

ABSTRACT
DNA methylation (DNAm) in blood (umbilical cord blood and capillary blood collected after birth on Guthrie cards) during the perinatal period is being increasingly studied with the aim of identifying epigenetic markers of in utero environmental exposures or later disease development. However, the comparability in DNAm between these two sources is unknown. To this end, DNAm from the cord blood and capillary blood of 34 subjects in the Isle of Wight 3rd Generation Birth Cohort (68 samples) were included to assess the comparability. Differences in average DNAm (overall agreement), correlations in DNAm, and intra-class correlation coefficients (ICC) in DNAm between the two sources, at each of the 430,742 CpG sites, were evaluated. The results showed that a high proportion (70.1%) of the CpGs DNAm agreed between cord blood and neonatal blood on Guthrie cards. A small portion of CpGs showed high correlation (correlation ≥0.5) or high ICC (ICC ≥0.5) in DNAm of the whole genome. This proportion increased dramatically in differentially methylated regions (DMRs) that are associated with exposure to maternal smoking, between the two sources.
KEYWORDS: DNA methylation, cord blood, Guthrie cards, Isle of Wight
Introduction
The early development of a child encompasses a period of profound changes in DNA methylation (DNAm) and may, as such, be a critical period for environmentally induced DNAm changes. Hence, this period is of great interest in DNAm studies in relation to specific exposures and long-term health outcomes. DNAm in blood, including umbilical cord blood and capillary blood collected after birth on Guthrie cards, is being increasingly studied for the purpose of detecting biomarkers of in utero environmental exposure and later disease development. However, the comparability in DNAm between these two sources is unknown, imposing difficulty in assessing the agreement between epigenetic findings based on cord blood and those based on capillary blood from Guthrie cards.
Blood samples are obtained from the umbilical cord after birth. DNAm in cord blood carries critical epigenetic information which is affected by both genetic variants and epigenetic information [1]. In addition, cord blood DNAm can include information of gestational exposures, such as maternal smoking [2–4] and in-utero exposures to toxins such as phthalates and heavy metals [5,6]. It has been shown that cord blood DNAm is associated with the child’s short- and long-term health outcomes [7].
Guthrie card samples are obtained from the newborn infant by puncturing the heel of the baby and collecting several drops of capillary blood on a pre-printed card [8]. In comparison to cord blood, collection of a blood sample on a Guthrie card is much easier and is usually performed routinely. In addition, Guthrie cards are easier to store than cord blood. Dried archived Guthrie blood spots have been identified as a novel source for analysing genome DNAm profiles of infants in the perinatal period, in addition to cord blood [9–12]. Like cord blood, the DNAm of newborn capillary blood is believed to be a biomarker of gestational exposure [13] and associated with health outcomes in later life [14]. However, without knowing the comparability in DNAm between these two sources, i.e., cord blood and Guthrie cards collected at birth, it is difficult to assess whether the detected biomarkers, based on DNAm measured using Guthrie cards, could be replicated using DNAm in cord blood.
To our knowledge, no study has yet compared DNAm profiles between these two sources, although studies have been conducted to evaluate the similarity and difference of white blood cell type proportions [15] and gene expression [16] between cord blood and dried blood samples from newborns. In the current investigation, we compare DNAm between the two types of blood samples using paired data collected in the Isle of Wight 3rd Generation Birth Cohort. For each CpG, we evaluate the average DNA methylation level (beta or M values) between the two sources using paired t-tests (overall agreement), Pearson correlation (consistency), and intra class correlation (stability). Findings from these assessments will improve our understanding of epigenetic changes shortly after birth, and our comprehension of epigenetic findings from different sources. Furthermore, given the convenience in sample collection and storage of Guthrie cards, at CpG sites with DNAm comparable between the two sources, we may significantly increase statistical testing power by utilizing both Guthrie cards and cord blood samples.
Methods
Isle of wight birth cohorts
The IoW Birth Cohort study was initiated at the David Hide Asthma and Allergy Research Centre on the Isle of Wight, UK, in 1989/1990 [17]. In a follow-up investigation of the children of both male and female cohort members, 431 offspring were recruited between 2010 and 2015. DNAm was measured in both cord blood samples and Guthrie cards from 34 neonates.
DNA methylation
DNA was isolated from dried blood spots on the Guthrie cards of 34 neonates using a method based on the procedure described by Beyan et al. [12]. For cord blood, the standard salting out procedure was used to isolate DNA and its concentration was determined by Qubit quantitation. One microgram of isolated DNA was bisulphite-treated for cytosine to thymine conversion using the EZ 96-DNA methylation kit (Zymo Research, CA, USA) according to the manufacturer’s standard protocol. The DNAm levels were obtained for 192 cord samples and 34 Guthrie samples. For the cord sample, 129 samples were measured by the Infinium HumanMethylation450 BeadChip and 63 by the HumanEPIC from Illumina (Illumina, San Diego, CA, USA). Among the 34 cord samples that matched with the Guthrie samples, 29 were by the HumanMethylation450 BeadChip and 5 by the HumanEPIC BeadChip. All the 34 Guthrie samples were analysed using the HumanEPIC BeadChip.
DNA methylation profile pre-processing
Quality control was undertaken with DNA methylation intensity level quantile normalized from a total of nine batches – seven from the cord blood samples and two from the Guthrie cards. The samples from different array platforms and batches were combined. The R package ComBat was used to remove batch effects in the combined dataset [18]. After pre-processing, DNAm at 430,742 CpGs of 34 subjects in both cord blood and Guthrie cards was included in subsequent analyses.
Statistical analyses
The demographic characteristics of the 34 subjects and the original Iow birth cohort were summarized and presented as percentages for categorical variables and mean ± standard deviation (SD) for continuous variables. The demographics of the sample were compared with the original cohort using one sample t test for continuous variables and proportion tests (Z tests) for categorical variables. The base 2 logit-transformation was used to convert DNAm beta-values to M-values. We assessed overall agreement, consistency, and stability between the two sources. Overall agreement was evaluated as the difference in M values between the two sources via paired t-tests at each CpG site. Multiple testing was adjusted by controlling a false discovery rate (FDR) of 0.05. Both Pearson correlations and Intraclass correlation coefficients (ICCs) were calculated between the 34 pairs for each CpG site. We used Pearson correlations to assess consistency between the two groups. CpGs with a correlation of 0.5 or higher were treated as consistent CpGs. ICCs were used to evaluate the stability. A CpG site with an ICC of 0.5 or higher was regarded as being stable. DNAm at some CpG sites might be due to mQTLs. We used the ARIES mQTL at birth database (cord.ALL.M.tab) downloaded from http://www.mqtldb.org [19]. In total, 89,302 of the 430,742 CpGs in our study were mQTL sites.
The identified CpGs were characterized by their allocation on the genes and their relationship to CpG Island on the chromosome. The location of the CpGs was provided by the Illumina Infinium MethylationEPIC v1.0 B4 Manifest File (https://support.illumina.com/downloads/infinium-methylationepic-v1-0-product-files.html). The categories are: 1) 1st Exon region; 2) 3ʹUTR, between the stop codon and the transcription termination site, poly A signal; 3) 5ʹUTR, within the 5ʹ untranslated region, between the TSS and the start codon, ATG; 4) Body, between the start and stop codon; irrespective of the presence of introns, exons, TSS, or promoters; 5) TSS200, 0–200 bases upstream of the transcriptional start site (TSS); 6) TSS1500, 200–1500 bases upstream of the TSS. CpGs not belonging to any of the six regions were considered as being in intergenic regions. In terms of relative location of a CpG site to a CpG island, we considered the following four categories: 1) Shore, upstream (5ʹ) and 0–2 kb from CpG island; 2) Shelf, upstream (5ʹ) and 2–4 kb from CpG island; 3) Island: on the CpG island; and 4) CpGs that are not in any of the three categories were considered as ‘other’.
The identification of differentially methylated regions was based on DNA methylation of 192 cord blood samples using maternal smoking during pregnancy as exposure. The R package DMRcate [20] was used to detect DMRs linked to smoking during pregnancy. The smoking status has two levels, Yes or No. In the analysis of DMR detection, we considered two criteria in the adjustment of multiple testing, FDR of 0.1 (more stringent) and FDR of 0.2 (covering a wider range of CpGs). For parameter setting in DMRcate, we used lamda = 1000 and C = 2, which is the default setting suggested by the author. We further summarized the agreement, consistency and stability between the two blood sources for the CpGs that are on the identified differentially methylated regions (DMRs).
Results
Demographics of study subjects
Table 1 shows the demographics for the original IoW second-generation cohort (n = 531) and the 34 newborns in the current study. In particular, the comparisons focused on gestational age, gender of the newborns, age of parents, and maternal smoking status during pregnancy. We did not observe any statistically significant differences between the sub-samples and the whole cohort except for the age of mothers. The average age of mothers in the subsamples when the baby was born was 23.55 (±1.35) years, while in the whole cohort, it was 24.32 (±3.70) years (p-value 0.0025). The average age of fathers in the sample was 26.17 (±4.40) years. For the 34 newborns, the capillary blood collection on Guthrie cards was during day 5 to day 7 after birth, with an average of 5.29 (±0.63) days.
Table 1.
Summary of the demographics for the original birth cohort and analytical sample.
| Original birth cohort |
Analytical sample with DNAm |
||
|---|---|---|---|
| n = 531 | n = 34 | p-value | |
| Gestational Age (week) | 38.51 (9.97) | 39.09 (2.02) | 0.1085 |
| Gender (Boy); n(%) | 281 (53.63%) | 17 (51.52%) | 0.8075 |
| Mother’s age (years) | 24.32 (3.70) | 23.55 (1.35) | 0.0025 |
| Father’s age (years) | 26.87 (4.48) | 26.17 (4.39) | 0.4012 |
| Maternal Smoking; n (%) | |||
| Yes | 164 (33.33%) | 15 (46.88%) | 0.1041 |
| No | 328 (66.67%) | 17 (53.13%) |
Overall agreement, consistency and stability between cord and Guthrie DNAm
Comparing DNAm between cord and Guthrie cards for their overall agreement using paired t-tests and adjusting for multiple testing by controlling for an FDR of 0.05, the mean level of DNAm at 128,807 CpGs (29.9% of total CpGs) was different between cord blood and Guthrie cards. Equivalently, DNAm at 70.1% (301,935 CpGs) of the 430,472 CpGs had no statistically significant differences, and these 301,935 CpGs were regarded as having overall agreement on the mean of the DNAm (Table 2).
Table 2.
Summary of the number and percentage of overall agreed, consistent and stable CpGs between DNAm of Cord blood and Guthrie Card among all the CpGs in the study (n = 430,647).
| Number of CpG (%) | |
|---|---|
| Overall Agreement | |
| Based on paired t test to compare the means | 301,935 (70.1%) |
| Consistency | |
| Correlation cut-off for consistency | |
| ≥0.3 | 33,513 (7.8%) |
| ≥0.5 | 94,293 (21.9%) |
| Stability | |
| ICC cut-off for stability | |
| ≥0.5 | 25,800 (6.0%) |
| ≥0.6 | 15,810 (3.7%) |
Regarding the assessment of consistency based on Pearson correlations, with Pearson correlation ≥0.5, DNAm at 33,128 CpGs (7.7% of total CpGs) showed a correlation at least 0.5 between cord blood and Guthrie card samples (Table 2). Among these consistent CpGs, 75.2% (24,914 CpGs) were also in the group of CpGs showing overall agreement on the mean (based on paired t-tests) (Figure 1). As noted in the literature, using correlation of 0.5 as an indicator for a relatively large association was likely to be overly stringent [21], and it was suggested to take 0.3 as a cut off for high correlations. Following this suggestion, the total number of CpGs that have a correlation equal to or larger than 0.3 between Cord and Guthrie Card increased to 94, 293, which is 21.9% of total CpGs.
Figure 1.
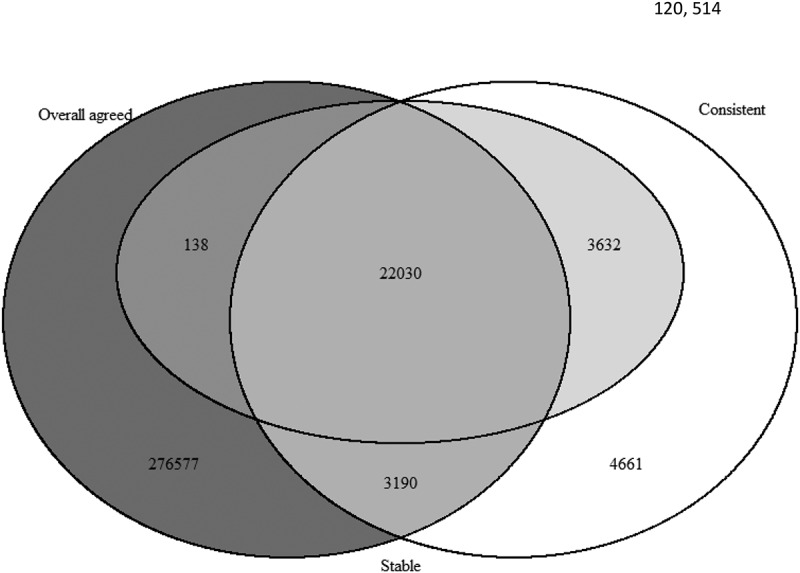
Overlap of identified overall agreed CpGs (P-value after adjusted by FDR>0.05, dark grey), consistent CpGs (Pearson correlation ≥0.5, white) and stable CpGs (ICC≥ 0.5, light grey).
For stability, as shown in Table 2, we identified 25,800 CpGs (6.0% of total CpGs) with ICC larger than or equal to 0.5 between cord blood and Guthrie card. In comparison to Pearson correlations, ICC is stricter and takes into account the difference in the means of DNAm between the two sources. Among these 25,800 stable CpGs, most of them (22,030 CpGs, 85.4% of 25,800) were in the group of CpGs showing overall agreement and consistency (Figure 1). If we increase the ICC cut-off to 0.6 following a suggestion in the literature [22], there are only 15, 810 CpGs (3.7%) showing such a high stability. To be consistent between different assessments (consistency and stability), in the following sections, we take the cut off value of 0.5 for both correlation and ICC. Among the 430,742 CpGs examined in our study, 89,302 are mQTL CpG sites as shown by Gaunt et al. [19]. Of the 89,302 sites, 61,789 CpGs (69.2%) have an overall agreement on the mean DNAm level, 14,989 CpGs (16.8%) have a Pearson correlation larger or equal to 0.5, and 11,834 CpGs (13.3%) have an ICC larger or equal to 0.5.
Gene locations of identified CpGs that show overall agreement, consistency or stability between cord and Guthrie DNAm
For the CpGs showing overall agreement, consistency, or stability between the two blood sources, we further examined the distribution of CpGs on a gene’s functional regions (Figure 2). Seven regions were considered, 1st exson, 3ʹ UTR, 5ʹ UTR, body, TSS200, TSS1500, and intergenic regions. We use the location 3ʹUTR as an example to demonstrate the calculation of relative frequencies. Out of the 430,742 CpGs included in our study, 17,429 were on the 3ʹUTR region. Among the 301,935 CpGs showing overall agreement, 12,705 were on the 3ʹUTR region, which was 72.9% of the 17,429 3ʹ UTR CpGs (first figure in Figure 2). This was the highest relative frequencies among all the different locations. Being in the region of 1st Exon showed the smallest percentages of CpGs with the overall agreement (63.1%). For both consistency and stability, intergenic regions showed the highest for both (11.3%, consistency and 8.83% satiability). On the other hand, CpGs in the TSS200 region had the smallest percentage of consistency (4.60%) and stability (3.54%).
Figure 2.

Summary of identified (a) CpGs that are overall agreed on the mean, (b) consistent CpGs, and (c) stable CpGs within each category of gene position. Each percentage was calculated as the number of identified CpGs divided by the total number of CpGs in that region.
Relation to CpG Island of identified CpGs showing overall agreement, consistency or stability between cord and Guthrie DNAm
Based on the relative frequencies of the identified CpGs according to their spatial relationship to a CpG Island (Figure 3), the distribution of overall agreed CpGs was comparable between shores and shelfs. The relative frequencies on the shores and shelfs were higher than the frequencies on the islands (62.9% on the islands but >70% on the shores and shelfs). The distribution of identified CpGs based on consistency (correlation) with respect to their relation to CpG islands was comparable to that based on stability (ICC) (last two figures in Figure 3). For instance, for both measurements (correlation and ICC), most identified CpGs were located on the shores.
Figure 3.
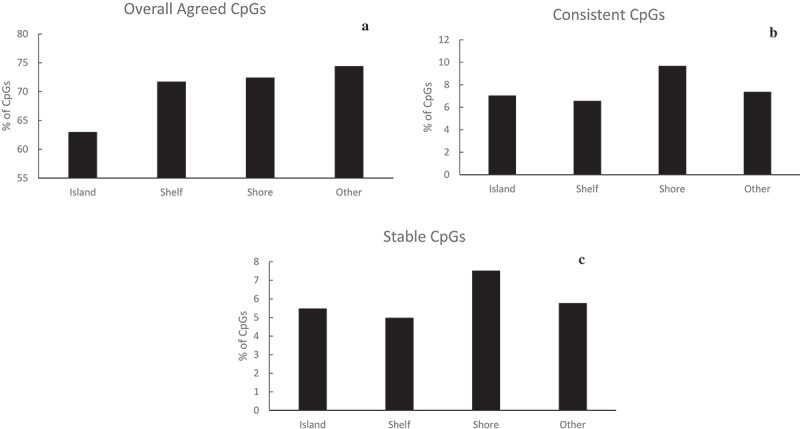
Summary of identified (a) CpGs that are overall agreed on the mean, (b) consistent CpGs, and (c) stable CpGs within each category of relation to CpG islands. Each percentage was calculated as the number of identified CpGs divided by the total number of CpGs in that region.
Grouping of consistent GpGs on DMRs
We further investigated the agreement, consistency and stability between the two blood sources on differentially methylated regions (DMRs). It has been shown that maternal smoking during pregnancy affects the DNAm of the newborn [23], we therefore detected DMRs based on maternal smoking status during pregnancy in the 192 cord samples using DMRcate [20]. With the more stringent multiple testing adjustment (FDR of 0.1), we identified two DMRs with a total of 17 CpGs. Within these 17 CpGs, 13 of them (76.47%) agreed between cord and Guthrie cards (p values larger than 0.05), 9 of them (52.94%) were consistent (Pearson correlation large or equal to 0.5), and 6 of the 17 CpGs (35.29%) were stable (ICC large or equal to 0.5). For broader DMRs identified based on an FDR of 0.2, we identified 6 DMRs with a total of 47 CpGs. Within these 47 CpGs, 35 of them (74.47%) agreed between cord and Guthrie cards, 17 (36.17%) of them were consistent, and 13 (27.66%) were stable. Although with a larger number of CpGs in these DMRs, the patterns of the statistics were similar to the situation under stringent DMRs.
Discussion
DNA methylation from cord blood and capillary blood on Guthrie cards shows a relatively high overall agreement, with 70% of CpGs (301,935 of the 430,472 CpGs) having similar mean DNAm levels. This offers an opportunity to conduct mean-based analyses with a higher testing power at those CpGs by combining the two sources (cord and Guthrie blood). The consistency measured by correlations (7.7%) and stability measured by ICC (6.0%) were both low. Thus, when the focus of a study is on association between DNAm and a continuous variable instead of comparison in means of DNAm between different groups or health conditions, simply combining DNAm from the two sources in the analysis is questionable and not recommended in general.
For CpGs that were found to be mQTL [19], the overall agreement on mean DNAm level is similar comparing to CpGs that are not mQTL, but higher percentages in consistency and stability were observed. This finding suggests that DNAm at mQTLs is affected more by genetic variants than other factors, e.g., cell compositions or time of blood sampling after birth.
On DMRs associated with maternal smoking during pregnancy, the percentage of CpGs having similar mean DNAm levels increased to 76% comparing to 70% on the whole genome. The percentage of consistent CpGs (high correlations) on DMRs increased to over 50%, which is 7 times of that for the whole genome. The percentage of stable CpGs also increased significantly from 6.0% in the whole genome to >35% on DMRs. The results suggest that DNAm in cord blood and in Guthrie cards was more comparable at CpG sites on DMRs. However, since the DMRs in our study were identified based on maternal smoking status during pregnancy, it is unclear if such a pattern persists with DMRs under different mechanisms, and thus further assessment on the comparability between the two sources is warranted.
The study has some other limitations. As a starting point, in this study, we assumed that CpGs were independent and examined one CpG at a time. DNAm at CpGs on islands tends to be correlated. To take this correlation into account, more advanced approaches, such as spatial modelling, are needed to further investigate the agreement, consistency, and stability between the two sources. In addition, the blood samples in the study were analysed using two different platforms for the measurement of DNA methylation. Some samples were analysed using the Infinium HumanMethylation450 BeadChip and others using the HumanEPIC BeadChip. In the stage of quality control, we have managed to minimize such platform effects along with other technical effects. However, the bias due to the differences between the platforms may still exit in the combined data. Finally, the small sample size in the present study could have introduced additional uncertainty in the comparisons. Further investigation in larger cohorts are certainly needed.
Conclusion
The findings suggested that DNAm in cord blood and in Guthrie cards was comparable at certain CpG sites. When the focus of a study is on the association between DNAm and a continuous variable instead of comparison in mean DNAm between different groups or health conditions, simply combining DNAm from the two sources in the analysis is questionable and not recommended in general.
Declarations
Ethics approval and consent to participate. We have received ethics approval from the Isle of Wight, Portsmouth and SE Hampshire Local Research Ethics Committee (Study Title: A Study of Epigenetic Driven Immunological Changes in the Development of Asthma and Allergy in Infancy).
Funding Statement
National Institutes of Health (NIH) R01AI121226 (PIs: Zhang, Holloway), R01HL132321 and R03HD092776 (PI: Karmaus), the start-up funds for Jiang from the School of Public Health at the University of Memphis
Acknowledgments
We are thankful for the computation support from the High Performance Computing (HPC) facility at the University of Memphis. The authors are the nurses and staff at the David Hide Asthma & Allergy Research Centre, Isle of Wight, UK, for their help in recruitment and sample collection. We are grateful to Mrs Sharon Matthews for her help with proof reading and editing.
Authors’ contributions
YJ and HZ conceived the biostatistical design of the study, analyzed and interpreted the data, and drafted the manuscript. JW conducted the statistical data analysis. FR conducted data cleaning and pre-processing of DNA methylation data. HA and JH supervised the data and sample collection and helped drafting the manuscript. SE conducted the DNA methylation analysis. All authors critically revised the manuscript for important intellectual content. The manuscript has been read and approved by all authors.
Availability of data and material
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Kile ML, Baccarelli A, Tarantini L, et al. Correlation of global and gene-specific DNA methylation in maternal-infant pairs. PloS One. 2010;5(10):e13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Joubert BR, Haberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Markunas CA, Xu Z, Harlid S, et al. Identification of DNA methylation changes in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2014;122(10):1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cardenas A, Koestler DC, Houseman EA, et al. Differential DNA methylation in umbilical cord blood of infants exposed to mercury and arsenic in utero. Epigenetics. 2015;10(6):508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Koestler DC, Avissar-Whiting M, Houseman EA, et al. Differential DNA methylation in umbilical cord blood of infants exposed to low levels of arsenic in utero. Environ Health Perspect. 2013;121(8):971–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Herbstman JB, Wang S, Perera FP, et al. Predictors and consequences of global DNA methylation in cord blood and at three years. PloS One. 2013;8(9):e72824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guthrie R, Susi A.. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:338–343. [PubMed] [Google Scholar]
- [9].Wong N, Morley R, Saffery R, et al. Archived Guthrie blood spots as a novel source for quantitative DNA methylation analysis. Biotechniques. 2008;45(4): 423–424. 6, 8 passim. [DOI] [PubMed] [Google Scholar]
- [10].Dugue PA, English DR, MacInnis RJ, et al. Reliability of DNA methylation measures from dried blood spots and mononuclear cells using the humanmethylation450k BeadArray. Sci Rep. 2016;6:30317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Joo JE, Wong EM, Baglietto L, et al. The use of DNA from archival dried blood spots with the infinium humanmethylation450 array. BMC Biotechnol. 2013;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beyan H, Down TA, Ramagopalan SV, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012;22(11):2138–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Marioni RE, Suderman M, Chen BH, et al. Tracking the epigenetic clock across the human life course: a meta-analysis of longitudinal cohort data. J Gerontol A Biol Sci Med Sci. 2018;74(1):57-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mohandas N, Bass-Stringer S, Maksimovic J, et al. Epigenome-wide analysis in newborn blood spots from monozygotic twins discordant for cerebral palsy reveals consistent regional differences in DNA methylation. Clin Epigenetics. 2018;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Olin A, Henckel E, Chen Y, et al. Stereotypic immune system development in newborn children. Cell. 2018;174(5):1277–92 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gillam-Krakauer M, Cochran CM, Slaughter JC, et al. Correlation of abdominal rSO2 with superior mesenteric artery velocities in preterm infants. J Perinatol. 2013;33(8):609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Arshad SH, Holloway JW, Karmaus W, et al. Cohort profile: the Isle Of Wight Whole Population Birth Cohort (IOWBC). Int J Epidemiol. 2018;47(4):1043–4i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. [DOI] [PubMed] [Google Scholar]
- [19].Gaunt TR, Shihab HA, Hemani G, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Bio. 2016;17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Peters TJ, Buckley MJ, Statham AL, et al. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gignac GE, Szodorai ET. Effect size guidelines for individual differences researchers. Pers Indiv Differ. 2016;102:74–78. [Google Scholar]
- [22].Hallgren KA. Computing inter-rater reliability for observational data: an overview and tutorial. Tutor Quant Methods Psychol. 2012;8(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98(4):680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.