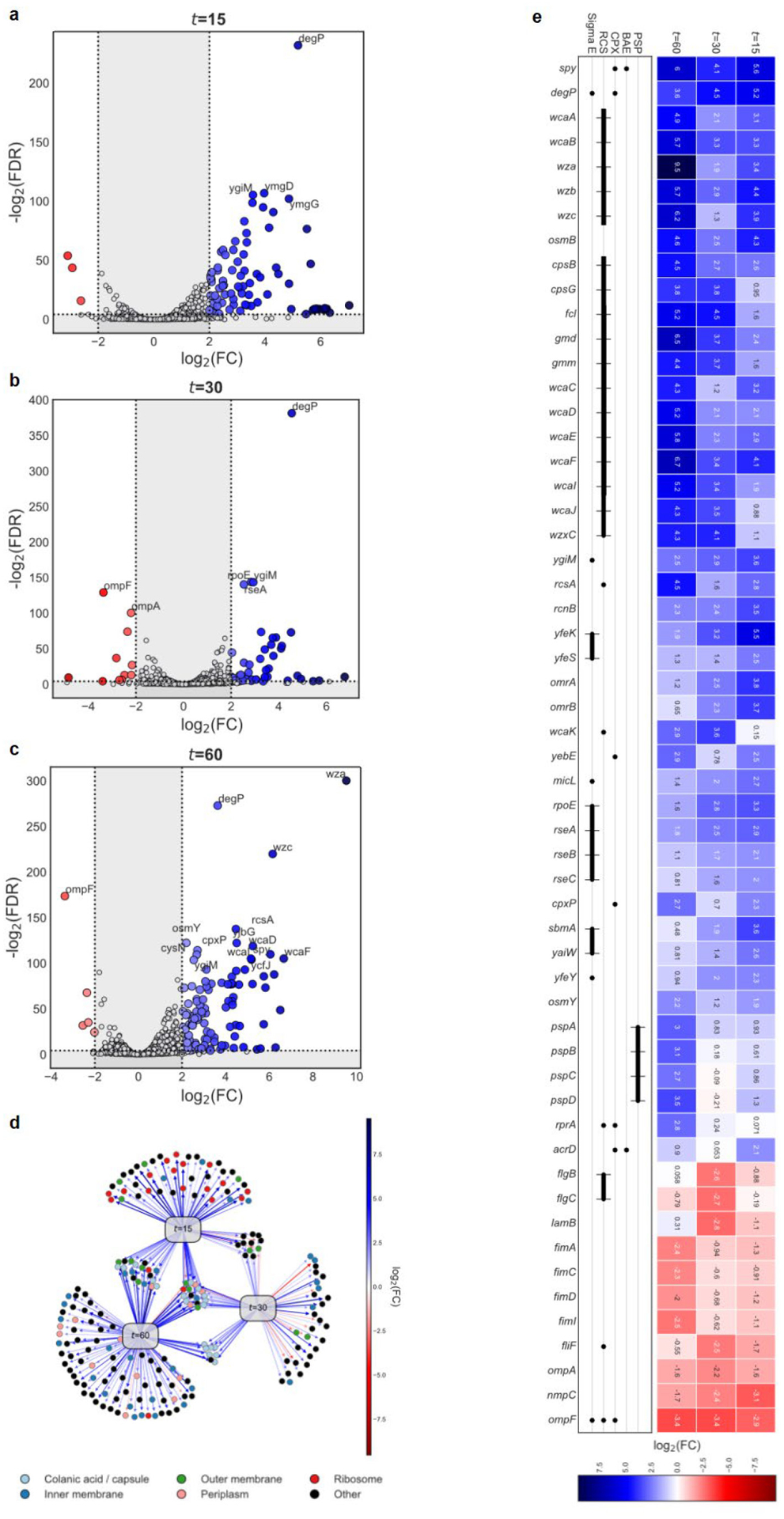
E. coli BW25113 were treated with 1xMIC darobactin, RNA isolated, and sequenced. a,b,c, Volcano plots illustrating differential gene expression (edgeR’s Fisher’s Exact Test; significance |log2FC| ≥ 2 and FDR < 0.001; n=3 biologically independent samples for each control/treatment) at time points a, t=15, b, t=30, and c, t=60 minutes after exposure. Gray, not significant. d, Network visualization of differentially expressed genes at each time point. Nodes include genes (colored circles) and time points (gray rectangle). Gene node colors represent relevant functional categories. Directed edges radiating from a time point node represent differentially expressed genes with respect to the given time point with weights reflecting the |log2FC|. e, (Top) Heatmap showing the differential expression (|log2FC|) of genes of interest and (bottom) assignment to envelope stress pathways. Solid lines depict members of the same operon. In all panels, red indicates down-regulation (lower expression in treatment relative to control) and blue indicates up-regulation.
Extended Data Figure 8. Two-dimensional thermal proteome profiling (2D-TPP) of darobactin.
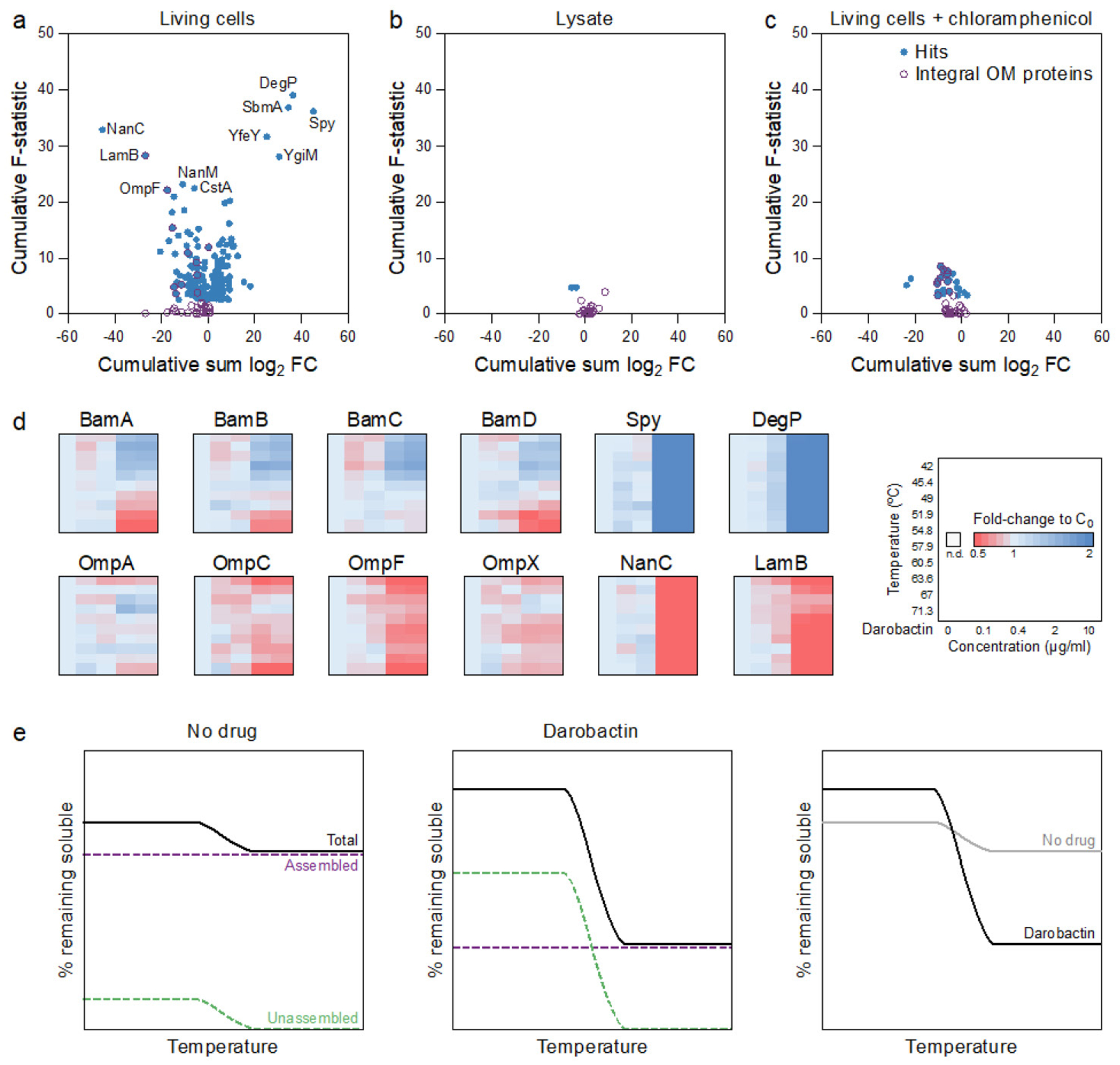
a,b,c, Pseudo-volcano plots for 2D-TPP experiments of darobactin treatment (10 min) of E. coli BW25113 in a, living cells, b, lysate, and c, living cells pre-treated with chloramphenicol to inhibit protein synthesis (n=1 at each concentration, heated to 10 different temperatures, for each experiment). Significant hits (false discovery rate <1%, calculated with a functional analysis of dose-response, requiring stabilization effects at n>1 temperatures as described in Sridharan et al. (2019)52) are highlighted in blue and integral outer membrane proteins are highlighted in purple. d, Heatmaps for selected proteins in the experiment with living cells. For each protein and temperature (key on right), the signal intensity was normalized to the vehicle control. e, Schematic of putative thermally stable assembled versus labile unassembled populations of BAM machinery with darobactin treatment.
Extended Data Figure 9. Darobactin single-dose pharmacokinetics and mouse thigh models.
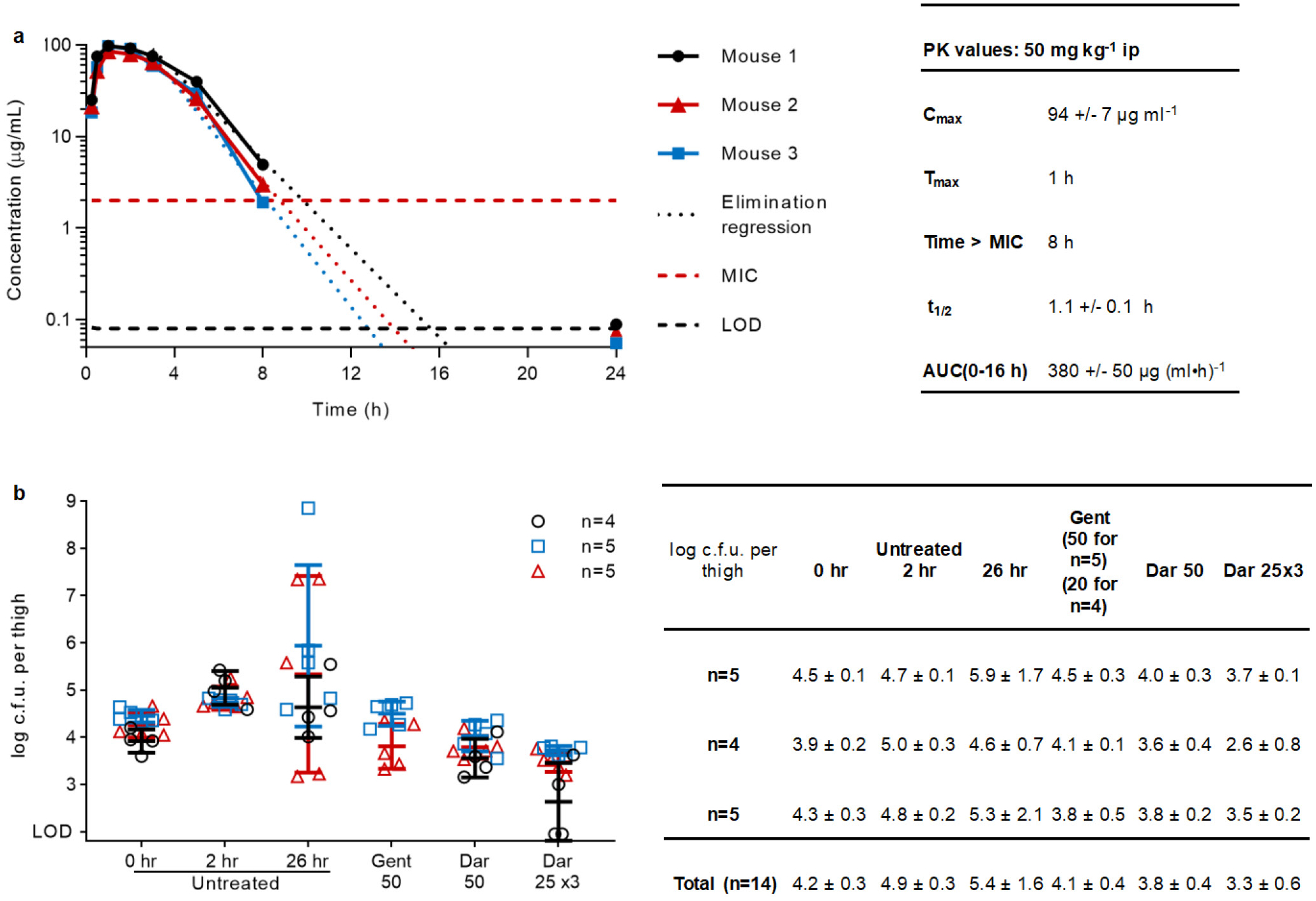
a, Three mice were injected with 50 mg kg−1 darobactin ip, and blood samples were collected by tail snip over 24 h. Samples (n=1 per timepoint and mouse) were analyzed for darobactin content by LC-MS/MS, and concentrations calculated using a standard curve created by linear regression on the log(AUC peak) to log(concentration) of standards. Pharmacokinetic values were calculated in Excel; t1/2 and Time>MIC assuming first order elimination and using linear regression on time points 3 – 8 h; AUC (0–16 h) using the trapezoid rule. Limit of detection (LOD) was 0.08 μg ml−1. b, A mouse thigh model was repeated three times testing the efficacy of darobactin against E. coli AR350. Mice were injected with bacteria in their right thigh at 0 hr, then dosed with no drug, gentamicin, or darobactin starting at 2 hr (50 mg kg−1 once, 25 mg kg−1 given three times every 6 h, or 20 mg kg−1 once). At 26 hr mice were sacked and thighs collected and plated for c.f.u. Centre lines are mean, error bars are SD.
Extended Data Table 1.
Photorhabdus and Xenorhabdus species.
| Photorhabdus sp. | # Strains in screen | Source | Xenorhabdus sp. | # Strains in screen | Source |
|---|---|---|---|---|---|
| P. akhurstii | 1 | DSMZ* | X. beddingii | 1 | HGB |
| P. caribbeanensis | 1 | DSMZ | X. bovienii | 12 | HGB |
| P. cinerea | 1 | DSMZ | X. doucetiae | 1 | HGB |
| P. hainanensis | 1 | DSMZ | X. indica | 5 | DSMZ |
| P. heterorhabditis | 2 | DSMZ | X. innexi | 3 | HGB and DSMZ |
| P. kayaii | 1 | DSMZ | X. ishibashi | 1 | DMZ |
| P. khanii | 2 | HGB† and DSMZ | X. japonica | 2 | HGB and DSMZ |
| P. kleinii | 1 | DSMZ | X. japonicus | 1 | HGB |
| P. laumondii subsp. laumondii | 1 | DSMZ | X. khoisanae | 4 | DSMZ |
| P. luminescens | 3 | HGB | X. miraniensis | 2 | HGB |
| P. noenieputensis | 1 | DSMZ | X. nematophila | 2 | HGB |
| P. stackebrandtii | 1 | DSMZ | X. poinarii | 3 | HGB |
| P. tasmaniensis | 1 | DSMZ | X. szentirmaii | 1 | HGB |
| P. temperata | 6 | HGB and DSMZ | |||
| P. thracensis | 1 | DSMZ | |||
| Photorhabdus sp. | 5 | HGB |
Number of strains and species of Photorhabdus and Xenorhabdus included in the screen.
DSMZ, Deutsche Sammlung von Mikroorganismen und Zellkulturen;
HGB, Heidi Goodrich-Blair.
Supplementary Material
Acknowledgements
This work was supported by NIH grant P01 AI118687 to K.L. and K.N. A.M. was supported by a fellowship from the EMBL Interdisciplinary Postdoc (EI3POD) Programme under Marie Skłodowska‐Curie Actions COFUND (grant number 664726). S.H. was supported by the Swiss National Science Foundation via the NFP 72 (407240_167125).
N.N. was supported by NIH grants GM127896 and GM127884. We thank Heidi Goodrich-Blair for providing strains of Photorhabdus and Xenorhabdus. We thank Michael Kagan for help in isolating darobactin; the Northeastern University Barnett Institute MS Core Facility for access to its LC-MS resources; and Dr. Donna Baldisseri from Bruker Biospin Corporation for recording some of the NMR data of darobactin. We thank Nils Kurzawa for the help with the data analysis of thermal proteome profiling data. We thank William Fowle for assistance with SEM and Yuan Su for assistance with the ITC experiments; and Ricardo Machado for help with taxonomy of Photorhabdus.
Footnotes
The authors declare no competing financial interests.
References
- 1.Payne DJ, Gwynn MN, Holmes DJ & Pompliano DL Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6, 29–40 (2007) [DOI] [PubMed] [Google Scholar]
- 2.Lewis K Platforms for antibiotic discovery. Nat Rev Drug Discov 12, 371–387 (2013) [DOI] [PubMed] [Google Scholar]
- 3.Lomovskaya O & Lewis K Emr, an Escherichia coli locus for multidrug resistance. Proc Natl Acad Sci U S A 89, 8938–8942 (1992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li XZ & Nikaido H Efflux-mediated drug resistance in bacteria. Drugs 64, 159–204 (2004) [DOI] [PubMed] [Google Scholar]
- 5.Tacconelli E et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18, 318–327 (2018) [DOI] [PubMed] [Google Scholar]
- 6.Brown ED & Wright GD Antibacterial drug discovery in the resistance era. Nature 529, 336–343 (2016) [DOI] [PubMed] [Google Scholar]
- 7.Crawford JM & Clardy J Bacterial symbionts and natural products. Chem Commun 47, 7559–7566 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tobias NJ, Shi YM & Bode HB Refining the natural product repertoire in entomopathogenic bacteria. Trends Microbiol 26, 833–840 (2018) [DOI] [PubMed] [Google Scholar]
- 9.Tambong JT Phylogeny of bacteria isolated from Rhabditis sp. (Nematoda) and identification of novel entomopathogenic Serratia marcescens strains. Curr Microbiol 66, 138–144 (2013) [DOI] [PubMed] [Google Scholar]
- 10.Yokoyama K & Lilla EA C-C bond forming radical SAM enzymes involved in the construction of carbon skeletons of cofactors and natural products. Nat Prod Rep 35, 660–694 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schramma KR, Bushin LB & Seyedsayamdost MR Structure and biosynthesis of a macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat Chem 7, 431–437 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Embley TM & Stackebrandt E The molecular phylogeny and systematics of the actinomycetes. Annu Rev Microbiol 48, 257–289 (1994) [DOI] [PubMed] [Google Scholar]
- 13.Lloyd-Price J, Abu-Ali G & Huttenhower C The healthy human microbiome. Genome Med 8, 51 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bokulich NA et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Shea R & Moser HE Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 51, 2871–2878 (2008) [DOI] [PubMed] [Google Scholar]
- 16.Ling LL et al. A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konovalova A, Kahne DE & Silhavy TJ Outer membrane biogenesis. Annu Rev Microbiol 71, 539–556 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakelar J, Buchanan SK & Noinaj N The structure of the beta-barrel assembly machinery complex. Science 351, 180–186 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghequire MGK, Swings T, Michiels J, Buchanan SK & De Mot R Hitting with a BAM: selective killing by lectin-like bacteriocins. mBio 9 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storek KM et al. Monoclonal antibody targeting the beta-barrel assembly machine of Escherichia coli is bactericidal. Proc Natl Acad Sci U S A 115, 3692–3697 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hart EM et al. “A small-molecule inhibitor of BamA impervious to efflux and the outer membrane permeability barrier.” Proc Natl Acad Sci U S A 116, 21748–21757 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann JB, et al. Sequence-specific solution NMR assignments of the beta-barrel insertase BamA to monitor its conformational ensemble at the atomic level. J Am Chem Soc 140, 11252–11260 (2018) [DOI] [PubMed] [Google Scholar]
- 23.Kaur H et al. Identification of conformation-selective nanobodies against the membrane protein insertase BamA by an integrated structural biology approach. J Biomol NMR 9, 1–10 (2019) [DOI] [PubMed] [Google Scholar]
- 24.Ramos-Castaneda JA et al. Mortality due to KPC carbapenemase-producing Klebsiella pneumoniae infections: Systematic review and meta-analysis: Mortality due to KPC Klebsiella pneumoniae infections. J Infect 76, 438–448 (2018) [DOI] [PubMed] [Google Scholar]
- 25.Xu L, Sun X & Ma X Systematic review and meta-analysis of mortality of patients infected with carbapenem-resistant Klebsiella pneumoniae. Annals of clinical microbiology and antimicrobials 16, 18 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun J, Zhang H, Liu YH & Feng Y Towards understanding MCR-like colistin resistance. Trends Microbiol 26, 794–808 (2018) [DOI] [PubMed] [Google Scholar]
- 27.Levasseur P et al. Efficacy of a ceftazidime-avibactam combination in a murine model of septicemia caused by Enterobacteriaceae species producing ampc or extended-spectrum beta-lactamases. Antimicrob Agents Chemother 58, 6490–6495 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wunderink RG et al. Effect and safety of meropenem-vaborbactam versus best-available therapy in patients with carbapenem-resistant Enterobacteriaceae infections: the TANGO II randomized clinical trial. Infect Dis Ther 7, 439–455 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King AM et al. Aspergillomarasmine A overcomes metallo-beta-lactamase antibiotic resistance. Nature 510 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Smith PA, Steed DB & Romesberg F Efforts toward broadening the spectrum of arylomycin antibiotic activity. Bioorg Med Chem Lett 23, 5654–5659 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith PA et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 561 (2018) [DOI] [PubMed] [Google Scholar]
- 32.Richter MF et al. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crits-Christoph A, Diamond S, Butterfield CN, Thomas BC & Banfield JF Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558, 440 (2018) [DOI] [PubMed] [Google Scholar]
- 34.Tobias NJ et al. Natural product diversity associated with the nematode symbionts Photorhabdus and Xenorhabdus. Nat Microbiol 2, 1676–1685 (2017) [DOI] [PubMed] [Google Scholar]
- 35.Pantel L et al. Odilorhabdins, antibacterial agents that cause miscoding by binding at a new ribosomal site. Mol. Cell 70, 83–94.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Racine E et al. In vitro and in vivo characterization of NOSO-502, a novel inhibitor of bacterial translation. Antimicrob. Agents Chemother 62, e01016–e01018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poinar G Jr Origins and phylogenetic relationships of the entomophilic rhabditids, Heterorhabditis and Steinernema. Fundam Appl Nematol 16, 333–338 (1993) [Google Scholar]
- 38.Antipov D, Korobeynikov A, McLean JS & Pevzner PA hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32, 1009–1015 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blin K et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res 45, 36–41 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lassak J, Henche AL, Binnenkade L, & Thormann KM ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol 76, 3263–3274 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Datsenko KA & Wanner BL One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang X, et al. Identification of thiotetronic acid antibiotic biosynthetic pathways by target-directed genome mining. ACS Chem Biol 10, 2841–2849 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gust B, Challis GL, Fowler K, Kieser T, & Chater KF PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A, 100, 1541–1546 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy KC & Campellone KG Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC molecular biology 4, 11 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mateus A et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol Syst Biol 14, e8242 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Becher I et al. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat Chem Biol 12, 908–910 (2016) [DOI] [PubMed] [Google Scholar]
- 49.Hughes CS et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol 10, 757 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes CS et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc 14, 68–85 (2019) [DOI] [PubMed] [Google Scholar]
- 51.Sridharan S et al. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat Commun 10, 1155 (2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franken H et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat Protoc 10, 1567–1593 (2015) [DOI] [PubMed] [Google Scholar]
- 53.Alvarez FJD, Orelle C, & Davidson AL Functional reconstitution of an ABC transporter in nanodiscs for use in electron paramagnetic resonance spectroscopy. J Am Chem Soc 132, 9513–9515 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritchie TK et al. Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Meth Enzymol 464, 211–231 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roman-Hernandez G, Peterson JH, & Bernstein HD Reconstitution of bacterial autotransporter assembly using purified components. Elife 3, e04234 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hagan CL, Kim S & Kahne D Reconstitution of outer membrane protein assembly from purified components. Science 328, 890–892 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the findings of this study are available within the paper and its Supplementary Information or in databases. The genome of P. khanii HGB1456 has been deposited to Genbank with identifier WHZZ00000000.The transcriptomic dataset (Extended Data Figure 7) has been deposited to NCBI Sequence Read Archive with identifier PRJNA530781.The mass spectrometry proteomics (Extended Data Figure 8; Supplementary Table 2) data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013319. Source Data for Figures 2c, 4 and Extended Data Figures 5b, 9 are provided with the paper. All other data available from corresponding author.