

ABSTRACT
Endocannabinoids (eCB) modulate growth cone dynamics and axonal pathfinding through the stimulation of cannabinoid type-1 receptors (CB1R), the function of which depends on their delivery and precise presentation at the growth cone surface. However, the mechanism involved in the axonal transport of CB1R and its transport role in eCB signaling remains elusive. As mutations in the kinesin-1 molecular motor have been identified in patients with abnormal cortical development and impaired white matter integrity, we studied the defects in axonal pathfinding and fasciculation in mice lacking the kinesin light chain 1 (Klc1−/−) subunit of kinesin-1. Reduced levels of CB1R were found in corticofugal projections and axonal growth cones in Klc1−/− mice. By live-cell imaging of CB1R-eGFP we characterized the axonal transport of CB1R vesicles and described the defects in transport that arise after KLC1 deletion. Cofilin activation, which is necessary for actin dynamics during growth cone remodeling, is impaired in the Klc1−/− cerebral cortex. In addition, Klc1−/− neurons showed expanded growth cones that were unresponsive to CB1R-induced axonal elongation. Together, our data reveal the relevance of kinesin-1 in CB1R axonal transport and in eCB signaling during brain wiring.
KEY WORDS: Actin, Axonal growth, Axonal transport, Axonal pathfinding, Cannabinoid, Endocannabinoids, Kinesin, Trafficking
Summary: This study reveals that kinesin-1 plays a key role in the endocannabinoid signaling that regulates activity at the axonal growth cones of cortical neurons for actin remodeling, axonal pathfinding and brain wiring.
INTRODUCTION
During the process of brain wiring, environmental guidance cues control the navigation of axonal growth cones via signaling cascades that regulate cytoskeletal dynamics (Dent and Gertler, 2003). Endocannabinoids (eCB) have emerged as regulators of brain wiring by modulating pathfinding and fasciculation of long-range connections (Argaw et al., 2011; Berghuis et al., 2007; Keimpema et al., 2010; Mulder et al., 2008; Wu et al., 2010; Zuccarini et al., 2020). The acquisition of neuronal connectivity with remarkable precision requires proper targeting of receptors for axon elongation and guidance on the surface of growth cones of navigating axons (Bashaw and Klein, 2010; Rozbesky and Jones, 2020). Precise levels and localization of receptors on the plasma membrane are maintained by a specific transport system (Winckler and Mellman, 2010; Stoeckli, 2018). In neurons, the anterograde axonal transport of vesicles that deliver molecules and receptors is mediated by microtubule-dependent motor proteins, i.e. kinesins (Goldstein, 2001; Guedes-Dias and Holzbaur, 2019). Although it is known that anterograde motors are essential for neuronal function, little is known about whether axonal transport of guidance receptors plays a modulatory role in signaling responses.
eCB, such as 2-arachidonoylglycerol (2-AG) as well as plant-derived cannabinoids, induce local intracellular signaling by the activation of cannabinoid type-1 receptors (CB1R; also known as Cnr1). The activation of this pathway results in rearrangement of the actin cytoskeleton that leads to the collapse of growth cones (Berghuis et al., 2007; Njoo et al., 2015; Roland et al., 2014). Defects in eCB signaling, such as those in CB1R-null mutant mice, result in axonal guidance errors with impaired pathfinding of corticofugal axons (Wu et al., 2010). The subcellular localization and membrane stabilization of CB1R is highly dynamic, and its distribution changes from brain development to adulthood (Keimpema et al., 2010; Fletcher-Jones et al., 2019). In the developing brain, CB1R are localized on the surface of axons and growth cones (Berghuis et al., 2007). In mature neurons, CB1R are specifically localized at the presynaptic terminal and exert a neuromodulatory role by regulating synaptic function (Castillo et al., 2012; Wickert et al., 2018). However, CB1R are also present in dendritic spines of mature neurons, where activation modulates the assembly of actin filaments (Njoo et al., 2015). Therefore, the intracellular mechanisms controlling the trafficking of CB1R during neuronal development may play a regulatory role, with significant effects on eCB signaling.
Axonal transport of vesicles depends on microtubule-associated molecular motors such as the kinesin superfamily (Guedes-Dias and Holzbaur, 2019). Kinesin-1 is an anterograde motor complex composed of a tetramer of two heavy chains (KIF5A, KIF5B or KIF5C) and two light chains (KLC1 or KLC2) that transport a variety of membrane cargos loaded with synaptic scaffolding proteins and receptors throughout the axon (Hirokawa et al., 2009). Although it is well established that kinesin-1 is essential for synapse formation and function (Nakajima et al., 2012; Lipton et al., 2018), axonal specification (Jacobson et al., 2006) and axonal growth (Deng et al., 2014; Watt et al., 2015), its role in axonal pathfinding remains poorly understood. Next-generation sequencing studies have found several mutations in kinesin-1 subunits associated with human neurodevelopmental disorders that are related to abnormal white matter formation (de Ligt et al., 2012; Guerrini and Dobyns, 2014; Jamuar and Walsh, 2014, 2015; Poirier et al., 2013; Shao et al., 2016; Willemsen et al., 2014). Mutations in the motor domain of KIF5C (p.Glu237Lys and p.Ala268Ser) lead to developmental cortical malformations, microcephaly, ventriculomegaly and callosal abnormalities (Cavallin et al., 2016; Poirier et al., 2013) that result in infantile onset epilepsy and intellectual disability (Michels et al., 2017). Moreover, a de novo mutation in KIF5A also causes microcephaly, ventriculomegaly and white matter dysgenesis (Michels et al., 2017). Studies in mouse models with mutations in subunits of the kinesin-1 motor support these defects. Our previous studies have identified white matter dysgenesis, expanded ventricles and defects in membrane presentation of key receptors in kinesin-1 mutants (Falzone et al., 2009; Cromberg et al., 2019). These studies highlight the relevance of understanding the contribution of different molecular motors to axonal pathfinding and how transport defects lead to brain wiring impairments. Therefore, we hypothesized that kinesin-1 is required for correct axonal pathfinding during early neural development.
In this work, we show that Klc1−/− mice exhibit hyperfasciculation and pathfinding defects of corticofugal axons with a specific reduction in the level of CB1R. Electron microscopy experiments revealed the expansion of axonal fascicles within the striatum, with a reduced number of axons displaying an enlarged caliber. Using high resolution live imaging of fluorescent CB1R vesicles we characterized the axonal transport properties of CB1R and the defects observed in Klc1−/− neurons, revealing the dependency on kinesin-1 in the delivery of CB1R. Moreover, cerebral cortex from Klc1−/− mice showed increased cofilin phosphorylation and defects in actin remodeling at axonal growth cones of cortical neurons. We show evidence that CB1R is transported by KLC1, which is essential for the eCB signaling pathway, as Klc1−/− neurons were unresponsive to axonal elongation and growth cone remodeling induced by CB1R stimulation. Taken together, our results suggest that eCB signaling requires kinesin-1-mediated axonal transport of CB1R to growth cones for proper actin remodeling, axonal pathfinding and brain wiring.
RESULTS
Axonal hyperfasciculation and pathfinding defects in Klc1−/− mice
We have previously shown white matter abnormalities with a significant reduction in several axonal tracts in the aged Klc1−/− brain (Falzone et al., 2009). To characterize the morphology of long-range projections in Klc1−/− brains during development we performed immunostaining for the axonal marker L1 neural cell adhesion molecule (L1-NCAM; L1cam) in serial coronal brain sections at postnatal day (P) 0 (Fig. 1A). Compared with wild-type mice (Klc1+/+), Klc1−/− mice exhibited a significant reduction in thickness of the internal capsule, a major axonal tract composed of corticofugal axons (CFA), which include the corticothalamic axons (CTA), and the reciprocal thalamocortical axons (TCA) that connect the neocortex and subcortical regions (Fig. 1A,B). To identify whether KLC1 is required for normal axonal fasciculation, we evaluated the morphology of L1-NCAM-positive fascicles in the caudal striatum next to the corticostriatal boundary (Fig. 1A′). The average number of individual L1-NCAM-positive fascicles was significantly reduced in Klc1−/− mice, although the mean fascicle diameter was increased when compared with Klc1+/+ (Fig. 1C). The diameter distribution analysis revealed a significant reduction in the proportion of small fascicles, with an increase in the proportion of bigger fascicles (>25 µm) in Klc1−/− mice compared with Klc1+/+ mice (Fig. 1D). The total area covered by dorsal striatal fascicles was smaller in Klc1−/− mice, with a significant reduction observed in the dorsolateral region (Fig. 1E,F), which receives inputs from the primary somatosensory cortex (Pan et al., 2010). A smaller number of fascicles, but larger striatal fascicle size, in Klc1−/− suggests that axon pathfinding of CFA and TCA is impaired. Moreover, we also observed ectopic axonal bundles in the corticostriatal boundary in all Klc1−/− mice analyzed. Together, these observations suggest that KLC1 is required for axonal pathfinding and guidance.
Fig. 1.

Axonal hyperfasciculation and pathfinding defects in Klc1−/− mice. (A) Epifluorescence images of L1-NCAM immunostaining of coronal brain sections in Klc1+/+ and Klc1−/− mice at P0. White arrows indicate internal capsule thickness. (A′) Higher magnifications of panel A showing CFA and TCA in the caudal striatum. Yellow arrowheads indicate ectopic axonal bundle. (B) Internal capsule thickness covered by L1-NCAM+ axons. (C) Number (left) and diameter (right) of individual L1-NCAM+ fascicles quantified in the area enclosed by the dashed line in A′. (D) L1-NCAM+ fascicle diameter distribution between Klc1+/+ and Klc1−/− mice. (E) L1-NCAM immunostaining of fascicles in dorsal striatum of Klc1+/+ and Klc1−/− mice at P0. Dashed line indicates dorsolateral striatum. (F) Striatal fascicle area relative to whole striatum area. Data in B, C and F are mean±s.e.m. n=8 animals/genotype from three independent experiments. *P<0.025, ***P<0.001, ****P<0.0001; Student's t-test. Data in D show probability density function; dashed lines indicate median. Klc1+/+, n=668; Klc1−/−, n=362 fascicles from eight animals/genotype from three independent experiments. ***P<0.0001; Kolmogorov–Smirnov test. Cx, cerebral cortex; cSt, caudal striatum; dls, dorsolateral striatum; ic, internal capsule; St, striatum; Th, thalamus. Scale bars: 500 μm (A); 100 μm (A′,E).
To further understand whether the deletion of KLC1 induced axons to coalesce or to become dystrophic within abnormal fascicles, we performed transmission electron microscopy (TEM) to obtain images from the caudal striatum in Klc1+/+ and Klc1−/− mice. Quantification of fascicle area by TEM again revealed enlarged fascicles in Klc1−/− compared with Klc1+/+ (Fig. 2A,B). We then performed higher magnification TEM imaging to quantify the number and diameter of individual axons within fascicles (Fig. 2A′). Interestingly, we found a significant reduction in the number of axons per area in Klc1−/− compared with Klc1+/+ mice (Fig. 2C). However, Klc1−/− axons were significantly enlarged or dystrophic when compared with Klc1+/+ (Fig. 2D). Together, these experiments suggest that fewer axons reach the cortex and/or the striatum because of pathfinding defects and that there is a significant axonal dystrophic phenotype in Klc1−/− striatal fascicles.
Fig. 2.

Klc1−/− brains display enlarged caliber but reduced axon number in striatal fascicles. (A) Transmission electron microscopy (TEM) images from caudal striatum of Klc1+/+ and Klc1−/− mice at P0. Klc1−/− mice display enlarged axon fascicles (area enclosed in red dashed line). (A′) Higher magnifications of TEM images of panel A showing reduced axon density but enlarged calibers in Klc1−/− mice. (B,C) Quantification of axon fascicle area (B) and axon number (C) per 30 µm2 area. (D) Quantification of axon diameter distribution. Data in B and C are mean±s.e.m. Klc1+/+, n=11-12; Klc1−/−, n=12-14 fascicles. *P<0.03, **P<0.01; Student's t-test. Data in D show probability density function analysis of pooled axon diameters; dashed lines indicate median. Klc1+/+, n=1536; Klc1−/−, n=750 axons. ***P<0.001; Kolmogorov–Smirnov test. Scale bars: 2 µm (A); 0.5 µm (A′).
We then performed axonal tracing experiments using a lipophilic carbocyanine dye (DiI) to determine whether CTA and/or TCA of Klc1−/− failed to reach their proper target. CTA were specifically labeled by placing DiI into the primary somatosensory cortex to determine whether CTA failed to reach the thalamus in the Klc1−/− brain (Fig. 3A-B′,F). Serial coronal slices of DiI-injected brains showed reduced staining from fewer CTA crossing the internal capsule and projecting towards the thalamus in Klc1−/− (Fig. 3A′). Quantitative assessment of DiI-labeled axons that reached the thalamus revealed a significant reduction in the internal capsule thickness of Klc1−/− mice compared with Klc1+/+ mice (Fig. 3C). In addition, at dorsal striatum levels, DiI-labeled CTA stopped before crossing the corticostriatal boundary in Klc1−/− mice (Fig. 3B′,F), a process that is necessary to generate the reciprocal connections between the thalamus and the cortex (Molnár and Butler, 2002). TCA that were labeled by placing DiI into the dorsal thalamus (Fig. 3D-D″,G) showed similar reductions in the internal capsule thickness in Klc1−/− mice (Fig. 3D′,E). Moreover, misrouted axons were observed at the level of the cortical boundary (Fig. 3G), which resulted in less DiI staining reaching the cortical layers (Fig. 3G). Altogether, our results suggest that KLC1 might be involved in the development of CTA and TCA reciprocal connections. Interestingly, given that the abnormal fasciculation and misrouted axon phenotype is reminiscent of the defects observed in CB1R knockout mice and that CB1R signaling modulates the pathfinding and fasciculation of CTA and TCA (Wu et al., 2010), we hypothesize that KLC1 is involved in the intracellular trafficking of CB1R.
Fig. 3.

Reciprocal connection between the cortex and the thalamus is impaired in Klc1−/− cemice. (A-B′) Axonal tracing with a lipophilic dye DiI placed into the primary somatosensory cortex in P0 fixed brain (inset) to label CTA. (A′) Higher magnification images of panel A showing reduced internal capsule thickness (white arrows) in Klc1−/− compared with Klc1+/+ mice. (B′) Higher-magnification images of panel B showing misrouted CTA in Klc1−/− mice (yellow dashed lines). (C) Quantification of internal capsule thickness of CTA. (D,D″) Axonal tracing with DiI placed into the dorsal thalamus in P0 mice to label TCA. (D′) Higher magnification images of panel D showing reduced internal capsule thickness (white arrows) in Klc1−/− mice. (E) Quantification of internal capsule thickness of TCA. (F,G) Ectopic CTA bundle (F) and misrouted TCA (G) were observed in Klc1−/− mice at the corticostriatal boundary (yellow arrowheads). TCA failed to reach the cerebral cortex in Klc1−/− compared with Klc1+/+ (yellow asterisks in D′ and G). Data are mean±s.e.m; n=4 animals/genotype in C, n=5 animals/genotype i E. **P<0.01, ***P<0.0001; Student's t-test. cx, cerebral cortex; ic, internal capsule; st, striatum; th, thalamus. Scale bars: 500 μm (in B for A,B,D,D″); 200 μm (A′,B′,D′,F,G).
Klc1 deletion reduced the axonal and growth cone levels of CB1R in cortical neurons.
CB1R is strongly expressed in CFA during embryonic and early postnatal periods, but then its axonal levels decline and it is enriched in the presynaptic terminal towards adulthood (Castillo et al., 2012; Njoo et al., 2015). Therefore, proper levels and localization of CB1R in these axons may be required for normal fasciculation and axonal pathfinding during development. To test whether Klc1 deletion impairs the levels of CB1R in CFA, we performed double immunofluorescence staining for CB1R and L1-NCAM in brain coronal sections at P0 (Fig. 4A). Hyperfasciculated CFA that project throughout the caudal striatum were positive for CB1R staining in Klc1+/+ and Klc1−/− (Fig. 4A,A′). However, CB1R fluorescence intensity normalized to L1-NCAM revealed a significant reduction in CB1R levels in Klc1−/− when compared with Klc1+/+ (Fig. 4B). Moreover, Klc1−/− showed a significant reduction of CB1R staining in axons that project to the thalamus when compared with Klc1+/+ (Fig. 4A″,C). To further examine whether Klc1 deletion impairs the subcellular localization of CB1R, we performed primary cortical cultures from Klc1+/+ and Klc1−/− mice. CB1R staining at growth cones of cortical neurons revealed a significant reduction of CB1R levels in Klc1−/− neurons when normalized to the intensity of actin filaments (F-actin) stained with phalloidin (Fig. 4D,E). These results revealed that abnormal CFA in Klc1−/− contain reduced axonal levels of CB1R, suggesting that Klc1 deletion impairs the axonal distribution of CB1R and highlighting a relevant role for KLC1 in the navigation of CB1R+ axons.
Fig. 4.

Reduced CB1R levels in CFA and growth cones of Klc1−/−. (A) Confocal images of coronal brain sections showing CB1R and L1-NCAM double immunostaining in Klc1+/+ and Klc1−/− mice at P0. (A′,A″) Higher magnification confocal images of boxed areas in panel A showing reduced CB1R levels in CFA at the caudal striatum (A′) and in thalamic regions next to the internal capsule (A″) of Klc1−/− mice (yellow asterisks indicate thalamic region). (B,C) Quantification of CB1R fluorescence intensity normalized to L1-NCAM in in single confocal sections of CFA (B) and thalamus (C). (D) High-resolution spinning disk confocal microscopy image of primary cortical neurons showing CB1R and phalloidin immunostaining in Klc1+/+ and Klc1−/− axonal growth cone (DIV 4). Dashed lines indicate growth cone membrane limit. (E) Quantification of fluorescence intensity of CB1R normalized to phalloidin in growth cones. Data are mean±s.e.m. Klc1+/+, n=4; Klc1−/−, n=5 (B,C) from 3-4 sections; n=60-70 growth cones/genotype from two independent experiments (E). *P<0.01, ****P<0.0001; Student's t-test. cSt, caudal striatum; ic, internal capsule; th, thalamus. Scale bars: 500 μm (A); 100 μm (A′,A″); 20 μm (D).
Cortical misrouted axons in Klc1−/− brain are positive for CB1R staining
To further evaluate whether CB1R-expressing CFA are misrouted in the absence of KLC1, we performed immunostaining for CB1R in Klc1−/− at P0. Abnormal misplaced axon bundles observed in deeper cortical layers separated from the corpus callosum were positive for CB1R staining in Klc1−/− mice (Fig. 5A). In addition, CB1R immunostaining in Klc1−/− brain sections allowed the identification of misrouted axon bundles in the corticostriatal boundary (Fig. 5B). Together, the aberrant guidance defects and misrouted projections observed in CB1R+ axons suggest a relevant role for KLC1 in the navigation of CFA.
Fig. 5.
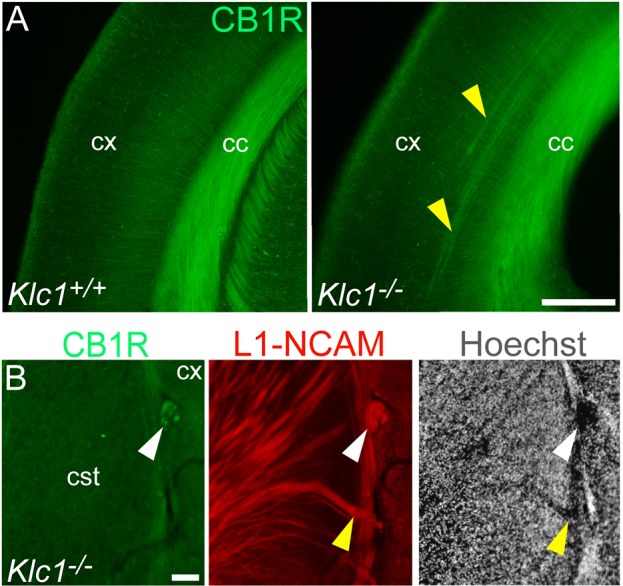
Misrouted axon fascicles in Klc1−/− brain are positive for CB1R. (A) Epifluorescence images of cerebral cortex showing CB1R immunostaining in Klc1+/+ and Klc1−/− mice at P0. (B) Higher magnification of CB1R and L1-NCAM immunostaining and Hoechst in the corticostriatal boundary in Klc1−/− mice at P0. Yellow arrowheads indicate ectopic cortical axon bundles positive for CB1R. White arrowhead indicates CB1R-positive misrouted axon bundles devoid of nuclear staining. cc, corpus callosum; cSt, caudal striatum; cx, cerebral cortex. Scale bars: 200 μm (A); 50 μm (B).
CB1R-containing vesicles colocalize with kinesin heavy chain subunits
CB1R is directly trafficked to the axon through the secretory pathway and is retained longer at the surface of axons than of dendrites (Fletcher-Jones et al., 2019). To test whether kinesin-1 may interact with CB1R vesicles to promote its axonal transport, we performed double immunofluorescence staining for endogenous CB1R and the heavy chain subunits of kinesin-1 (KIF5; hereafter referred to as KHC) in cortical wild-type neurons treated with saponin before fixation (Fig. 6A). Single optical sections of high resolution confocal images showed that CB1R vesicles partially co-localized with KHC in axons (Fig. 6A). Colocalization was also evident at growth cones, where both CB1R and KHC accumulate. A Pearson correlation coefficient analysis revealed a moderate degree of colocalization (median 0.48, percentiles 25/75=0.367/0.545) between CB1R vesicles and KHC-positive vesicles in axons (Fig. 6B), suggesting that kinesin-1 associates with CB1R-containing vesicles and mediates their transport.
Fig. 6.
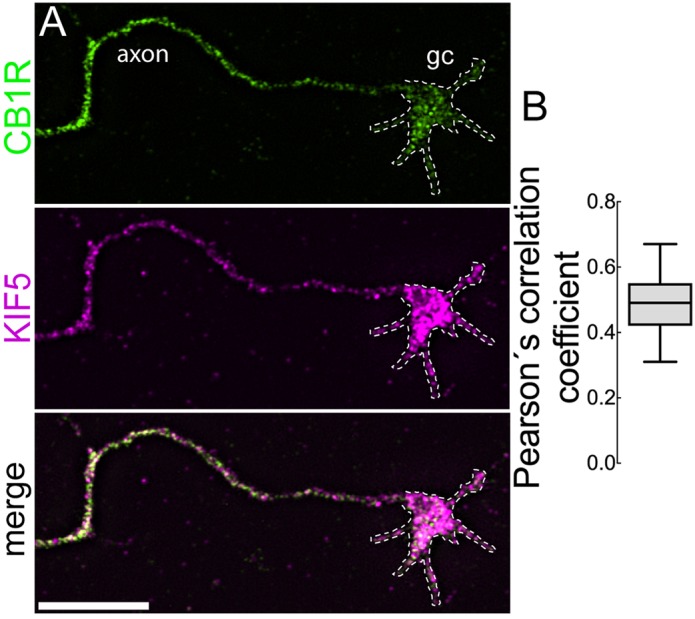
CB1R vesicles colocalize with kinesin heavy chain subunits. (A) High-resolution spinning disk confocal microscopy images of primary cortical neurons immunostained for endogenous CB1R and KIF5 (DIV 4). Merged image shows CB1R and KIF5 colocalizing in vesicles from axons and growth cones (gc). (B) Quantification of colocalization between CB1R and KHC using Pearson's correlation coefficient. Data indicate median ±25th/75th percentile (box) and 5th/95th percentile (whiskers). n=14 neurons. Scale bar: 10 μm.
Axonal transport of CB1R is impaired in Klc1−/− neurons
To characterize the axonal transport properties of CB1R vesicles and unravel whether they depend on kinesin-1 function, we performed live-cell imaging of CB1R tagged with the enhanced green fluorescent protein (eGFP) at the C-terminal region (CB1R-eGFP) in primary hippocampal cultures from Klc1+/+ and Klc1−/− mice. CB1R-eGFP is functional and was shown to display normal signaling and trafficking in HEK293 cells (Leterrier et al., 2004). Transfected neurons showed a vesicular distribution of CB1R-eGFP along the axons (Fig. 7A) and live-cell imaging showed bidirectional movement of fluorescent vesicles (Movies 1, 2 and 3). Kymographs obtained from movies revealed a significant decrease in the proportion of anterograde and retrograde moving CB1R-eGFP vesicles, whereas there were more stationary vesicles in Klc1−/− neurons compared with Klc1+/+ (Fig. 7C). CB1R-eGFP vesicles moved at fast average velocities (anterograde: 0.82 ±0.08 μm/s; retrograde: 0.76±0.09 μm/s) in Klc1+/+ axons (Fig. 7D). Moreover, anterograde-moving CB1R-eGFP vesicles in Klc1−/− neurons displayed reduced average velocities compared with Klc1+/+ neurons (Fig. 7E), suggesting that deletion of KLC1 leads to severe defects in the axonal transport of moving CB1R vesicles. To test whether CB1R transport defects were specifically induced by KLC1 deletion, we performed a rescue experiment using the overexpression of KLC1 in Klc1−/− neurons. CB1R transport dynamics were analyzed after co-transfecting CB1R-eGFP and pcDNA-KLC1 (a vector driving KLC1 expression) vectors in Klc1−/− neurons. Co-transfected neurons were immunostained for KLC1 antibody to confirm the recovery of KLC1 expression (Fig. 7B). The overexpression of KLC1 in neurons depleted of endogenous KLC1 restored the movement of CB1R vesicles by increasing anterograde and retrograde vesicle proportion and by reducing the proportion of stationary vesicles to Klc1+/+ levels (Fig. 7D).
Fig. 7.

CB1R axonal transport is impaired in Klc1−/− neurons. (A) Epifluorescence image obtained from a live-cell movie of 7-8 DIV hippocampal neurons expressing CB1R-eGFP. (B) Hippocampal neurons co-transfected with CB1R-eGFP+pcDNA-KLC1 and immunostained for KLC1 showing KLC1 overexpression. (C) Kymographs of CB1R-eGFP transfected neurons obtained from live-cell imaging of Klc1+/+, Klc1−/−, and Klc1−/−+pcDNA-KLC1 hippocampal cultures. (D,E) Quantification of anterograde (Anter), stationary (Stat) and retrograde (Retro) proportion of CB1R-eGFP vesicles (D), and average velocities for anterograde and retrograde CB1R-eGFP vesicles (E). Data are mean±s.e.m. Klc1+/+, n=52; Klc1−/−, n=43; Klc1−/−+pcDNA-KLC1, n=34 neurons from three independent experiments. *P<0.01, **P<0.001, ****P<0.0001; Student's t-test. Scale bars: 20 μm (A); 50 μm (B).
To obtain detailed information on the axonal transport properties of CB1R that had not been previously described, we analyzed dynamics by tracking single particle trajectories of moving CB1R-eGFP vesicles using MATLAB routines (Alloatti et al., 2018; Lacovich et al., 2017). Interestingly, KLC1 deletion induced a significant decrease in the run lengths of anterograde and retrograde CB1R vesicles, indicating that processive activity was reduced (Fig. 8A). Moreover, overall flux of CB1R-eGFP vesicles was impaired in Klc1−/− because of the increase in the duration of stationary behavior (Fig. 8B). The plot and distribution analysis of segmental velocities by MATLAB routines also revealed a significant shift towards an increased number of slower CB1R-eGFP vesicles and a reduction in the frequency of faster velocities in Klc1−/− neurons (Fig. 8C). Together, these results demonstrate that KLC1 deletion significantly impairs the processive activity, speed and overall axonal transport dynamics of CB1R, suggesting that kinesin-1 function is essential for proper bidirectional distribution of CB1R-vesicles during neuronal development.
Fig. 8.

Dynamic properties of CB1R vesicles are impaired in Klc1−/− neurons. (A,B) Anterograde (Ante), stationary (Stat) and retrograde (Retro) average run lengths (A) and run length duration (B) obtained from CB1R-eGFP vesicle single trajectories using MATLAB. Data are mean±s.e.m. Klc1+/+, n=1499 (ante), n=1487 (retro), n=1897 (stat); Klc1−/−, n=595 (ante), n=638 (retro), n=911 (stat). *P<0.01, **P<0.001, ***P<0.0001; Student's t-test. (C) Distribution of segmental velocities for anterograde and retrograde CB1R-eGFP vesicle trajectories. Insets show relative frequency distribution of segmental velocities. Data show probability density function analysis of pooled segmental velocities. Klc1+/+, n=1821 (ante), n=1729 (retro); Klc1−/−, n=482 (ante), n=556 (retro). ***P<0.00001; Kolmogorov–Smirnov test.
Abnormal growth cones in Klc1−/− cortical neurons are insensitive to CB1R inhibition
CB1R activation stimulates axonal growth by inducing growth cone collapse in developing pyramidal neurons primarily by the modulation of actin filament dynamics (Njoo et al., 2015; Roland et al., 2014). Therefore, we analyzed the morphology of growth cones of Klc1+/+ and Klc1−/− cortical neurons in control conditions and after inhibiting CB1R activation with the CB1R inverse agonist AM251 (1 µM). Of note was that Klc1−/− neurons in DMSO showed a significant (30%) increase in growth cone area covered with actin filaments when compared with Klc1+/+, resembling an expanded and static state of growth cone behavior due to the absence of KLC1 (Fig. 9A,B). When Klc1+/+ neurons were treated with AM251, a significant expansion of the growth cone area (an increase of 50% compared with Klc1+/+ in DMSO) was induced owing to the stabilization of actin filaments, suggesting a role for CB1R in actin filament regulation (Fig. 9A,B). However, AM251 incubation in Klc1−/− did not modify the growth cone area compared with Klc1−/− in DMSO (Fig. 9A,B), suggesting that Klc1−/− growth cones were insensitive to further expansion and actin filament stabilization induced by a CB1R inverse agonist. This result supports a role for KLC1 in the CB1R-mediated signaling that regulates actin dynamics.
Fig. 9.

Abnormal growth cone rearrangement and cofilin activation in Klc1−/− cortex. (A) Epifluorescence images of axonal growth cones stained for tubulin and phalloidin (F-actin) in Klc1+/+ and Klc1−/− cortical neurons (DIV 3) treated with vehicle (DMSO) or AM251 (1 μM) for 1 h. (B) Quantification of the axonal growth cone area covered by F-actin in DMSO- or AM251-treated neurons. Data are mean±s.e.m. Klc1+/+, n=141 neurons (DMSO), n=100 (AM251); Klc1−/−, n=98 (DMSO), n=96 (AM251) from five independent experiments. *P<0.01, ***P<0.001; two-way ANOVA followed by Bonferroni post-test. (C) Immunoblot of KLC1, RhoA, ROCK1, cofilin, p-cofilin and tubulin from primary somatosensory cortex homogenates of Klc1+/+ and Klc1−/− mice at P0. (D) Quantification of protein levels of RhoA and ROCK1 normalized to tubulin, and p-cofilin normalized to total cofilin. Data are mean±s.e.m. Klc1+/+, n=5; Klc1−/−, n=6. *P<0.05; Student's t-test. Scale bar: 10 μm.
CB1R activation results in RhoA- and ROCK-dependent modulation of actin dynamics operating over a large range of actin modifying proteins, such as Wave1 (Wasf1), allowing actin polymerization (Njoo et al., 2015), or by inducing actin contraction through actomyosin II (Roland et al., 2014). We found enlarged growth cones with stabilized actin filaments in Klc1−/− neurons, suggesting an impaired mechanism of filamentous actin breakdown. Cofilin, an important regulator of actin filament severing, is also downstream of the RhoA/ROCK intracellular pathway; however, it has not been linked to CB1R-mediated regulation. Cofilin severs actin filaments when active, but is inactivated by a RhoA/ROCK-dependent phosphorylation process (Pertz, 2010). We tested whether KLC1 deletion changes the levels of proteins that modulate actin dynamics by performing western blots against total protein levels of RhoA, ROCK and cofilin from the cerebral cortex at P0. No changes in total RhoA, ROCK and cofilin levels were observed in Klc1−/− compared with Klc1+/+ (Fig. 9C,D). However, when tested for cofilin activation by performing western blots against phosphorylated cofilin (p-cofilin) we observed significant increases (40%) in p-cofilin in Klc1−/− cortical neurons compared with Klc1+/+ (Fig. 9C,D). As cofilin is inactivated by phosphorylation, increased p-cofilin in Klc1−/− neurons suggests that actin filament breakdown is impaired, leading to enhanced stabilization of the actin cytoskeletal network. These results reinforce the proposition that CB1R promotes rearrangements of actin filaments at growth cones and show that deletion of KLC1 impairs the neuronal ability to respond to CB1R-mediated actin remodeling.
CB1R-mediated axonal elongation is impaired in Klc1−/− cortical neurons
eCB guide and promote axonal extension through specific intracellular pathways that modulate actin filament dynamics in growth cones during development (Mulder et al., 2008). To test whether KLC1 is relevant for CB1R-dependent axonal elongation, we analyzed the axonal length of cortical neurons in culture after treatment with DMSO or the CB1R selective agonist ACEA for 72 h. Interestingly, axonal extension evaluated in neurons at 4 days in vitro (DIV) were similar in both Klc1+/+ and Klc1−/− treated with DMSO, suggesting a normal intrinsic axonal growth process for Klc1−/− neurons without the stimulation of external guiding cues (Fig. 10A,B). The stimulation of CB1R with ACEA (300 nM) induced a significant increase (40%) in axonal elongation compared with DMSO in Klc1+/+ neurons (Fig. 10A,B), reinforcing the knowledge that eCB promote axonal extension. However, Klc1−/− neurons were unable to respond to the treatment with ACEA, showing similar axonal length as Klc1−/− neurons in DMSO conditions (Fig. 10A,B). Axonal branching and dendritic arborization using Sholl analysis was measured in Klc1+/+, and Klc1−/− after DMSO or ACEA treatment, without revealing significant morphological changes (Fig. 10C,D). Together, we show that CB1R activity involves regulation that drives the axonal elongation process. Moreover, Klc1−/− neurons display a normal intrinsic program of axonal extension; however, they are unable to respond to cannabinoid signaling that further promote axonal elongation through the specific stimulation of CB1R.
Fig. 10.

Klc1−/− neurons are unresponsive to CB1R-mediated axonal elongation. (A) Epifluorescence images of Klc1+/+ and Klc1−/− primary cortical neurons transfected with pcDNA3-Cherry and treated for 72 hours with DMSO (vehicle) or CB1R selective agonist ACEA (300 mM). (B) Axonal length quantified in pcDNA-Cherry transfected neurons. Data are mean±s.e.m. Klc1+/+, n=80 (DMSO), n=64 (ACEA); Klc1−/−, n=61 (DMSO), n=61 (ACEA) from three independent experiments. ***P<0.001; two-way ANOVA followed by Bonferroni post-test. (C) Quantification of the number of primary axonal branches. Data indicate median ±25th/75th percentile (box) and 5th/95th percentile (whiskers). (D) Sholl analysis to quantify the average number of dendrite intersections. Data are mean±s.e.m. Klc1+/+, n=84 (DMSO), n=66 (ACEA); Klc1−/−, n=83 (DMSO), n=94 (ACEA) neurons from three independent experiments. Scale bar: 200 μm.
DISCUSSION
Our study highlights the role of kinesin-1 in axonal pathfinding and fasciculation of cortical axons specifically by mediating the axonal transport, localization and signaling of CB1R. Beyond the cell intrinsic polarization process, kinesin-1 motor function is required for CB1R-mediated regulation of actin filament stability, growth cone morphology, and axonal elongation. Our data strongly suggest that dysfunction of CB1R axonal transport can trigger defects in eCB-dependent axonal pathfinding by impairing cofilin activation and actin remodeling at growth cones.
We have previously described in aged Klc1−/− mice abnormal Tau (Mapt) phosphorylation, axonopathies with cytoskeletal disorganization, and cargo accumulation within axons associated with neurodegeneration (Falzone et al., 2009). However, the early observation of reduced brain size, enlarged ventricles and decreased corpus callosum thickness in Klc1−/− mice (Falzone et al., 2010, 2009) suggests the presence of neurodevelopmental defects. Here, by performing detailed analysis of the Klc1−/− developing brain, we revealed an early onset of axonal guidance and pathfinding defects. Moreover, reduced white matter and brain size in Klc1−/− mice are reminiscent of human neurodevelopmental disorders recently associated with somatic mutations in genes encoding kinesin-1 subunits. Patients with mutations in KIF5C showed diminished white matter area, abnormal corpus callosum and pachygyria (Jamuar and Walsh, 2014) that cause severe intellectual disability, epilepsy, microcephaly and cortical malformation (Willemsen et al., 2014). Also, a novel KIF5A mutation was found in a patient with progressive leukoencephalopathy, characterized by white matter volume loss and enlarged lateral ventricles (Rydzanicz et al., 2017). Therefore, kinesin-1 defects could drive to defective axonal transport of molecules involved in axonal guidance that may lead to severe impairments in early neuronal connectivity of the human brain.
The achievement of brain architecture is a complex process that depends on proper brain wiring. TCA navigate to the cortex by co-fasciculating with reciprocal early-born CTA after converging near the corticostriatal boundary as is proposed in the ‘handshake’ hypothesis (Molnár and Blakemore, 1995). According to the handshake model, developing CTA and TCA associate in specific fascicles at the pallial-subpallial boundary to follow the same axonal route but in opposite directions (Molnár et al., 2012). eCB signaling regulates the ‘handshake’ interaction between CTA and TCA by segregated expression of CB1R in CTA and sn-1-diacylglycerol lipases (DAGLα/β), 2-AG synthesizing enzymes, in TCA (Wu et al., 2010). Interestingly, we showed a significant hyperfasciculation phenotype in the striatum, plus aberrant axonal trajectories at the level of corticostriatal boundary in Klc1−/− brains, similar to those observed after Cb1r deletion in mice (Wu et al., 2010). Comparable axonal defects were observed during early neural development in utero in mice pharmacologically treated with CB1R antagonists (Mulder et al., 2008). TCA provide 2-AG to activate CB1R on CTA. The expression of monoacylglycerol lipase (MGL; Mgll), a 2-AG degrading enzyme, in both TCA and CFA can function as 2-AG-inactivating barrier to establish a 2-AG gradient and to promote CB1R-dependent directional CTA pathfinding (Keimpema et al., 2010). Given that proper localization of CB1R is essential for axon fasciculation, the hyperfasciculation phenotype with a reduced number of axons in Klc1−/− brains could be explained by reduced levels of CB1R in CTA axons because of impaired transport towards the growth cone. Reduced thickness of the internal capsule, a major highway to and from the neocortex, is probably due to failures in axonal crossing of the corticostriatal boundary in Klc1−/− mice. Moreover, DiI experiments showed that CTA originating from neurons at the primary somatosensory cortex and reciprocal thalamic axons fail to enter the dorsolateral striatum. Indeed, these axons deviate their way to the thalamus. In line with these results, Cb1r−/− mice showed several similar guidance defects at the level of the corticostriatal boundary (Wu et al., 2010). This region is relevant for the development of reciprocal CTA and TCA connection, acting as a choice point for axonal navigation (Molnár and Butler, 2002; Robichaux et al., 2014). Recent evidence demonstrates that growth cone responsiveness in commissural axons at the midline of the spinal cord depend on selective trafficking of guidance receptors (Alther et al., 2016). We found strong evidence for selective phenotypes consistent with CB1R-mediated defects in axonal guidance and fasciculation. We therefore propose that kinesin-1-mediated transport of vesicles to the growth cone surface, with specific cannabinoid guidance receptors as cargo, represents a molecular mechanism that underlies the switch in axonal responsiveness at the corticostriatal boundary zone.
The trafficking and subcellular localization of CB1R are essential for proper eCB signaling (Leterrier et al., 2006). Using live cell imaging in primary neurons we described the axonal transport properties of CB1R and revealed defects in the transport of CB1R vesicles induced by Klc1 deletion. The specificity of KLC1-dependent cargos was previously assessed, showing selective impairments for another cargo such as the amyloid precursor protein (APP) vesicle, whereas mitochondria axonal transport was not affected (Falzone et al., 2009), suggesting that overall microtubule-dependent dynamics are not impaired when KLC1 is deleted from neurons. A coordinated-competition model has been proposed to take place during axonal transport regulation of fast-moving vesicles (Lacovich et al., 2017). In agreement with this model, the deletion of Klc1 induced defects in anterograde and retrograde CB1R vesicle proportions, segmental velocities and run lengths. A possible explanation for the changes in CB1R retrograde movement when an anterograde motor is missing is supported by the kinesin-1-dependent recruitment of the dynein retrograde complex to the cargo, as was shown for other vesicles (Szpankowski et al., 2012; Twelvetrees et al., 2016). It is important to stress that cargo transport has many levels of regulation (Maday et al., 2014). For example, kinesin-1 associates with vesicles by interacting with adaptor proteins such as JNK-interacting protein (JIP) 1 and JIP3, which regulate cargo selectivity (Koushika, 2008). Interestingly, Jip3−/− and Jip1−/− mice also show severe defects in the pathfinding of telencephalic commissure and also hyperfasciculation of CTA and TCA (Cho et al., 2011; Ha et al., 2005; Kelkar et al., 2003), suggesting a strong link between axonal transport and molecules that mediate guidance. As the reduction of CB1R motility in Klc1−/− was not complete, it is possible that other molecular motors are also involved in CB1R transport. Further studies are required to elucidate how CB1R vesicles are linked to kinesin-1 or other motors that propel their axonal transport.
Previously, it has been reported that the kinesin-1/JIP1/Rab10 complex is required for axonal elongation (Deng et al., 2014). Hippocampal neurons transfected with dominant-negative mutants of KLC1 showed impairments in neuronal polarity and decreased axonal length (Deng et al., 2014). However, here we showed that Klc1−/− cortical neurons did not impair the basal program of axonal outgrowth. Moreover, cell intrinsic processes showed that Klc1−/− neurons without stimulation in culture achieved normal polarization and extend axons in a similar way to wild-type neurons. Consistent with the function of external guidance cues, it has been shown that axonal growth was promoted in cortical neurons exposed to anandamide (Mulder et al., 2008), 2-AG (Tapia et al., 2017) or a MGL inhibitor (Keimpema et al., 2010). However, other studies proposed that treatment with CB1R agonist WIN55 for 7 days reduced the axonal length of cortical neurons transfected with CB1-eGFP (Vitalis et al., 2008; Shum et al., 2020). Of note is that axonal elongation was precluded in Klc1−/− neurons stimulated with CB1R agonists, contrary to what was observed in control neurons. This functional inability to responds to cannabinoid signaling in Klc1−/− neurons could be explained by an abnormal presentation of membrane cargos due to defective transport of CB1R towards growth cones.
CB1R activates downstream effectors within axonal growth cones that are necessary for the cytoskeletal plasticity that allows growth cone build-up and collapse during axonal navigation (Berghuis et al., 2007; Njoo et al., 2015; Roland et al., 2014). The stimulation of CB1R induces contraction of the actomyosin cytoskeleton through Rho-associated kinase and triggers growth cone retraction (Roland et al., 2014). It has been proposed that CB1R activation affects actin polymerization and stability by inducing Rac1 phosphorylation and regulating WAVE1 and the ARP2/3 (Actr2/3) complex (Njoo et al., 2015). The observation of an expanded growth cone in Klc1−/− neurons raises the possibility that actin dynamics were impaired owing to abnormal eCB signaling. Consistent with this possibility, we found increased cofilin inactivation (p-cofilin) in the cerebral cortex of neonatal Klc1−/− mice. Cofilin activity is necessary for the severing of actin filaments (Andrianantoandro and Pollard, 2006) and this process regulates growth cone remodeling (Endo et al., 2003). The analysis of Klc1−/− neurons revealed a significant increase in cofilin phosphorylation that supports the observation of static expanded growth cone areas. In addition, neurons without KLC1 were unable to further respond to a CB1R antagonist, which expanded growth cones in Klc1+/+ neurons. Interestingly, the transport of the WAVE1 complex to the growth cone requires kinesin-1 via CRMP-2 (Dpysl2)-mediated binding to KCL1 (Kawano et al., 2005). Mass spectrometry analyses reinforced this association by identifying WAVE1 as a component of the APP vesicle as a kinesin-1 cargo (Almenar-Queralt et al., 2014). These observations, together with our results, suggest a role for motor proteins in the transport of relevant cargos that mediate eCB-dependent growth cone rearrangement.
Taken together, our study provides strong evidence that kinesin-1 mediates the axonal transport of CB1R, playing key roles in eCB signaling that regulate actin remodeling at the growth cone for the developing of TCA and CTA connections. Knowledge on the spatiotemporal regulation of CB1R axonal transport should contribute towards understanding the implications of proper subcellular localization of eCB signaling molecules during the establishment of neuronal connectivity and the molecular mechanisms underlying neurodevelopmental diseases associated with human brain wiring defects.
MATERIALS AND METHODS
Animals
Klc1+/− mice from a C57BL/6J background were crossed with each other to obtain wild-type and Klc1−/− littermates (Rahman et al., 1999). Both P0 males and females were used in experiments. Mice were housed in temperature- and 12 h light/dark-controlled rooms under approved university protocols (El Comité Institucional para el Cuidado y Uso de Animales de Laboratorio; UBA-FMED 2532/2019). Mice had access to food and water ad libitum. Genotyping of mice obtained from Klc1+/− crossing was performed by PCR amplification of wild-type and recombinant Klc1 alleles. The primer sequences were: wild-type allele forward 5′-GCATCGAGCTGGGTAATAAGCGTTGGCAAT-3′, and reverse 5′-GACACCAGACCAACTGGTAATGGTAGCGAC-3′; Klc1 mutant allele forward 5′-CGGGCTGTTTCTCTTGGCTTGCTC-3′, and reverse 5′-GGAGCGTGCGCAGCCTTGCAGGGA3′.
Antibodies
Monoclonal antibodies against the following proteins were used: anti-KIF5 (mouse, clone KHC-H2, Millipore, MAB1614, 1:800), anti-tubulin (mouse, clone DM1a, Covance, a5-829, 1:10,000), anti-L1-NCAM (rat, clone 324, Millipore, MAB5272, 1:2000). Polyclonal antibodies (rabbit) against the following proteins were used: anti-CB1R (C-term, Cayman, 10006590, 1:1000), anti-kinesin light chain 1 (Santa Cruz Biotechnology, sc25735, 1:1000), anti-cofilin (Cell Signaling Technology, 3318, 1:1000), anti-phospho-cofilin (ser3) (Cell Signaling Technology, 3313, 1:1000), RhoA (Cell Signaling Technology, 2117, 1:1000), ROCK1 (Cell Signaling Technology, 4035, 1:1000). The following secondary antibodies were used: anti-mouse Alexa Fluor 568 (Invitrogen, MA11004, 1:500), anti-rabbit Alexa Fluor 488 (Invitrogen, A11008, 1:500), anti-rat Alexa Fluor 594 (Invitrogen, A11001, 1:500), HRP goat anti-rabbit (Vector Laboratories, 111-035-144, 1:7000), HRP goat anti-mouse (Vector Laboratories, 115-035-146, 1:7000).
Tissue preparation, immunohistochemistry and quantification
Neonatal mice (P0) were transcardially perfused with saline solution followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) (pH 7.4). Brains were dissected and postfixed in fixation buffer overnight and then cryoprotected in 30% sucrose in 0.1 M phosphate buffer saline (PBS) (pH 7.4) for 48 h at 4°C. Brains were embedded in 4% agarose for vibratome coronal sectioning (50 µm thick). Immunofluorescence was performed on free-floating sections of positions 255 and 296 in Allen Developing Mouse Brain Atlas. Non-specific immunoreactivity was blocked by incubating the sections in 5% normal goat serum (NGS; Gibco) and 0.3% Triton X-100 (Sigma-Aldrich) in PBS for 1 h at room temperature. Sections were incubated with one or two primary antibodies diluted in 1% NGS and 0.1% Triton X-100 in PBS for 16 h at 4°C, followed by incubating with Alexa-Fluor-conjugated secondary antibodies for 2 h at room temperature. Sections were stained with Hoechst 33432 (Sigma-Aldrich), dehydrated and mounted with DPX (Sigma-Aldrich). Epifluorescence images were obtained using an Olympus IX-81 inverted microscope. For morphometric analysis of fascicle diameter and area covered, the diameter of all axon fascicles present in an area of 500×100 µm2 located in the caudal striatum next to the corticostriatal boundary was measured. For CB1R and L1-NCAM level analysis, the intensity of CB1R and L1-NCAM in CFA present in the caudal striatum next to the corticostriatal boundary was quantified using a spinning disk confocal microscope (IX-83, Olympus).
Electron microscopy and analysis
Klc1+/+ and Klc1−/− P0 mice were perfused transcardially with 4% PFA and 0.25% glutaraldehyde in 0.1 M PB (pH 7.4). The brain was removed and postfixed overnight at 4°C in the same fixative, and the following day sectioned on a vibratome at a thickness of 400 µm. The caudal striatum was dissected under a microscope and was postfixed in the same fixative solution for 2 h. Then tissue was blocked and fixed with 1% osmium tetroxide in 0.1 M PB before incubation with 1% uranyl acetate in PB 0.1 M (pH 6.0) and then embedded in epoxy Durcupan resin. Then 70 nm sections were obtained and stained with 2% uranyl acetate and lead citrate. Images were generated on a Zeiss EM 109T transmission electron microscope. The fascicle area was identified and counted in 3000× magnification and the number and diameter of axons were identified and counted in 12,000× magnification. Axon density and caliber was determined by manually counting and measuring axons in equal-sized boxes placed within the fascicles.
DiI tracing experiment
P0 mice were transcardially perfused with saline solution followed by 4% PFA in 0.1 M PB. Brains were dissected and postfixed in fixation buffer overnight. DiI crystals (D3911, Molecular Probe) were placed in the presumptive primary somatosensory cortex (S1) to label CTA and in the dorsal thalamus to label TCA. Labeled brains were incubated in 4% PFA in 0.1 M PB with 0.1% sodium azide at 37°C in darkness for 21 days. Brains were embedded in 4% agarose in PBS and sectioned at 100 µm in the coronal plane using a vibratome. Sections were washed and incubated with Hoechst 33432. Epifluorescence images were obtained using an Olympus IX-81 microscope.
Vectors
The CB1R-eGFP was a kind gift from Dr Lenkei (ESPCI-ParisTech, CNRS, Paris, France). pcDNA3-mCherry was provided by Nathan Shaner (The University of California, San Diego, USA). The pcDNA3-KLC1-TAP-TAG (pcDNA-KLC1) vector was used in axonal transport recovery experiments to overexpress KLC1 in Klc1−/− neurons.
Primary neuronal culture and cell transfection
Primary hippocampal and cortical neurons were obtained from P0 Klc1+/+ (wild-type) and Klc1−/− mice as described previously (Falzone et al., 2009). Mouse hippocampi or cerebral cortex were dissected in Hanks balanced salt solution (Invitrogen), dissociated with 45 U papain and 0.05% DNase in PBS for 20 min at 37°C and then triturated by gentle pipetting in 10% fetal bovine serum (FBS) in DMEM (Invitrogen). Cells were suspended in DMEM with 10% FBS, then changed to Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen) and 2 mM L-glutamine (Sigma-Aldrich). Cells were plated on 1 mg/ml poly-L-lysine (Sigma-Aldrich)-coated coverslips and grown at 37°C under 5% CO2. Cells were transfected using a mix of Lipofectamine 2000 (2 μl, Life Technologies) with plasmid DNA (0.8 μg) in optimum. After the addition of complete Neurobasal medium (Thermo Scientific, 21103049), the mix was applied onto the neuronal culture for 2 h at 37°C. Hippocampal neurons transfected with CB1R-eGFP or co-transfected with CB1R-eGFP and pcDNA3-KLC1-TAP-TAG at DIV 7-10 were used for movie acquisition. For axonal growth assay, cortical neurons were incubated with DMSO and 300 nM ACEA for 72 h at DIV 1, transfected with pcDNA3-Cherry at DIV 3, and fixed at DIV 4 to evaluate the axonal length.
Immunocytochemistry on primary cultured neurons
Primary cortical cultured neurons were rinsed with ice-cold PBS and fixed in 4% PFA with 4% sucrose in PB for 30 min at 4°C. For KHC and CB1R immunostaining, cells were pre-permeabilized by incubating for 5 min at 4°C in 80 mM PIPES (pH 6.8), 5 mM EGTA, 1 mM MgCl2 containing 0.1% bovine serum albumin (BSA) and 0.01% saponin (Sigma-Aldrich). After three washes in ice-cold PBS, cells were fixed in 4% PFA for 30 min at 4°C. After three washes cells were permeabilized and blocked with 0.3% Triton X-100 and 5% NGS in PBS for 1 h. Cells were incubated with one or two primary antibodies or phalloidin Alexa Fluor 568 diluted in 1% NGS and 0.1% Triton X-100 in PBS for 16 h at 4°C, followed by incubating with Alexa-Fluor-conjugated secondary antibodies (1:500, Molecular Probe) for 2 h at room temperature. Coverslips were mounted in Mowiol mounting medium (Sigma-Aldrich). The images were acquired either using an inverted epifluorescent microscope (Olympus, IX-81) or a spinning disk confocal microscope (Olympus, IX-83). For growth cone assay, cortical neurons at DIV 2 were treated with vehicle (DMSO) or 1 µM AM251 (Cayman) for 1 h, fixed and stained with phalloidin Alexa Fluor 568 and tubulin to measure growth cone area.
Movies and kymograph analysis
Live-cell imaging and kymograph analysis of CB1-eGFP movement was registered as previously described (Falzone and Stokin, 2012). Briefly, primary hippocampal neurons at DIV 7-8 were transfected with CB1-eGFP using Lipofectamine 2000 to achieve low transfection efficiency. At 16 h post-transfection, CB1R-eGFP vesicle movements were recorded. Continuous 30 s stacks (224 frames) at a speed of 125 ms/frame were collected on an inverted epifluorescent microscope (Olympus IX-81) connected to a CCD camera (Olympus DP71/12.5 megapixels) with a 60× lens (1.4 NA). Cells were maintained at 37°C, 5% CO2 and 10% humidity using a heating stage and CO2 humid chamber (Olympus) (Otero et al., 2014). Movies were obtained from axons, identified by morphology, at least 200 µm away from the soma. The directionality of the vesicles was determined by either locating the cell body or the growth cone in isolated transfected neurons. Kymographs were generated from stacked images using the multiple kymographs plug-in from ImageJ (National Institutes of Health) and processed using Image-Pro Plus 5 (Media Cybernetics) (Otero et al., 2018). Segmental velocities and run lengths were obtained from individual trajectories tracked from the kymographs using customized algorithms written in MATLAB (MathWorks) as previously described (Lacovich et al., 2017). For the calculation of segmental velocities, processive trajectories were divided into segments and the mean velocity of each segment was computed from the slope of a linear approximation to the trajectory using the least-squares method. Points at which movement is equal to 0 defined the limits of segments considered anterograde or retrograde if they showed positive or negative velocities, respectively, or pauses if they fitted a stationary criteria (<0.16 μm/s). The duration of these segments was also computed and pauses were presented as stationary in the analysis.
Western blot
Primary somatosensory cortex at P0 was homogenized in 100 μl lysis buffer [40 mM Tri-HCl (pH 7.5), 150 mM NaCl, 1% Igepal (Sigma-Aldrich) and 1× protease inhibitor cocktail (Sigma-Aldrich)] and centrifuged for 10 min at 10,000 rpm (8000 g) at 4°C. Protein concentration of the supernatant was measured using BCA Protein Assay Kit (Pierce, 23225). Equal amounts of protein (30 µg) in 20 µl of 1× Laemmli sample buffer (0.5% bromophenol blue, 10% glycerol and 2% β-mercaptoethanol) were loaded onto 12% SDS-polyacrylamide gel and See-Plus 2 (Invitrogen) was used as a molecular-weight marker. Proteins were transferred onto nitrocellulose membranes using a wet system in 25 mM Tris base, 190 mM glycine and 20% methanol. Membranes were blocked in 5% BSA in 0.1% Tween-20 in TBS (TBS-T) for 1 h and incubated in 1% BSA in TBS-T with primary antibody overnight at 4°C. After washing, membranes were incubated with HRP-conjugated secondary antibody for 4 h at room temperature. Proteins were visualized using an ECL kit (Pierce, 32106). Scanned images were analyzed using ImageJ software.
Statistical analysis
Statistical differences between conditions were assessed using GraphPad Prism 8.2.1., MATLAB and R core team (www.graphpad.com; www.mathworks.com; www.r-project.org). The normal distribution of the samples was assessed before analysis of significance. Values are expressed as mean±s.e.m. or median±25/75 percentiles for non-parametric data for the indicated number of independent experiments or corresponding particles (n). The experimental setting, the n, and the number of independent experiments are described in figure legends. No data was excluded from analysis. Images for analysis of intensity, quantification of axonal tracks from movies, or in Sholl analysis, were double-coded so the experimenter was blind to genotype or treatment. A two-tailed Student's t-test was used to compare differences between groups. Two-way ANOVA was performed to identify the interaction between two independent variables and analysis was followed by a Bonferroni post-test. The Kolmogorov–Smirnov test was used to compare frequency distributions of fascicle diameter, segmental velocities and axon diameters. Differences that resulted in P<0.05 were considered significant.
Supplementary Material
Acknowledgements
We thank Dr Lenkei for providing the CB1R-eGFP expression vector; Ken Mackie for providing the CB1 antibody; A. Pecile, M. Ceol and M. Ponce for animal care; the Laboratorio Nacional de Investigación y Servicios de Microscopía Electrónica at the Instituto de Biología Celular y Neurociencia and L. Anton for transmission electron microscopy services; and N. Villalba for technical assistance. We acknowledge Fundación René Baron for general institutional support.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: T.M.M.S., D.M.G., T.L.F.; Methodology: T.M.M.S.; Software: I.F.B.; Validation: T.M.M.S., L.S.; Formal analysis: T.M.M.S., I.F.B., M.S.R., M.A., M.G.O., L.E.C., V.M.P.D., G.O.; Investigation: T.M.M.S., V.M.P.D., G.O.; Resources: T.M.M.S., M.G.B.; Writing - original draft: T.M.M.S., T.L.F.; Writing - review & editing: T.M.M.S., M.G.B., D.M.G., T.L.F.; Supervision: T.L.F.; Project administration: T.L.F.; Funding acquisition: T.L.F.
Funding
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) (PICT 2013-0402, 2016-1648 to T.L.F.; PICT 2012-1175 to M.G.B.); Secretaría de Ciencia y Técnica, Universidad de Buenos Aires (UBA) (UBACyT 2011-2014, 2014-2016 to T.L.F.; UBACyT 20020170200052BA to T.M.M.S.) and National Institutes of Health (R01TW008662 to M.G.B.). T.L.F. acknowledges support from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), UBA and ANPCyT. T.M.M.S., M.A., M.G.O., V.M.P.D. and L.E.C. received fellowships from CONICET; I.F.B. received a fellowship from UBA; M.S.R. received a fellowship from Consejo Interuniversitario Nacional. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.184069.supplemental
Peer review history
The peer review history is available online at https://dev.biologists.org/lookup/doi/10.1242/dev.184069.reviewer-comments.pdf
References
- Alloatti M., Bruno L. and Falzone T. L. (2018). Methods for quantitative analysis of axonal cargo transport. Methods Mol. Biol. 1727, 217-226. 10.1007/978-1-4939-7571-6_16 [DOI] [PubMed] [Google Scholar]
- Almenar-Queralt A., Falzone T. L., Shen Z., Lillo C., Killian R. L., Arreola A. S., Niederst E. D., Ng K. S., Kim S. N., Briggs S. P. et al. (2014). UV irradiation accelerates amyloid precursor protein (APP) processing and disrupts APP axonal transport. J. Neurosci. 34, 3320-3339. 10.1523/JNEUROSCI.1503-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alther T. A., Domanitskaya E. and Stoeckli E. T. (2016). Calsyntenin 1-mediated trafficking of axon guidance receptors regulates the switch in axonal responsiveness at a choice point. Development 143, 994-1004. 10.1242/dev.127449 [DOI] [PubMed] [Google Scholar]
- Andrianantoandro E. and Pollard T. D. (2006). Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24, 13-23. 10.1016/j.molcel.2006.08.006 [DOI] [PubMed] [Google Scholar]
- Argaw A., Duff G., Zabouri N., Cecyre B., Chaine N., Cherif H., Tea N., Lutz B., Ptito M. and Bouchard J.-F. (2011). Concerted action of CB1 cannabinoid receptor and deleted in colorectal cancer in axon guidance. J. Neurosci. 31, 1489-1499. 10.1523/JNEUROSCI.4134-09.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw G. J. and Klein R. (2010). Signaling from axon guidance receptors. Cold Spring Harbor Perspect. Biol. 2, a001941 10.1101/cshperspect.a001941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghuis P., Rajnicek A. M., Morozov Y. M., Ross R. A., Mulder J., Urban G. M., Monory K., Marsicano G., Matteoli M., Canty A. et al. (2007). Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316, 1212-1216. 10.1126/science.1137406 [DOI] [PubMed] [Google Scholar]
- Castillo P. E., Younts T. J., Chávez A. E. and Hashimotodani Y. (2012). Endocannabinoid signaling and synaptic function. Neuron 76, 70-81. 10.1016/j.neuron.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallin M., Hubert L., Cantagrel V., Munnich A., Boddaert N., Vincent-Delorme C., Cuvellier J. C., Masson C., Besmond C. and Bahi-Buisson N. (2016). Recurrent KIF5C mutation leading to frontal pachygyria without microcephaly. Neurogenetics 17, 79-82. 10.1007/s10048-015-0459-8 [DOI] [PubMed] [Google Scholar]
- Cho I.-H., Lee K.-W., Ha H.-Y. and Han P.-L. (2011). JNK/stress-activated protein kinase associated protein 1 is required for early development of telencephalic commissures in embryonic brains. Exp. Mol. Med. 43, 462-470. 10.3858/emm.2011.43.8.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromberg L. E., Saez T. M. M., Otero M. G., Tomasella E., Alloatti M., Damianich A., Pozo Devoto V., Ferrario J., Gelman D. M., Rubinstein M. et al. (2019). Neuronal KIF5b deletion induces striatum-dependent locomotor impairments and defects in membrane presentation of dopamine D2 receptors. J. Neurochem. 149, 362-380. 10.1111/jnc.14665 [DOI] [PubMed] [Google Scholar]
- de Ligt J., Willemsen M. H., van Bon B. W. M., Kleefstra T., Yntema H. G., Kroes T., Vulto-van Silfhout A. T., Koolen D. A., de Vries P., Gilissen C. et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921-1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Deng C.-Y., Lei W.-L., Xu X.-H., Ju X.-C., Liu Y. and Luo Z.-G. (2014). JIP1 mediates anterograde transport of Rab10 cargos during neuronal polarization. J. Neurosci. 34, 1710-1723. 10.1523/JNEUROSCI.4496-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent E. W. and Gertler F. B. (2003). Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron 40, 209-227. 10.1016/S0896-6273(03)00633-0 [DOI] [PubMed] [Google Scholar]
- Endo M., Ohashi K., Sasaki Y., Goshima Y., Niwa R., Uemura T. and Mizuno K. (2003). Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J. Neurosci. 23, 2527-2537. 10.1523/JNEUROSCI.23-07-02527.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone T. L. and Stokin G. B. (2012). Imaging amyloid precursor protein in vivo: an axonal transport assay. Methods Mol. Biol. 846, 295-303. 10.1007/978-1-61779-536-7_25 [DOI] [PubMed] [Google Scholar]
- Falzone T. L., Stokin G. B., Lillo C., Rodrigues E. M., Westerman E. L., Williams D. S. and Goldstein L. S. B. (2009). Axonal stress kinase activation and tau misbehavior induced by kinesin-1 transport defects. J. Neurosci. 29, 5758-5767. 10.1523/JNEUROSCI.0780-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzone T. L., Gunawardena S., McCleary D., Reis G. F. and Goldstein L. S. B. (2010). Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet. 19, 4399-4408. 10.1093/hmg/ddq363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher-Jones A., Hildick K. L., Evans A. J., Nakamura Y., Wilkinson K. A. and Henley J. M. (2019). The C-terminal helix 9 motif in rat cannabinoid receptor type 1 regulates axonal trafficking and surface expression. eLife 8, e44252 10.7554/eLife.44252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein L. S. B. (2001). Kinesin molecular motors: transport pathways, receptors, and human disease. Proc. Natl. Acad. Sci. USA 98, 6999-7003. 10.1073/pnas.111145298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes-Dias P. and Holzbaur E. L. F. (2019). Axonal transport: driving synaptic function. Science 366, eaaw9997 10.1126/science.aaw9997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R. and Dobyns W. B. (2014). Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 13, 710-726. 10.1016/S1474-4422(14)70040-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha H.-Y., Cho I.-H., Lee K.-W., Lee K.-W., Song J.-Y., Kim K.-S., Yu Y.-M., Lee J.-K., Song J.-S., Yang S.-D. et al. (2005). The axon guidance defect of the telencephalic commissures of the JSA-deficient brain was partially rescued by the transgenic expression of JIP1. Dev. Biol. 277, 184-199. 10.1016/j.ydbio.2004.09.019 [DOI] [PubMed] [Google Scholar]
- Hirokawa N., Noda Y., Tanaka Y. and Niwa S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell. Biol. 10, 682-696. 10.1038/nrm2774 [DOI] [PubMed] [Google Scholar]
- Jacobson C., Schnapp B. and Banker G. A. (2006). A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron 49, 797-804. 10.1016/j.neuron.2006.02.005 [DOI] [PubMed] [Google Scholar]
- Jamuar S. S. and Walsh C. A. (2014). Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 371, 2038 10.1056/NEJMc1411784 [DOI] [PubMed] [Google Scholar]
- Jamuar S. S. and Walsh C. A. (2015). Genomic variants and variations in malformations of cortical development. Pediatr. Clin. North. Am. 62, 571-585. 10.1016/j.pcl.2015.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano Y., Yoshimura T., Tsuboi D., Kawabata S., Kaneko-Kawano T., Shirataki H., Takenawa T. and Kaibuchi K. (2005). CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol. Cell. Biol. 25, 9920-9935. 10.1128/MCB.25.22.9920-9935.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keimpema E., Barabas K., Morozov Y. M., Tortoriello G., Torii M., Cameron G., Yanagawa Y., Watanabe M., Mackie K. and Harkany T. (2010). Differential subcellular recruitment of monoacylglycerol lipase generates spatial specificity of 2-arachidonoyl glycerol signaling during axonal pathfinding. J. Neurosci. 30, 13992-14007. 10.1523/JNEUROSCI.2126-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar N., Delmotte M.-H., Weston C. R., Barrett T., Sheppard B. J., Flavell R. A. and Davis R. J. (2003). Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3). Proc. Natl. Acad. Sci. USA 100, 9843-9848. 10.1073/pnas.1733944100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika S. P. (2008). “JIP”ing along the axon: the complex roles of JIPs in axonal transport. BioEssays 30, 10-14. 10.1002/bies.20695 [DOI] [PubMed] [Google Scholar]
- Lacovich V., Espindola S. L., Alloatti M., Pozo Devoto V., Cromberg L. E., Čarná M. E., Forte G., Gallo J.-M., Bruno L., Stokin G. B. et al. (2017). Tau isoforms imbalance impairs the axonal transport of the amyloid precursor protein in human neurons. J. Neurosci. 37, 58-69. 10.1523/JNEUROSCI.2305-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leterrier C., Bonnard D., Carrel D., Rossier J. and Lenkei Z. (2004). Constitutive endocytic cycle of the CB1 cannabinoid receptor. J. Biol. Chem. 279, 36013-36021. 10.1074/jbc.M403990200 [DOI] [PubMed] [Google Scholar]
- Leterrier C., Laine J., Darmon M., Boudin H., Rossier J. and Lenkei Z. (2006). Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J. Neurosci. 26, 3141-3153. 10.1523/JNEUROSCI.5437-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton D. M., Maeder C. I. and Shen K. (2018). Rapid assembly of presynaptic materials behind the growth cone in dopaminergic neurons is mediated by precise regulation of axonal transport. Cell Rep. 24, 2709-2722. 10.1016/j.celrep.2018.07.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S., Twelvetrees A. E., Moughamian A. J. and Holzbaur E. L. F. (2014). Axonal transport: cargo-specific mechanisms of motility and regulation. Neuron 84, 292-309. 10.1016/j.neuron.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels S., Foss K., Park K., Golden-Grant K., Saneto R., Lopez J. and Mirzaa G. M. (2017). Mutations of KIF5C cause a neurodevelopmental disorder of infantile-onset epilepsy, absent language, and distinctive malformations of cortical development. Am. J. Med. Genet. A 173, 3127-3131. 10.1002/ajmg.a.38496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z. and Blakemore C. (1995). How do thalamic axons find their way to the cortex? Trends Neurosci. 18, 389-397. 10.1016/0166-2236(95)93935-Q [DOI] [PubMed] [Google Scholar]
- Molnár Z. and Butler A. B. (2002). The corticostriatal junction: a crucial region for forebrain development and evolution. BioEssays 24, 530-541. 10.1002/bies.10100 [DOI] [PubMed] [Google Scholar]
- Molnár Z., Garel S., López-Bendito G., Maness P. and Price D. J. (2012). Mechanisms controlling the guidance of thalamocortical axons through the embryonic forebrain. Eur. J. Neurosci. 10, 1573-1585. 10.1111/j.1460-9568.2012.08119.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J., Aguado T., Keimpema E., Barabas K., Ballester Rosado C. J., Nguyen L., Monory K., Marsicano G., Di Marzo V., Hurd Y. L. et al. (2008). Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc. Natl. Acad. Sci. USA 105, 8760-8765. 10.1073/pnas.0803545105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima K., Yin X., Takei Y., Seog D.-H., Homma N. and Hirokawa N. (2012). Molecular motor KIF5A is essential for GABA(A) receptor transport, and KIF5A deletion causes epilepsy. Neuron 76, 945-961. 10.1016/j.neuron.2012.10.012 [DOI] [PubMed] [Google Scholar]
- Njoo C., Agarwal N., Lutz B. and Kuner R. (2015). The cannabinoid receptor CB1 interacts with the WAVE1 complex and plays a role in actin dynamics and structural plasticity in neurons. PLoS Biol. 13, e1002286 10.1371/journal.pbio.1002286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero M. G., Alloatti M., Cromberg L. E., Almenar-Queralt A., Encalada S. E., Pozo Devoto V. M., Bruno L., Goldstein L. S. B. and Falzone T. L. (2014). Fast axonal transport of the proteasome complex depends on membrane interaction and molecular motor function. J. Cell Sci. 127, 1537-1549. 10.1242/jcs.140780 [DOI] [PubMed] [Google Scholar]
- Otero M. G., Fernandez Bessone I., Hallberg A. E., Cromberg L. E., De Rossi M. C., Saez T. M., Levi V., Almenar-Queralt A. and Falzone T. L. (2018). Proteasome stress leads to APP axonal transport defects by promoting its amyloidogenic processing in lysosomes. J. Cell Sci. 131, jcs214536 10.1242/jcs.214536 [DOI] [PubMed] [Google Scholar]
- Pan W. X., Mao T. and Dudman J. T. (2010). Inputs to the dorsal striatum of the mouse reflect the parallel circuit architecture of the forebrain. Front. Neuroanat. 4, 147 10.3389/fnana.2010.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertz O. (2010). Spatio-temporal Rho GTPase signaling - where are we now? J. Cell. Sci. 123, 1841-1850. 10.1242/jcs.064345 [DOI] [PubMed] [Google Scholar]
- Poirier K., Lebrun N., Broix L., Tian G., Saillour Y., Boscheron C., Parrini E., Valence S., Pierre B. S., Oger M. et al. (2013). Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 45, 639-647. 10.1038/ng.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A., Kamal A., Roberts E. A. and Goldstein L. S. B. (1999). Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J. Cell Biol. 146, 1277-1288. 10.1083/jcb.146.6.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaux M. A., Chenaux G., Ho H.-Y. H., Soskis M. J., Dravis C., Kwan K. Y., Šestan N., Greenberg M. E., Henkemeyer M. and Cowan C. W. (2014). EphB receptor forward signaling regulates area-specific reciprocal thalamic and cortical axon pathfinding. Proc. Natl. Acad. Sci. USA 111, 2188-2193. 10.1073/pnas.1324215111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland A. B., Ricobaraza A., Carrel D., Jordan B. M., Rico F., Simon A., Humbert-Claude M., Ferrier J., McFadden M. H., Scheuring S. et al. (2014). Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. eLife 3, e03159 10.7554/eLife.03159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozbesky D. and Jones E. Y. (2020). Cell guidance ligands, receptors and complexes - orchestrating signalling in time and space. Curr. Opin. Struct. Biol. 61, 79-85. 10.1016/j.sbi.2019.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydzanicz M., Jagła M., Kosinska J., Tomasik T., Sobczak A., Pollak A., Herman-Sucharska I., Walczak A., Kwinta P. and Płoski R. (2017). KIF5A de novo mutation associated with myoclonic seizures and neonatal onset progressive leukoencephalopathy. Clin. Genet. 91, 769-773. 10.1111/cge.12831 [DOI] [PubMed] [Google Scholar]
- Shao L., Golbaz K., Honer W. G. and Beasley C. L. (2016). Deficits in axon-associated proteins in prefrontal white matter in bipolar disorder but not schizophrenia. Bipolar Disord. 18, 342-351. 10.1111/bdi.12395 [DOI] [PubMed] [Google Scholar]
- Shum C., Dutan L., Annuario E., Warre-Cornish K., Taylor S. E., Taylor R. D., Andreae L. C., Buckley N. J., Price J., Bhattacharyya S. et al. (2020). Δ9-tetrahydrocannabinol and 2-AG decreases neurite outgrowth and differentially affects ERK1/2 and Akt signaling in hiPSC-derived cortical neurons. Mol. Cell. Neurosci. 103, 103463 10.1016/j.mcn.2019.103463 [DOI] [PubMed] [Google Scholar]
- Stoeckli E. T. (2018). Understanding axon guidance: are we nearly there yet? Development 145, dev151415 10.1242/dev.151415 [DOI] [PubMed] [Google Scholar]
- Szpankowski L., Encalada S. E. and Goldstein L. S. B. (2012). Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proc. Natl. Acad. Sci. USA 109, 8582-8587. 10.1073/pnas.1120510109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia M., Dominguez A., Zhang W., Del Puerto A., Ciorraga M., Benitez M. J., Guaza C. and Garrido J. J. (2017). Cannabinoid receptors modulate neuronal morphology and AnkyrinG density at the axon initial segment. Front. Cell Neurosci. 11, 5 10.3389/fncel.2017.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twelvetrees A. E., Pernigo S., Sanger A., Guedes-Dias P., Schiavo G., Steiner R. A., Dodding M. P. and Holzbaur E. L. F. (2016). The dynamic localization of cytoplasmic dynein in neurons is driven by kinesin-1. Neuron 90, 1000-1015. 10.1016/j.neuron.2016.04.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitalis T., Lainé J., Simon A., Roland A., Leterrier C. and Lenkei Z. (2008). The type 1 cannabinoid receptor is highly expressed in embryonic cortical projection neurons and negatively regulates neurite growth in vitro. Eur. J. Neurosci. 28, 1705-1718. 10.1111/j.1460-9568.2008.06484.x [DOI] [PubMed] [Google Scholar]
- Watt D., Dixit R. and Cavalli V. (2015). JIP3 activates kinesin-1 motility to promote axon elongation. J. Biol. Chem. 290, 15512-15525. 10.1074/jbc.M115.651885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickert M., Hildick K. L., Baillie G. L., Jelinek R., Aparisi Rey A., Monory K., Schneider M., Ross R. A., Henley J. M. and Lutz B. (2018). The F238L point mutation in the cannabinoid type 1 receptor enhances basal endocytosis via lipid rafts. Front. Mol. Neurosci. 11, 230 10.3389/fnmol.2018.00230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen M. H., Ba W., Wissink-Lindhout W. M., de Brouwer A. P. M., Haas S. A., Bienek M., Hu H., Vissers L. E. L. M., van Bokhoven H., Kalscheuer V. et al. (2014). Involvement of the kinesin family members KIF4A and KIF5C in intellectual disability and synaptic function. J. Med. Genet. 51, 487-494. 10.1136/jmedgenet-2013-102182 [DOI] [PubMed] [Google Scholar]
- Winckler B. and Mellman I. (2010). Trafficking guidance receptors. Cold Spring Harb. Perspect. Biol. 2, a001826 10.1101/cshperspect.a001826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.-S., Zhu J., Wager-Miller J., Wang S., O'Leary D., Monory K., Lutz B., Mackie K. and Lu H.-C. (2010). Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur. J. Neurosci. 32, 693-706. 10.1111/j.1460-9568.2010.07337.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarini G., D'Atri I., Cottone E., Mackie K., Shainer I., Gothilf Y., Provero P., Bovolin P. and Merlo G. R. (2020). Interference with the cannabinoid receptor CB1R results in miswiring of GnRH3 and AgRP1 axons in zebrafish embryos. Int. J. Mol. Sci. 21, 168 10.3390/ijms21010168 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.