

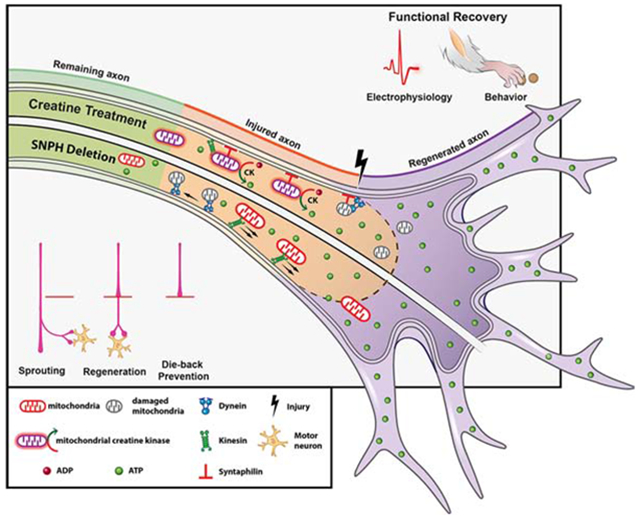
SUMMARY
Axonal regeneration in the central nervous system (CNS) is a highly energy demanding process. Extrinsic insults and intrinsic restrictions lead to an energy crisis in injured axons, raising the question of whether recovering energy deficits facilitates regeneration. Here, we reveal that enhancing axonal mitochondrial transport by deleting syntaphilin (Snph) recovers injury-induced mitochondrial depolarization. Using three CNS injury mouse models, we demonstrate that Snph−/− mice display enhanced corticospinal tract (CST) regeneration passing through a spinal cord lesion, accelerated regrowth of monoaminergic axons across a transection gap, and increased compensatory sprouting of uninjured CST. Notably, regenerated CST axons form functional synapses and promote motor functional recovery. Administration of the bioenergetic compound creatine boosts CST regenerative capacity in Snph−/− mice. Our study provides mechanistic insights into intrinsic regeneration failure in CNS and suggests that enhancing mitochondrial transport and cellular energetics is a promising strategy to promote regeneration and functional restoration after CNS injuries.
Keywords: CNS injury, axon regeneration, axonal transport, energy metabolism, energy deficits, mitochondria, syntaphilin, creatine, spinal cord injury
Graphical Abstract
eTOC:
Using three CNS injury mouse models, Han et al reveal new mechanistic insights into intrinsic regenerative failure of axons in the CNS and demonstrate that enhancing mitochondrial transport and energetic metabolism represents a promising therapeutic direction to stimulate axonal regeneration and functional recovery after spinal cord injury.
INTRODUCTION
Mature central nervous system (CNS) neurons typically fail to regenerate after injury, leading to permanent neurological impairments. Prior studies have elucidated genetic programs, signaling mechanisms, and extracellular inhibitory factors affecting axonal regeneration (Bradbury and McMahon, 2006; Case and Tessier-Lavigne, 2005; Filbin, 2006; Harel and Strittmatter, 2006; Kaplan et al., 2015; Onishi et al., 2014; Silver and Miller, 2004; Yiu and He, 2006). These studies suggest that the limited regrowth capacity of mature CNS is due to an intrinsic decline of permissive conditions. While stimulating intrinsic growth potential can promote axonal regeneration (Anderson et al., 2018; Liu et al., 2010; Liu et al., 2017; Moore et al., 2009), the underlying mechanisms accounting for regeneration failure in spinal cord injury (SCI) models remain largely unknown (Sofroniew, 2018).
For successful regeneration, injured axons must reseal injured terminals, reconstruct cytoskeleton, synthesize and transport building materials, assemble axon components, and form growth cones (Bradke et al., 2012; He and Jin, 2016; Lu et al., 2014). All of these events require energy in the form of adenosine triphosphate (ATP), most of which is supplied by mitochondria in neurons. Given their polarized structures and extended axons, neurons face the exceptional challenge of maintaining mitochondrial distribution and energy homeostasis in distal axons and growth cones (Sheng, 2014; Sheng and Cai, 2012). Thus, activating mitochondrial transport in injured axons is critical for boosting regeneration (Cartoni et al., 2016; Han et al., 2016). We previously revealed that syntaphilin (Snph), a static anchor protein, holds axonal mitochondria stationary via microtubule interactions (Chen and Sheng, 2013; Kang et al., 2008). Snph expression is robustly increased in mature neurons, thereby holding the majority of axonal mitochondria stationary in mature CNS (Lewis et al., 2016; Sun et al., 2013; Takihara et al., 2015; Vagnoni and Bullock, 2018; Zhou et al., 2016a). Deleting Snph in mouse robustly increases axonal mitochondrial motility both in vitro and in vivo (Zhou et al., 2016a). Mitochondria anchored in distal axons provide local energy, thus mitochondrial damage due to axon injury (Cavallucci et al., 2014; O’Donnell et al., 2013) leads to insufficient ATP supply. Combined with the increased energy demand of regeneration, injured axons face a net energy deficit. We hypothesize that recovery of local energy supply may be critical to support regeneration of injured long-projection axons, such as the corticospinal tract (CST). Enhancing axonal mitochondrial transport by turning off Snph-mediated anchoring would help to remove damaged mitochondria and replenish healthy ones in injured axons, thus meeting heightened energy demand during regeneration. Testing this hypothesis in in vivo SCI models will conceptually advance our knowledge regarding the effects of local energetics on axonal regeneration, synapse reconnection, and motor function restoration.
In this study, we took advantage of three murine CNS injury models to study axon regeneration in Snph−/− mice, in which axonal mitochondrial transport is robustly increased. First, we used a 5th cervical (C5) dorsal hemisection (DH) model to study CST regeneration. Second, we used a unilateral pyramidotomy model to transect one side of the CST at the medullary pyramid to assess sprouting of intact CST axons across the midline to innervate the denervated side of the spinal cord. Lastly, we performed a thoracic 8 (T8) spinal complete transection to investigate regeneration of both CST and monoaminergic pathways. We demonstrated that regenerated CST axons in Snph−/− mice form functional synapses, transmit electrophysiological signals, and promote motor function recovery after SCI. We provided in vivo evidence that enhancing mitochondrial transport recovers axonal mitochondrial integrity by delivering healthy mitochondria from neurons in the motor cortex to the injured CST axons and by removing damaged mitochondria. Our hypothesis of “injury-induced energy crisis” was further supported by systemic administration of creatine, an energy facilitator. Our study establishes that injury-induced “energy crisis” contributes to CNS regeneration failure after SCI. Recovering local energy by either enhancing mitochondrial transport or by increasing energy metabolism promotes axonal sprouting and regeneration after SCI. Therefore, our study suggests a promising new cellular target for stimulating regeneration and functional recovery after CNS injuries, particularly in the spinal cord.
RESULTS
Enhancing Axonal Mitochondrial Transport in Snph−/− Mice Promotes CST Axonal Regeneration after a C5 DH
Using adult Snph−/− mice, we investigated whether enhancing axonal mitochondrial transport promotes CST regeneration after SCI. First, we performed a spinal DH at the 5th cervical vertebra (C5) level to bilaterally eliminate descending CST projections. We injected an anterograde tracer, biotinylated dextran amine (BDA), into the sensorimotor cortex to trace descending CST axons 8 weeks post injury (wpi) (Figures 1A and 1B). Since axonal retraction or dieback is typical for injured CST axons (Liu et al., 2008; Pallini et al., 1988), we assessed CST axons rostral to the lesion. BDA-labeled CST axons in wild-type (WT) mice died back dramatically from the rostral lesion border, defined by astrocytic gliosis (GFAP-labeled), for several hundred micrometers (Figures 1C and 1D). In contrast, BDA-labeled CST axons in Snph−/− mice remained at the rostral lesion border. Quantitative analysis of CST axon number index revealed a significant increase in CST axon number in Snph−/− mice within −0.5 to 0 mm of the rostral lesion border compared to WT mice (Figures 1E and 1F). However, die-back in WT mice was more evident in parasagittal sections of CST main tracts, but less obvious in sections lateral to the main CST, where both WT and Snph−/− mice showed little dieback from the rostral lesion border (Figures 1G–1J).
Figure 1. Snph−/− Mice Display Enhanced CST Axonal Regeneration after the C5 DH.
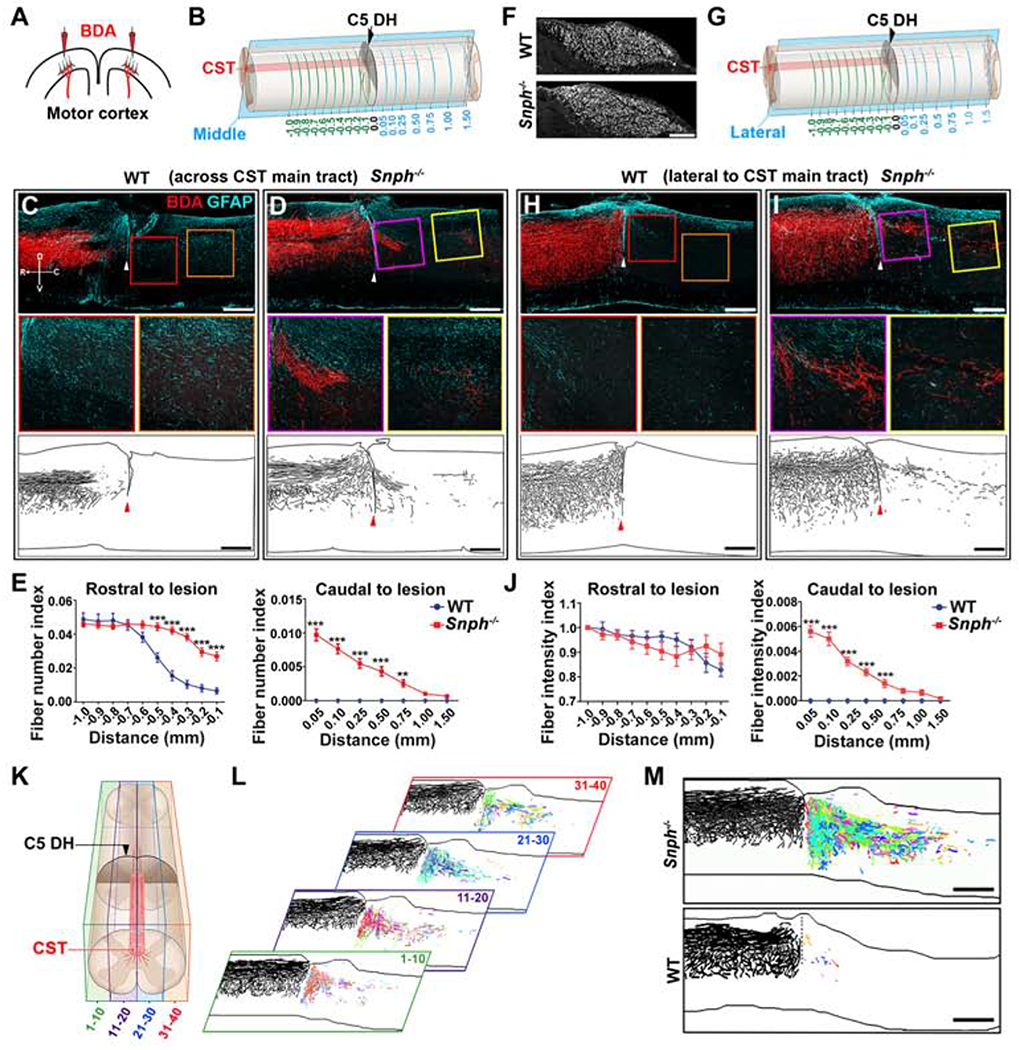
(A) Schematic diagram of BDA injections into bilateral sensorimotor cortices to anterogradely label the CST axons.
(B) Schematic diagram of a C5 DH SCI model. CST axonal density was measured at various distances rostral (green lines, up to −1 mm) and caudal (blue lines, up to 1.5 mm) to the C5 DH (0 mm). The parasagittal rectangle indicates a cut through the BDA-labelled CST main tract.
(C, D) BDA labeling of the CST main tract in the dorsal column after the C5 DH in WT and Snph−/− mice, 8 wpi. Arrowheads indicate the lesion sites. Middle panels: high magnifications of boxed areas. Bottom panels: Imaris reconstruction images from WT and Snph−/− mice. Compass in C shows the dorsal (D), ventral (V), rostral (R), and caudal (C) orientation.
(E) Quantification of CST axon (fiber) number index at various distances rostral (left) and caudal (right) to the C5 DH.
(F) BDA-labeled CST axons through transverse sections of the medullary pyramids.
(G) Schematic showing BDA-labeled axons lateral to CST main tract.
(H, I) Lateral CST distribution after the C5 DH (arrowheads). Middle panels: high magnifications of boxed areas showing BDA-labeled regenerated axons caudal to the injury 8 wpi. Bottom panels: Imaris reconstruction images from WT and Snph−/− mice.
(J) Quantitative analysis of BDA-labeled CST fiber intensity index at various distances rostral (left) and caudal (right) to the lesion.
(K, L) Schematic showing a spinal cord segment divided into four longitudinal blocks, each containing 10 sections (K). BDA-labeled CST axons in each section were traced and color-coded using Imaris (L). Each of the four color-coded blocks on the right was generated by overlaying 10 consecutive sections. Each color represents a trace of BDA-labeled axons caudal to the lesion in a single section of one block.
(M) Reconstruction of a stack of all color-coded images shows robust CST regeneration across and beyond the lesion in a Snph−/− mouse at 8 wpi.
Data were presented as mean ± sem. n = 10-11 mice/group. Two-way ANOVA with Bonferroni post hoc test. **P < 0.01, ***P < 0.001. Scale bars: 100 μm (F), 400 μm (C, D, H, I), and 500 μm (M). (Also see Figure S1)
We next assessed CST axonal regeneration caudal to the lesion. A complete C5 DH produced by a vibraknife device stereotaxically transected both the dorsal/main tract and lateral tract of the CST; any axons passing through the lesion were considered as regeneration. WT mice displayed no CST axon regeneration through or beyond the lesion in all parasagittal sections (Figure 1C). In contrast, Snph−/− mice exhibited robust regeneration, with many BDA-labeled CST axons growing through the lesion and extending to the caudal spinal cord (Figure 1D). In all 11 injured Snph−/− mice, CST axons extended caudally a great distance beyond the lesion, with the longest regenerating axons tapering off around 1.5 mm (Figure 1E). Despite a large number of CST axons remaining at the rostral lesion border in lateral parasagittal sections in WT mice, no BDA-labeled CST axons were found past the lesion gap (Figures 1H and 1J). However, CST axons in Snph−/− mice grew through the lesion and extended into the caudal spinal cord (Figures 1I and 1J), consistent with the CST axonal regeneration in medial sagittal sections. These results indicate that increasing mitochondrial transport in Snph−/− mice facilitates CST axonal regeneration through and beyond the lesion in SCI.
To achieve a 3-D view of the entire spinal cord segment, all serial sections were reconstructed and CST axon regeneration was examined in a double-blinded manner by an independent analyst (Figure 1K). From the stacked sagittal sections, Snph−/− mice exhibited robust axon regeneration caudal to the lesion; with the longest axons gradually tapering off ~2 mm from the lesion (Figures 1L and 1M). In contrast, WT mice showed little axonal regrowth beyond the lesion. In addition to examining astrocytic response, several measures were taken to ensure that CST axons caudal to the lesion in Snph−/− mice were regenerated as opposed to spared (uncut). First, most regenerating CST axons grew directly through the lesion, extending into the caudal spinal cord and many showed ectopic trajectories, suggesting that these are not spared axons. Second, reconstruction revealed that CST axons caudal to the lesion ended around 1.5 - 2.0 mm in the caudal spinal cord (Figures 1L and 1M), while spared axons would be expected to continue much further. Third, the CST main tract was located in the ventral portion of the dorsal funiculus in cross sections rostral to the lesion, but not in sections 2.0 mm caudal to the lesion in both WT and Snph−/− mice (Figures S1A–S1D), confirming complete transection at the C5 level. Importantly, a few BDA-labeled axons were found in the gray matter of Snph−/− mice, but not in WT mice (Figures S1B and S1D). Together, these data suggest that CST axons regenerated through and beyond the lesion in Snph−/− mice. Thus, our first SCI model demonstrates that enhancing axonal mitochondrial transport facilitates robust CNS axonal regeneration after SCI.
Regenerated CST Axons Reconstitute Cortico-Motoneuronal Circuits
SCI disconnects CST axons from their postsynaptic targets and disrupts the original cortico-motoneuronal circuit. Thus, motor function restoration after SCI is dependent on CST axon regeneration and subsequent reestablishment of cortico-motoneuronal circuitry. Robust axonal regeneration observed in Snph−/− mice prompted us to investigate the capacity of regenerated CST axons to form functional synapses with their targets. We first examined whether regenerated CST axons are incorporated into the caudal spinal circuits in Snph−/− mice. BDA was injected into the motor cortex to anterogradely label the CST axons and cholera toxin B subunit (CTB) was separately injected into the biceps muscle of the forelimb to retrogradely label cervical motoneurons 8 wpi (Figure 2A). A parasagittal section shows that BDA-labeled regenerating CST axons passed through and beyond the C5 DH (Figure 2B). In the gray matter of the caudal spinal cord, where CTB-labeled motoneurons and their dendrites reside, many regenerating axons colocalized with the presynaptic maker vGlut1, suggesting formation of new glutamatergic presynaptic terminals (Figures 2C and 2D). Importantly, some BDA-labeled regenerating axons with vGlut1 puncta were in close apposition with CTB-labeled motoneurons. To further validate synaptic formation in Snph−/− mice, the spinal cord segment located 1.5-mm caudal to the lesion was sectioned into 100 μm-thick cross-sections. Following the histochemical reaction for BDA and examination by electron microscopy (EM), BDA-labeled CST axons were shown to form morphologically mature presynaptic terminals with synaptic vesicle clusters at the active zone and in close connection with the electron-dense postsynaptic density (Figure 2E). Mitochondria were enriched within BDA-labeled regenerated axons caudal to the injury in Snph−/− mice; some axons were wrapped by new myelin. Altogether, both light and EM images consistently showed re-established cortico-spinal motor connections in the spinal cord caudal to the injury in Snph−/− mice.
Figure 2. Regenerated CST Axons Form Functional Cortico-Motoneuron Connections.
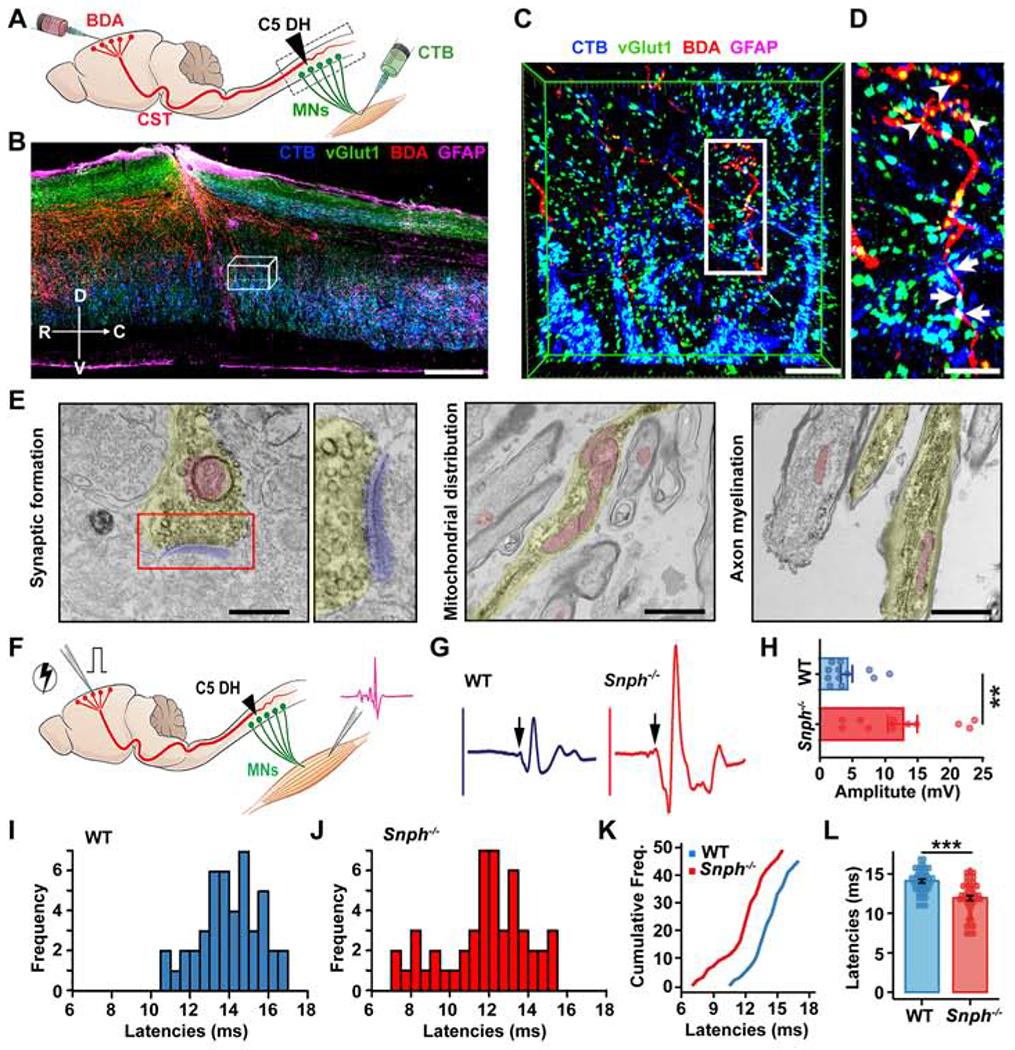
(A) Illustration showing bilateral injections of BDA into the motor cortex to label CST axons and CTB into the bicep’s muscles to label cervical motoneurons (MNs) after the C5 DH.
(B-D) BDA-labeled regenerated CST axons (red) formed synapse-like connections (vGlut1, green) with CTB retrogradely labeled MNs (blue) caudal to the C5 DH (GFAP-IR, magenta) in Snph−/− mice. (C) 3D magnification of boxed area in B. (D) High magnification of boxed area in C. Arrowheads (in C) indicate vGlut1 distribution along BDA-labeled CST axons. Arrows indicate vGlut1 co-labeled with both BDA-labeled CST axons and CTB-labeled MNs. Compass: D, dorsal; V, ventral; R, rostral; C, caudal.
(E) Electron micrographs showing synaptic formation of regenerated CST axons in caudal spinal cords. Left: BDA-labeled CST axon (yellow colored) caudal to the lesion site that formed a presynaptic terminal containing synaptic vesicle clusters in the active zone and contacts on a postsynaptic density (blue colored). Middle: mitochondria were enriched and clustered within BDA-labeled newly growing axon tip in sections caudal to the injury. Right: BDA-labeled myelinated CST axons below the lesion.
(F-L) Schematic diagram (F) and electromyography (EMG) analysis (F-L) showing signals recorded from forelimb biceps in response to motor cortex single-pulse stimulation. Sample EMG traces (G) from WT (blue) and Snph−/− (red) mice were recorded in response to single-pulse stimulations after the C5 DH. Arrows indicate evoked-potential onset. (H) Average peak-to-peak EMG amplitude in WT (n = 12) and Snph−/− (n = 11) mice, P < 0.01. (I, J) Frequency distributions of EMG latencies from WT and Snph−/− mice, respectively. (K) Cumulative frequency distribution plot for WT (blue) and Snph−/− mice (red). (L) EMG latencies in WT (14.08 ± 0.23, n = 45 stimulation sites from 11 animals) and Snph−/− mice (11.95 ± 0.31, n = 49 stimulation sites from 10 animals).
Data were presented as mean ± sem. Unpaired two-tailed Student’s t-test. **P < 0.01, ***P < 0.001). Scale bars: 400 μm (B), 50 μm (C), 25 μm (D), 400 nm (E, left), 1200 nm, (E, middle and right). (Also see Figure S1).
We next examined whether regenerated CST axons could functionally relay cortical commands to forelimb motor neurons by detecting the electromyography (EMG) signals recorded from forelimb biceps in response to single-pulse stimulation in the motor cortex (Figure 2F). Electrical stimulation elicited motor evoked potentials with shorter latency (P < 0.001) and larger amplitude (P < 0.01) in forelimb muscles of Snph−/− mice than those recorded in WT mice after the C5 DH (Figures 2G–2L). To validate regeneration-driven synaptic transmission, animals from both groups were subjected to a C5 re-lesion (Figures S1E and S1N). The increased EMG responses in Snph−/− mice vanished after the re-lesion; instead, much smaller amplitudes similar to those seen in WT mice were detected after the re-lesion (Figures S1F–S1K). These data indicate that higher EMG response in Snph−/− mice after SCI was dependent on CST axon regeneration. Notably, rather than completely abolishing the EMG responses, both WT and Snph−/− mice exhibited similarly small, yet detectable, reflex responses to motor cortex stimulation after the C5 re-lesion, a phenomenon previously reported (Azim and Alstermark, 2015; Wang et al., 2017b). We hypothesized that the cortico-motoneuronal circuit may also involve a subset of motoneurons at C2-4 above the C5 DH, thus contributing to forelimb motor control. To test this, we performed a second C2 DH injury in the C5 DH injured mice to eliminate the CST projection at the C2 level (Figures S1L and S1N). As expected, acute C2 DH almost completely abolished EMG activity in both WT and Snph−/− animals (Figure S1M). These results consistently suggest that regenerated CST axons in Snph−/− mice reestablish cortico-motoneuron connections and thereby conduct synaptic transmission caudal to the injury to restore forelimb connectivity.
Enhancing Mitochondrial Transport Facilitates Motor Functional Recovery after SCI
To assess the contribution of regenerated CST axons to motor function restoration in Snph−/− mice, we performed three behavioral tests, including the footprint, grid walk, and sticker removal assessments after the C5 DH. To our surprise, enhanced CST axon regeneration in Snph−/− mice was not associated with locomotion recovery in footprint analysis. We did not detect any difference in stride length or width between WT and Snph−/− mice at any time point after injury (Figures S2A–S2C). In the grid walk test, which assesses descending fine motor control (Metz et al., 2000), there was no difference between WT and Snph−/− mice up to 8 wpi (Figure S2D). Sensitivity and dexterity were assessed using the sticker removal test (Bouet et al., 2009). Both WT and Snph−/− mice showed a similarly increased time-to-removal of the sticker after injury at all time points examined (Figure S2E).
Since these behavioral tests failed to reveal any motor functional improvements in Snph−/− mice after SCI, we questioned whether these assessments lacked the necessary sensitivity to detect long-term motor recovery or whether the regenerated CST axons in Snph−/− mice were insufficient to support functional recovery. To address this, we performed the single-pellet retrieval task, a more stringent behavioral test for higher discrimination of forelimb dexterity in mice with CST lesion (Hurd et al., 2013; Whishaw et al., 1993). We delineated two categories, success and fail (including dropping, missing and knocking off) to assess task scores (Figure S2F). To evaluate this behavior further, we analyzed paw movement at each time-interval post-injury using manual kinematic tracing, where successful trials were colored and unsuccessful attempts were grey Figures 3A–3D and S2G). Before the C5 DH, both WT and Snph−/− mice displayed comparable high success and similar paw movement at baseline (Figures 3A, 3B, 3E and 3F), indicating no dexterity deficits in mice with Snph deletion. In contrast, success rates dramatically dropped to ~ 0% in both WT and Snph−/− mice at 2 wpi (Figures 3I and S2G). However, Snph−/− mice exhibited significantly better skilled motor improvement, with more accurate retrieval and higher success rate at 6 (P < 0.05) and 8 wpi (P < 0.01) compared to WT mice. The Snph−/− mice displayed a more focused paw position in close proximity to the pellet when compared with that of WT mice at 6 and 8 wpi (Figures 3G, 3H and S2H); and grasped the pellet significantly faster (P < 0.05) than WT mice at 2, 4 and 6 wpi (Figure 3J). Furthermore, the success rate in Snph−/− mice nicely correlated with the regenerated CST number index caudal to the lesion (P < 0.01) (Figure 3K), indicating that CST axon regeneration contributes to the improvement of fine forelimb motor functions after SCI.
Figure 3. Regenerated CST Axons Contribute to Recovery in Manual Dexterity.
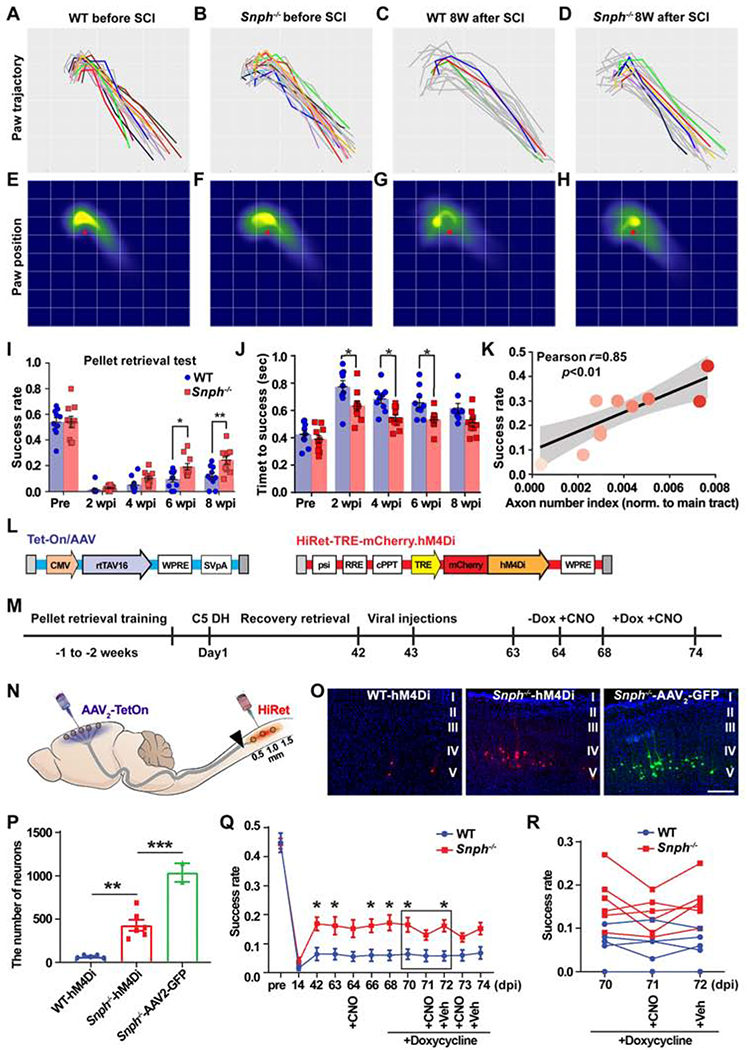
(A-D) Paw motion trajectories in the reaching phase of pellet retrieval test in WT and Snph−/− mice before and 8 weeks after the C5 DH (8 wpi). Successful reaches are colored and unsuccessful attempts are grey.
(E-H) Heat maps of paw spatial positions in the grasping phase relative to the food pellet (red spot) in WT and Snph−/− mice before and at 8 wpi.
(I-K) Analysis of the success rate of skilled reaching (I) and time to successful retrieval (J) shows that Snph−/− mice outperformed WT mice in the dexterity task. (K) Correlation between success rate and BDA-labeled CST axon number index.
(L-N) Schematics showing dual viral vectors (L) and experimental design (M) of DREADDs-mediated silencing of CST neurons (N).
(O) Labeling of corticospinal neurons in the motor cortex that received dual viral or AAV2-GFP infection.
(P) Total number of dual-virus infected corticospinal neurons in WT and Snph−/− mice, as well as corticospinal neurons infected by AAV2-GFP virus in Snph−/− mice.
(Q, R) Changes of forelimb dexterity in pellet retrieval test via administration of CNO either before or after induction of hM4Di expression with doxycycline treatment.
Data were presented as mean ± sem. n = 10 mice/group in pellet retrieval test (A-K); n = 5-6 mice/group in DREADDs study (N-R). Two-way ANOVA with Bonferroni post hoc test. * P < 0.05, ** P < 0.01, *** P < 0.001. Scale bar: 200 μm (O). (Also see Figure S2).
To test if regenerated CST axons account for forelimb dexterity recovery, we performed a pathway-selective silencing study via an inhibitory designer receptor exclusively activated by designer drugs (DREADDs) (Sheikh et al., 2018), which selectively inhibits neurotransmission in the corticospinal pathway. To target regenerated CST axons after the C5 DH, HiRet-TRE-hM4Di-mCherry was injected into the caudal spinal cord (within 1.5 mm caudal to the injury), where most regenerated CST axons were located (Figures 3L–3N). Meanwhile, Tet-On/AAV was stereotaxically injected into bilateral motor cortices where the corticospinal neurons reside. To induce expression of hM4Di, doxycycline was administered for 7 days and Clozapine-n-oxide (CNO) was injected on day 4 and 6 after the doxycycline administration. The Snph−/− mice expressed hM4Di in a greater number of corticospinal neurons compared to WT mice (P < 0.01) (Figures 3O and 3P) and showed a significant decrease in the success rate of recovered dexterity in the pellet retrieval test after administration of CNO (P < 0.05) (Figures 3Q and 3R). These results clearly indicate that regeneration of CST axons and their synaptic formation in the spinal cord contribute to dexterous improvement in Snph−/− mice after the C5 DH.
Enhancing Mitochondrial Transport Facilitates the CST Sprouting Following a Unilateral Pyramidotomy
In addition to axonal regeneration from the severed end, axonal growth could also be stimulated from fiber tracts that are not damaged but rather positioned adjacent or parallel to an injury (Cafferty et al., 2008; Tuszynski and Steward, 2012). We hypothesized that enhanced mitochondrial transport in Snph−/− mice would stimulate this compensatory sprouting response. To test this, we utilized a unilateral pyramidotomy model in which CST axons were severed unilaterally in the medulla of the brainstem, leading to complete loss of CST innervation on the contralesional spinal cord (Figures S3A–S3C). Pyramidotomy in WT mice caused no significant change in the termination pattern of BDA-labeled CST collaterals, with most axons remaining ipsilateral to the lesion at cervical, thoracic and lumbar spinal cord levels (Figures 4A–4C). In contrast, pyramidotomy in Snph−/− mice substantially enhanced CST sprouting across the midline and into the denervated side at cervical, thoracic, and lumbar spinal cord levels (Figures 4D–4H). This difference of injury-induced sprouting between WT and Snph−/− mice was appreciated by color-coded heatmaps representing the density of CST sprouting in the contralaterally denervated spinal hemi-cord (Figures 4D–4F).
Figure 4. Snph−/− Mice Display Enhanced Compensatory CST Spouting after Unilateral Pyramidotomy.
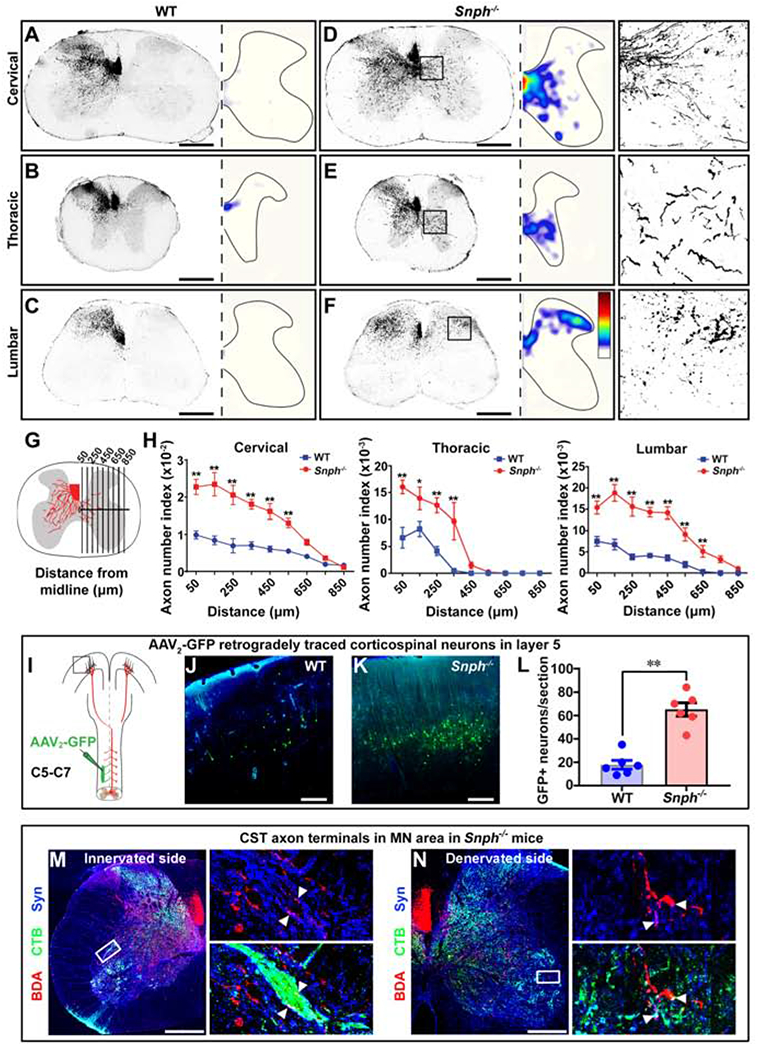
(A-F) Cross-sectional images showing midline-crossing of BDA-labeled CST axons at cervical, thoracic, and lumbar levels following unilateral (left) pyramidotomy. The distribution of axonal sprouting into the denervated side was converted to heatmaps; red represents the highest numbers of axon pixels, blue represents the lowest, and white represents background (D-F). Right panels in D-F: details of boxed areas showing BDA-labeled CST terminal sprouting at denervated regions of the cervical, thoracic, and lumbar cords, respectively, in a Snph−/− mouse.
(G, H) Schematic (G) and analysis (H) showing BDA-labeled midline-crossing CST axons.
(I) Unilateral injection of AAV2-GFP into the denervated (right) side of the C5-C7 spinal intermediate gray matter to retrogradely label corresponding corticospinal neurons in the motor cortex.
(J-L) Images of boxed area in I (J, K) and analysis (L) showing GFP-labeled CST neurons in layer V of WT and Snph−/− mice.
(M, N) Close apposition of BDA-labeled CST (red), synaptophysin (Syn)-labeled presynaptic terminals (blue), and CTB-labeled MNs (green) in both innervated (M) and denervated (N) spinal cord in Snph−/− mice. Left panels: high magnifications of boxed areas in M and N, respectively. Arrowheads indicate triple-positive appositions.
Data were presented as mean ± sem. n = 6 mice/group (H, L). Unpaired two-tailed Student’s t-test. * P < 0.05, ** P < 0.01. Scale bars: 500 μm (A-F), 250 μm (J, K), 400 μm (M, N). (Also see Figure S3)
This striking difference was supported by the evidence that more GFP-expressing corticospinal neurons were identified in contralesional motor cortex in Snph−/− mice than those in WT mice after injecting a small amount of the retrograde virus AAV2-GFP into the denervated C5-C6 spinal hemi-cord (Figures 4I–4L and S3D). Furthermore, the trans-midline axons colocalized with synaptophysin puncta in Snph−/− mice were in close contact with the dendrites of CTB-labeled motoneurons, similar to the synaptic apposition observed in innervated side of spinal cord (Figures 4M and 4N). Notably, intact WT and Snph−/− mice showed comparable minimum midline-crossing CST termination throughout the whole cervical, thoracic and lumbar spinal cord (Figure S3E), suggesting that genetically deleting Snph did not affect the normal CST projection in the spinal cord. Taken together, these data indicate that enhancing axonal mitochondrial transport by deleting Snph in mice is sufficient to induce robust CST sprouting to the denervated contralesional hemi-cord after a unilateral pyramidotomy.
Enhancing Mitochondrial Transport Stimulates Monoaminergic Axon regeneration in a T8 Complete Transection Model
The majority of human spinal cord injuries are severe, resulting in complete and permanent loss of function below the lesion (Fawcett et al., 2006). Although it is a clinically important problem, CST regeneration following a complete SCI remains unattainable in the field. Thus, a complete SCI animal model would mimic the most severe SCI in human (Steward and Willenberg, 2017). Given the robust CST regeneration in Snph−/− mice, we next explored whether enhancing mitochondrial transport could promote CST axon regeneration after complete transection of the spinal cord at the 8th thoracic vertebral level (T8). In both WT and Snph−/− mice, a large GFAP-negative lesion cavity was bordered rostrally and caudally by GFAP-immunoreactive (IR) astrogliosis. CST axons remained in the rostral spinal cord without penetration into the lesion gap in both genotypes (Figures S4A and S4B). However, CST axons dramatically died back from the rostral lesion border in all WT mice examined. Notably, such typical retraction did not occur in Snph−/− mice. Instead, BDA-labeled CST axon terminals in Snph−/− mice were sustained at the rostral lesion border. Within 0.5 mm of the rostral lesion border, CST fiber intensity index was significantly higher in Snph−/− mice as compared with WT mice (Figure S4C).
While the C5 DH injury in Snph−/− mice elicited robust CST axon regeneration across the lesion gap (Figure 1), a complete T8 transection was not sufficient to induce CST regeneration beyond the injury gap despite the prevention of axonal dieback (Figure S4). One difference between the two injury models is that GFAP-labeled astrocytes in the C5 DH model were capable of sealing off the injured tissue to regain tissue integrity (Anderson et al., 2016; Dias et al., 2018; Faulkner et al., 2004; Liu et al., 2010). Such tissue sealing may be necessary to permit CNS axonal regrowth after injury. Indeed, in the C5 DH, regenerated CST axons aligned with GFAP-labeled astrocytic processes and extended together through and beyond the lesion site. In contrast, a large lesion gap devoid of host tissue and astrocytes was found after the T8 complete transection, thus providing no permissive substrates for CST axons to regenerate via the injury gap. Thus, we speculate that enhanced axonal mitochondrial transport can promote CST regeneration only if the physical lesion gap is bridged with permissive substrates.
We further examined axon regeneration of the monoaminergic pathways after the same T8 complete spinal transection. Descending monoaminergic pathways include the serotonergic (5-HT) immunoreactivity (IR) and dopaminergic (TH IR) tracts, which are known to possess a high growth capacity following injury (Gowrishankar et al., 2015; Saruhashi et al., 1996). We reasoned that by focusing on monoaminergic axons in this injury model we might detect any pro-regenerative effect of enhanced axonal mitochondrial transport (Filli et al., 2011; Lee et al., 2010). Indeed, in Snph−/− mice, both 5-HT IR and TH IR axons grew substantially into the large lesion gap, with some extending far into the caudal spinal cord after the T8 complete transection (Figures 5A–5E). Despite no axonal retraction, 5-HT IR and TH IR axons in WT mice often stopped in the rostral penumbra of the lesion and did not penetrate deeply into the lesion gap nor regenerated beyond the lesion. Heatmaps revealed an enhanced density of 5-HT IR and TH IR axons caudal to the lesion in Snph−/− mice as compared with WT mice (Figure 5F). Remarkably, Snph−/− mice exhibited significantly higher BMS scores (Basso et al., 2006) compared to WT mice starting from 5 wpi and continuing up to 8 wpi, despite their failure to regain locomotor coordination, which requires a minimun BMS score of 5 (Figure 5G). 5-HT and TH fiber intensities were positively correlated with BMS scores in Snph−/− mice after the T8 transection (r=0.73, r=0.66, respectively) (Figure 5H).
Figure 5. Monoaminergic Axon Regeneration after T8 Complete Transection in Snph−/− Mice.
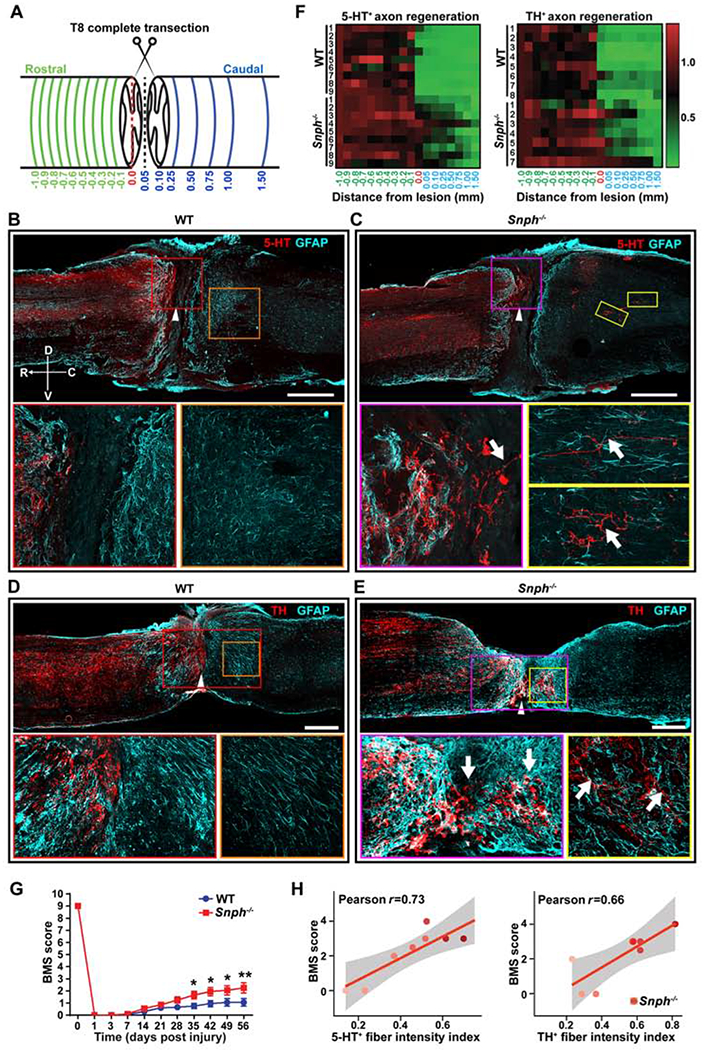
(A) Diagram illustrates a complete thoracic (T8) transection model. Green lines indicate various cross-sections spaced 0.1 mm apart up to 1 mm rostral to the lesion border; blue lines indicate cross-sections at various distances caudal to the lesion border up to 1.5 mm caudal to the rostral lesion border. The red-dashed line indicates the edge of the rostral lesion border, which is set to 0 mm for both rostral and caudal directions.
(B, C) Images of 5-HT-immunoreactive (IR) serotonergic axons (red) costained for GFAP (cyan) in sagittal sections following a complete T8 transection in WT (B) and Snph−/− mice (C). Arrowheads indicate the lesion center, which was magnified in lower left panels. Lower right panels: high magnifications of boxed areas in B or C respectively. Compass: D, dorsal; V, ventral; R, rostral; C, caudal.
(D, E) TH-IR dopaminergic axons (red) costained for GFAP (cyan) in sagittal sections following T8 transection in WT (D) and Snph−/− mice (E). Arrowheads indicate the lesion centers, which were magnified in lower left panels, respectively. Lower right panels: high magnifications of boxed areas of the caudal spinal cord in D or E, respectively.
(F) Color-coded density heatmap indicates the mean intensity of 5-HT-IR and TH-IR fibers after T8 transection. The horizontal axis indicates the distance rostral and caudal to the rostral lesion border (0.0). The vertical axis indicates individual mice in WT or Snph−/− group, respectively. n = 7-9 mice/group.
(G, H) BMS scores in Snph−/− mice were significantly higher than WT mice from 35 to 56 days post-injury (*P < 0.05; ** P < 0.01) (G) and correlated to 5-HT and TH fiber intensity (r = 0.73, r = 0.06, respectively) (H). Scale bars: 500 μm (B-E). (Also see Figure S4).
We next determined the substrates through which monoaminergic axons regrow after injury. Unlike GFAP-immunoreactive astrocytes (Figures S4A and S4B), the substrate molecules laminin, fibronectin, and collagen substantially deposited in the non-neuronal lesion core and intertwined with regenerating serotoninergic axons (Figure S4D), suggesting a permissive contribution of these extracellular components to neural plasticity and regeneration.
Enhancing Mitochondrial Transport Recovers Injury-Insulted Mitochondria Damage
In mature CNS axons, the majority of mitochondria remain stationary (Lewis et al., 2016; Smit-Rigter et al., 2016; Sun et al., 2013). Axonal injury is an acute stress that induces mitochondrial dysfunction (Cavallucci et al., 2014). We hypothesized that enhancing mitochondrial transport would effectively remove damaged mitochondria from and replenish healthy ones to injured axons to ensure sufficient ATP production in order to meet increased energy demand during regeneration. To test this, we measured axonal mitochondrial membrane potential (Δψm) in CST axons before and after the C5 DH injury. We applied a fixable Δψm-dependent dye, MitoTraker Orange CMTMRos (De Vos et al., 2007; Kang et al., 2008; Lin et al., 2017; Zhou et al., 2016a), to lesioned regions and monitored fluorescence intensity of CMTMRos in the dorsal funiculus, where the main CST is located rostral to the lesion. At 3- and 6-hour post-injury, we observed a dramatic reduction in CMTMRos intensity rostral to the lesion as compared to non-injured counterparts in both WT and Snph−/− mice, indicating that SCI is an acute insult that damages local mitochondria (Figure 6A). Snph−/− mice exhibited progressive Δψm recovery at 12 and 24 hours and up to 7 days after injury (Figure 6B), indicating that injury-induced mitochondrial damage can be effectively reversed by enhancing axonal mitochondrial transport in Snph−/− mice. As a control for total mitochondrial density, mitochondrial protein TOM20 showed no significant change between WT and Snph−/− mice with or without C5 DH (Figure 6A).
Figure 6. Snph−/− Mice Recover Local CST Axonal Mitochondrial Integrity after SCI.
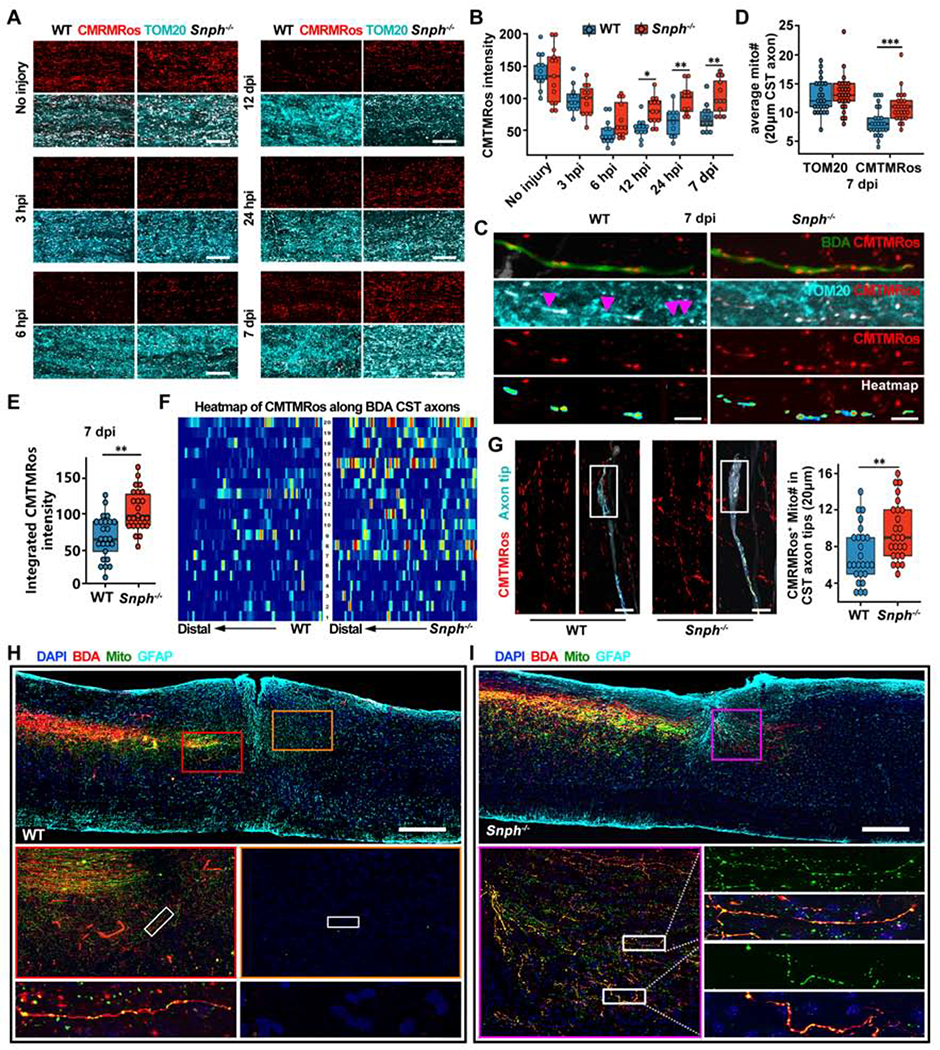
(A, B) Images (A) and analysis (B) of CMTMRos-labeled polarized mitochondria over TOM20-immunostained total mitochondria in the spinal cord dorsal white matter at different times following C5 DH. Data were presented as mean ± sem; n = 3-4 mice/group at each time.
(C) Distribution of polarized mitochondria along individual BDA-labeled CST axons in the rostral lesion site vicinity at 7 days after the C5 DH (7 dpi). Arrowheads represent TOM20-positive but CMTMRos-negative depolarized mitochondria along WT axons, indicating injury-induced damage of mitochondrial integrity. CMTMRos-labeled mitochondria along CST axons were isolated and converted to color-coded heatmaps, in which red and blue indicate the highest and lowest CMTMRos fluorescent intensity, respectively.
(D) Average number of TOM20-labeled or CMTMRos-labeled mitochondria along 20 μm BDA-labeled CST axons rostral to the lesion in WT (blue) and Snph−/− mice (red) at 7 dpi. Data were presented as mean ± sem; n = 25 axons from 4 mice/group.
(E) Analysis of integrated CMTMRos fluorescence intensity within TOM20 masked area along individual BDA-labeled CST axons in WT (blue) and Snph−/− mice (red) at 7 dpi. Data were presented as mean ± sem. n = 25 axons from 4 mice/group.
(F) Pseudo-colored heatmaps represent CMTMRos-labeled mitochondria along BDA-labeled CST axons rostral to the injury in WT and Snph−/− mice at 7 dpi. In heatmaps, red and blue indicate the highest and lowest CMTMRos fluorescent intensity, respectively. n = 20 axons from 4 mice/group.
(G) CMTMRos-labeled mitochondria were distributed within growing tips of BDA-labeled CST axons 7 days after the C5 DH in WT and Snph−/− mice. Boxplot shows the average number of CMTMRos-labeled mitochondria in the distal 20 mm of CST axon tips. n = 25 axons from 4 mice/group.
(H, I) Relative distribution of motor cortex-derived mitochondria, labeled by AAV9-mito-GFP, in BDA-labeled CST axons at 8 weeks following the C5 DH in WT (H) and Snph−/− mice (I). Middle panels (H): magnification of boxed area in the top panel; Bottom panels (H): high magnification of boxed area in middle panels; lower left panel (I): magnification of boxed area in the upper panel, lower right panels (I): high magnification of boxed area in left panels.
Differences were analyzed by two-way ANOVA with Bonferroni post hoc correction (B, D) or unpaired two-tailed Student’s t-test (E, G). *P < 0.05; ** P < 0.01. ***P < 0.001. Scale bars: 20 μm (A), 5 μm (C), 10 μm (G), 500 μm (H, I). (Also see Figure S5).
We next assessed Δψm in single BDA-labeled CST axons rostral to the lesion site at 7 days following the C5 DH. In WT mice, TOM20-positive but CMTMRos-negative dysfunctional mitochondria were observed along BDA-labeled CST axons after injury, while healthy mitochondria co-labeled with TOM20 and CMTMRos were primarly found in Snph−/− injured CST axons (Figure 6C). We found no difference in the average number of TOM20-labeled total mitochondria along BDA-labeled CST axons between WT and Snph−/− mice. However, a significantly larger number of healthy mitochondria labeled with CMTMRos was observed in Snph−/− axons at 7 days (P < 0.001) after injury (Figures 6D and 6E), suggesting that enhancing mitochondrial transport effectively recovers mitochondrial integrity in injured CST axons after SCI.
Mitochondria ATP production depends upon Δψm maintenance and dissipation of Δψm leads to loss of ATP production capacity. We next examined mitochondrial quality and distribution in single CST axons after SCI. CMTMRos-labeled mitochondria were isolated from CST axons and color-coded heatmaps were generated to represent mitochondrial Δψm and their relative distribution in injured axons rostral to the lesion. An analysis of 20 individual axons exhibited higher Δψm intensity distribution within Snph−/− axons when compared within WT axons at 7 days post-injury (Figure 6F). CMTMRos-labeled mitochondria number in the tips of severed CST axons was significantly larger in Snph−/− mice (P < 0.01) at 7 days post-injury (Figure 6G). Some lesioned axon terminals in Snph−/− mice formed a slim growth cone with a filopodia-like structure. In contrast, lesioned axonal terminals in WT mice rarely exhibited such filopodia-like structures. Moreover, at 8 wpi, motor cortex-derived mitochondria, labeled by injection of AAV9-mito-GFP into the motor cortex, were found to be abundantly distributed in BDA-labeled regenerated CST axons caudal to the lesion site in Snph−/− mice (Figures 6H and 6I), indicating effective long-distance delivery of healthy mitochondria from the motor cortex into regenerated CST axons in the spinal cord. In contrast, BDA-labeled CST axons in WT mice were only present in the rostral lesion border where cortex-derived mitochondria were much less abundant at 8 wpi. Mitochondrial fission was also reported at early stages of nerve injury (Cavallucci et al., 2014; Kiryu-Seo et al., 2016). However, there was no significant difference in mitochondrial length distribution between WT and Snph−/− mice with or without the C5 DH injury, although mitochondria became smaller in the injured spinal cord (Figures S5A). This finding suggests that Δψm recovery in Snph−/− mice is not attributed to altered mitochondrial fusion and fission. These data indicate that enhancing mitochondrial transport helps to recover mitochondrial integrity and thus rescue energy deficits, thereby facilitating axon regeneration.
As mitochondrial dysfunction is accompanied by enhanced generation of reactive oxygen species (ROS), we next examined whether enhancing mitochondrial transport affects ROS levels after injury. Given the technical challenge of examining in vivo ROS levels using the redox probe Grx1-roGFP2, which requires assessment of ratiometric integrated intensity (405/488 nm) (Gutscher et al., 2008), we applied microfluidic devices to study injury-triggered mitochondrial ROS response. While both WT and Snph−/− neurons displayed increased redox potential 1 hour after axotomy, enhanced mitochondrial transport in Snph−/− neurons significantly reduced the redox level 5 hours after injury when compared to WT neurons (Figures S5B and S5C). Therefore, enhancing transport also reduces ROS-related toxicity by removing damaged mitochondria from the injury.
Increased Energy Supply by Creatine Treatment Promotes Axonal Regeneration
Given that regeneration is a highly energy-demanding process, regenerating axons face an energy crisis due to injury-induced mitochondrial dysfunction. However, it was not clear whether reversing this energy deficit could facilitate CNS regeneration after SCI. We aimed to directly target energy metabolism using creatine, an FDA approved blood-brain-barrier permeable ergogenic compound that rapidly regenerates ATP from ADP independent of mitochondrial transport (Tarnopolsky and Beal, 2001). Creatine monohydrate (2 g/kg) was administered to mice via gavage twice per day up to 8 wpi (Figure 7A). In vertebrates, creatine is converted into phosphocreatine for rapid ATP generation by creatine kinase (CK); CK activity correlates with creatine content and energy demands in tissue (Wyss and Kaddurah-Daouk, 2000). To measure creatine metabolism, CK activity was monitored throughout the experiments. Creatine treatment significantly increased CK activity in the CNS compared to saline treatment (Figure 7B). The increase in CK activity in spinal cords was more pronounced than that in the brains of creatine-treated mice. We next assessed the effect of creatine on axon regeneration after C5 DH. Saline-treated WT mice showed no CST axon regeneration into or beyond the lesion site (Figures 7C–7E). In contrast, creatine-treated WT mice exhibited significantly greater regeneration past the lesion site (Figures 7F–7H). Although axonal regrowth in creatine-treated WT mice was similar to that in untreated Snph−/− mice after injury, CST fiber number index revealed that most regenerated axons in creatine-treated WT mice stopped at 0.5 mm caudal to the lesion (Figure 7I). In contrast, regenerating axons in untreated Snph−/− mice extended further up to 1.5 mm, in the caudal spinal cords (Figures 1E and 1J), implying variable efficacies between creatine-induced limited recovery of energy metabolism in injury-damaged mitochondria and long-term enhanced delivery of healthy mitochondria into injured axons by Snph deletion. We further assessed motor skill dexterity of creatine-treated WT mice by the single-pellet retrieval test. WT mice treated with creatine showed a higher success rate of pellet retrieval by forelimb than saline-treated mice at 6- and 8-week post-injury (P < 0.05) (Figure 7J).
Figure 7. Systemic Administration of Creatine Promotes CST Axonal regeneration after the C5 DH.
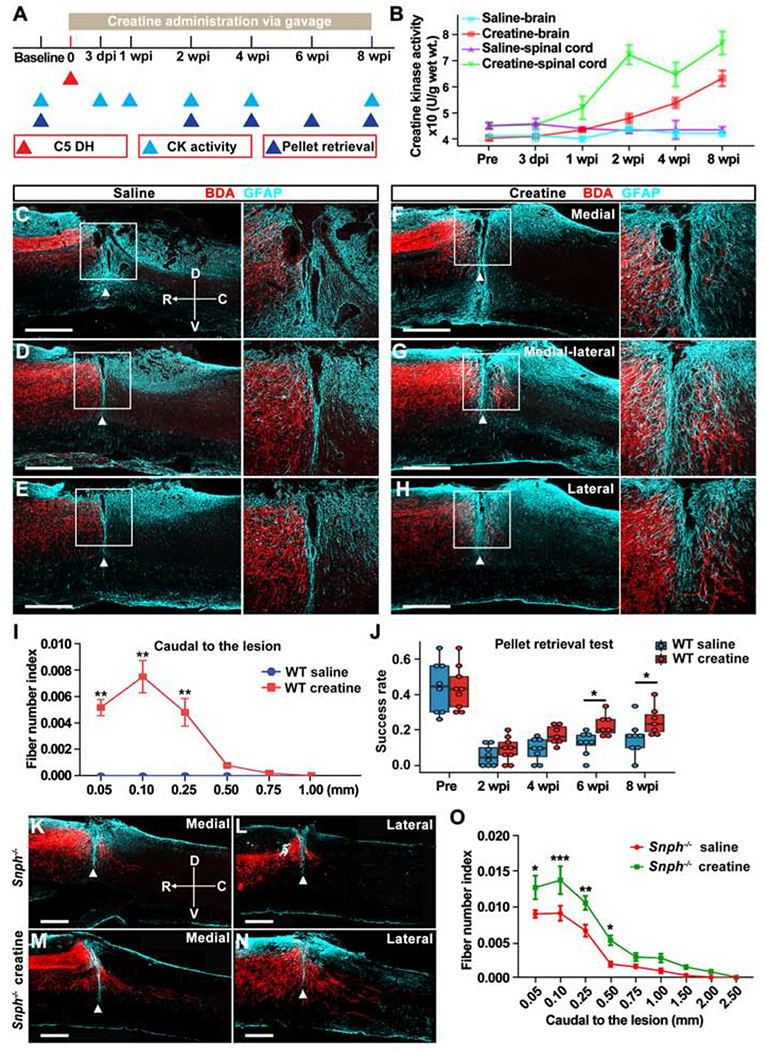
(A) Schematic of experimental design for creatine administration and analysis.
(B) Creatine kinase (CK) activity in the brain and spinal cord before and after the C5 DH.
(C-H) BDA-labeled CST axons in sagittal sections at medial (100 μm, C, F), medial-lateral (400 μm, D, G), and lateral (700 μm, E, H) distances from the midline in saline-treated (C-E) and creatine-treated (F-H) WT mice at 8 wpi. Arrowheads indicate lesion sites (GFAP, cyan). Right panels: magnification of boxed area. Compass: D, dorsal; V, ventral; R, rostral; C, caudal.
(I) Analysis of BDA-labeled CST axon number index caudal to the lesion.
(J) Analysis of the success rate in a single pellet retrieval test between saline-treated and creatine-treated WT mice over 8 wpi.
(K-N) BDA-labeled CST axons in sagittal sections at medial (K, M) and lateral (L, N) distances from the midline in saline-treated (K and L) and creatine-treated (M and N) Snph−/− mice at 8 wpi.
(O) Analysis of BDA-labeled CST axonal number index caudal to the lesion.
Data were presented as mean ± sem. n = 3-4 mice/group at each timepoin (B), n = 8 mice/group (I), n = 7-9 mice/group (J), and n = 7 mice/group (O). Two-way ANOVA with Bonferroni post hoc test. * P < 0.05; ** P < 0.01; *** P < 0.001. Scale bars: 500 mm (C-H, K-N). (Also see Figure S6).
To determine whether enhancing mitochondrial transport combined with elevating bioenergetic processes boosts axonal regeneration, we conducted the same protocol used for creatine treatment in Snph−/− mice. At 8 weeks after SCI, saline-treated Snph−/− mice showed regenerated CST axons crossing the lesion site and extending into the caudal spinal cord with various distances. Snph−/− mice that received creatine treatment exhibited even greater CST axon regeneration through and beyond the C5 DH (Figures 7K–7N). Quantitative analysis revealed a significant increase in the total number and total growing length of regenerated axons in the caudal spinal cord in creatine-treated Snph−/− mice as compared to the saline-treated Snph−/− mice (Figure 7O). In addition, we assessed the capacity of creatine to promote growth of other CNS axons. Creatine treatment significantly increased the density of 5-HT-positive axons in the ventral horn of the spinal cord as compared to the saline group at 8 wpi (Figures S6A and S6B). 5-HT IR fiber intensity from the ventral horn of spinal cord was significantly higher (P < 0.05) than saline-treated mice (Figures S6C–S6F). Astrocytes within the lesion site serve as a permissive bridge for axonal regrowth (Anderson et al., 2016; Liu et al., 2010). GFAP-positive astrocytes were found to bridge the lesion gap and in close association with regenerated CST axons, providing morphological evidence that astrocytes could serve as a growth permissive substrate for CST axonal regeneration across the lesion gap (Figures S6G–S6I). These results suggest that reversing injury-induced energy deficits by either genetic deletion of Snph or by administration of creatine can promote regeneration of CNS axons. The combination of the two offers even greater effects to boost axon regeneration and functional improvement after SCI.
DISCUSSION
In this study, we revealed that enhancing axonal mitochondrial transport via genetic Snph deletion in mice (1) recovered injury-induced mitochondrial damage, (2) promoted axon regeneration and synaptic connection following SCIs, and (3) restored motor functions. We further showed that systemic administration of creatine facilitated axonal regeneration. Our study establishes for the first time, to the best of our knowledge, that SCI-induced mitochondrial dysfunction and the associated energy deficit is an intrinsic mechanism linked to declined CNS regeneration. Thus, reversing energy deficits by enhancing mitochondrial transport and/or by elevating local energy metabolism are new therapeutic directions to stimulate axonal regeneration and functional recovery after CNS injuries.
Injury-Induced Mitochondria Damage Contributes to Regeneration Failure
CNS injury in adult mammals is characterized by an initial trauma followed by a progressive secondary injury cascade over weeks (Ahuja et al., 2017). The primary injury ruptures the axon plasma membrane, elevates intracellular calcium, and induces mitochondrial depolarization (Bradke et al., 2012). Widespread secondary injury includes inflammation, excitotoxicity, and oxidative response, which may cause chronic mitochondrial dysfunction (Scholpa and Schnellmann, 2017; Zhou et al., 2016a). Previously, we revealed that axonal injury acutely damages mitochondria, leading to local mitochondrial depolarization and impaired ATP production (Zhou et al., 2016a). Herein, we revealed that SCI triggers acute depolarization of local mitochondria within injured axons, starting 3 hours after injury, and persisting up to 7 days after SCI in WT mice. Several possibilities may explain this long-lasting mitochondrial dysfunction. First, the majority of axonal mitochondria are held stationary in mature neurons by the ‘static anchor’-Snph, which becomes highly expressed as neurons mature. This means that mitochondria damaged by injury are likely to be held stationary in adult axons. Second, damaged mitochondria may release harmful ROS, causing long-term cumulative stress. Using the redox probe Grx1-roGFP2, we found that WT and Snph−/− neurons displayed higher redox production in injured axons, however enhanced mitochondrial transport in Snph−/− neurons significantly reduced redox level 5 hours after injury when compared to WT neurons.
Mitochondria-derived ATP production provides the majority of axonal energy (Sheng, 2017). Considering that axonal injury induces acute mitochondrial damage in distal axons (Cavallucci et al., 2014; O’Donnell et al., 2013), it is likely that local energy supply is insufficient to power highly energy-demanding nerve regeneration. Enhanced mitochondrial transport in Snph−/− mice effectively recovers axonal mitochondrial integrity by removing damaged mitochondria and replenishing healthy mitochondria in injured axons in order to power regeneration. We tested this notion by labeling mitochondria through injection of AAV9-mito-GFP into the motor cortex. Motor cortex-derived mitochondria were widely distributed in BDA-labeled regenerated CST axons caudal to the lesion in Snph−/− mice, indicating long-distance delivery of healthy mitochondria from the motor cortex into regenerated CST axons in the spinal cord. These data indicate that enhancing mitochondrial transport recovers distal mitochondrial integrity and rescues energy deficits. Thus, mature CNS neurons can regain their regrowth capacity by replacing damaged mitochondria in injured axons. Enhancing mitochondrial transport was also sufficient to activate intrinsic signaling pathways and overcome extrinsic inhibitory pathways, thereby promoting regeneration.
Snph-Mediated Anchoring Accounts for Declined Regeneration in Mature CNS
While young neurons possess robust axon growth, mature CNS neurons typically fail to regrow after injury. This raises a fundamental question of whether mitochondrial dysfunction and the associated energy crisis serve as an intrinsic mechanism accounting for regeneration failure in mature CNS. Snph expression is robustly increased in adult brains and axonal mitochondrial motility progressively declines as neurons mature. Our study demonstrates that Snph expression in adult mice is not changed at 3, 7, or 14 days post SCI when compared to uninjured mice (data not shown), providing one clue as to why adult CNS axons fail to regenerate. Snph is a neuron-specific and axon-sorted protein that is integrated into mitochondrial membranes via its carboxyl-terminal transmembrane domain where it serves to anchors axonal mitochondria through its microtubule-binding domain (Kang et al 2008). We recently confirmed specific targeting of Snph to axonal mitochondrial surface at ultrastructural levels and at super-resolution imaging levels with STED nanoscopy (Lin et al., 2017). Neurons lacking Snph do not display observable differences in mitochondrial morphology or integrity, and neuronal morphology and cell survival are comparable to WT neurons (Kang et al., 2008; Lin et al., 2017; Zhou et al., 2016). Thus, an indirect effect of Snph deletion on axonal regeneration from non-neuronal cells seems unlikely. This is further supported by a previous report demonstrating that enhancing mitochondrial transport by expressing motor adaptor Miro1 in mature cortical neurons similarly boosts axonal regrowth after injury in vitro (Zhou et al., 2016).
Enhancing Mitochondrial Transport Promotes CST Axon regeneration
We hypothesize that maintaining axonal energy supply by enhancing mitochondrial transport is crucial to meet the metabolic demands of regeneration after CNS injuries. To test this hypothesis in vivo, we utilized three CNS injury models in adult mice. We found that Snph deletion promotes both compensatory sprouting of intact CST axons in the pyramidotomy model and regenerative growth of CST axons in the C5 dorsal hemisection model. This increased regrowth was not specific to CST axons, but could be applied to other CNS pathways, such as monoaminergic axons, which show greater regenerative capacity than CST axons in the complete spinal cord transection model. Restored CNS regeneration, combined with our previous findings in the PNS (Zhou et al., 2016a), indicate that enhancing mitochondrial transport via Snph deletion can enable a broader effective regenerative response to injury in both the PNS and CNS.
Our findings on SCI are supported by recent reports in other models. In C. elegans mutant ric-7, which exhibits impaired mitochondrial transport to distal axons, injured axons degenerate rapidly, a phenotype that is ameliorated by forcing mitochondria into axons (Rawson et al., 2014). In both Drosophila and mouse models, axonal mitochondria are a key target for WldS, a protein that protects axons from Wallerian degeneration after injury. WldS enhances mitochondrial flux into axons, which is essential for maximal protection after injury (Avery et al., 2012). In C. elegans, axotomy increases axonal mitochondrial density to supply ATP for sustained axon regeneration (Han et al., 2016). Enhanced mitochondrial transport also protects axotomized neurons from cell death and promote axonal regeneration of retinal ganglion cells (Cartoni et al., 2016).
While increased mitochondrial transport is required for successful axonal regeneration, whether these regenerating axons re-establish proper synaptic connectivity at the circuit level remains unknown. By supplying energy and buffering calcium, mitochondria support synaptic formation and sustain synaptic transmission; both of these processes are highly energy-demanding and tightly regulated by synaptic mitochondria (Devine and Kittler, 2018; Sheng and Cai, 2012). We revealed that newly regenerated CST axons in Snph−/− mice are able to form functional synapses, transmit electrophysiological signals, and promote functional recovery after SCI. Our findings suggest that enhancing mitochondrial transport serves as an intrinsic facilitator of axon regeneration and also supports synaptic activity and functional restoration.
Reversing Local Energy Deficits Boosts Axonal Regeneration
The injured CNS is expected to face local energy deficits due to (1) injury-induced mitochondrial damage (Bradke et al., 2012; Dumont et al., 2001); (2) limited diffusion capacity of ATP into distal axons (Hubley et al., 1996; Sun et al., 2013); (3) reduced mitochondrial mobility in mature neurons with elevated Snph expression (Zhou et al., 2016a); (4) polarized structures with long axons; and (5) the highly energy-consuming processes required for regeneration. Therefore, local energy crisis may serve as an intrinsic mechanism restricting regeneration of mature CNS neurons after injury. Our “energy crisis” model is a new concept in the field, particularly regarding SCI. Our model is further supported by systemic administration of the bioenergetic compound creatine, which elevated creatine kinase activity and promoted axon regeneration after SCI, similar to our findings in Snph−/− mice. The significant, but rather limited, axonal regrowth after creatine treatment when compared to untreated Snph−/− mice suggests that future development of a more effective energy facilitator would be beneficial (Kaasik, 2016). Moreover, as SCI is a multifaceted problem, reversing energy crisis would be expected to further boost axon regeneration when combined with other interventions known to enhance regrowth following CNS injuries (Hilton and Bradke, 2017; O’Shea et al., 2017; Ramer et al., 2014; Tran et al., 2018).
Regenerated CST Axons Contribute to Recovery in Forelimb Dexterity After C5 DH Injury
Forelimb dexterity is primarily controlled by motor cortex via the CST derived from layer 5 pyramidal neurons that make synaptic connections with lower interneurons and motor neurons in the spinal cord (Gu et al., 2017; Heffner and Masterton, 1983; Ueno et al., 2018). Forelimb dexterity deficits are caused by disruption of CST projections. The C5 DH model transects all CST projections to caudal spinal cord without interrupting motor cortex, and thereby has little influence on brain motor learning and memory. The single pellet retrieval task is considered the most sensitive behavioral assessment of deficits and recovery of forelimb digital skills (Farr Tracy and Whishaw Ian, 2002; Wang et al., 2017a), indicative of CST axon regeneration and reconstruction of forelimb motor circuitry. Our study revealed that Snph−/− mice, in which CST axon regeneration is enhanced, exhibited significantly better recovery of skilled forelimb motor function as compared to WT mice. In addition, specific silencing of the regenerated CST axons via DREAADs in Snph−/− mice further confirmed the dependence of skilled forelimb motor recovery on axon regeneration within the CST.
In summary, our data support the concept that reversing injury-induced energy deficits, by enhancing axonal mitochondrial transport or by applying an energy buffer, facilitates axonal regeneration after CNS injuries. Enhanced mitochondrial transport helps remove damaged mitochondria and replenishes healthy ones to injured axons, thereby recovering local mitochondrial integrity and energy deficits. Enhanced local ATP supply is critical for meeting the metabolic requirements of axon regeneration. Thus, activating an intrinsic CNS “growth program” requires coordinated recovery of energy supply by enhanced mitochondrial transport and/or by elevated bioenergetic metabolism. Such coordinated regulation may represent a new therapeutic strategy to facilitate axonal regeneration and functional restoration of the CNS after injury or in other neurological disorders.
Limitations of Study
C5 DH is an ideal SCI model to study CST axon regeneration and assess functional recovery of forelimb dexterity. However, this injury model likely maintains CST innervation of motoneurons at C2-C4 spinal levels and spares other descending pathways. To address this limitation, we performed behavioral assessments by specific silencing of the regenerated CST axons via DREAADs in Snph−/− mice to confirm the dependence of skilled forelimb motor recovery on axon regeneration within the CST. Given the technical challenge of monitoring in vivo ATP and ROS levels in injured spinal cord using ATP and redox probes, we alternatively applied microfluidic devices to study injury-triggered ATP reduction and mitochondrial ROS response in live neurons. Systemic administration of creatine has rather limited effect on facilitating axonal regeneration after SCI when compared to enhanced mitochondrial transport in Snph−/− mice. Future development of a more effective energy facilitators with higher blood-brain barrier permeability would be beneficial to quickly reverse energy deficits following traumatic CNS injuries.
STAR ★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead corresponding author Zu-Hang Sheng (shengz@ninds.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
The Snph−/− mouse line was generated by targeted gene replacement in embryonic stem (ES) cells as previously described (Kang et al., 2008). The Snph−/− mouse strain has been backcrossed to the inbred strain, C57BL/6 (Snph+/+) wild mice, more than 20 times. All mouse surgery and regeneration procedures were carried out in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine (IACUC #11011 and #18081) and Institutional Biosafety Committee (IBC #1556); and mouse breeding and maintenance were approved by the NIH, NINDS/NIDCD Animal Care and Use Committee (ASP1303-18). Animals were group housed (4-5 mice per cage) under at a constant temperature (24 ± 0.5 °C) and a 12 h/12 h light-dark circadian cycle (7 am – 7 pm) with access to food and water. The housing cages were generally changed once weekly. The health status of the mice, especially the post-surgery ones, was monitored by both veterinarians and experimenter daily. The mouse genotype did not cause changes in general weight and health. Littermates from both sexes were randomly assigned to experimental groups, unless stated otherwise.
Surgeries
With acquiring baseline values for the behavioral tests described below, 8-10-week-old wild-type (Snph+/+) or Snph−/− mice (either male or female) were subjected to one of the following three types of surgeries, the C5 dorsal hemisection, the unilateral pyramidotomy, and T8 complete transection. The anesthesia and analgesia in all subjects were accomplished with the use of a ketamine/xylazine mixture (120 mg/3.3 mg) and buprenorphine solution, respectively. The anesthetized animals were placed on a heating pad to maintain body temperature at 37°C ± 0.5°C after surgery. All the surgical procedures an d post-operative care were followed by the regulations of the Institutional Animal Care and Use Committee of Indiana University School of Medicine.
C5 dorsal Hemisection Model
With adequate sedation, a vertical incision was made from the posterior skull down to the 2nd thoracic (T2) spinous process which serves as a reliable landmark. Using a U shape mouse spinal stabilizer to stabilize the C5 vertebra, a laminectomy was performed on the 5th cervical vertebral lamina (C5) and the underlying dura was exposed. A 32-gauge needle was used to perform a transverse durotomy across the width of the C5 spinal cord. Then, a 1.0 mm-deep DH lesion was made by using a 2.4-mm flat blade attached to the Louisville Injury System Apparatus (LISA) (Walker et al., 2015). Because the oscillating blade was controlled by high precision micro-driver, the depth of the lesion using the LISA is accurate to 0.01 mm. Spinal cord laceration at depths of 1.0 mm would extend incision down through the central canal (~0.9 mm in depth from the dorsal spinal surface), which ensure complete transection of the dorsal (main) and lateral corticospinal tracts (CSTs). After laceration, the mouse was released from spinal stabilizer and the muscle and skin were then closed in layers.
Unilateral Pyramidotomy Model
Once anesthetized, the mouse was placed in a supine position and a midline incision was made to ventral neck to expose trachea. Blunt dissection was performed to reach the ventral surface of the skull. To gain access to the underlying pyramid, a left craniotomy was made in basioccipital bone by using a micro drill which was attached to steel burr with a ball-size of 0.6 mm (Stoelting, Item No. 58610). The dura was incised longitudinally by a 32-gauge needle. A cut was then made in left pyramid approximately 1.0 mm wide spanning the width of the pyramid and 0.5 mm deep with a modified fine scalpel perpendicular to the basilar artery to interrupt the descending CST axons. The muscle and skin were closed in layers after pyramidotomy. For assessing the injury completeness, BDA was injected bilaterally into sensorimotor cortex at 1 day after pyramidotomy and the mice were terminated 2 weeks later. For assessing CST axonal sprouting, BDA was injected unilaterally into the contralesional (right side) sensorimotor cortex at 6 weeks after the left pyramidotomy and the mice were terminated 2 weeks later.
T8 complete Spinal Cord Transection Model
The procedure for T8 complete spinal cord transection is similar to what was described previously (Deng et al., 2016; Xu et al., 1995) with minor modifications. A laminectomy was performed in T7 thoracic vertebrae and the cord segments T8 were exposed. Noyes spring scissor (Fine science tools, item No. 15013-12) was used to transected the entire depth of spinal cord followed by a no.11 scalpel blade drawing bilaterally across the lesion to confirm completeness of the lesion. Bladders of injured animals were manually squeezed to eliminate urine twice per day for the entire duration of the experiment.
METHOD DETAILS
Corticospinal Tract Tracing
The procedure of tracing the corticospinal tract (CST) is similar to what we published previously (Al-Ali et al., 2017; Wang et al., 2014). Briefly, A vertical midline incision was made from between the eyes to the posterior skull. Using the bregma as the center in both the x and y planes, bilateral windows (5 mm in length and 2 mm in width) were created with the medial edges of the windows 0.5 mm lateral to the bregma. Using a digital stereotactic injector (Item: 51709, stoelting Co. USA), 0.5 μl of biotin dextran amine (BDA; MW 10,000; 10% in PBS; Molecular Probes) was injected into one of the 10 total sites (5 sites/side). Mediolateral (ML) coordination: 1.5 mm lateral to the bregma; anteroposterior (AP) coordination from the bregma: −1.0, −0.5, 0, −0.75 and 1.5 mm; dorsoventral (DV) coordination: 0.5 mm from the cortical surface; rate: 0.1 μl/minute. After each injection was completed, the injector tip was left in place for an additional 5 minutes to ensure that the BDA solution was adequately penetrated the tissue. Two weeks later, mice were anesthetized and perfused with 4% paraformaldehyde for detecting CST distribution in the spinal cord.
Corticospinal Neuron Tracing
To retrogradely label the corticospinal neurons in the sensorimotor cortex, AAV2-GFP (1.2X1011 TU/μl, provided by Dr. George Smith in Temple University) was injected into the intermediate zone of right side of the C5-C7 spinal cord (0.5 mm lateral to the midline; 0.75 mm below spinal surface) after laminectomy of multilevel vertebra. AAV injections were applied to 3 sites (one on each segment of the cord) using glass micropipettes connected to 10-μl Hamilton syringes under the control of digital stereotactic injectors. Mice were kept for an additional 2 weeks before sacrificing for histological analysis.
Motoneuron Tracing
After adequate sedation, the mouse was secured to a mount with the forelimbs perpendicular to the body. A horizontal incision was made at the intersection of a vertical line down from the lateral aspect of the skull and a horizontal line from the forelimbs. The overlying muscle was dissected and retracted until the brachioplexus was visualized and isolated. A 0.5 μl of Cholera Toxin Subunit B (CTB, 1% wt per vol, List Biological Laboratories, Campbell, CA) was bilaterally delivered in the upper trunk of brachial plexus using a Hamilton syringe as we described previously (Gowrishankar et al., 2015). The overlying muscle was then re-approximated and the skin closed using suture. Mice were kept for an additional 3 days to allow tracer transport before sacrificing for histological analysis.
CST Axonal Reconstruction
To reconstruct the regenerating CST axons, high-resolution images were taken blindly with regard to genotypes from each of the longitudinal spinal cord sections by using the Hamamatsu ORCA-R2 digital camera attached to a Zeiss microscope. The Adobe Photoshop software was used to align all of the sections with respect to the lesion site, as well as the dorsal and ventral boundaries caudal to the lesion. The subsequent aligned images were imported to Imaris software (Bitplane, Switzerland) and all of the regenerated axons caudal to the lesion site were manually traced. In each of the series, one section was chosen to trace the axons rostral to the lesion site in order to create a representative image of axon regeneration. Afterward, the axonal reconstructive tracings were merged together using Adobe Photoshop.
Stimulus-triggered Electromyography (EMG) Recordings
Mice were anesthetized with a mixture of ketamine-xylazine and subjected to ketamine anesthesia to maintain adequate muscle tone for EMG recordings. Animals were shaved, disinfected and secured completely in a stereotaxic apparatus and the body temperature was maintained with a heating pad. A craniotomy was performed to expose the M1 region of the motor cortex. Electrode penetrations were guided by stereotaxic apparatus to a depth of 700-1000 μm from the brain surface to target corticospinal neurons in the sensorimotor cortex. Single tungsten electrodes were used for microsimulation (Item: WE30030.5A3, Microprobes). Electrical stimulation was initially delivered at 13 bipolar pulse stimuli with every 200 μs in duration at a frequency of 333 Hz and repeated every two seconds to identify the forelimb motor cortex. Penetration sites that failed to produce any observed EMG signals within the range of current intensity, 0-100 μA, were defined as non-responsive. For stimulus-triggered averaging, single pulse bipolar stimuli with 200 μs in duration at every 10 seconds with a total of 30 sweeps was used and EMG signals were recorded and amplified using a differential AC amplifier (model 1700, A-M system, USA).
Behavioral Tests
The behavioral tests described below, including grid walking, adhesive paper removal, footprint analysis and pellet retrieval test, were completed prior to surgery for baseline values, and every two-week interval up to 8 weeks post-injury with the help of a behavioral test specialist. These assessments were performed in a blinded fashion, i.e. the behavioral evaluation and analysis were done by a behavioral specialist with an independent assistant who are unaware of the genotypes or treatment/non-treatment groups. Animals with the incomplete injury identified by histological analysis were excluded from the dataset.
Basso Mouse Scale
Basso Mouse Scale (BMS) for locomotion was performed weekly up to 8 weeks following the T8 complete transection by two observers blinded to the experimental groups according to a method published previously (Basso et al., 2006; Wang et al., 2018). Briefly, mice were placed in an open field (diameter: 42 inches) for 4 min to identify all visually discernable attributes of locomotor recovery for severely injured mice. The scores were on a scale of 0–9 (0, complete hind limb paralysis; 9, normal locomotion), which is based on hind limb movements made in an open field including hind limb joint movement, weight support, plantar stepping, coordination, paw position, and trunk and tail control.
Grid Walking
A (12 mm x 12 mm) mesh metal grid setup was used for this test (Wang et al., 2015). Prior to each measurement, the bladder of mouse was expressed. The mouse was placed in the center of the mesh grid and its walking on the grid was observed for 3 minutes. Two observers blinded to experimental groups assessed the test with one person counted the total number of steps while the other person counted the number of times that each of the front limbs fell through the grid holes. Two runs were recorded for each mouse.
Adhesive Paper Removal
Prior to testing, the bladder of each mouse was expressed to empty the urine. The mouse was primed by putting it in a clear container with no bedding for at least 5 minutes. Each mouse was then tested one at a time by placing a small adhesive tape strips (3 mm x 4 mm) to each front paw and placing the mouse in a separate transparent container (Bouet et al., 2009). Time began once the mouse was placed in the container and the time at first contact (Tc) and time at removal (Tr) were recorded for both the left and right sides. A first trial was completed for all of the mice before the second trial began in order to prevent the mice from becoming fatigued. If there was a large disparity between the first and second trials, then a third trial was introduced following the same procedures as the first 2 trials.
Footprint Analysis
Footprint analysis was conducted at every two-week-intervals after the C5 DH according to our existing protocol (Titsworth et al., 2007) with modifications. Briefly, the animal’s forelimbs were coated with nontoxic paint. The mice moved from a brightly illuminated starting box to a darkened box by walking through a narrow-custom-built plexiglass trough (5 cm wide by 40 cm long), leaving a trace of their paw prints on the white sheet which was affixed to the trough. Trials were excluded and repeated if the animal turned around at any point. The footprints were scanned, and digitized images were measured using Photoshop CC (Adobe Systems, Canada). Stride length was measured by the distance between the center of subsequent left and right forelimb. Stride width was determined by drawing two lines connecting two consecutive forepaw prints and then measuring the distance between these two lines at the midpoint. At least four footprints per side from three sessions per animal per group were measured to determine the mean values of each parameter assessed.
Pellet Retrieval Test
The procedures for pellet retrieval followed those described by Chen et al (Chen et al., 2014) and our previous publication (Al-Ali et al., 2017). The first 2 days were food deprivation days where mice were weighed and given food at 10% of their body weight. We attempted to keep the food at 10% body weight throughout the experiment; however, some of the mice became fatigued and sick. So, the decision was made to use food at 10% body weight 1 week prior to the week when behavior testing was performed. Except for that time frame, food was given at 20% of their body weight. Following food deprivation, we began to shape the mice. Shaping days 1 and 2 were acclimatization days where the mice were placed for 20 minutes in the custom Plexiglas chambers. On shaping day 3, each mouse was placed in the chambers with a feeding well insert in order to train the mice to reach through the slit and grab the millet seeds. During this day, both the total number of reaches as well as number of reaches with each paw was recorded in order to determine paw dominance, which was defined by at least 70% of the total attempts. Shaping was considered complete when a total number of reaches was at least 20 attempts and paw dominance was determined. After shaping was completed, each mouse was trained for 7 days with the training/testing well insert, which has 6 divots. Small millet was placed in 1 of 6 wells. The wells were arranged in such a way that there were 3 on the right and 3 on the left. The well that was chosen for each subject was decided based on dominant handedness and animal preference, which were revealed during the training procedures. The preferred divot was determined and recorded on training day 1 and used for the rest of the experiment. However, occasionally, the preferred divot for some subjects changed after surgery and had to be changed.
Each trial was categorized as a success or fail during the actual test. The trial was considered a success if the mouse grabbed the millet and ate it. The trial was considered a failure if the mouse grabbed the pellet but dropped it outside of the chamber, or if the mouse failed to grab the pellet or knocked the pellet from the well. The performance was evaluated by the rate of successful reaches, which was calculated by the number of successful reaches divided by the total number of reaches (successes + fails). The average time the mice took to grasp the pellet was also calculated from the onset of the first passage of the paw through the opening slit to successful retrieval of that pellet.
To assess kinematic features of the forelimb trajectory, the high-speed camera was used to record reaching behaviors during all pellet retrieval trails. We then used Tracker (Open source physics) to manually trace the most distal tip of the paw frame by frame as the moment the paw lifted from the ground until it succeeded or failed to grasp the pellet. With a known reference (5 x 5 cm), the paw location was converted as x-y coordinates during grasp phase. We aligned all traces to the constant pellet position and averaged the data from ≥6 independent trails in RStudio for graphical representation. Paw positions were also represented by heatmaps to show their spatial probabilities relative to pellet during grasp phase.
Immuno-transmission EM (iTEM)
To detect synapse formation in regenerated axons, BDA was injected into the sensorimotor cortex of Snph−/− mice to trace the CST axons 8 weeks after the C5 DH. Two weeks later, mice were anesthetized and perfused with freshly made EM fixative (4% PFA and 0.05% glutaraldehyde in PBS, pH 7.4). Both rostral and caudal spinal cords were carefully dissected from the lesion site and sectioned into 100-μm sections using a VT1000S vibratome (Leica Biosystems). Sections were washed with PBS and then permeabilized and blocked with 0.1% saponin / 5% normal goat serum in PBS for 1 hour. BDA was labeled with the AB complex (Vectastain ABC kit, PK-6100) for 30 minutes at RT with gentle shaking. After three washes with PBS, the BDA substrate (Sigma, D6190-100ML) was added and sections were monitored under a dissecting microscope until the desired stain intensity develops. After the wash with PBS, samples were fixed with 2% glutaraldehyde in PBS for at least 30 minutes and then sent to the EM facility of NINDS, NIH for processing. Slices of the spinal cord were washed in 0.1N cacodylate buffer (pH 7.4) and treated with 1% osmium tetroxide in 0.1N cacodylate buffer at pH 7.4 for 1 hour on ice, dehydrated with a series of graded ethanol (50%, 70%, 80%, 90%, 95%, and 100%, 3 times each), and embedded in epoxy resins. Thin sections (70 nm) were counterstained with lead citrate for 5 minutes, examined under a JEOL200CX transmission electron microscope, and photographed with a bottom-mounted digital CCD camera (AMT XR-100, Danvers, MA, USA).
DREADDs Silencing Experiments
HiRet-TRE-hM4Di-mCherry (2.19 x 107 TU/mL) and AAV2-CMV-rtTAV16 (AAV-Tet/On, 1.2 x 1013 GC/mL) were provided by Dr. George Smith at Temple University and tested following previously described methods (Sheikh et al., 2018). To selectively target regenerating CST axons after the C5 DH, HiRet-TRE-hM4Di-mCherry was injected into the caudal spinal cord with three injection sites (0.5, 1.0, 1.5 mm caudal to the lesion site) where the most regenerated CST axons were present based on the BDA tracing results. Meanwhile, AAV-Tet/On was stereotaxically injected into the motor cortex at five sites (AP coordinates from bregma in mm: 1.0/1.5, 0.5/1.5, 0.0/1.5, −0.75/1.5, −1.5/1.5, all at a depth of 0.5 mm into cortex). All injections were done using a digital mouse stereotaxic instrument (Stoelting, USA) fitted with a beveled, pulled glass needle, at a speed of 0.1 μl/min, the needle was held in place for 5 mins before being slowly retracted. After tracer injections, all animals were maintained for an additional 3 weeks before drug administration. To induce expression of hM4Di, Dox (30 mg/kg, s.c.) was administered for 7 days and Clozapine-n-oxide (CNO; 4 mg/kg, i.p.) was injected on day 4 and 6 after Dox administration. Pellet retrieval assessment was performed daily during Dox administration but 20-30 minutes after CNO administration to determine the effect of inhibitory DREADD activation on forelimb function. Dox was also administered prior to euthanasia to visualize the mCherry expression in corticospinal neurons upon histological analysis.
In Vivo Labeling of Axonal Mitochondria
After adequate sedation, mice received a C5 DH as described above. Two percent liquefied agarose was then applied around the lesion site to build the agarose well. The lesion area was dried by removing the injury-generated fluid and blood using cotton Q-tips. A 5 μl CMTMros mitotracker (5 μM, ThermoFisher Scientific, M7510) was injected into the lesion center 0.75 mm below the surface at 0.1 μl per minute using glass micropipette connected to 10-μl Hamilton syringes under the control of microinfusion pumps. Gelform fully soaked with CMTMRos solution was then applied on top of the lesion center for a 2-hour incubation in the dark. After the dye application, mice were immediately perfused and the spinal cord was dissected out and post-fixed in 4% paraformaldehyde for 30 minutes. Then 30 μm-thick cryosections were prepared for immunohistochemistry and analysis. Five time points (i.e. 3, 6, 12, 24 hours, and 7 days post-injury) were chosen to demonstrate injury-induced mitochondria integrity changes. With each time point, the dye was applied for 2 hours before the ending of the experiment. For mitochondria labeling in intact mice, the same labeling protocol was followed except for the injection of 5 μl CMTMros mitotracker directly into the dorsal funiculus of the intact spinal cord (0.45 mm below the surface). Mice were excluded from the study if significant CST axons were damaged by dye injection alone. To detect the distribution of axonal mitochondrial, BDA was injected into the motor cortex to label the CST projection in the spinal cord at 2 weeks prior to the mitotracker application.
To label distribution of motor cortex-derived axonal mitochondrial in regenerating CST axons, we conducted two injections into bilateral motor cortices in Snph−/− mice after SCI: (1) injection of AAV9-mito-GFP to label mitochondria in the motor cortex 4 weeks post-C5 DH, and (2) injection of BDA in the same motor cortex to label the CST axons 6 weeks post-C5 DH. Both injections in the motor cortex were separated at five sites on each side with the following coordinations. Mediolateral (ML) coordination: 1.5 mm lateral to the bregma; anteroposterior (AP) coordination from the bregma: −1.0, −0.5, 0, −0.75 and 1.5 mm; dorsoventral (DV) coordination: 0.5 mm from the cortical surface; rate: 0.1 μl/minute. Mice were kept for an additional 2 weeks for further histological analysis.
Creatine Treatment and Creatine Kinase Activity Assay
The C57BL/6J or Snph−/− mice (8-10 weeks old, both sexes) were randomly assigned into 2 groups: (1) SCI + saline; and (2) SCI + creatine. The mouse in each group received the C5 DH as described above. SCI mice fed with either saline or creatine monohydrate (2g/kg, Sigma) that dissolved in sterile water via gavage twice per day up to 8 weeks post-injury. To examine creatine kinase (CK) activity in the CNS over time after creatine treatment, fresh samples from motor cortex and spinal cord (a 5-mm-long segment including the C5 lesion site) were lysed prior to SCI and on days 3, week(s) 1, 2, 4 and 8 post-injury using the CK Activity Assay Kit (ab155901, Abcam) according to the protocol provided by the manufacturer. The relative CK activity in each sample was calculated based on NADH standard calibration curve.
Immunofluorescence Staining
Mice were deeply anesthetized and transcardially perfused sequentially with 0.09% saline and 4% paraformaldehyde in PBS. Tissues were harvested followed by post-fixation in 4% paraformaldehyde overnight and soaked in 30% sucrose 2 days for dehydration. Serial sagittal or cross cryostat-sections were stained with primary antibodies in blocking buffer (3% goat serum and 5% donkey serum in 0.3% PBST) overnight at 4 °C at least for 18 hours. Detection was accomplished by incubation with Alexa Fluor-coupled secondary antibodies (Life Technologies/Abcam) diluted in blocking buffer for 2 hours at room temperature. The Hamamatsu ORCA-R2 digital camera attached to a Zeiss microscope was used to acquire low magnification (10X) images. A confocal microscope, in conjunction with the Olympus FV10-ASW 4.2 software, was used to visualize the immunostaining at higher magnifications (20X and 60X). The antibodies used in the study were the following: mouse anti-GFAP (1:2000, Abcam, ab8049), guinea anti-vGlut1 (1:1000, Millipore, AB5905), goat anti-CTB (1:3000, List biological laboratories, 703B), mouse anti-synaptophysin (1:1000, Abcam, ab8049), rabbit anti-tyrosine hydroxylase (TH, 1:1000, Abcam, ab112), rabbit anti-serotonin (5-HT, 1:500, Sigma-Aldrich, S5545), goat anti-serotonin (5-HT, 1:1000, Abcam, ab66047), rabbit anti-TOM20 (1:200, Santa Cruz, sc-11415), rabbit anti-laminin (1:1000, Abcam, ab11575), rabbit anti-fibronectin (1:1000, Abcam, ab2413), rabbit anti-collagen I (1:1000, Abcam, ab34710), donkey anti-mouse (Alexa Fluor® 405, 1:1000, Abcam, ab175658), goat anti-guinea pig (Alexa Fluor® 647, 1:1000, Thermo Fisher Scientific, A-21450), donkey anti-rabbit (Alexa Fluor® 488, 1:1000, Thermo Fisher Scientific, A-21206), donkey anti-rabbit (Alexa Fluor®594, 1:1000, Thermo Fisher Scientific, R37119), and donkey anti-goat (Alexa Fluor®594,1:1000, Thermo Fisher Scientific, A-11055).
BDA Staining
To detect BDA-labeled CST axons/fibers, BDA staining was performed by incubating the sections in PBS containing streptavidin–horseradish peroxidase (Vectastain R.T.U. Elite ABC Reagent, Cat. No: PK-7100) for 1 hour at room temperature. PerkinElmer Biotinyl tyramide (1:100 in amplification diluent) was then added to the sections for another 1 hour. Detection was accomplished by incubation with Extra-Avidin@ TRITC (1:200, Sigma) in 0.1% PBS-T for 2 hours.
Microfluidic Neuron Culture and Axotomy
For neuronal cultures, mouse brain tissues were dissected out from E18–19 WT or Snph KO mouse embryos (both sexes randomly selected) and kept in ice-cold Hibernate buffer supplemented with 2% B27 and Antimycotic (1X, Thermo Fisher Scientific, 15240062). After the embryos were genotyped by direct tail PCR, cortical neuron cultures were prepared with papain as described previously (Kang et al., 2008). After preparing a single-cell suspension, 1 × 105/10 μl dissociated neurons were added into the cell body chamber of the house-made microfluidic devices as previous described (Zhou et al., 2016b). After cells were attached, 100 μl culture medium was added into each well of the device. For axotomy, axons in terminal chambers were axotomized by vacuum aspiration at DIV12.
Redox Measurement
Both WT and Snph KO cortical neurons were infected with lenti-virus containing redox-sensitive Grx1-roGFP2 probe at DIV7, followed by imaging of the distal 150-μm microgrooves before or 1, or 5 h after axotomy at DIV12. For redox probe imaging, Grx1-roGFP2 signal was excited by 405 nm and 488 nm laser line. Pseudo-color ratiometric images and the 405/488 nm ratiometric integrated intensity of Grx1-roGFP2 signal were calculated by ImageJ.
Data Analysis
To quantify the fiber/axon number index of regenerated CST axons growing through and beyond the lesion site at 8 weeks after the C5 DH, we drew multiple vertical lines across sagittal spinal cord sections at irregular distance starting from the lesion center to caudal spinal cord in every third series of sagittal sections from each mouse. The number of axons intercepting lines was counted by observer blind to experimental groups. Total intercepts were normalized to the total number of BDA-labeled CST axons in the medulla and presented as a percentage of CST regeneration. For correlation analyses between regenerated CST axons and the success rate of pellet retrieval test, the average axon number index from each animal was plotted.
To quantify the fiber/axon intensity index of BDA-labeled CST axons rostral to the lesion border, every third series of sagittal sections from each mouse was analyzed using the ImageJ software (Huang et al., 2016). Ten consecutive rectangular boxes with the same size (100 × 100 μm2) were drawn on the parasagittal sections at regular distances, −1.0~−0.1 mm relative to the rostral lesion border (0 mm). The fiber intensity index was presented as a percentage of the density of BDA-labeled axons at a specific position versus the density of axons 1 mm rostral to the lesion border.
To quantify the fiber number index of sprouted CST axons, we followed the established protocol (Geoffroy et al., 2015; Liu et al., 2010) with minor modifications. Briefly, we drew a horizontal line through the central canal and across the lateral rim of the gray matter. Vertical lines were drawn at 50 μm from the central canal, then every 100 μm lateral to the rim. Only fibers crossing these vertical lines were counted in each of five sections for each mouse and numbers of axons were normalized against total axon count in contralateral CST main tract.
To determine the distribution of sprouted CST axons, images were imported and analyzed in ImageJ by using the skeletonize function to erode all CST axons to a single pixel width (Asante and Martin, 2013). The total number of pixels in a given area thus corresponds to the overall density of CST label in a given area of the section. The pixelated data were further processed in the Matlab (Mathworks) to generate heatmaps with red color represents the highest axon density and blue color represents the lowest density and white color represents the background of the images. To define the margin of gray matter, dorsal columns from representative images were used as a fiduciary maker to create a gray matter border in the Matlab. For each mouse, images of at least 3 sections from each cervical, thoracic and lumbar levels were converted and analyzed.
To quantify the intensity index of the TH- and 5-HT-positive axons rostral and caudal to the lesion site after the T8 spinal complete transection, every third section of all sagittal sections from each animal was analyzed using the ImageJ. In each sagittal section, we drew a (dorsoventral) line starting from the rostral lesion border (0.0 mm), then ten vertical lines at regular distance from −1.0 ~ −0.1 mm rostral to lesion border, and seven vertical lines at irregular distance from 0.05 ~ 1.5 mm caudal to lesion border. The axon intensity index was presented as a ratio of axon density at a specific region versus the axon density −1.0 mm rostral to the lesion border. The averaged axon intensity per animal was further processed in GraphPad to generate regional density maps at certain distances from the starting line.
To analyze mitochondrial membrane potential (Δψm) rostral to the C5 DH, total mitochondria in sagittal sections were immunostained with antibody TOM20 and healthy mitochondria were examined by loading MitoTracker Orange-CMTMRos, a fixable Δψm-dependent dye. The mitochondrial mean intensity was assessed by the integrated MitoTracker CMTMRos fluorescence intensity against the area of total TOM20-positive mitochondria in a given region using ImageJ particle analysis. To analyze mitochondria number in the injured CST axons after the C5 DH, individual BDA-labelled CST axons with CMTMRos-positive puncta were immunostained with TOM20. The total number of TOM20-positive and CMTMRos-positive mitochondria along a single axon in 50 μm-length was counted using Photoshop software. To quantify the changes in mitochondrial length before and after SCI, images with TOM20-positive staining were processed using the ImagePro Plus software 6.0 (Media Cybernetics, Silver Spring, MD) (Koopman et al., 2006). In this software, mitochondrial length was measured by the option called Feret diameter which is the maximum distance between two parallel planes enclosing a mitochondrial object. The data profiles were further processed in Rstudio to generate distribution histograms. To quantify the number of CMTMRos positive mitochondria in the injured CST axonal tips at 1 week after the C5 DH, axonal tips with a length of 20 μm rostral to the injury were selected and CMTMRos positive mitochondria within the tips were manually counted in ImageJ.
We developed a quantitative method to determine axonal mitochondria Δψm after the C5 DH. In sagittal sections, BDA-labelled CST axons with CMTMRos positive puncta rostral to lesion site were captured using a confocal microscope (60x). To compare axonal Δψm between groups, single axon with a length of 50 μm was selected followed by alignment to a standard rostrocaudal position in Photoshop CC (Adobe Systems). Then, background noise and all mitochondria outside of the axon were manually removed in Photoshop such that only mitochondria within the CST axon remained. Intensities of axonal mitochondria were then calculated in the ImageJ software by using the functional called “plot profile”. In the profile, the values of x-axis represent the position of all individual mitochondria along single axon, and the matching values in y-axis represent the pixel intensity of each mitochondria. To enable a consistent quality of intensity analysis, a global automatic threshold was chosen for a background-noise cutoff in all images. All the mitochondria Δψm profiles were further transferred to the RStudio to generate intensity heat maps between experimental groups after SCI.
The percentage of BDA-labeled CST axons presenting retraction bulbs at 7 days after C5 DH were quantified in sagittal sections from the medial portion of the spinal cord containing the main dorsal CST (3-5 sections per animal) (Dias et al., 2018). The sum of retraction bulbs was normalized with total number of CST main tract in each section and expressed as a percentage of fiber retraction. To quantify the intensity of 5-HT axons in the caudal spinal cord in mice with C5 DH, spinal cord 5 mm away from the lesion site was sectioned into 35-μm cross-sections and stained with antibody against serotonin. Monochrome images were imported to ImageJ and analyzed for 5-HT fiber density. A region of interest (ROI) was drawn around the grey matter of the ventral horn. The intensity of 5-HT IR signal was thresholded to background for each image and analyzed per μm2.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as mean ± SEM. Statistical parameters including the definitions and the exact value of n (e.g. number of animals, number of slice sections, number of axons, number of images, etc), deviations, P values, and types of statistical tests are reported in the Figures and corresponding Figure Legends. Statistical analyses were carried out using Prism 7 (Graphpad Software) or R programming language. Comparisons between WT (control) and treatment (Snph−/− or creatine) groups were performed by Unpaired two-tailed Student’s t-tests or a Mann-Whitney test, Two-way ANOVA with Bonferroni post hoc correction were used to analyze interactions between groups. P < 0.05 was considered statistically significant. Samples sizes were not pre-determined by power analysis but instead based on our previous axonal regeneration studies following SCI. In this study, we utilized three different CNS injury models. For assessing axon regeneration following C5 DH, 2 mice with unsuccessful BDA tracing and 2 mice with incomplete lesions were exclude from a total of 14 WT animals for further analysis. In Snph−/− group, 2 out of 13 mice were excluded due to limited BDA labeling and incomplete lesion. For forelimb dexterity assessment after the C5 DH, 2 out of 12 WT mice and 1 out of 11 Snph−/− mouse were excluded from the study due to their reluctance to grasp the food pellet during 8 weeks post injury. In other two injury models (unilateral pyramidotomy and T8 complete transection), no data were excluded.
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or codes.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFAP | Abcam | ab8049 |
| Guinea anti-vGlut1 | Millipore | AB5905 |
| Goat anti-CTB | List biological laboratories | 703B |
| Mouse anti-synaptophysin | Abcam | ab8049 |
| Rabbit anti-tyrosine hydroxylase | Abcam | ab112 |
| Rabbit anti-serotonin | Sigma-Aldrich | S5545 |
| Goat anti-serotonin | Abcam | ab66047 |
| Rabbit anti-TOM20 | Santa Cruz | sc-11415 |
| Rabbit anti-laminin | Abcam | ab11575 |
| Rabbit anti-fibronectin | Abcam | ab2413 |
| Rabbit anti-collagen I | Abcam | ab34710 |
| Donkey anti-mouse (Alexa Fluor® 405) | Abcam | ab175658 |
| Goat anti-guinea pig (Alexa Fluor® 647) | Thermo Fisher Scientific | A-21450 |
| Donkey anti-rabbit (Alexa Fluor® 488) | Thermo Fisher Scientific | A-21206 |
| Donkey anti-rabbit (Alexa Fluor®594) | Thermo Fisher Scientific | R37119 |
| Donkey anti-goat (Alexa Fluor®594) | Thermo Fisher Scientific | A-11055 |
| Bacterial and Virus Strains | ||
| AAV2-GFP | Dr. George Smith | Temple University |
| AAV9-mito-GFP | Dr. George Smith | Temple University |
| HiRet-tre-hM4Di-mCherry | Dr. George Smith | Temple University |
| AAV2-CMV-rtTAV16 | Dr. George Smith | Temple University |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Grx-1-roGFP2 | Dr. Chang Chen | Chinese Academy of Science |
| Cholera Toxin B Subunit (Choleragenoid) from Vibrio cholerae (CTB) | List biological laboratories | 103B |
| CMTMros mitotracker | Thermo Fisher Scientific | M7510 |
| Antimycotic | Thermo Fisher Scientific | 15240062 |
| Dextran, biotin, 10000MW, lysine fixable (BDA-10000) | Invitrogen | D1956 |
| Clozapine-n-oxide (CNO) | Sigma-Aldrich | C0832-5MG |
| Doxycycline (Dox) | Sigma-Aldrich | D1822-500MG |
| Paraformaldehyde | Sigma-Aldrich | P6148-5KG |
| Creatine monohydrate | Sigma-Aldrich | C3630-100G |
| DAPI | Sigma-Aldrich | D9542-1MG |
| ExtrAvidin® TRITC | Sigma-Aldrich | E3011-1ML |
| ExtrAvidin® FITC | Sigma-Aldrich | E2761-1ML |
| Critical Commercial Assays | ||
| Creatine Kinase Activity Assay Kit | Abcam | ab155901 |
| TSA Biotin Tyramide Reagent Pack | PerkinElmer | SAT700001EA |
| Vectastain R.T.U. Elite ABC Reagent | Vector Laboratories | PK-7100 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice | The Jackson Laboratory | Stock No: 000664 |
| snph+/+ adult mice | (Kang et al., 2008) | N/A |
| snph−/− adult mice | (Kang et al., 2008) | N/A |
| REAGENT or RESOURCE | ||
| Mouse WT and snph KO forward primer GGAAGCCCCGAACCACGTGCC |
(Kang et al., 2008) | N/A |
| Mouse WT reverse primer ATCCGGCAGAGGCTCGTCTAC |
(Kang et al., 2008) | N/A |
| Mouse snph KO reverse primer ACGAGACTAGTGAGACGTGCT |
(Kang et al., 2008) | N/A |
| Software and Algorithms | ||
| Adobe Illustrator CC 2018 | Adobe Systems | https://www.adobe.com/products/illustrator.html |
| Adobe Photoshop CC 2018 | Adobe Systems | https://www.adobe.com/products/photoshop.html |
| Fiji | Schindelin et al., 2012 | http://fiji.sc/ |
| GraphPad Prism version 7.0 | GraphPad Software, Inc | https://www.graphpad.com/scientific-software/prism/ |
| Image-Pro Plus 6.0 | Media Cybernetics | http://www.mediacy.com/imageproplus |
| Imaris | Bitplane | http://www.bitplane.com/ |
| Matlab | Mathworks | https://www.mathworks.com/products/matlab.html |
| RStudio | N/A | www.r-project.org |
Context and Significance.
Nerve fibers or axons of the central nervous system (CNS) in adult mammals regenerate poorly after injury, often leading to permanent neurological impairments. Axonal regeneration is a highly energetic process, suggesting that enhancing energy metabolism may be a way to facilitate axonal regrowth after injury. Using three CNS injury mouse models, this collaborative study by the Xu and Sheng groups reveals that enhancing the transport of mitochondria, the power supply of the cell, by deleting one of its protein anchors rescues injury-induced cellular damage, facilitates axonal regeneration and its connections, while restoring motor functions. Administration of the bioenergetic compound creatine also facilitates regeneration. This study establishes that mitochondrial dysfunction and energy deficits contribute to poor CNS axonal regeneration and that supplementing the energy supply can be ameliorative.
Highlights:
Injury-induced mitochondrial dysfunction contributes to CNS axonal regenerative failure
Enhancing its transport recovers mitochondrial integrity after spinal cord injury (SCI)
Removing a mitochondrial anchor protein enhances functional recovery after SCI
Increasing energy metabolism via creatine treatment promotes axon regeneration after SCI
ACKNOWLEDGMENTS
We thank members of the Sheng and Xu labs for constructive discussion; C. Chen (Institute of Biophysics, Chinese Academy of Science) for Grx1-roGFP2K, H. Dai performed behavioral assessments, G.M. Smith (Temple University) provided AAV9-mito-GFP, HiRet-tre-hM4Di-mCherry and AAV2-CMV-rtTAV16 vectors and technical consultation for the silencing study of regenerated CST axons, and the NINDS EM Facility. This work was supported in part by NIH 1R01 100531, 1R01 NS103481, Merit Review Award I01 BX002356, I01 BX003705, I01 RX002687 from the U.S. Department of Veterans Affairs, Indiana Spinal Cord and Brain Injury Research Foundation (No.19919), Mari Hulman George Endowment Funds (X-M. Xu); and the Intramural Research Program of NINDS, NIH ZIA NS003029, and ZIA NS002946 (Z-H. Sheng).
Abbreviations:
- 5-HT
serotonin
- ATP
adenosine triphosphate
- BDA
biotinylated dextran amine
- C5
5th cervical
- CK
creatine kinase
- CNO
Clozapine-n-oxide
- CNS
central nervous system
- CST
corticospinal tract
- CTB
cholera toxin subunit B
- DH
dorsal hemisection
- dpi
days post injury
- DREADDs
designer receptor exclusively activated by designer drugs
- EM
electron microscopy
- EMG
electromyography
- GFAP
glial fibrillary acidic protein
- ROS
reactive oxygen species
- Snph
syntaphilin
- SCI
spinal cord injury
- WT
wild-type
- TH
tyrosine hydroxylase
- Δψm
mitochondrial membrane potential
- wpi
weeks post injury
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Ahuja CS, Wilson JR, Nori S, Kotter MRN, Druschel C, Curt A, and Fehlings MG (2017). Traumatic spinal cord injury. Nature Reviews Disease Primers 3, 17018. [DOI] [PubMed] [Google Scholar]
- Al-Ali H, Ding Y, Slepak T, Wu W, Sun Y, Martinez Y, Xu XM, Lemmon VP, and Bixby JL (2017). The mTOR Substrate S6 Kinase 1 (S6K1) Is a Negative Regulator of Axon Regeneration and a Potential Drug Target for Central Nervous System Injury. J Neurosci 37, 7079–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, and Sofroniew MV (2016). Astrocyte scar formation aids central nervous system axon regeneration. Nature 532, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, Kim JH, James ND, Rogers A, Kato B, et al. (2018). Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asante CO, and Martin JH (2013). Differential Joint-Specific Corticospinal Tract Projections within the Cervical Enlargement. Plos One 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, and Freeman MR (2012). Wld(S) Prevents Axon Degeneration through Increased Mitochondrial Flux and Enhanced Mitochondrial Ca2+ Buffering. Current Biology 22, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azim E, and Alstermark B (2015). Skilled forelimb movements and internal copy motor circuits. Current Opinion in Neurobiology 33, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, and Popovich PG (2006). Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23, 635–659. [DOI] [PubMed] [Google Scholar]
- Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, and Freret T (2009). The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc 4, 1560–1564. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, and McMahon SB (2006). Spinal cord repair strategies: why do they work? Nature Reviews Neuroscience 7, 644–653. [DOI] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW, and Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 13, 183–193. [DOI] [PubMed] [Google Scholar]
- Cafferty WB, McGee AW, and Strittmatter SM (2008). Axonal growth therapeutics: regeneration or sprouting or plasticity? Trends Neurosci 31, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartoni R, Norsworthy MW, Bei F, Wang C, Li S, Zhang Y, Gabel CV, Schwarz TL, and He Z (2016). The Mammalian-Specific Protein Armcx1 Regulates Mitochondrial Transport during Axon Regeneration. Neuron 92, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case LC, and Tessier-Lavigne M (2005). Regeneration of the adult central nervous system. Current Biology 15, R749–R753. [DOI] [PubMed] [Google Scholar]
- Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, et al. (2014). Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death &Amp; Disease 5, e1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CC, Gilmore A, and Zuo Y. (2014). Study motor skill learning by single-pellet reaching tasks in mice. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, and Sheng Z-H (2013). Kinesin-1–syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. The Journal of Cell Biology 202, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau K-F, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, et al. (2007). Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Human Molecular Genetics 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Ruan Y, Chen C, Frye CC, Xiong W, Jin X, Jones K, Sengelaub D, and Xu X-M (2016). Characterization of dendritic morphology and neurotransmitter phenotype of thoracic descending propriospinal neurons after complete spinal cord transection and GDNF treatment. Exp Neurol 277, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, and Kittler JT (2018). Mitochondria at the neuronal presynapse in health and disease. Nature Reviews Neuroscience 19, 63. [DOI] [PubMed] [Google Scholar]
- Dias DO, Kim H, Holl D, Werne Solnestam B, Lundeberg J, Carlen M, Goritz C, and Frisen J (2018). Reducing Pericyte-Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, and Dumont AS (2001). Acute Spinal Cord Injury, Part I: Pathophysiologic Mechanisms. Clinical Neuropharmacology 24, 254–264. [DOI] [PubMed] [Google Scholar]
- Farr Tracy D, and Whishaw Ian Q (2002). Quantitative and Qualitative Impairments in Skilled Reaching in the Mouse (Mus musculus) After a Focal Motor Cortex Stroke. Stroke 33, 1869–1875. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, and Sofroniew MV (2004). Reactive Astrocytes Protect Tissue and Preserve Function after Spinal Cord Injury. The Journal of Neuroscience 24, 2143–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Curt A, Steeves JD, Coleman WP, Tuszynski MH, Lammertse D, Bartlett PF, Blight AR, Dietz V, Ditunno J, et al. (2006). Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal Cord 45, 190. [DOI] [PubMed] [Google Scholar]
- Filbin MT (2006). Recapitulate development to promote axonal regeneration: good or bad approach? Philosophical transactions of the Royal Society of London Series B, Biological sciences 361, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filli L, Zorner B, Weinmann O, and Schwab ME (2011). Motor deficits and recovery in rats with unilateral spinal cord hemisection mimic the Brown-Sequard syndrome. Brain 134, 2261–2273. [DOI] [PubMed] [Google Scholar]
- Geoffroy CG, Lorenzana AO, Kwan JP, Lin K, Ghassemi O, Ma A, Xu N, Creger D, Liu K, He Z, et al. (2015). Effects of PTEN and Nogo Codeletion on Corticospinal Axon Sprouting and Regeneration in Mice. J Neurosci 35, 6413–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, De Camilli P, and Ferguson SM (2015). Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kalambogias J, Yoshioka S, Han W, Li Z, Kawasawa YI, Pochareddy S, Li Z, Liu F, Xu X, et al. (2017). Control of species-dependent cortico-motoneuronal connections underlying manual dexterity. Science 357, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP (2008). Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5, 553–559. [DOI] [PubMed] [Google Scholar]
- Han SM, Baig HS, and Hammarlund M (2016). Mitochondria Localize to Injured Axons to Support Regeneration. Neuron 92, 1308–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel NY, and Strittmatter SM (2006). Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nature Reviews Neuroscience 7, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, and Jin Y (2016). Intrinsic Control of Axon Regeneration. Neuron 90, 437–451. [DOI] [PubMed] [Google Scholar]
- Heffner RS, and Masterton RB (1983). The Role of the Corticospinal Tract in the Evolution of Human Digital Dexterity. Brain, Behavior and Evolution 23, 165–183. [DOI] [PubMed] [Google Scholar]
- Hilton BJ, and Bradke F (2017). Can injured adult CNS axons regenerate by recapitulating development? Development 144, 3417–3429. [DOI] [PubMed] [Google Scholar]
- Huang Z, Gao Y, Sun Y, Zhang C, Yin Y, Shimoda Y, Watanabe K, and Liu Y (2016). NB-3 signaling mediates the cross-talk between post-traumatic spinal axons and scar-forming cells. EMBO J 35, 1745–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubley MJ, Locke BR, and Moerland TS (1996). The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochimica et Biophysica Acta (BBA) - General Subjects 1291, 115–121. [DOI] [PubMed] [Google Scholar]
- Hurd C, Weishaupt N, and Fouad K (2013). Anatomical correlates of recovery in single pellet reaching in spinal cord injured rats. Experimental neurology 247, 605–614. [DOI] [PubMed] [Google Scholar]
- Kaasik A (2016). Mitochondrial Mobility and Neuronal Recovery. N Engl J Med 375, 1295–1296. [DOI] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, and Sheng ZH. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A, Ong Tone S, and Fournier AE (2015). Extrinsic and intrinsic regulation of axon regeneration at a crossroads. Front Mol Neurosci 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu-Seo S, Tamada H, Kato Y, Yasuda K, Ishihara N, Nomura M, Mihara K, and Kiyama H (2016). Mitochondrial fission is an acute and adaptive response in injured motor neurons. Scientific Reports 6, 28331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman WJH, Visch H-J, Smeitink JAM, and Willems PHGM (2006). Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry Part A 69A, 1–12. [DOI] [PubMed] [Google Scholar]
- Lee JK, Chow R, Xie F, Chow SY, Tolentino KE, and Zheng BH (2010). Combined Genetic Attenuation of Myelin and Semaphorin-Mediated Growth Inhibition Is Insufficient to Promote Serotonergic Axon Regeneration. J Neurosci 30, 10899–10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon S-K, Losonczy A, and Polleux F (2016). Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Current biology : CB 26, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MY, Cheng XT, Tammineni P, Xie Y, Zhou B, Cai Q, and Sheng ZH (2017). Releasing Syntaphilin Removes Stressed Mitochondria from Axons Independent of Mitophagy under Pathophysiological Conditions. Neuron 94, 595–610 e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. (2010). PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang X, Li W, Zhang Q, Li Y, Zhang Z, Zhu J, Chen B, Williams PR, Zhang Y, et al. (2017). A Sensitized IGF1 Treatment Restores Corticospinal Axon-Dependent Functions. Neuron 95, 817–833 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang X, Lu C-C, Sherman-Kermen R, Steward O, Xu X-M, and Zou Y (2008). Repulsive Wnt Signaling Inhibits Axon Regeneration after CNS Injury. The Journal of Neuroscience 28, 8376–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P, Woodruff G, Wang Y, Graham L, Hunt M, Wu D, Boehle E, Ahmad R, Poplawski G, Brock J, et al. (2014). Long-distance axonal growth from human induced pluripotent stem cells after spinal cord injury. Neuron 83, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz GAS, Merkler D, Dietz V, Schwab ME, and Fouad K (2000). Efficient testing of motor function in spinal cord injured rats. Brain Research 883, 165–177. [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, and Goldberg JL (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell KC, Vargas ME, and Sagasti A (2013). WldS and PGC-1alpha regulate mitochondrial transport and oxidation state after axonal injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 14778–14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea TM, Burda JE, and Sofroniew MV (2017). Cell biology of spinal cord injury and repair. The Journal of clinical investigation 127, 3259–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi K, Hollis E, and Zou Y (2014). Axon guidance and injury—lessons from Wnts and Wnt signaling. Current Opinion in Neurobiology 27, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallini R, Fernandez E, and Sbriccoli A (1988). Retrograde Degeneration of Corticospinal Axons Following Transection of the Spinal-Cord in Rats - a Quantitative Study with Anterogradely Transported Horseradish-Peroxidase. Journal of Neurosurgery 68, 124–128. [DOI] [PubMed] [Google Scholar]
- Ramer LM, Ramer MS, and Bradbury EJ (2014). Restoring function after spinal cord injury: towards clinical translation of experimental strategies. Lancet Neurology 13, 1241–1256. [DOI] [PubMed] [Google Scholar]
- Rawson RL, Yam L, Weimer RM, Bend EG, Hartwieg E, Horvitz HR, Clark SG, and Jorgensen EM (2014). Axons degenerate in the absence of mitochondria in C. elegans. Current biology : CB 24, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruhashi Y, Young W, and Perkins R (1996). The recovery of 5-HT immunoreactivity in lumbosacral spinal cord and locomotor function after thoracic hemisection. Exp Neurol 139, 203–213. [DOI] [PubMed] [Google Scholar]
- Scholpa NE, and Schnellmann RG (2017). Mitochondrial-Based Therapeutics for the Treatment of Spinal Cord Injury: Mitochondrial Biogenesis as a Potential Pharmacological Target. Journal of Pharmacology and Experimental Therapeutics 363, 303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh IS, Keefe KM, Sterling NA, Junker IP, Eneanya CI, Liu Y, Tang X-Q, and Smith GM (2018). Retrogradely Transportable Lentivirus Tracers for Mapping Spinal Cord Locomotor Circuits. Frontiers in Neural Circuits 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z-H (2014). Mitochondrial trafficking and anchoring in neurons: New insight and implications. The Journal of Cell Biology 204, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z-H (2017). The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends in Cell Biology 27, 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, and Cai Q (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver J, and Miller JH (2004). Regeneration beyond the glial scar. Nat Rev Neurosci 5, 146–156. [DOI] [PubMed] [Google Scholar]
- Smit-Rigter L, Rajendran R, Silva, Catia AP, Spierenburg L, Groeneweg F, Ruimschotel, Emma M, van Versendaal D, van der Togt C, Eysel Ulf T., Heimel JA, et al. (2016). Mitochondrial Dynamics in Visual Cortex Are Limited In Vivo and Not Affected by Axonal Structural Plasticity. Current Biology 26, 2609–2616. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV (2018). Dissecting spinal cord regeneration. Nature 557, 343–350. [DOI] [PubMed] [Google Scholar]
- Steward O, and Willenberg R (2017). Rodent spinal cord injury models for studies of axon regeneration. Exp Neurol 287, 374–383. [DOI] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan P-Y, Chen Y, and Sheng Z-H (2013). Motile Axonal Mitochondria Contribute to the Variability of Presynaptic Strength. Cell Rep 4, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takihara Y, Inatani M, Eto K, Inoue T, Kreymerman A, Miyake S, Ueno S, Nagaya M, Nakanishi A, Iwao K, et al. (2015). In vivo imaging of axonal transport of mitochondria in the diseased and aged mammalian CNS. Proceedings of the National Academy of Sciences 112, 10515–10520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky MA, and Beal MF (2001). Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol 49, 561–574. [PubMed] [Google Scholar]
- Titsworth WL, Onifer SM, Liu NK, and Xu XM (2007). Focal phospholipases A2 group III injections induce cervical white matter injury and functional deficits with delayed recovery concomitant with Schwann cell remyelination. Exp Neurol 207, 150–162. [DOI] [PubMed] [Google Scholar]
- Tran AP, Warren PM, and Silver J (2018). The Biology of Regeneration Failure and Success After Spinal Cord Injury. Physiological reviews 98, 881–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, and Steward O (2012). Concepts and methods for the study of axonal regeneration in the CNS. Neuron 74, 777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Nakamura Y, Li J, Gu ZR, Niehaus J, Maezawa M, Crone SA, Goulding M, Baccei ML, and Yoshida Y (2018). Corticospinal Circuits from the Sensory and Motor Cortices Differentially Regulate Skilled Movements through Distinct Spinal Interneurons. Cell Rep 23, 1286-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnoni A, and Bullock SL (2018). A cAMP/PKA/Kinesin-1 Axis Promotes the Axonal Transport of Mitochondria in Aging Drosophila Neurons. Current Biology 28, 1265–1272.e1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MJ, Walker CL, Zhang YP, Shields LB, Shields CB, and Xu XM (2015). A novel vertebral stabilization method for producing contusive spinal cord injury. J Vis Exp, e50149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu NK, Zhang YP, Deng L, Lu QB, Shields CB, Walker MJ, Li J, and Xu XM (2015). Treadmill training induced lumbar motoneuron dendritic plasticity and behavior recovery in adult rats after a thoracic contusive spinal cord injury. Exp Neurol 271, 368–378. [DOI] [PubMed] [Google Scholar]
- Wang X, Hu J, She Y, Smith GM, and Xu X-M (2014). Cortical PKC Inhibition Promotes Axonal Regeneration of the Corticospinal Tract and Forelimb Functional Recovery After Cervical Dorsal Spinal Hemisection in Adult Rats. Cerebral Cortex 24, 3069–3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Liu Y, Li X, Zhang Z, Yang H, Zhang Y, Williams PR, Alwahab NSA, Kapur K, Yu B, et al. (2017). Deconstruction of Corticospinal Circuits for Goal-Directed Motor Skills. Cell 171, 440–455.e414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu W, Wu X, Sun Y, Zhang YP, Deng L-X, Walker MJ, Qu W, Chen C, Liu N-K, et al. (2018). Remodeling of lumbar motor circuitry remote to a thoracic spinal cord injury promotes locomotor recovery. eLife 7, e39016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whishaw IQ, Pellis SM, Gorny B, Kolb B, and Tetzlaff W (1993). Proximal and distal impairments in rat forelimb use in reaching follow unilateral pyramidal tract lesions. Behavioural Brain Research 56, 59–76. [DOI] [PubMed] [Google Scholar]
- Wyss M, and Kaddurah-Daouk R (2000). Creatine and creatinine metabolism. Physiol Rev 80, 1107–1213. [DOI] [PubMed] [Google Scholar]
- Xu XM, Guenard V, Kleitman N, and Bunge MB (1995). Axonal regeneration into Schwann cell-seeded guidance channels grafted into transected adult rat spinal cord. The Journal of comparative neurology 351, 145–160. [DOI] [PubMed] [Google Scholar]
- Yiu G, and He Z (2006). Glial inhibition of CNS axon regeneration. Nature Reviews Neuroscience 7, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Yu P, Lin M-Y, Sun T, Chen Y, and Sheng Z-H (2016). Facilitation of axon regeneration by enhancing mitochondrial transport and rescuing energy deficits. The Journal of Cell Biology 214, 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or codes.