

Abstract
Objective
FMF is an inherited autoinflammatory syndrome caused by mutations in the MEFV gene. MEFV variants are still largely classified as acvariant of uncertain significance, or with unresolved classification, posing significant challenges in FMF diagnosis. Rare Exome Variant Ensemble Learner (REVEL) is a recently developed variant metapredictor tool. To reduce the number of MEFV variants with ambiguous classification, we extracted REVEL scores for all missense variants present in the INFEVERS database, and analysed its correlation with expert-based classification and localization in the MEFV-encoded pyrin functional domains.
Methods
The data set of 216 MEFV missense variants was divided into four categories (likely benign, variant of uncertain significance, likely pathogenic and unresolved). Variants were plotted onto the pyrin protein, the distribution of REVEL scores in each category was computed and means, confidence intervals, and area under the receiver operating curve were calculated.
Results
We observed a non-random distribution of pathogenic variants along the pyrin functional domains. The REVEL scores demonstrated a good correlation with the consensus classification of the International Study Group for Systemic Autoinflammatory Diseases. Sensitivity, specificity and accuracy were calculated for different cut-off values of REVEL scores and a gene-specific-threshold of 0.298 was computed with confidence boundary limits. This cut-off value allowed us to propose a reclassification of 96 MEFV gene variants, thus reducing the variant of uncertain significance proportion from 61.6% to 17.6%.
Conclusion
The combination of available expert information with sensitive predictor tools could result in a more accurate interpretation of clinical consequences of MEFV gene variants, and to a better genetic counselling and patient management.
Keywords: familial Mediterranean fever, MEFV, genetics, variant interpretation
Rheumatology key messages
Variant interpretation in FMF can be simplified.
MEFV pathogenic variants are non-randomly distributed.
Introduction
FMF is the most common of the hereditary recurrent fevers with an estimated prevalence of 1 in 1–5/10 000. FMF was first described more than sixty years ago [1, 2], and since the identification of the causative gene (MEFV, NM_000243.2) has been intensively studied [3, 4]. The MEFV gene encodes for an 86 kDa protein (pyrin or marenostrin, TRIM20) prevalently expressed in immune cells (neutrophils, eosinophils, monocytes, dendritic cells and synovial fibroblasts), and is related to abnormal response of the innate immune system and IL-1β during periodic attacks of fever and serositis [5, 6]. Despite several reports describing large FMF patients’ datasets, a complete understanding of the pathogenic mechanisms responsible for this disease has not been reached yet. Recent studies suggested the possibility of dominant inheritance, at least for some mutations. This hypothesis is casting doubts on the traditional autosomal recessive mode of inheritance [7–10], a paradigm in FMF for long time. For FMF, a well-curated locus-specific database is available at INFEVERS (https://infevers.umai-montpellier.fr/web/), containing genotype and clinical information for >500 variants. Recently, the International Study Group for Systemic Autoinflammatory Diseases (INSAID) has critically reviewed the clinical information for 858 variants in the four genes (MEFV, TNFRSF1A, NLRP3 and MVK) responsible for hereditary recurrent fevers reaching a consensus classification for 94% of variants analysed [11]. The notable exception was represented by the MEFV gene, where only 55.7% of the variants could be successfully classified with almost a third of them being variants of uncertain significance (VOUS). As a result, clinicians face the harsh reality of frequently receiving an inconclusive genetic referral for FMF diagnosis. Internationally recognized guidelines have been reported by the American College of Medical Genetics (ACMG) and the Association for Molecular Pathology (AMP) [12], providing a five-tier classification of genetic variants in: pathogenic, likely pathogenic, VOUS (or VUS), likely benign and benign. However, because even the mode of inheritance in FMF families has been recently jeopardized by several reports of apparently dominant mutations, the ACMG/AMP guidelines might be of limited value in FMF. In this work, we used REVEL (a recently developed ensemble method that has been demonstrated to outperform most commonly used web-based predictors) [13], to improve the classification of many unsolved, unclassified or of uncertain significance MEFV gene variants. We also report a non-random distribution of variants along the coding sequence of the MEFV gene. This observation possibly identifies novel hotspots for variants with a putative pathogenic role.
Methods
Dataset of missense variants
The dataset consisted of all the 216 missense variants reported in the INFEVERS database (https://infevers.umai-montpellier.fr/web/) last accessed on 28 February 2019. We excluded from the following statistical analyses all the synonymous changes, terminating and noncoding variants, insertions, duplications and indels. We merged the ‘pathogenic’ and ‘likely pathogenic’, ‘benign’ and ‘likely benign’, ‘unsolved’ and ‘not classified’ variants (as classified at the INFEVERS locus specific database) in the ‘LIKELY PATHOGENIC’, ‘LIKELY BENIGN’ and ‘UNRESOLVED’ categories, respectively. Notably, of the 216 missense variants listed at INFEVERS only one ‘benign’ and six ‘pathogenic’ variants were present. The VOUS category corresponded to variants classified as ‘VOUS’ at INFEVERS. Therefore, our dataset was represented by 41 LIKELY PATHOGENIC, 89 VOUS, 41 LIKELY BENIGN and 45 UNRESOLVED variants. All variants were mapped along the MEFV coding sequence and its functional domains using Lollipops [14].
Statistical comparison
Rare Exome Variant Ensemble Learner (REVEL) is an ensemble method for predicting the pathogenicity of missense variants. It combines 18 individual prediction scores (eight based on functional scores and 10 on residues conservation scores) from 13 different in silico predictors, therefore can be considered a metapredictor tool. Differently from other computational tools, REVEL has been specifically trained and tested on rare missense variants [13].
We extracted the REVEL score of each variant and the mean (s.d.) and the 95% confidence intervals were determined for each category. The statistical significance of the differences between categories was evaluated by ordinary one-way ANOVA, and unpaired t test between each category.
To evaluate the predictive potential of REVEL scores in the FMF context, we built a classifier on the dataset represented by the LIKELY BENIGN and LIKELY PATHOGENIC variants (n = 82). Five different algorithms were tested to identify the best model for predicting variants’ pathogenicity potential based on their REVEL score, namely, Linear Discriminant Analysis, Classification and Regression Trees, k-Nearest Neighbours, Support Vector Machines with a linear kernel, and Random Forest. This set of algorithms was chosen as it represents a good combination of linear and non-linear models to test. The dataset was split into two parts, 80% was used to train the models through a 10-fold cross validation, and 20% was used as a validation dataset. All models were weighed using the ‘accuracy’ metric. Computations were performed in R using the Caret package.
Finally, the specificity, sensitivity, accuracy, Matthews’ correlation coefficient and Youden index for each REVEL score value in correctly identifying LIKELY BENIGN and LIKELY PATHOGENIC variants were calculated. Where not otherwise stated, GraphPad Prism v.8.0.1 (GraphPad Software, San Diego, CA, USA) was used for all statistical analyses.
Results
Non-random distribution of variants
The REVEL score was extracted for all the 216 missense variants present at the INFEVERS database. Each variant was mapped along the coding sequence of the MEFV gene, and with respect to the functional domains of the pyrin protein (Fig. 1). We observed that LIKELY PATHOGENIC variants (as defined in the methods) clustered in the SPRY and zf-B_box domains. In contrast, LIKELY BENIGN variants were markedly underrepresented in functional domains of the pyrin protein being localized, in large majority, in the exon 2 of the MEFV gene. Finally, VOUS occurred along the entire coding sequence of the MEFV, whereas distribution of UNRESOLVED variants appeared to mostly overlap with the LIKELY PATHOGENIC one.
Fig. 1.

Distribution of INFEVERS variants along pyrin protein
Distribution of 216 MEFV INFEVERS missense variants onto the pyrin protein. Pops size is proportional to the number of variants at a single aminoacid. The exons of the MEFV gene are shown in light blue.
REVEL variants score
The means of REVEL scores for LIKELY BENIGN (n = 41, 0.18), VOUS (n = 89, 0.25), LIKELY PATHOGENIC (n = 41, 0.39) and UNRESOLVED (n = 44, 0.31) variants showed statistically significant differences among the four categories (F 21.04, P < 0.0001, ordinary one-way ANOVA) demonstrating an appreciable level of correspondence between the INSAID consensus-based variant classification and an in silico computational scoring method such as REVEL (Fig. 2). The results of unpaired t-tests among each category demonstrated statistically significant differences between LIKELY BENIGN and UNRESOLVED, LIKELY BENIGN and LIKELY PATHOGENIC, and VOUS vs LIKELY PATHOGENIC (Fig. 2). Only one variant classified by INSAID as benign or likely benign had an unexpectedly high pathogenic score (E230Q, 0.737, identified as an outlier using ROUT method). In contrast, 28 of 41 INSAID pathogenic or likely pathogenic variants had a score below the REVEL threshold for pathogenicity (<0.4) [15]. Worthy of note, the M694V variant, considered to be responsible for the most severe FMF phenotype, barely reached the threshold for pathogenicity (REVEL score 0.403). Finally, the area under the ROC curve was 0.879 (P < 0.0001, Fig. 3)
Fig. 2.
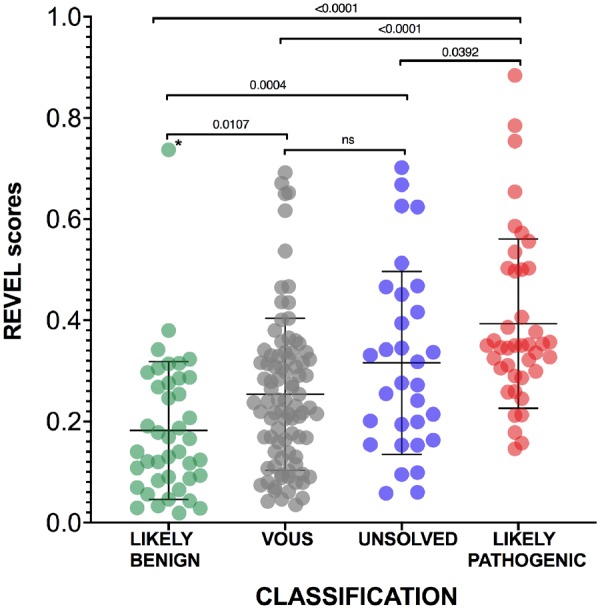
REVEL scores of MEFV variants with different classification
The mean and standard deviations are reported for each category. Unpaired t-tests were performed among all categories, two-tailed P-values after Tukey’s post hoc analysis for multiple comparison are shown. An asterisk denotes the only variant identified as outlier (E230Q). REVEL: Rare Exome Variant Ensemble Learner.
Fig. 3.
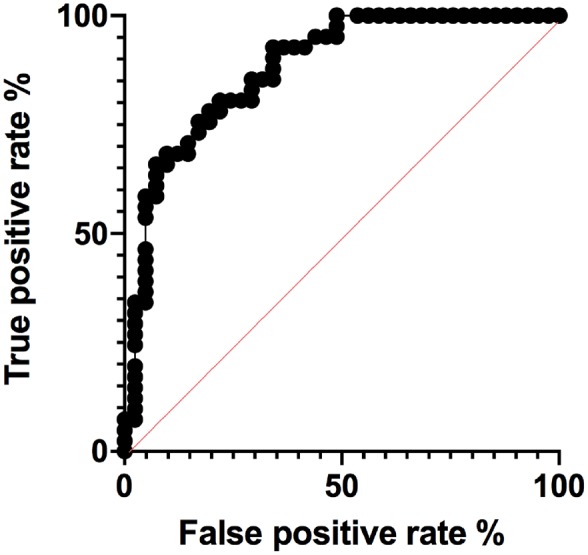
AUC for REVEL scores
The AUC is shown based on REVEL scores for the 41 BENIGN and 41 PATHOGENIC MEFV gene variants corresponding to the INFEVERS classes benign and likely benign, pathogenic and likely pathogenic, respectively. AUC: area under the ROC curve; REVEL: Rare Exome Variant Ensemble Learner.
Development of a new classifier and reclassification of MEFV variants
Because the REVEL precision in scoring variants was markedly different for the LIKELY BENIGN and LIKELY PATHOGENIC categories, we hypothesized that MEFV gene variants with putative pathogenic effects may act more subtly than variants used to train REVEL. Therefore, we decided to build a model trained on a dataset formed exclusively by the MEFV variants classified by INSAID panel as ‘benign’ and’ likely benign’, or ‘pathogenic’ and ‘likely pathogenic’.
Of the five algorithms tested, Linear Discriminant Analysis had the best performance, and was therefore further explored on the validation set. Prediction results on this set demonstrated an accuracy of 75%, with a specificity of 87.5%, and a sensitivity of 62.5%, and identified an approximate cut-off value of 0.29.
Next, we focused on the unclassified variants and aimed to assess their pathogenicity based on this previously trained model. To further optimize the performance of this new cut-off, we calculated the sensitivity and the specificity for different REVEL score thresholds (Fig. 4).
Fig. 4.
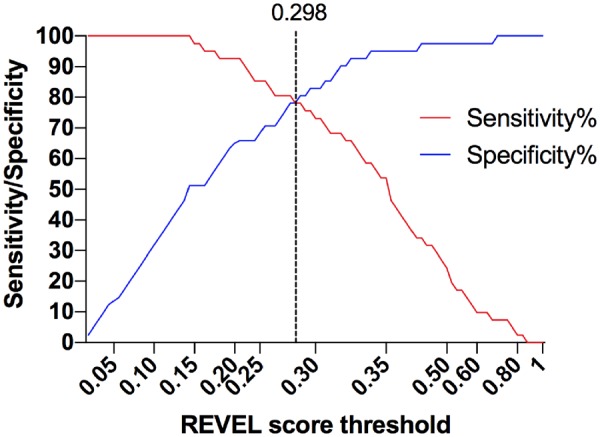
Sensitivity and specificity of different REVEL scores thresholds
Different REVEL score values were used as thresholds to assess 41 BENIGN and 41 PATHOGENIC variants of the pyrin protein. The dotted line indicates the best performing REVEL threshold. REVEL: Rare Exome Variant Ensemble Learner.
A nearly identical REVEL score of 0.298 yielded the best performance as cut-off, showing the highest accuracy (80.2%), positive predictive value (83.8%), Matthews’ correlation coefficient (60.8%) and Youden index (58.2%) values, with a sensitivity of 76% and a specificity of 83%, estimated on the entire dataset of LIKELY BENIGN and LIKELY PATHOGENIC variants (Supplementary material, available at Rheumatology online). Thus, when using 0.298 as the cut-off value, 34 of 41 LIKELY BENIGN variants had a score under the threshold for pathogenicity, while 31 of 41 LIKELY PATHOGENIC variants had a score above the cut-off. The classifier we developed had an overall area under the ROC curve of 0.878 (P < 0.0001).
We then re-classified all of the 89 ‘variant of uncertain significance’, 31 ‘unsolved’ and 13 ‘not classified’ variants from the INFEVER database with more precision, using the information deriving from the descriptive statistics of the LIKELY BENIGN and LIKELY PATHOGENIC REVEL scores variants to narrow the VOUS to a smaller fraction. To move from binary to graded classifications, all variants with a REVEL score lower than 0.225 (upper 95% confidence interval of the LIKELY BENIGN mean) were classified as LIKELY BENIGN, while variants with a score higher than 0.340 (lower 95% confidence interval of the LIKELY PATHOGENIC mean) were classified as LIKELY PATHOGENIC. The remaining variants (0.225⩽ REVEL score ⩽0.340) were considered VOUS.
Applying these criteria to the 133 VOUS and UNRESOLVED variants, 61 and 35 could be reclassified as LIKELY BENIGN and LIKELY PATHOGENIC, respectively (Supplementary Table S1, available at Rheumatology online, and Fig. 5). The same criteria changed the classification of three likely benign variants into LIKELY PATHOGENIC (including the E230Q variant) and five likely pathogenic into LIKELY BENIGN (Supplementary Table S1, available at Rheumatology online). Notably, all but one of these eight variants were only provisionally classified by INSAID experts [11].
Fig. 5.
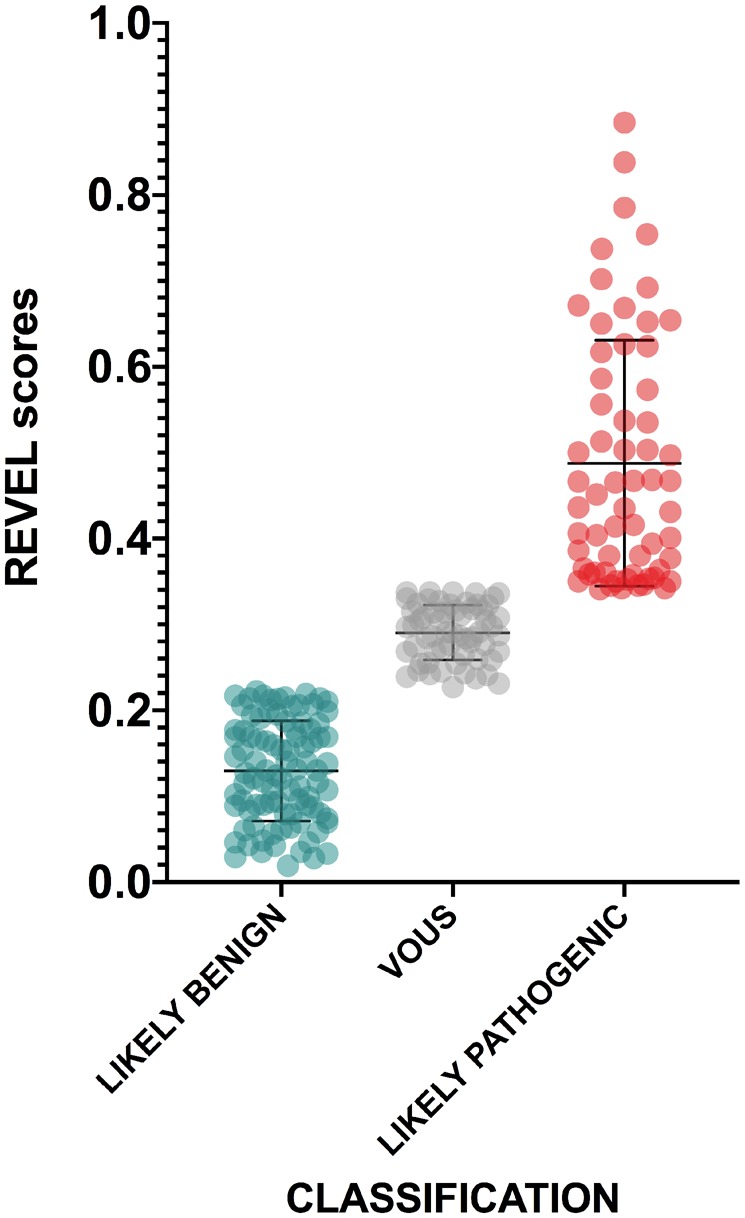
Distribution of MEFV variants’ REVEL scores after reclassification
The mean and standard deviations are reported for each category. Unpaired t-tests were performed among all categories, two-tailed P-values after Tukey’s post hoc analysis for multiple comparison are shown. REVEL: Rare Exome Variant Ensemble Learner.
Finally, we mapped the newly classified variants along the MEFV protein amminoacid sequences, which confirmed the preferential distribution of pathogenic variants prevalently in the PRY and SPRY domains (Fig. 6).
Fig. 6.

Distribution of MEFV gene variants after reclassification
Distribution of 216 MEFV missense variants onto the pyrin protein after reclassifying VOUS and UNSOLVED variants. Pops size is proportional to number of variants at single aminoacid positions. VOUS: variant of uncertain significance.
Discussion
The advent of massive parallel next generation sequencing technology has greatly increased the power and the sensitivity of genetic testing in the clinical setting. However, the vast amount of genomic data generated poses the significant challenge of accurately assessing the pathogenicity of identified variants. While many bioinformatic resources are currently available to aid sequence variant interpretation, several inconsistencies are present in variant classification across laboratories and reference databases [16, 17]. The ultimate diagnosis of FMF is linked to life-time therapy to prevent the complications due to secondary amyloidosis, and to definitively improve patients’ quality of life [18]. Thus, FMF presents all the above-mentioned challenges in addition to still developing effective clinical criteria for identifying truly affected patients [19, 20].
In fact, of the 119 MEFV missense variants present in ClinVar, only 24 have an ACMG classification different from VOUS or conflicting interpretations. Therefore, geneticists dealing with post-test counselling in FMF find little help in the largest available database of clinical variation in the human genome. Recently, an expert consensus panel classified the clinical significance of gene variants identified in four genes causing hereditary recurrent fevers (MEFV, TNFRSFS1A, NLRP3 and MVK) [11]. However, 30% of MEFV gene variants remained unclassified or with uncertain significance.
With this initial limitation in mind, we designed the present analysis. First, by mapping variants along the coding sequence of the MEFV, we confirmed that pathogenic changes cluster in the SPRY domain. Two further domains, the PYRIN domain (also denoted as PYD) and the zf-B_box domain, appeared to host an excess of likely pathogenic variants, although differences did not reach statistically significant difference. The N-terminal PYRIN domain is present in >20 proteins modulating apoptosis and inflammation and is responsible for interaction, among others PYRIN domain-containing proteins, with the apoptosis-associated speck-like protein with a CARD [21, 22]. The zf-B_box domain is necessary and sufficient for binding the proline serine threonine phosphatase-interacting protein [PSTPIP1, or CD2-binding protein 1 (CD2BP1)]. This protein is involved in cytoskeletal organization, and its mutations cause pyogenic arthritis, pyoderma gangrenosum and acne, a different autoinflammatory disorder [23]. In contrast, benign variants occur largely in non-functional regions of the pyrin protein.
Next, we used REVEL, which has been demonstrated to outperform most of the available predictors, to score both classified and unclassified variants of the MEFV gene. When using the originally reported best threshold [13], REVEL scores were almost perfect in identifying the benign and likely benign variants (97.2%) while only 13 pathogenic/likely pathogenic variants (31.7%) demonstrated a score above the threshold for pathogenicity. We hypothesized that mechanisms conferring pathogenic effects to MEFV variants are subtler than mechanisms of disease-causing variants used to train many prediction algorithms including REVEL.
Indeed, the reasons why MEFV variants cause symptoms onset are poorly understood. In a mouse model, human MEFV knock-in variants act as gain-of-function mutations, although severity varied inversely with respect to human phenotypes [24]. More recently, in FMF patients a small set of MEFV pathogenic variants have been shown to act as hypermorphic mutations with a gene dosage effect [25]. Surprisingly, in vitro experiments have demonstrated that FMF mutations (including M694V) render pyrin inflammasome insensitive to colchicine, while this drug is the mainstay treatment in FMF patients [26]. Although many variants with a putatively pathogenic role lie in the C-terminus of the pyrin protein, no clear genotype-phenotype correlations have emerged so far.
A further proof of the complex mechanisms leading to pathogenicity in FMF is demonstrated by the markedly different REVEL scores variants interesting the same aminoacid. For instance, the well-known polymorphism E148Q has a REVEL score of 0.274 while for the variant E148V the computed score of 0.617 has a clearly pathogenic value. Similar differences recur at five different positions along the MEFV coding sequence. Notably, the four most common MEFV variants on FMF chromosomes (M680I, M694V, M694I and V726A) would be all classified as neutral should the most stringent REVEL threshold score be used [27, 28].
Therefore, using a classifier trained exclusively on MEFV variants unambiguously classified by INSAID as ‘pathogenic’, likely pathogenic’ and ‘likely benign’, ‘benign’, we computed a novel REVEL threshold for pathogenicity that allowed us to propose a novel classification for all the MEFV variants previously considered as VOUS or UNSOLVED. Indeed, a larger dataset of variants with a definitive classification as pathogenic or benign would contribute to developing more accurate statistical models to predict the pathogenicity of variants of uncertain significance or newly identified.
The development of reliable functional assays able to assess precisely the effects of MEFV gene variants on the pyrin inflammasome should also help in expanding the set of variants with a robust classification.
The ACMG/AMP 2015 guidelines recommend that the usage of in silico predictors should have only a supporting role in establishing the pathogenic role of variants identified during genetic testing. In addition, these widely followed guidelines suggest that concordance among multiple algorithm-based predictions should be sought. However, while several metapredictors incorporating many of these older algorithms have been demonstrated to perform robustly in large heterogeneous datasets, first-generation algorithms such as SIFT, Polyphen, Mutation Taster, CADD and Provean are still used even in recent work [29–31].
A further consideration should be mentioned about the thresholds used from the in silico tools to predict functional consequences. Most of these predictors return both a score and a prediction for the submitted variant. However, many others simply return a numeric value without any suggestion on the forecasted impact. Therefore, it is up to the submitters to frame the returned score in a putative pathogenic classification and translate it in clinical recommendations for specialists and patients. REVEL belongs to the second class of predictors, and two different threshold values for pathogenicity have been proposed by authors using this metapredictor in their studies, while a threshold was not given in the original paper [13, 15, 28].
In this work, we identified a MEFV gene-specific threshold, as causative mutations in FMF appear to act through a mechanism that cannot be considered either loss-of-function and gain-of-function. Variants leading to either loss-of-function or gain-of-function represent the large majority in the training datasets used to develop prediction algorithms. In contrast, MEFV variants seem to be hypermorphic mutations conferring to the mutant pyrin protein an enhanced reactivity to yet unknown triggers of the inflammasome [25]. Therefore, variant interpretation in FMF appears to be the prototypical case for a gene-specific calibration of prediction algorithms, as suggested by recent work [28].
In addition, based on recent analyses of mutations present in locus-specific databases, we found that erroneous interpretations of the mechanisms responsible for the causative role of several variants can lead to misuse of prediction tools [32]. This appears to be the also the case for FMF, where nine different frameshifting or nonsense variants are currently classified at INFEVERS as pathogenic or likely pathogenic in apparent contrast to the described hypermorphic nature of MEFV mutations [25]. Thus, critical assessment of genetic information is fundamental for translating precision medicine in everyday practice.
A limitation of our study is the small number of variants with a validated classification used to train our classifier. However, this constraint could only be overcome with an expanding set of variants identified in patients with a firm clinical evaluation that would certainly help in further narrowing the number of VOUS variants in the MEFV gene.
Concerning the possibility of establishing functional assays to experimentally validate variants interpretation, no studies for FMF have been reported as so far being capable of faithfully reproducing the clinical phenotypes. Further, it is possible that the extreme variation in the severity of symptoms observed even in individuals with a matching genotype would not be simply recapitulated in an in vitro assay.
A possible caveat could stem from the three categories scheme we have used, which is different from the five-tier ACMG/ACP classification. However, in FMF, clinical symptoms appear to manifest themselves in a continuum ranging from severe and at early onset to mild with late onset. This is somehow expected, given the current hypothesis of a gene dosage model combined with environmental and genetic modifiers. Given the present knowledge, it would be extremely difficult to identify a rationale to subdivide pathogenic from likely pathogenic variants and benign from likely benign.
Most in silico predictors have been trained on thousands of gene variants, possibly underscoring the diverse molecular mechanisms responsible for pathogenicity in many genes. Thus, using a locus-specific scoring system may result in improved specificity and sensitivity at least for some disease-causing genes.
In summary, combining validated clinical information with the development of a gene-specific variant classifier, we aimed to provide an improved framework for variant interpretation that should help in reducing the uncertainty in FMF diagnostic and would benefit genetic counsellors for better patient management.
Supplementary Material
Acknowledgements
This work was supported by a FFABR (Fund for Individual Basic Research) grant from the Italian Ministry of Higher Education and Research (MIUR) to A.S.
Funding: No specific funding was received from any funding bodies in the public, commercial or not-for-profit sectors to carry out the work described in this manuscript.
Disclosure statement: The authors have declared no conflicts of interest.
References
- 1. Reimann HA, Moadie J, Semerdjian S, Sahyoun PF.. Periodic peritonitis; heredity and pathology: report of seventy-two cases. JAMA 1954;154:1254–9. [DOI] [PubMed] [Google Scholar]
- 2. Heller H, Sohar E, Sherf L.. Familial Mediterranean fever. AMA Arch Intern Med 1958;102:50–71. [DOI] [PubMed] [Google Scholar]
- 3.French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997;17:25–31. [DOI] [PubMed] [Google Scholar]
- 4.The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997;90:797–807. [DOI] [PubMed] [Google Scholar]
- 5. Centola M, Wood G, Frucht DM. et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 2000;95:3223–31. [PubMed] [Google Scholar]
- 6. Seshadri S, Duncan MD, Hart JM, Gavrilin MA, Wewers MD.. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1 processing and release. J Immunol 2007;179:1274–81. [DOI] [PubMed] [Google Scholar]
- 7. Stoffels M, Szperl A, Simon A. et al. MEFV mutations affecting pyrin amino acid 577 cause autosomal dominant autoinflammatory disease. Ann Rheum Dis 2014;73:455–61. [DOI] [PubMed] [Google Scholar]
- 8. Sarrabay G, Touitou I.. Dominant familial Mediterranean fever. Rheumatology 2017;56:173–5. [DOI] [PubMed] [Google Scholar]
- 9. Rowczenio DM, Iancu DS, Trojer H. et al. Autosomal dominant familial Mediterranean fever in Northern European Caucasians associated with deletion of p.M694 residue-a case series and genetic exploration. Rheumatology 2017;56:209–13. [DOI] [PubMed] [Google Scholar]
- 10. Stella A, Cortellessa F, Scaccianoce G. et al. Familial Mediterranean fever: breaking all the (genetic) rules. Rheumatology 2019;58:463–7. [DOI] [PubMed] [Google Scholar]
- 11. Van Gijn ME, Ceccherini I, Shinar Y. et al. New workflow for classification of genetic variants' pathogenicity applied to hereditary recurrent fevers by the International Study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet 2018;55:530–7. [DOI] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ioannidis NM, Rothstein JH, Pejaver V. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99:877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jay JJ, Brouwer C.. Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS One 2016;11:e0160519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Zhao T, Zhang Y. et al. Performance evaluation of pathogenicity-computation methods for missense variants. Nucleic Acids Res 2018;46:7793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amendola LM, Jarvik GP, Leo MC. et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet 2016;99:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pepin MG, Murray ML, Bailey S. et al. The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med 2016;18:20–4. [DOI] [PubMed] [Google Scholar]
- 18. Bonfrate L, Scaccianoce G, Palasciano G, Ben-Chetrit E, Portincasa P.. A novel cluster of patients with Familial Mediterranean Fever (FMF) in southern Italy. Eur J Clin Invest 2017;47:622–9. [DOI] [PubMed] [Google Scholar]
- 19. Giancane G, Ter Haar NM, Wulffraat N. et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis 2015;74:635–41. [DOI] [PubMed] [Google Scholar]
- 20. Federici S, Sormani MP, Ozen S. et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis 2015;74:799–805. [DOI] [PubMed] [Google Scholar]
- 21. Martinon F, Hofmann K, Tschopp J.. The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr Biol 2001;11:R118–20. [DOI] [PubMed] [Google Scholar]
- 22. Richards N, Schaner P, Diaz A. et al. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem 2001;276:39320–9. [DOI] [PubMed] [Google Scholar]
- 23. Shoham NG, Centola M, Mansfield E. et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci USA 2003;100:13501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chae JJ, Cho YH, Lee GS. et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity 2011;34:755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jamilloux Y, Lefeuvre L, Magnotti F. et al. Familial Mediterranean fever mutations are hypermorphic mutations that specifically decrease the activation threshold of the Pyrin inflammasome. Rheumatology 2018;57:100–11. [DOI] [PubMed] [Google Scholar]
- 26. Van Gorp H, Saavedra PH, de Vasconcelos NM. et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc Natl Acad Sci USA 2016;113:14384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Touitou I. Standardized testing for mutations in familial Mediterranean fever. Clin Chem 2003;49:1781–2. [DOI] [PubMed] [Google Scholar]
- 28. Ghosh R, Oak N, Plon SE.. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol 2017;18:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seifi M, Footz T, Taylor SA, Walter MA.. Comparison of bioinformatics prediction, molecular modeling, and functional analyses of FOXC1 mutations in patients with axenfeld-rieger syndrome. Hum Mut 2017;38:169–79. [DOI] [PubMed] [Google Scholar]
- 30. Riera C, Padilla N, de la Cruz X.. The complementarity between protein-specific and general pathogenicity predictors for amino acid substitutions. Hum Mut 2016;37:1013–24. [DOI] [PubMed] [Google Scholar]
- 31. Ernst C, Hahnen E, Engel C. et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med Genomics 2018;11:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stella A, Lastella P, Loconte DC. et al. Accurate classification of NF1 gene variants in 84 Italian patients with neurofibromatosis type 1. Genes 2018;9:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.