

Abstract
After subarachnoid haemorrhage, prolonged exposure to toxic extracellular haemoglobin occurs in the brain. Here, we investigate the role of haemoglobin neurotoxicity in vivo and its prevention. In humans after subarachnoid haemorrhage, haemoglobin in cerebrospinal fluid was associated with neurofilament light chain, a marker of neuronal damage. Most haemoglobin was not complexed with haptoglobin, an endogenous haemoglobin scavenger present at very low concentration in the brain. Exogenously added haptoglobin bound most uncomplexed haemoglobin, in the first 2 weeks after human subarachnoid haemorrhage, indicating a wide therapeutic window. In mice, the behavioural, vascular, cellular and molecular changes seen after human subarachnoid haemorrhage were recapitulated by modelling a single aspect of subarachnoid haemorrhage: prolonged intrathecal exposure to haemoglobin. Haemoglobin-induced behavioural deficits and astrocytic, microglial and synaptic changes were attenuated by haptoglobin. Haptoglobin treatment did not attenuate large-vessel vasospasm, yet improved clinical outcome by restricting diffusion of haemoglobin into the parenchyma and reducing small-vessel vasospasm. In summary, haemoglobin toxicity is of clinical importance and preventable by haptoglobin, independent of large-vessel vasospasm.
Keywords: subarachnoid haemorrhage, haptoglobin, haemoglobin, neurofilament light chain, outcome
Extracellular haemoglobin is toxic; its contribution to neuronal damage in vivo was unknown. Garland et al show that exposing the brain to haemoglobin recapitulates the clinicopathological features of subarachnoid haemorrhage. These features are preventable by administration of haptoglobin, which keeps haemoglobin out of the brain tissue, limiting neuronal damage.
Graphical Abstract
Graphical Abstract.
Introduction
After subarachnoid haemorrhage (SAH), blood is released into the subarachnoid space (Macdonald and Schweizer, 2017). As the blood clot in the subarachnoid space is resorbed, red blood cell lysis leads to the release of cell-free haemoglobin (Hb) over a protracted period exerting a further secondary, and thus potentially reversible, injury. There is a sound biological rationale to hypothesize that prolonged exposure to Hb in the subarachnoid compartment impacts long-term outcome (Bulters et al., 2018). Outside of the controlled environment of the erythrocyte, Hb and its breakdown products (haem, bilirubin and free iron) are involved in toxic redox reactions via the iron atom (Reeder, 2010, 2017), leading to oxidation of DNA, protein and lipids, and hence cellular dysfunction, and brain injury.
Focal intracortical injection of a high concentration of Hb causes seizures (Rosen and Frumin, 1979). Similar injections of whole blood in the striatum of mice cause neurologic deficits such as posturing, incoordination and/or paresis (Zhao et al., 2009). Less is known about the neurological consequences of prolonged exposure to lower concentrations of Hb distributed throughout the CSF space, as happens during clot lysis after SAH. Repeated cisterna magna injections of washed autologous red blood cells in rabbits caused iron deposition and a microglial reaction in the cerebellum, but neuronal integrity and behavioural deficits were not studied (Koeppen and Borke, 1991). A single intraventricular Hb injection in 7-day-old neonatal rats caused neuronal damage (Garton et al., 2016), so a similar process could occur after prolonged exposure to subarachnoid Hb in adults.
Haptoglobin (Hp) is the body’s natural defence against extracellular Hb (Andersen et al., 2017). Hp lowers the redox potential of Hb to prevent damaging peroxidative reactions, and chaperones Hb to prevent haem release and its degradation to free iron. Intrathecal Hp concentration is significantly lower compared with plasma, and most of the Hb after SAH is not bound to Hp (Galea et al., 2012). Therefore, direct supplementation with exogenous Hp, which would be expected to increase CNS Hp concentration more rapidly and to a higher level than drug-induced upregulation of Hp synthesis, might be expected to be of therapeutic benefit after SAH.
This project consists of human, in vitro and in vivo animal studies, presented in this order. First, we sought to identify whether tissue damage was linked to Hb after SAH in humans. Then, in experimental models, we tested whether prolonged intrathecal exposure to pure Hb is neurotoxic and causes behavioural abnormalities similar to those seen after human SAH. To model prolonged intrathecal Hb exposure, the mouse ventricular system was infused with a clinically relevant Hb concentration for 2 weeks. We studied behavioural deficits, large-vessel and parenchymal vasospasm, the astrocytic and microglial/macrophage reaction to Hb, and synaptic loss. Finally, we assessed if these changes were reversed by administration of Hp.
Materials and methods
Human study
Human studies were performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki. Nineteen control participants were patients with non-inflammatory non-haemorrhagic conditions who underwent lumbar puncture and were subsequently found to have normal CSF (National Research Ethics Committee approval number 11/SC/0204). Forty-four Fisher grade III–IV non-traumatic SAH patients, requiring an external ventricular drain (EVD) as part of their clinical management (to manage acute hydrocephalus), were recruited (National Research Ethics Committee approval number 12/SC/0666). CSF was obtained from the EVD on alternate days until removal of the EVD. CSF was drawn from a three-way tap connecting the ventricular catheter (∼30 cm long) to the tubing leading to an external CSF drainage and monitoring system (Becker®, Medtronic). For sampling, the tap was opened to the ventricular catheter and closed to the drainage system. The first 3 ml of CSF (representing dead space) was discarded to ensure fresh CSF was obtained. Only 42 patients contributed to this study due to early removal of the EVD. CSF was centrifuged at 1500 rcf for 10 min at 21°C and frozen within 1 h of sampling.
Ultra-performance liquid chromatography (UPLC) was used to separate CSF components in a tris-saline mobile phase, coupled to absorbance measurement at 415 nm to identify haem-containing species. Full details are in Supplementary material. Hb was measured in CSF before and after saturating with Hp (Bio Products Laboratory Limited, Elstree, UK) to measure Hp-bindable and Hp-unbindable uncomplexed Hb (Fig. 1). CSF neurofilament light chain (NFL) was measured by enzyme-linked immunosorbent assay (UmanDiagnostics, Umea, Sweden; Supplementary material).
Figure 1.

Human study: methodology. Assay of Hb binding by Hp. (A) UPLC method. CSF alone, or CSF incubated with excess Hp, was run through a C18 column to achieve separation of proteins. The absorbance of the eluate was measured at 415 nm to detect haem-containing species. (B) A representative CSF chromatogram demonstrating peaks of Hb complexed to dimeric and polymeric Hp, haem complexed to haemopexin and albumin, and uncomplexed Hb. Peak area under the curve was quantified using a Hb standard curve. (C) Uncomplexed Hb able to bind Hp was measured by running CSF alone (orange), and separately CSF to which excess Hp was added (grey). The increase in complexed Hb peak area after adding excess Hp represents uncomplexed Hb that can bind Hp. The uncomplexed Hb peak remaining after adding excess Hp represents uncomplexed Hb unable to bind Hp.
In vitro study
Primary hippocampal neurones were cultured according to a modified method from the Ittner lab (Fath et al., 2009; Garland et al., 2012; Supplementary material). Cultured neurones were challenged with Hb at day 7 until day 14. Over the 7 days, Hb concentration was maintained during media changes. An equimolar-binding ratio of Hp (see Supplementary material) was added concomitantly for Hp treatment experiments. Hb and Hp preparations, immunocytochemistry for microtubule-associated protein-2 and quantification of viable neurones are detailed in Supplementary material.
In vivo animal model
Mouse model of prolonged intrathecal Hb exposure
To model prolonged intrathecal Hb exposure, a 2-week intracerebroventricular infusion of Hb was used. Locally bred male C57BL/6 mice were housed at 21°C under a 12-h light/12-h dark cycle in a conventional animal research facility and allowed access to food and water ad libitum; surgery was performed at 10–12 weeks of age. All procedures were performed under UK Home Office licence 30-3057 and all applicable international, national and/or institutional guidelines for the care and use of animals were followed. Alzet osmotic minipumps (Model 2002, #296, 2-week infusion, 0.5 µl/h, Charles River, Harlow, Essex) were loaded with vehicle (0.9% saline), Hb, Hb + Hp or Hp. Treatment allocation was random. The infusate Hb concentration was 20 mg/ml to achieve a clinically relevant CSF Hb concentration of 10 µM as detailed in Supplementary material. The Hp concentration was 14 mg/ml to achieve 1:1 Hb to Hp binding stoichiometry, guided by data from empirical determination of Hb to Hp binding ratio (Supplementary material). The minipump was attached to a 30-gauge ventricular cannula using vinyl tubing (Brain Infusion Kit 3, #8851, Charles River, Harlow, Essex) and primed with test solution according to the manufacturer’s instructions. Mice were anaesthetized using a ketamine (Bayer, Reading, UK) and xylazine (Pfizer, Tadworth, Surrey, UK) mixture (100 and 10 mg/kg, respectively). The head was immobilized in a stereotactic frame. Mice body temperature was thermostatically controlled with a rectal probe and a heated pad. An aseptic technique was used. Blunt dissection was used to create a subcutaneous pocket in the interscapular region for insertion of the minipump. A burr hole was drilled and the ventricular cannula inserted at the following coordinates from bregma: anteroposterior, −0.4 mm; lateral, 1 mm; depth, 2.5 mm. Cyanoacrylate was used to secure the cannula holder to the skull surface. Buprenorphine (Centaur, Castle Cary, Somerset, UK) was administered immediately following surgery (0.5 mg/kg subcutaneously) and the next morning in 5 ml fruit jelly (300 μg/45 ml). Mice were housed individually following surgery for 14 days so the experimental unit was a single animal. Mice were checked twice in the first 24 h and daily thereafter. Residual volume in the minipumps was systematically examined after explantation, at the end of the 2 weeks, to ensure the appropriate volume of 200 µl was delivered while in situ. Correct placement of the ventricular cannula was confirmed during tissue sectioning by observing the cannula track.
Behavioural, histological and biochemical readouts
Open-field behavioural analysis was performed in a laser-monitored behaviour arena (Med Associates, Fairfax, VA, USA) over a 5-min period, twice a day at the same time in the morning and afternoon, as detailed in Supplementary material. At 14 days post-surgery, mice were transcardially perfused with heparinized saline (1 unit/ml, LEO Pharma, Hurley, Berkshire, UK), followed by 4% paraformaldehyde in 0.1 M phosphate buffer. Brains were removed and post-fixed in 4% paraformaldehyde overnight at 4°C and then embedded in paraffin wax. Coronal sections were cut at 10 µm, dewaxed in xylene and rehydrated through graded ethanol/water solutions. Immunohistochemistry and imaging for glial fibrillary acidic protein (GFAP), ionized calcium-binding adapter molecule 1 (Iba1), synaptophysin, Hb, CD163 and smooth-muscle actin (SMA), and Perl’s histochemistry was performed as detailed in Supplementary material. To ensure blinding, all samples were coded prior to image capture and analysis. All analyses were performed by two-dimensional quantification, averaged over two sections per animal within each area studied, and data are presented per mm2, unless otherwise indicated; further details are in Supplementary material. For biochemistry, the hemisphere ipsilateral to cannula insertion was homogenized in buffer, after transcardial perfusion with heparinized saline only and tissue processed for Western blotting as detailed in Supplementary material.
Fluorescent imaging of Circle of Willis in situ
The method for in situ imaging was established in the Zipfel group (Aum et al., 2017). Mice were anaesthetized using a ketamine (Bayer, Reading, UK) and xylazine (Pfizer, Tadworth, Surrey, UK) mixture (100 and 10 mg/kg, respectively) and transcardially perfused through the left ventricle with 10 mM glucose–physiological-buffered saline (PBS) followed by 20 ml 20 μM 5-(6)-carboxy-X-rhodamine, succinimidyl ester (Sigma-Aldrich, Gillingham, Dorset, UK) dye in 10 mM glucose-PBS prior to perfusions with 4% paraformaldehyde (in PBS). All perfusions were performed with solutions at 21°C, at a constant pressure of 80 ± 2 mmHg using a GE Druck DP1705. Animals were decapitated, the calvaria removed and post-fixed in 4% paraformaldehyde in the dark at 4°C for 24 h. Brains were removed under a dissection microscope to preserve the basal arteries. Then, brains were placed en bloc on a glass coverslip and gently covered in PBS before placing on the stage of a confocal laser scanning microscope (SP8, Leica, Wetzlar, Germany). Measurements of anterior and middle cerebral artery (MCA) diameters were made at the narrowest point across the first millimetre of the vessel, as detailed in Supplementary material.
Surface plasmon resonance
Surface plasmon resonance analysis was carried out as described (Madsen et al., 2004) using a Biacore 3000 instrument (Biacore, Uppsala, Sweden). Mouse or human CD163, produced using recombinant technology in-house (Kristiansen et al., 2001; Etzerodt et al., 2013), was immobilized in 10 mM sodium acetate (pH 4) and remaining binding sites were blocked with 1 M ethanolamine (pH 8.5). The resulting densities were: human CD163 0.0468 pmol/mm2 and murine CD163 0.0320 pmol/mm2. Sensorgrams were generated using the running buffer CaHBS with 2 mM free Ca2Cl2 (10 mM Hepes, 150 mM NaCl, 3.0 mM CaCl2, 1.0 mM EGTA + 0.005% P20, and pH 7.4) and the protein concentrations: 5 µg/ml of mouse and human Hb (Sigma-Aldrich, Gillingham, Dorset, UK); 7.5 µg/ml of mouse Hp (MyBioSource, San Diego, Ca) and human Hp1-1 (Sigma-Aldrich, Gillingham, Dorset, UK). The flow cell was regenerated in 10 mM glycine, 20 mM EDTA, 500 mM NaCl + 0.005% P20, pH4.0 between the runs. All experiments were conducted at least in triplicate and data was evaluated using the BiaEvaluation ver. 4.1 software (Biacore, Uppsala, Sweden).
Statistical analysis
Statistical analysis and graph preparation were performed using SPSS (v24) and GraphPad Prism (v7.01), with data expressed as mean ± standard error of the mean (SEM), median ± inter-quartile range or 95% confidence intervals. Normality and heteroscedasticity were routinely determined across all data sets. Where necessary, logarithmic transformation was used to normalize data. Alpha (α), the probability of a Type I error, was 0.05. Two-tailed hypotheses were considered throughout. Details of individual statistical analyses are available in Supplementary material. Animals were randomly selected and sequentially assigned to treatment groups. Animal in vivo experiments were reported according to ARRIVE guidelines (Kilkenny et al., 2010). Supplementary Table 1 summarizes all the analyses performed and the results.
Data availability
Datasets are available from the corresponding author on reasonable request in accordance with the University of Southampton’s data-sharing policies, ethical approvals and contracts with the co-authors and their institutions.
Results
Human study
NFL concentration in CSF is predicted by Hb
The ideal test of the hypothesis that exposure of the brain to Hb affects outcome after SAH in humans is to measure Hb in the CSF serially and relate this to outcome. However, this poses a significant practical challenge since it is difficult to sample CSF with sufficient frequency and duration to arrive at a reliable estimate of total Hb exposure over the course of clot lysis, a process taking up to a month (Naff et al., 2001). Therefore, we focussed on an initial 2-week time interval, during which we serially sampled CSF and investigated whether there was a temporal relationship between Hb and NFL, a marker of neuronal damage (Siedler et al., 2014). We hypothesized that within the 2-week sampling window, peak NFL in the CSF could be predicted by the preceding peak Hb. Serial CSF samples were collected from 42 patients with Fisher grade III–IV SAH via an EVD. Control CSF was collected by lumbar puncture in patients with non-inflammatory non-haemorrhagic neurological symptoms. Demographic and clinical characteristics are shown in Table 1. NFL measured by enzyme-linked immunosorbent assay was higher in SAH CSF versus control CSF (Fig. 2A). This difference occurred even though there is a ventriculo-lumbar gradient in CSF NFL, with lumbar CSF concentration being higher (Jeppsson et al., 2013). CSF NFL levels after SAH had a wide distribution, some within the reference range (Fig. 2A). CSF samples were analysed using UPLC for Hb species as detected by absorbance in the Soret band at 415 nm to quantify total Hb, irrespective of oxidation state (Fig. 1A and B). Total Hb increased gradually from the third-day post-ictus onwards, peaking at day 11 (Fig. 2B) and reaching a plateau between days 11 and 13. There was a small initial spike in CSF NFL level, followed by a gradual rise (Fig. 2B). The gradual rise in NFL (day 5 onwards) followed the rise in Hb (day 3 onwards; Fig. 2B). When comparing patients with high versus normal peak CSF NFL levels, the former had significantly higher CSF Hb levels preceding the peak CSF NFL (Fig. 2C). Peak NFL concentration in serial CSF samples was predicted by the peak Hb level preceding it, controlling for initial neurological state as determined by the World Federation of Neurological Surgeons (WFNS; Teasdale et al., 1988), age and sex (Fig. 2D).
Table 1.
Demographic and clinical characteristics of the clinical cohort
| Non-traumatic SAH patients | Control patients | |
|---|---|---|
| Number | 44 | 19 |
| Age (years)a | 59.8 ± 12.3 | 55.2 ± 25.9 |
| Genderb | ||
| Male | 15 (34%) | 5 (26%) |
| Female | 29 (66%) | 14 (79%) |
| Hypertensionb | ||
| Yes | 23 (52.3%) | |
| No | 21 (47.7%) | |
| Initial WFNS scoreb | ||
| 1 | 5 (11.4%) | |
| 2 | 10 (18%) | |
| 3 | 6 (13.6%) | |
| 4 | 15 (29.5%) | |
| 5 | 8 (13.6%) | |
| Fisher gradeb | ||
| 3 | 2 (4.5%) | |
| 4 | 42 (95.5%) | |
| Aneurysmal managementb | ||
| Coiled | 32 (72.7%) | |
| Clipped | 5 (11.4%) | |
| Supportive | 6 (13.6%) | |
| Aneurysm locationb | ||
| Anterior circulation | 32 (72.7%) | |
| Posterior circulation | 7 (15.9%) | |
| Perimesencephalic | 4 (9.1%) | |
| Unknown | 1 (2.3%) |
The WFNS score describes the initial neurological condition.
Mean and standard deviation.
Number and %.
Figure 2.

Human study. Hb predicts NFL concentration in the CSF of SAH patients. (A) Peak NFL concentration is significantly higher in the CSF of SAH patients (n = 42) versus a non-inflammatory and non-haemorrhagic control population (n = 19). Upper and lower limits were derived from the Ln normal distribution of the control population (mean ± 95% CI, unpaired t-test). (B) The CSF concentration of total Hb and NFL increase over a 2-week period post-ictus [n = 42; median ± inter-quartile range (IQR, shaded zone)]. (C) Peak Hb concentration preceding peak NFL concentration is significantly higher in SAH patients with high NFL (n = 30) versus SAH patients with normal NFL (n = 12; mean ± 95% CI, unpaired t-test). (D) Multivariable linear regression of peak NFL on preceding peak total Hb, controlling for WFNS, age and sex [n = 42, B = 0.364 (95% CI: 0.156–0.573), P = 0.001]. (E) Uncomplexed Hb (Hb not bound to Hp). (F) Uncomplexed Hb that can bind Hp (E, F) n = 42, median with IQR). (A, C) P-values: **P < 0.01, ****P < 0.0001.
An opportunistic therapeutic window for intrathecal Hp supplementation after SAH
Hp is present at low levels in CSF (Galea et al., 2012), so we set out to find the percentage of Hb complexed to Hp after SAH. Hb–Hp complexes could be differentiated from uncomplexed Hb, due to the difference in retention time on the column, and a corresponding difference in peak size (Fig. 1B). Most of the Hb was uncomplexed [median 96.3%, inter-quartile range (IQR): 83.3–99.4%, from third day onwards, Fig. 2E].
The prolonged presence of Hb in a pro-inflammatory acidotic environment is particularly conducive to denaturing modifications of Hb, rendering it less able to bind Hp (Vallelian et al., 2008). Modified Hb forms have been detected in CSF after SAH (Reeder et al., 2002) and their formation has the potential to interfere with the therapeutic potential of Hp. Therefore, we asked whether the uncomplexed Hb in the CSF after SAH could be bound by exogenous Hp. CSF was first run on UPLC on its own. Then exogenous Hp was added in excess of Hb-binding stoichiometry and the mixture run on UPLC to determine the proportion of uncomplexed Hb which could be bound by Hp (Fig. 1C). When exogenous Hp was added to the CSF samples, most of the uncomplexed Hb formed a complex with Hp: 90.6% (IQR: 65.8–96.5%) from the third day onwards (Fig. 2F). Similarly, 76.4% (IQR: 67.7–96.4%) of uncomplexed Hb could form a complex with Hp during the day 11–13 plateau. Therefore, most uncomplexed Hb in the CSF was able to bind Hp in the first 2 weeks after ictus. This represents a therapeutic opportunity for intrathecal Hp supplementation after SAH.
In vitro study
Hp attenuates the toxicity of Hb towards primary mouse neurones
Next, we wanted to confirm that Hp can protect against Hb toxicity. The toxicity of Hb to cortical neurones in mixed glial cultures from foetal mice has been observed previously (Regan and Panter, 1993). We used a protocol that allows culturing of primary hippocampal neurones from mouse pup brain at low density (Fath et al., 2009; Garland et al., 2012; Fig. 3A). Microtubule-associated protein 2 (MAP2) positive neurones with 4ʹ,6-diamidino-2-phenylindole, dihydrochloride (DAPI)-positive non-apoptotic nuclei (hence viable neurones) were counted following a 1-week challenge with increasing concentrations of mouse Hb. The number of viable neurones was found to be reduced after exposure to Hb concentrations above 1 μM (Fig. 3A–C). Concomitant addition of an equimolar amount of Hp completely neutralized this Hb toxicity (Fig. 3D).
Figure 3.
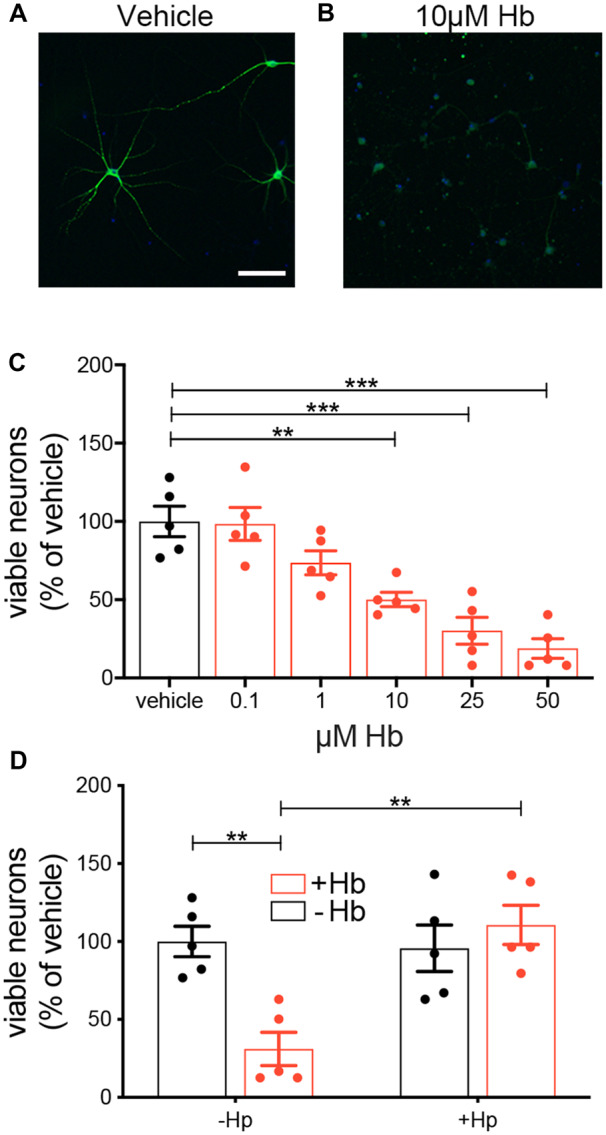
In vitro study. Hp reverses Hb toxicity towards mouse hippocampal neurones. (A, B) Representative images of MAP2-positive neurones challenged for 1 week with vehicle or 10 µM mouse Hb. (C) Increasing concentration of Hb reduces the number of MAP2-positive neurones (n = 5 experiments each with duplicate wells per condition, mean ± SEM, one-way ANOVA P < 0.0001, P-values for multiple comparisons are from Dunnett’s test). (D) An equimolar-binding ratio of Hp fully reverses a 1-week 15 µM Hb challenge (n = 5 experiments each with duplicate wells per condition, mean ± SEM). Two-way ANOVA: Hb × Hp interaction P = 0.0032. (A) Scale bar = 100 μm. (C, D) P-values: **P < 0.01, ***P < 0.001.
In vivo animal model
Prolonged intrathecal exposure to Hb causes behavioural deficits, which are attenuated by Hp
A therapeutic potential for Hp has been strongly suggested by experiments in an animal model of focal intraparenchymal cerebral haemorrhage, in which Hp overexpression reversed some of the acute neurological deficits after a large (12 µl) intrastriatal injection of whole blood (Zhao et al., 2009). This study provided valuable information, yet the mechanical and inflammatory consequences of a large intraparenchymal injection, and the associated plasma proteins, cell membranes, and white cell constituents, meant that it could not be definitely concluded that Hp targeted the Hb component. Moreover, in the same study Hp expression was manipulated indirectly in vivo (Zhao et al., 2009), making it hard to be absolutely certain that Hp was the neuroprotective agent. In order to model the chronic Hb exposure aspect of SAH in isolation, as opposed to the whole SAH condition, adult male C57BL6 mice underwent insertion of an intraventricular cannula attached to a subcutaneously implanted osmotic minipump; this protocol delivered Hb slowly over 2 weeks (Fig. 4A and B). Male mice were used since (i) sex did not affect Hb neurotoxicity in the human studies [Fig. 2D, n = 42, B = 0.044 (95% CI:−0.742 to 0.822), P = 0.918] and (ii) variability in planned behavioural measures is greater in females, due to the oestrous cycle (Gray and Cooney, 1982). The concentration of Hb was selected to achieve a clinically relevant concentration of 10 μM, based on the median total Hb observed in CSF from SAH patients. In vitro incubation of Hb or Hb + Hp complexes at 37°C for 2 weeks showed that Hb did not degrade to release iron, as measured by a free iron assay (Supplementary Fig. 1). The general health of animals in this model was assessed, including daily measurements of weight (Fig. 4C). Perls’ staining of brain sections showed iron deposition within the parenchyma and over the convexities (Fig. 4D), the latter confirming circulation of Hb out of the ventricles, into the subarachnoid space, and over the outer surface of the brain.
Figure 4.

In vivo animal model: behaviour. Hp reverses behavioural deficits resulting from intra-cerebroventricular infusion of Hb. (A) Experimental timeline. Mice were habituated to the open-field and weighing apparatus for 3 days prior to surgery. Data from the day before surgery were taken as baseline. Post-surgery weight and open-field behaviour were assessed from day 2 until day 13. (B) Ventricular cannula and osmotic minipump placement. (C) Daily assessment of animal weights following surgery over the 2-week experiment (saline: n = 10, Hb: n = 13, Hb + Hp: n = 13, Hp: n = 9). (D) Representative images of Perls’ staining, showing iron deposition. (E) Representative traces following a 5-min open-field assessment. (F) Linear-mixed modelling analysis of time-series data for ambulation performance in the open field (saline: n = 11, Hb: n = 16, Hb + Hp: n = 15, Hp: n = 9). Hb × Hp interaction P = 0.049. (G) Linear-mixed modelling analysis of time-series data for freezing, i.e. period spent immobile in the periphery of the open-field arena in the first minute (saline: n = 11, Hb: n = 16, Hb + Hp: n = 15, Hp: n = 9). Hb × Hp interaction P = 0.065. (C, F, G) Mean ± SEM. (F, G) P-values: *P < 0.05, ***P < 0.001. Scale bar = 500 μm.
An automated open-field arena was used to assess mouse ambulatory activity; animals were tested between days 2 and 13 post-surgery and compared relative to their pre-surgery baseline (Fig. 4E–G). Linear-mixed modelling of this time-series data revealed a significant reduction in ambulation in animals challenged with 10 μM Hb compared with the vehicle control (Fig. 4F, Supplementary Fig. 2). However, concomitant infusion of Hp with Hb recovered this behavioural deficit (Fig. 4F, Supplementary Fig. 2).
Longer periods of immobility, so-called freezing, in the periphery of the open-field have been described previously as a symptom of anxiety (Simon et al., 1994). Therefore, zonal analysis of the open-field data was performed to measure the time mice spent freezing in the periphery over the first minute (Fig. 4G). A significant increase in freezing was observed in animals challenged with 10 μM Hb compared with vehicle. This recovered following concomitant treatment with Hp (Fig. 4G).
Prolonged intrathecal exposure to Hb causes a glial reaction and synaptic loss, which are attenuated by Hp
Following behavioural assessment, brain tissue was collected for immunohistochemistry. Two main brain parenchymal regions were selected for quantification (Fig. 5A), close to and distal from the infusion site (cortical and hippocampal regions respectively). The microglial/macrophage response to intrathecal Hb was assessed using immunohistochemistry for Iba1, a cytoplasmic protein constitutively expressed by microglia and upregulated during inflammation (Ohsawa et al., 2004). In the cortical region, Iba1-positive cells were more numerous after Hb challenge compared with vehicle challenge (Fig. 5B, C and E). Concomitant infusion of an equimolar-binding amount of Hp with Hb reduced the number of Iba1-positive cells (Fig. 5D and E). A similar pattern was observed caudally, in the glia-rich hippocampal molecular region (Fig. 5F–I) and throughout the hippocampus (Supplementary Fig. 3A).
Figure 5.

In vivo animal model: histology. Hp reverses changes in immunohistochemical markers of inflammation, injury and synapse loss following prolonged intrathecal Hb exposure. (A) Sampling sites (blue squares). (B–E) Log(10) transformed counts of Iba1-positive cells in the cortex show an increase following Hb infusion, which was reduced to control levels with Hp treatment (n = 7–13 per condition). Two-way ANOVA: Hb × Hp interaction P = 0.003. (F–I) Iba1-positive cells in the hippocampal molecular region (n = 7–11 per condition). Two-way ANOVA: main effect for Hb P = 0.04. (J–M) GFAP-positive cells in the hippocampal molecular region show an increase following Hb infusion, which is reduced to control levels by Hp treatment (n = 8–13 per condition). Two-way ANOVA: Hb × Hp interaction P = 0.026. (N–Q) Sy38 (synaptophysin) staining across the hippocampal layers shows a reduction after Hb, which is reversed by Hp treatment (n = 8–11 per condition). Two-way ANOVA: Hb × Hp interaction P = 0.07. (B, F, J) Scale bar = 50 µm. (N) Scale bar = 100 µm. (E, I, M, Q) Mean ± SEM, P-values: *P < 0.05, **P < 0.01, ***P < 0.001.
The astrocytic response to injury from intrathecal Hb was assessed using GFAP. Compared with vehicle, animals challenged with Hb had more GFAP-positive cells in the hippocampal molecular region; this was reversed by concomitant treatment with Hp (Fig. 5J–L). A similar trend was present throughout the hippocampus (Supplementary Fig. 3B) and cortex (Supplementary Fig. 3C).
We investigated hippocampal synaptic integrity using Sy38, an antibody against the pre-synaptic protein synaptophysin (Fig. 5N–Q). Synaptic loss was observed in Hb animals, relative to vehicle (Fig. 5N, O and Q). Concomitant infusion of Hp with Hb increased Sy38 staining back to control levels (Fig. 5P and Q). A trend towards an interaction between Hb and Hp was found for this marker (P = 0.07); significant main effects were observed (Supplementary Table 1). A similar picture was observed when tissue was analysed for postsynaptic density protein 95 (PSD-95) by western blotting (Supplementary Fig. 4).
CD163 expression
CD163 is the Hb receptor (Kristiansen et al., 2001) and its expression is increased after uptake of Hb (Boyle et al., 2009; Liu et al., 2017). CD163 expression in the brain is very limited (Galea et al., 2012), so it was important to determine whether the Hp therapeutic effect relied on CD163. In order to determine whether the therapeutic effect of Hp was mediated via upregulation of CD163 expression, we used immunohistochemistry. CD163 expression was seen in the meninges and perivascular spaces, as is usual in normal brain (Galea et al., 2008). We did not observe robust changes in CD163 expression after infusion of Hb or Hb–Hp complexes, at either 3 or 14 days in the parenchyma or convexity (Fig. 6A). Also, surface plasmon resonance demonstrated that human Hp does not increase binding of mouse Hb to mouse CD163 (Fig. 6B), in contrast to the human scavenging system (Fig. 6C). In summary, the Hp therapeutic effect did not rely on CD163.
Figure 6.

In vivo animal model: CD163 and iron. Top panel—Immunohistochemical analysis of CD163 expression. (A) At 6 and 14 days post-surgery, both parenchymal and convexity staining for CD163 was unchanged between experimental groups (n = 8–13, median ± IQR). Middle panel—Assessment of mixed-species Hp to Hb binding. (B, C) Surface plasmon resonance analysis shows human Hp (hHp) does not augment binding of mouse Hb (mHb) to mouse CD163 (B), but hHp does increase binding of human Hb (hHb) to human CD163 (C). Bottom panel—Hp reverses Perls’ staining following intrathecal infusion of Hb. (D) Perls’ staining in the ipsilateral parenchyma at 14 days is significantly increased following Hb infusion, which is fully reversed by Hp (n = 8–12, Kruskal–Wallis P = 0.0002, median ± IQR). (E) Perls’ staining on the brain surface and outer layers of the cortex on the ipsilateral side is significantly increased following Hb infusion (n = 8–13, Kruskal–Wallis P = 0.0003, median ± IQR), which is decreased by Hp. (D, E) P-values: *P < 0.05, ***P < 0.001.
Hp prevents iron deposition
When Hb is oxidized and subsequently degrades, free chelatable iron is released (Asleh et al., 2005; Reeder, 2010). Deposition of this iron in tissue can be visualized using Perls’ stain, which we have observed to be increased in post-mortem brain tissue from SAH patients (Garland et al., 2016). Similarly, iron deposition was observed in the parenchyma (Fig. 6D) and on the brain surface and outer layers of the cortex (Fig. 6E) in Hb-challenged animals relative to vehicle. In both locations, Hp treatment reduced iron deposition (Fig. 6D and E). Positive correlations were observed between the Perls’ staining and: microglia (Iba1, Supplementary Fig. 5A, B, G and H), astrocytes (GFAP, Supplementary Fig. 5C and I), and freezing (Supplementary Fig. 5F and L). Negative correlations were observed between the Perls’ staining and Sy38 staining in the hippocampus (Supplementary Fig. 5D and J) and ambulation (Supplementary Fig. 5E and K).
Hp prevents diffusion of Hb into the parenchyma and attenuates small-vessel, but not large-vessel, vasospasm
Angiographic large-vessel vasospasm and long-term outcome are dissociated (Diringer, 2013), interventions which attenuate large-vessel vasospasm have proved unsuccessful (Cho et al., 2019) and interventions which improve long-term outcome do not affect large-vessel vasospasm (Pickard et al., 1989). However, dysregulation of microcirculation following SAH is an emerging area of study (Terpolilli et al., 2015). As Hb is a known mediator of vasospasm, we hypothesized that the Hb challenge in our model would promote large and small-vessel vasospasm, and this would be attenuated by Hp. Following a 3-day infusion of Hb, anaesthetized mice were transcardially perfused with 5-(6)-carboxy-X-rhodamine, succinimidyl ester for in situ imaging of the Circle of Willis (Fig. 7A, C and F–H). A reduction in MCA diameter was observed with Hb versus vehicle and this was not reversed by Hp (Fig. 7C). A similar picture (Hb-induced vasospasm, not reversed by Hp) was seen with other cerebral arteries (Supplementary Fig. 6). To study parenchymal microcirculatory changes, the same brains used for large-vessel imaging were then prepared for immunohistochemistry and stained for SMA (Fig. 7B, D, I and K). The ratio of vessel lumen diameter to wall thickness was used as a metric of vasospasm (Sabri and Macdonald, 2012; Fig. 7B). Hb caused vasospasm which was reversed by Hp. Two-way ANOVA revealed a significant interaction between Hb and Hp (Fig. 7D). Posthoc analysis revealed significant vasospasm of SMA+ vessels with Hb compared with vehicle, and a significant recovery with Hp (Fig. 7D and I–K).
Figure 7.

In vivo animal model: vasospasm. Large vessel—Intrathecal infusion of Hb for 3 days results in vasospasm of the MCA. (A) Illustration of Circle of Willis and sampling area. (C) Hb decreases ipsilateral MCA diameter, and this is unaffected by Hp. Two-way ANOVA: main effect Hb = 0.0058 (n = 11, mean ± SEM). (F, G, H) Representative images of MCA in situ. Scale bar = 100 μm. (C) P-value: *P < 0.05. Small vessel—Hp reverses small-vessel vasospasm resulting from intrathecal infusion of Hb. (B) Illustration of parenchymal sampling areas and method to determine vasoconstriction. (D) Hp significantly reverses vasospasm in ipsilateral SMA+ vessels caused by a 3-day intrathecal Hb infusion. Two-way ANOVA: Hb × Hp interaction P = 0.0182 (n = 11, mean ± SEM). (E) Immunohistochemical staining for Hb reveals a significant increase in the ipsilateral parenchyma after a 3-day Hb infusion, which is fully reversed by Hp (n = 10–11, Kruskal–Wallis P = 0.0001, median ± IQR). (I–K) Representative confocal images of SMA+ vessels in the ipsilateral parenchyma. Inset images are confocal slices at higher magnification (×20). Scale bar = 50 μm. (D, E) P-values: *P < 0.05, **P < 0.01.
Hb may cause vasospasm via decreasing nitric oxide (Kanamaru et al., 1987; Lin et al., 2001), yet Hb–Hp complexes bind nitric oxide with similar affinity to Hb alone (Azarov et al., 2008). After Hb infusion, vasospasm occurred in both large and parenchymal vessels but Hp only reversed vasospasm in parenchymal vessels. Hence, it was possible that Hp was influencing the distribution of Hb in the brain. Complexation by Hp, especially the polymeric form, results in sequestration of Hb in moieties with a molecular weight of up to 1760 kDa, which would severely restrict entry of Hb into the brain tissue. The movement of solutes into the brain is size-selective; single-domain antibody (17 kDa) penetrates brain tissue better than whole immunoglobulin G (150 kDa; Pizzo et al., 2018), and movement of ovalbumin-immunoglobulin G immune complexes (500–2000 kDa; Mosconi et al., 2010) within brain tissue is significantly limited (Carare et al., 2013). To test the hypothesis that Hp was sequestering Hb outside brain tissue, we assessed the Hb content of the brain parenchyma using immunohistochemistry. At 3 days post-surgery, the same time point as the observed microcirculatory vasospasm, Hb staining in the ipsilateral parenchyma was significantly increased after Hb infusion, compared with vehicle. Hp reversed this parenchymal increase in Hb content (Fig. 7E).
Discussion
SAH results in the prolonged presence of Hb in the subarachnoid space. In humans after SAH, we followed the course of Hb and a neuronal damage marker over an initial discrete observation window. NFL level in the CSF rose gradually, indicating a prolonged stimulus, as opposed to an acute rise immediately after ictus. CSF Hb concentration was low in the first 2 days, but subsequently increased to a plateau after 10 days, likely due to the cumulative effect of red blood cell lysis. CSF NFL level followed this rise in Hb with a lag of 2 days. CSF NFL and preceding Hb peaks were significantly related. NFL was used as a marker for Hb-induced tissue damage, hence the mechanisms underlying NFL release after exposure to Hb were not investigated here; these may include direct neuronal damage or secondary consequences such as inflammation.
While it is well-known that SAH results in the prolonged presence of Hb in the subarachnoid space, the pathophysiological consequences of prolonged in vivo exposure to Hb have not been investigated in isolation. We confirmed previous observations that Hb is toxic to cultured neurones at clinically relevant concentrations (Regan and Panter, 1993; Zhao et al., 2009). In order to establish a clinically relevant model of prolonged Hb exposure, one needs to consider that SAH survivors experience cognitive, behavioural and emotional deficits (Al-Khindi et al., 2010). Modelling these functional outcomes in pre-clinical research is important (Turan et al., 2017). In both the endovascular perforation and blood injection models of SAH, the open-field paradigm has shown significant changes in ambulation and anxiety (Boyko et al., 2013; Fanizzi et al., 2017). However, it was not known whether prolonged exposure to Hb alone—without the mechanical and ischaemic tissue injury, and potential effects of other blood constituents—could independently contribute to, or suffice for, the occurrence of these behavioural deficits. In this study, we have shown that a 2-week intrathecal exposure to Hb does in fact lead to behavioural deficits similar to those seen in clinic, evidenced by reduced ambulation and increased anxiety. A loss of synaptic integrity, as suggested by immunohistochemistry for synaptophysin, accompanied these behavioural deficits. In response to Hb exposure, accumulation of macrophages/microglia was observed in this model, as happens after human and experimental SAH (Schneider et al., 2015). The increase in GFAP-positive cells seen here parallels the high levels of astrocyte-derived serum GFAP and S100β after human SAH, especially observed in patients with poor outcome (Vos et al., 2006; Lai and Du, 2016). Vasospasm was observed in both large cerebral arteries and arterioles in the parenchyma, as seen in human SAH (Terpolilli et al., 2015; Macdonald and Schweizer, 2017). In summary, we observed behavioural, vascular, cellular and molecular changes strikingly similar to those after SAH, by purely modelling one single aspect of SAH: the prolonged intrathecal exposure to uncomplexed Hb.
This study provides robust and conclusive evidence that uncomplexed Hb is a promising therapeutic target in haemorrhagic brain conditions. Hp can reverse Hb toxicity (Zhao et al., 2009), yet Hp concentration is very low in the brain (Galea et al., 2012). We found that most Hb present in human CSF after SAH was uncomplexed. Most of this uncomplexed Hb retained the capacity to bind exogenously added Hp throughout the first 2 weeks. Therefore, there is a wide therapeutic window for treatments based on administering or upregulating Hp in the CNS after SAH. In an in vivo model, Hp supplementation was well-tolerated and reversed the neurological deficit occurring in response to Hb. In addition, Hp reduced the microglial and astrocytic reaction to Hb and synaptic loss.
We did not observe robust changes in CD163 expression accompanying the therapeutic effect of Hp in this model. In mouse, Hp is not required for binding of Hb to CD163 (Etzerodt et al., 2013), unlike in humans, where the Hb–Hp complex has a 10-fold higher affinity to the scavenging receptor CD163 relative to Hb alone (Kristiansen et al., 2001). Human Hp employed in these experiments did not increase the affinity of mouse Hb to mouse CD163. Taken together these findings suggest that the therapeutic effect of Hp was not mediated by increased Hb uptake. Instead, Hp stabilized Hb, preventing its degradation and subsequent release of iron. Iron deposition correlated with behavioural deficits, immunohistochemical markers of an astrocytic and microglial reaction and synaptic loss. Hence, clinically relevant Hp protection can be achieved solely by stabilizing Hb in solution, independent of clearance by CD163. This is particularly encouraging since there is evidence that CD163 binding sites in the human brain are much reduced when compared with the periphery, and fully saturated after SAH (Galea et al., 2012), such that Hb–Hp complexes queue for clearance. Endogenous clearance of CSF into the bloodstream and extracerebral lymphatics occurs via arachnoid granulations, cribriform plate and cranial/spinal nerve roots (Kida et al., 1993; Aspelund et al., 2015). The results in this study confirm that there is therapeutic potential for Hp supplementation despite saturation of CD163 binding sites after SAH.
Hp treatment reduced parenchymal Hb staining, suggesting that another mechanism of action of Hp is to reduce the bioavailability of Hb by sequestering it in large complexes in the subarachnoid space and therefore limiting its diffusion into brain tissue. Hp treatment did not attenuate large-vessel vasospasm, yet improved clinical outcome. Instead, Hp attenuated small-vessel vasospasm. Hence, we experimentally confirm that clinical outcome can be improved independently of large-vessel vasospasm, in keeping with the clinically observed dissociation between clinical outcome and angiographic vasospasm.
Haem released by Hb degradation is bound by haemopexin, and the complex is cleared by CD91 receptor-mediated endocytosis (Hvidberg et al., 2005), providing a natural defence mechanism against haem. Both haemopexin and CD91 are expressed by neurones and glia (Moestrup et al., 1992; Morris et al., 1993). Haemopexin, in contrast to Hp, has a much higher concentration in CSF than anticipated from its plasma concentration (Garland et al., 2016). This suggests that haemopexin may mediate protection from haem. However a higher CSF haemopexin level was associated with a worse outcome after SAH (Garland et al., 2016), and it has been shown in vitro that haemopexin increases the neurotoxicity of Hb when Hp is absent (Chen-Roetling et al., 2018). It may be possible to improve the therapeutic efficacy of Hp by co-administration with haemopexin.
Future work is needed to determine whether Hp can produce a clinically relevant improvement after SAH in animal models such as prechiasmatic blood injection (Sabri et al., 2009), cisternal blood injection or endovascular perforation models (Marbacher et al., 2010). Ideally, this modelling is performed in a CD163-humanized mouse model, in view of the marked differences in CD163 biology between mouse and man (Etzerodt et al., 2010; Etzerodt et al., 2013). Another suitable animal model would be the pig, in which Hp increases the affinity of Hb to CD163, as in the human system (personal communication, J.H. Graversen and S.K. Moestrup). A Phase I trial is needed to ensure that Hp administration is safe in humans. It has been suggested that Hp increases the vulnerability of murine CD163-expressing neurones to Hb in vitro (Chen-Roetling and Regan, 2016), but this was not observed in our in vitro experiments. In a mouse model of intracerebral haemorrhage, CD163 appeared to be detrimental in the first 4 days after the bleed but then conferred protection after 4 days (Leclerc et al., 2018). This inflection may represent a change in the ratio of CD163 expression by neurones versus glia/macrophages (Jing et al., 2018), since myelomonocytic ingress into the CNS is delayed by a few days after an inflammatory stimulus (Andersson et al., 1992).
There are several possible Hp-based therapeutic approaches. Direct intrathecal delivery of Hp after high-grade SAH would be feasible by the time Hb starts rising in the CSF, since by then patients are usually in a tertiary neurological centre, and likely to have an EVD in situ. Hence, Hp and Hb would also be present at the same time in the human situation, as modelled with the osmotic minipump here. The challenge in the human situation relates to ensuring that Hp and Hb are present in the same place, i.e. that intrathecal Hp dosing is such that it reaches the far corners of the subarachnoid space, to ensure that Hb is bound by Hp as soon as it is released. The mouse is not the right species to model the intricacies of Hp dosing in humans in view of different subarachnoid space anatomy, CSF turnover rate and drainage pathways. High purity clinical-grade Hp is available, though all preparations available at the moment are a mixture of dimeric and polymeric Hp. In these experiments, we used a similar preparation, though it was enriched in dimeric Hp. Future work needs to address the issue of which Hp isoform is most efficacious, especially in view of reported differences in clinical outcome between SAH patients with different HP genotypes (Gaastra et al., 2019). It may be preferable to determine an individual’s Hp genotype and administer the same Hp type, to prevent unwanted immunological rejection in a brain compartment (the subarachnoid space) which is not immunologically privileged (Galea et al., 2007). Spectrophotometric Hb assays which can be easily adapted to achieve a quantitative measure of Hb concentration (Duiser et al., 2001) are clinically available in most hospitals, and using serial CSF Hb assays, it may be possible to alter the Hp dose on a patient-to-patient and day-to-day basis, to deliver the right amount of Hp at the right time. Our observations that Hp–Hb binding is sufficient for therapeutic efficacy, independent of CD163 or brain penetration, suggest that an alternating intrathecal Hp infusion and CSF drainage protocol may be appropriate. Other possible therapeutic avenues are drugs which upregulate Hp production such as nuclear factor-erythroid 2 (NF-E2)-related factor 2 (nrf2) inducers (Zolnourian et al., 2019).
In summary, this study provides several novel lines of insight into SAH pathophysiology. In CSF, the time course of the tissue damage marker NFL followed that of Hb. We confirm experimentally that Hb on its own is sufficient to produce an array of behavioural, vascular, cellular and molecular changes strikingly similar to those after human SAH. Robust evidence is presented here that Hp protects against Hb toxicity in vivo, mediated via stabilization of Hb and reduction in iron deposition, and by preventing the entry of Hb into brain tissue. We confirm experimentally the clinically observed dissociation between angiographic vasospasm and clinical outcome, in favour of an effective treatment. As treatment options are limited for SAH, further work is needed to establish Hp as a therapy. The observations regarding haptoglobin efficacy and mechanism of action may apply to other haemorrhagic conditions.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
For help with this work, we thank Sarah Kingsland, Richard Chester and Joan Dalton (Bio Products Laboratory Limited); Scott Harris (University of Southampton); and Diane Aum, Ananth Vellimana and Gregory Zipfel (Washington University School of Medicine).
Funding
We thank the Medical Research Council (Grant no. MR/L01453X/1), Engineering and Physical Sciences Research Council, Bio Products Laboratory Limited, Smile for Wessex, Royal College of Surgeons of Edinburgh, University of Southampton, The Novo Nordisk Foundation and the Independent Research Foundation Denmark. H.Z. is a Wallenberg Academy Fellow supported by grants from the UK Dementia Research Institute at UCL and the Swedish and European Research Councils.
Competing interests
A.I.O. is a former employee and J.M. is a current employee of Bio Products Laboratory Limited, a plasma-derived therapeutics manufacturing company.
Glossary
- CI
confidence interval
- EVD
external ventricular drain
- GFAP
glial fibrillary acidic protein
- Hb
haemoglobin
- Hp
haptoglobin
- Iba1
ionized calcium-binding adapter molecule 1
- IQR
inter-quartile range
- MCA
middle cerebral artery
- NFL
neurofilament light chain
- PBS
physiological-buffered saline
- SAH
subarachnoid haemorrhage
- SEM
standard error of the mean
- SMA
smooth-muscle actin
- UPLC
ultra-performance liquid chromatography
- WFNS
World Federation of Neurological Surgeons
References
- Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010; 41: e519–36. [DOI] [PubMed] [Google Scholar]
- Andersen CBF Stodkilde K Saederup KL Kuhlee A Raunser S Graversen JH, et al. Haptoglobin. Antioxid Redox Signal 2017; 26: 814–31. [DOI] [PubMed] [Google Scholar]
- Andersson PB, Perry VH, Gordon S. The acute inflammatory response to lipopolysaccharide in CNS parenchyma differs from that in other body tissues. Neuroscience 1992; 48: 169–86. [DOI] [PubMed] [Google Scholar]
- Asleh R, Guetta J, Kalet-Litman S, Miller-Lotan R, Levy AP. Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ Res 2005; 96: 435–41. [DOI] [PubMed] [Google Scholar]
- Aspelund A Antila S Proulx ST Karlsen TV Karaman S Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med 2015; 212: 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aum DJ Vellimana AK Singh I Milner E Nelson JW Han BH, et al. A novel fluorescent imaging technique for assessment of cerebral vasospasm after experimental subarachnoid hemorrhage. Sci Rep 2017; 7: 9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azarov I He X Jeffers A Basu S Ucer B Hantgan RR, et al. Rate of nitric oxide scavenging by hemoglobin bound to haptoglobin. Nitric Oxide 2008; 18: 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko M Azab AN Kuts R Gruenbaum BF Gruenbaum SE Melamed I, et al. The neuro-behavioral profile in rats after subarachnoid hemorrhage. Brain Res 2013; 1491: 109–16. [DOI] [PubMed] [Google Scholar]
- Boyle JJ Harrington HA Piper E Elderfield K Stark J Landis RC, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol 2009; 174: 1097–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulters D Gaastra B Zolnourian A Alexander S Ren D Blackburn SL, et al. Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat Rev Neurol 2018; 14: 416–32. [DOI] [PubMed] [Google Scholar]
- Carare RO Teeling JL Hawkes CA Puntener U Weller RO Nicoll JA, et al. Immune complex formation impairs the elimination of solutes from the brain: implications for immunotherapy in Alzheimer’s disease. Acta Neuropathol Commun 2013; 1: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Ma SK, Cao Y, Shah A, Regan RF. Hemopexin increases the neurotoxicity of hemoglobin when haptoglobin is absent. J Neurochem 2018; 145: 464–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Roetling J, Regan RF. Haptoglobin increases the vulnerability of CD163-expressing neurons to hemoglobin. J Neurochem 2016; 139: 586–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SS, Kim SE, Kim HC, Kim WJ, Jeon JP. Clazosentan for aneurysmal subarachnoid hemorrhage: an updated meta-analysis with trial sequential analysis. World Neurosurg 2019; 123: 418–24.e3. [DOI] [PubMed] [Google Scholar]
- Diringer MN. Controversy: does prevention of vasospasm in subarachnoid hemorrhage improve clinical outcome? Stroke 2013; 44 (6 Suppl 1): S29–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duiser HJ, Roelandse FW, Lentjes EG, van Loon J, Souverijn JH, Sturk A. Iterative model for the calculation of oxyhemoglobin, methemoglobin, and bilirubin in absorbance spectra of cerebrospinal fluid. Clin Chem 2001; 47: 338–41. [PubMed] [Google Scholar]
- Etzerodt A, Kjolby M, Nielsen MJ, Maniecki M, Svendsen P, Moestrup SK. Plasma clearance of hemoglobin and haptoglobin in mice and effect of CD163 gene targeting disruption. Antioxid Redox Signal 2013; 18: 2254–63. [DOI] [PubMed] [Google Scholar]
- Etzerodt A, Maniecki MB, Moller K, Moller HJ, Moestrup SK. Tumor necrosis factor alpha-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J Leukoc Biol 2010; 88: 1201–5. [DOI] [PubMed] [Google Scholar]
- Fanizzi C, Sauerbeck AD, Gangolli M, Zipfel GJ, Brody DL, Kummer TT. Minimal long-term neurobehavioral impairments after endovascular perforation subarachnoid hemorrhage in mice. Sci Rep 2017; 7: 7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath T, Ke YD, Gunning P, Gotz J, Ittner LM. Primary support cultures of hippocampal and substantia nigra neurons. Nat Protoc 2009; 4: 78–85. [DOI] [PubMed] [Google Scholar]
- Gaastra B Ren D Alexander S Bennett ER Bielawski DM Blackburn SL, et al. Haptoglobin genotype and aneurysmal subarachnoid hemorrhage: individual patient data analysis. Neurology 2019; 92: e2150–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol 2007; 28: 12–8. [DOI] [PubMed] [Google Scholar]
- Galea I, Felton LM, Waters S, van Rooijen N, Perry VH, Newman TA. Immune-to-brain signalling: the role of cerebral CD163-positive macrophages. Neurosci Lett 2008; 448: 41–6. [DOI] [PubMed] [Google Scholar]
- Galea J Cruickshank G Teeling JL Boche D Garland P Perry VH, et al. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem 2012; 121: 785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland P Broom LJ Quraishe S Dalton PD Skipp P Newman TA, et al. Soluble axoplasm enriched from injured CNS axons reveals the early modulation of the actin cytoskeleton. PLoS One 2012; 7: e47552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland P Durnford AJ Okemefuna AI Dunbar J Nicoll JA Galea J, et al. Heme-hemopexin scavenging is active in the brain and associates with outcome after subarachnoid hemorrhage. Stroke 2016; 47: 872–6. [DOI] [PubMed] [Google Scholar]
- Garton TP, He Y, Garton HJ, Keep RF, Xi G, Strahle JM. Hemoglobin-induced neuronal degeneration in the hippocampus after neonatal intraventricular hemorrhage. Brain Res 2016; 1635: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray P, Cooney J. Stress-induced responses and open-field behavior in estrous and nonestrous mice. Physiol Behav 1982; 29: 287–92. [DOI] [PubMed] [Google Scholar]
- Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005; 106: 2572–9. [DOI] [PubMed] [Google Scholar]
- Jeppsson A, Zetterberg H, Blennow K, Wikkelso C. Idiopathic normal-pressure hydrocephalus: pathophysiology and diagnosis by CSF biomarkers. Neurology 2013; 80: 1385–92. [DOI] [PubMed] [Google Scholar]
- Jing C, Zhang H, Shishido H, Keep RF, Hua Y. Association of brain CD163 expression and brain injury/hydrocephalus development in a rat model of subarachnoid hemorrhage. Front Neurosci 2018; 12: 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamaru K, Waga S, Kojima T, Fujimoto K, Niwa S. Endothelium-dependent relaxation of canine basilar arteries. Part 2: inhibition by hemoglobin and cerebrospinal fluid from patients with aneurysmal subarachnoid hemorrhage. Stroke 1987; 18: 938–43. [DOI] [PubMed] [Google Scholar]
- Kida S, Pantazis A, Weller RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 1993; 19: 480–8. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 2010; 8: e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen AH, Borke RC. Experimental superficial siderosis of the central nervous system. I. Morphological observations. J Neuropathol Exp Neurol 1991; 50: 579–94. [DOI] [PubMed] [Google Scholar]
- Kristiansen M Graversen JH Jacobsen C Sonne O Hoffman HJ Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature 2001; 409: 198–201. [DOI] [PubMed] [Google Scholar]
- Lai PM, Du R. Association between S100B levels and long-term outcome after aneurysmal subarachnoid hemorrhage: systematic review and pooled analysis. PLoS One 2016; 11: e0151853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclerc JL Lampert AS Loyola Amador C Schlakman B Vasilopoulos T Svendsen P, et al. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J Cereb Blood Flow Metab 2018; 38: 262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G, Macdonald RL, Marton LS, Kowalczuk A, Solenski NJ, Weir BK. Hemoglobin increases endothelin-1 in endothelial cells by decreasing nitric oxide. Biochem Biophys Res Commun 2001; 280: 824–30. [DOI] [PubMed] [Google Scholar]
- Liu R, Cao S, Hua Y, Keep RF, Huang Y, Xi G. CD163 expression in neurons after experimental intracerebral hemorrhage. Stroke 2017; 48: 1369–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet 2017; 389: 655–66. [DOI] [PubMed] [Google Scholar]
- Madsen M Moller HJ Nielsen MJ Jacobsen C Graversen JH van den Berg T, et al. Molecular characterization of the haptoglobin.hemoglobin receptor CD163. Ligand binding properties of the scavenger receptor cysteine-rich domain region. J Biol Chem 2004; 279: 51561–7. [DOI] [PubMed] [Google Scholar]
- Marbacher S, Fandino J, Kitchen ND. Standard intracranial in vivo animal models of delayed cerebral vasospasm. Br J Neurosurg 2010; 24: 415–34. [DOI] [PubMed] [Google Scholar]
- Moestrup SK, Gliemann J, Pallesen G. Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res 1992; 269: 375–82. [DOI] [PubMed] [Google Scholar]
- Morris CM, Candy JM, Edwardson JA, Bloxham CA, Smith A. Evidence for the localization of haemopexin immunoreactivity in neurones in the human brain. Neurosci Lett 1993; 149: 141–4. [DOI] [PubMed] [Google Scholar]
- Mosconi E Rekima A Seitz-Polski B Kanda A Fleury S Tissandie E, et al. Breast milk immune complexes are potent inducers of oral tolerance in neonates and prevent asthma development. Mucosal Immunol 2010; 3: 461–74. [DOI] [PubMed] [Google Scholar]
- Naff NJ, Williams MA, Rigamonti D, Keyl PM, Hanley DF. Blood clot resolution in human cerebrospinal fluid: evidence of first-order kinetics. Neurosurgery 2001; 49: 614–9; discussion 619–21. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Sasaki Y, Kohsaka S. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J Neurochem 2004; 88: 844–56. [DOI] [PubMed] [Google Scholar]
- Pickard JD Murray GD Illingworth R Shaw MD Teasdale GM Foy PM, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ 1989; 298: 636–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo ME Wolak DJ Kumar NN Brunette E Brunnquell CL Hannocks MJ, et al. Intrathecal antibody distribution in the rat brain: surface diffusion, perivascular transport and osmotic enhancement of delivery. J Physiol 2018; 596: 445–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder BJ. The redox activity of hemoglobins: from physiologic functions to pathologic mechanisms. Antioxid Redox Signal 2010; 13: 1087–123. [DOI] [PubMed] [Google Scholar]
- Reeder BJ. Redox and peroxidase activities of the hemoglobin superfamily: relevance to health and disease. Antioxid Redox Signal 2017; 26: 763–76. [DOI] [PubMed] [Google Scholar]
- Reeder BJ, Sharpe MA, Kay AD, Kerr M, Moore K, Wilson MT. Toxicity of myoglobin and haemoglobin: oxidative stress in patients with rhabdomyolysis and subarachnoid haemorrhage. Biochem Soc Trans 2002; 30: 745–8. [DOI] [PubMed] [Google Scholar]
- Regan RF, Panter SS. Neurotoxicity of hemoglobin in cortical cell culture. Neurosci Lett 1993; 153: 219–22. [DOI] [PubMed] [Google Scholar]
- Rosen AD, Frumin NV. Focal epileptogenesis after intracortical hemoglobin injection. Exp Neurol 1979; 66: 277–84. [DOI] [PubMed] [Google Scholar]
- Sabri M Jeon H Ai J Tariq A Shang X Chen G, et al. Anterior circulation mouse model of subarachnoid hemorrhage. Brain Res 2009; 1295: 179–85. [DOI] [PubMed] [Google Scholar]
- Sabri M, Macdonald RL. Vasospasm: Measurement of Diameter, Perimeter, and Wall Thickness In: Chen J, Xu X-M, Xu ZC, Zhang JH, editors. Animal models of acute neurological injuries II: injury and mechanistic assessments. Vol. 1 Totowa, NJ: Humana Press; 2012. p. 473–9. [Google Scholar]
- Schneider UC Davids AM Brandenburg S Muller A Elke A Magrini S, et al. Microglia inflict delayed brain injury after subarachnoid hemorrhage. Acta Neuropathol 2015; 130: 215–31. [DOI] [PubMed] [Google Scholar]
- Siedler DG, Chuah MI, Kirkcaldie MT, Vickers JC, King AE. Diffuse axonal injury in brain trauma: insights from alterations in neurofilaments. Front Cell Neurosci 2014; 8: 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon P, Dupuis R, Costentin J. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav Brain Res 1994; 61: 59–64. [DOI] [PubMed] [Google Scholar]
- Teasdale GM Drake CG Hunt W Kassell N Sano K Pertuiset B, et al. A universal subarachnoid hemorrhage scale: report of a committee of the World Federation of Neurosurgical Societies. J Neurol Neurosurg Psychiatry 1988; 51: 1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terpolilli NA, Brem C, Buhler D, Plesnila N. Are we barking up the wrong vessels? Cerebral microcirculation after subarachnoid hemorrhage. Stroke 2015; 46: 3014–9. [DOI] [PubMed] [Google Scholar]
- Turan N Miller BA Heider RA Nadeem M Sayeed I Stein DG, et al. Neurobehavioral testing in subarachnoid hemorrhage: a review of methods and current findings in rodents. J Cereb Blood Flow Metab 2017; 37: 3461–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallelian F Pimenova T Pereira CP Abraham B Mikolajczyk MG Schoedon G, et al. The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic Biol Med 2008; 45: 1150–8. [DOI] [PubMed] [Google Scholar]
- Vos PE, van Gils M, Beems T, Zimmerman C, Verbeek MM. Increased GFAP and S100beta but not NSE serum levels after subarachnoid haemorrhage are associated with clinical severity. Eur J Neurol 2006; 13: 632–8. [DOI] [PubMed] [Google Scholar]
- Zhao X Song S Sun G Strong R Zhang J Grotta JC, et al. Neuroprotective role of haptoglobin after intracerebral hemorrhage. J Neurosci 2009; 29: 15819–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolnourian A, Galea I, Bulters D. Neuroprotective role of the Nrf2 pathway in subarachnoid haemorrhage and its therapeutic potential. Oxid Med Cell Longev 2019; 2019: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets are available from the corresponding author on reasonable request in accordance with the University of Southampton’s data-sharing policies, ethical approvals and contracts with the co-authors and their institutions.