

Abstract
Bile acids are synthesized from cholesterol only in hepatocytes. Bile acids circulating in the enterohepatic system act as physiological detergent molecules to help solubilize biliary cholesterol and emulsify dietary lipids and fat-soluble vitamins in small intestine. Bile acids are signaling molecules that activate nuclear receptor farnesoid X receptor (FXR) and cell surface G protein-coupled receptor TGR5. FXR critically regulates bile acid homeostasis by mediating bile acid feedback inhibition of hepatic bile acid synthesis. In addition, bile acid-activated cellular signaling pathways regulate metabolic homeostasis, immunity, and cell proliferation in various metabolically active organs. In the small and large intestine, gut bacterial enzymes modify primary bile acids to generate secondary bile acids to help shape the bile acid pool composition and subsequent biological effects. In turn, bile acids exhibit anti-microbial properties and modulate gut microbiota to influence host metabolism and immunity. Currently, bile acid-based therapies including systemic and intestine-restricted FXR agonists, TGR5 agonists, fibroblast growth factor 19 analogue, intestine FXR antagonists, and intestine apical sodium-bile acid transporter (ASBT) inhibitors have been developed as promising treatments for non-alcoholic steatohepatitis (NASH). These pharmacological agents improved metabolic and inflammatory disorders via distinct mechanisms of action that are subjects of extensive research interest. More recently, human and experimental alcoholic liver disease (ALD) has been associated with disrupted bile acid homeostasis. In additional, new findings showed that targeting bile acid metabolism and signaling may be promising therapeutic approaches for treating ALD.
Keywords: Bile acid, farnesoid X receptor (FXR), microbiota, non-alcoholic steatohepatitis (NASH), alcoholic liver disease (ALD)
Introduction
Bile acids are physiological detergent molecules produced exclusively by hepatocytes from cholesterol (1). After synthesis, bile acids are efficiently conjugated to glycine or taurine and present in the form of bile salts under physiological pH (2). In liver, conversion of cholesterol to bile acids is the major cholesterol catabolic pathway. Secretion of bile acids across the apical membrane of hepatocytes into the bile generates bile flow and facilitates cholesterol solubilization (3). Bile acids in bile are further concentrated in gallbladder during fasting and released into the small intestine postprandially. One of the classic functions of bile salts is to help emulsify dietary lipids in mixed micelles and facilitate their digestion and absorption in the small intestine. Majority of the bile acids are re-absorbed mainly in the terminal ileum and transported back to the liver for re-secretion into bile. Bile acids circulates between liver and intestine a few times a day, which is referred to as the enterohepatic circulation of bile acids. Although the classic physiological functions of bile acids depend on their physiochemical property as amphipathic detergent molecules, bile acids also act as endogenous ligands for a few intracellular nuclear receptors and cell surface G protein coupled receptors (4-6). These bile acid-activated receptors play important roles in regulating various biological functions ranging from metabolic homeostasis to immune responses and cell proliferation. Disrupted bile acid homeostasis has been associated with various forms of genetic and acquired human diseases, while therapeutic approaches targeting bile acid metabolism, transport and signaling have been developed as promising therapies of liver and metabolic diseases (7). In this review we will briefly summarize the basic knowledge of bile acid chemistry and biology, and then discuss the recent advances in bile acid-based therapies for cholestasis and non-alcoholic steatohepatitis (NASH) and the emerging roles of bile acid signaling in alcoholic liver disease (ALD).
Bile acid synthesis and regulation
Enzymes in the endoplasmic reticulum, mitochondria, cytosol, and peroxisomes mediate the multi-step conversion of cholesterol to bile acids (8). Current evidence suggests that hepatocyte is the only cell type that express all enzymes required for de novo bile acid synthesis, while certain oxysterol intermediates produced in extrahepatic tissues can enter the bile acid synthesis pathway in hepatocytes. As shown in Figure 1A, bile acid synthesis converts the C27 cholesterol molecule to C24 bile acid molecules with several hydroxyl groups positioning on one side of the steroid nucleus, which renders bile acids amphipathic properties. After synthesis, the side chain of bile acids is linked to either glycine or taurine to form conjugated bile acids (Figure 1B). Under physiological pH, conjugated bile acids are ionized and soluble. Conjugated bile acids are membrane impermeable and their transport is mediated by various transporters in the enterohepatic system. The human bile acid pool consists of two primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA), which accounts for ~80% of the total bile acid pool. In mice, majority of CDCA is further converted to isoforms of muricholic acids (MCA). CA and MCAs are therefore the predominant primary bile acid species in mice. Bile acid pool contains numerous secondary bile acids that are formed from primary bile acids in small and large intestine by bacterial enzymes. Deoxycholic acid (DCA) is the major secondary bile acid in humans and mice and can account for up to 20% of the total bile acid pool. Other common secondary bile acids such as lithocholic acid (LCA) and ursodeoxycholic acid (UDCA) are present in trace amount under physiological conditions.
Figure 1.
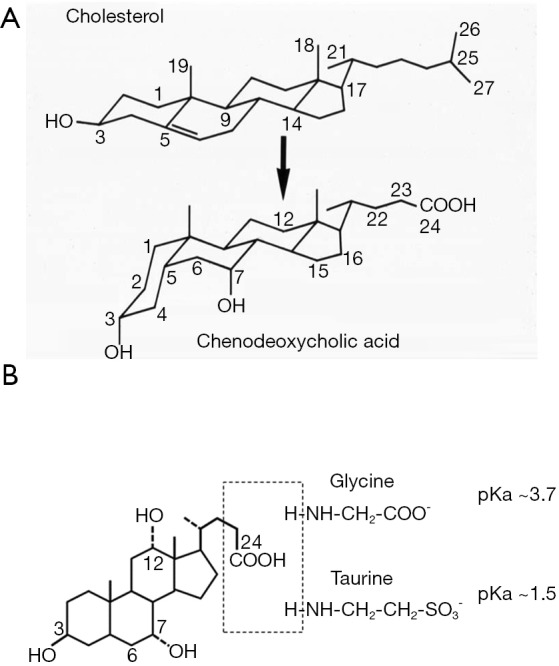
The biochemical property of bile acids. (A) Cholesterol and cholic acid structure. Compared to cholesterol, cholic acid has hydroxyl groups at the C7 and C12 position, a shortened and oxidized sterol side chain, and C5-C6 double bond saturation; (B) illustration of glycine and taurine conjugated bile acids. Bile acids conjugation is mediate by the bile acid CoA ligase (BAL) and the bile acid CoA: amino acid N-acetyltransferase (BAAT).
De novo bile acid synthesis in hepatocytes
Hepatocytes synthesize primary bile acids via two pathways, namely the classic pathway and the alternative pathway (Figure 2). It is believed that the classic bile acid synthesis pathway accounts for about 80% of the total bile acid production in humans, while in rodents such as mice and rats the alternative pathway may contribute to half of the total bile acid synthesis. In the classic bile acid synthesis pathway cholesterol is hydroxylated at the C-7 position by the rate-limiting cholesterol 7α-hydroxylase (CYP7A1), a cytochrome p450 enzyme located in the endoplasmic reticulum, to produce 7α-hydroxycholesterol (9). The downstream intermediate 7α-hydroxy-4-cholestene-3-one (C4) is the common precursor of CDCA and CA and has been used as a surrogate marker for hepatic bile acid synthesis rate (10,11). Microsomal sterol 12α-hydroxylase (CYP8B1)-mediated C-12 hydroxylation of C4 results in the production of CA. Alternatively, C4 will be converted to CDCA if the C-12 position is intact. The mitochondrial sterol 27-hydroxylase (CYP27A1) catalyzes the hydroxylation and oxidation of the sterol side chain (12). The subsequent enzymatic sterol side chain cleavage to produce C-24 bile acids and bile acid CoA: amino acid N-acyltransferase (BAAT)-mediated conjugation to taurine or glycine occurs in the peroxisomes (2,13). In the alternative pathway, CYP27A1 mediates the hydroxylation and oxidation of the cholesterol side-chain to produce 27-hydroxycholesterol and then 3β-hydroxy-5-cholestenoic acid before other enzymatic modifications of the steroid nucleus occur. The alternative pathway is also referred to as “the acidic pathway” because the intermediates in this pathway contain a C27-carboxylic acid group, while the classic pathway is sometimes referred to as “the neutral pathway”. The oxysterol 7α-hydroxylase (CYP7B1) mediates the C-7 hydroxylation reaction in the alternative pathway. It was originally thought that the alternative pathway only produces CDCA. However, recent studies showed that in Cyp7a1 deficient mice hepatic CYP7B1 and CYP8B1 were induced and the bile acid pool in Cyp7a1 deficient mice still contained a substantial amount of CA (14), suggesting that the alternative pathway produces both CDCA and CA.
Figure 2.

Bile acid synthesis pathways. The classic and alternative bile acid synthesis pathways and major enzymes involved in bile acid synthesis. Major enzymes in the pathways are shown. CYP7A1 is the rate-limiting enzyme in the classic bile acid synthesis pathway. In humans, hepatic bile acid synthesis produces the primary bile acid cholic acid and CDCA. In the small and large intestine, primary bile acids can be deconjugated by BSH and then modified by bacterial enzymes to produce secondary bile acids. Bacterial 7α-dehydroxylase removes C7 hydroxyl group to convert cholic acid to DCA and CDCA to LCA. Other secondary bile acids also exist at very low abundance in the bile acid pool. CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; BSH, bile salt hydrolases.
Bacterial transformation of bile acids: synthesis of secondary bile acids
Primary bile acids undergo bacterial enzyme-mediated biotransformation to produce secondary bile acids in the ileum and large intestine (15). Bacterial bile salt hydrolases (BSH) convert conjugated bile acids to free bile acids, which further undergo 7α de-hydroxylation reaction by bacterial 7α-dehydroxylase. This reaction converts CA to DCA and CDCA to LCA (15-17). Unconjugated primary and secondary bile acids are passively re-absorbed in the ileum and large intestine and conjugated in hepatocytes (18). The fecal bile acids consist of predominantly unconjugated bile acids. Other minor secondary bile acids are also produced through deconjugation, de-hydroxylation, hydroxylation, and epimerization reactions, which has been discussed in detail elsewhere (7).
Species-dependent differences in bile acid synthesis and composition
Because biomedical research is primarily conducted in rodent models, it is important to point out the species-dependent differences in the bile acid pool composition. The de novo bile acid synthesis pathways are highly conserved in humans, rats and mice, but the majority of the CDCA is converted to isoforms of MCA (α-MCA, β-MCA and ω-MCA) in mice and rats. The α-MCA has a 7α-hydroxyl group while β-MCA has a 7β-hydroxyl group. Both α-MCA and β-MCA have a 6β-hydroxyl group. The ω-MCA contains a 7β-hydroxyl group and a 6α-hydroxyl group. The C-6 hydroxylation renders MCAs more hydrophilic bile acids with significantly reduced signaling properties. Therefore, changes in bile acid pool composition are expected to have differential impacts on bile acid-regulated biological effects in humans and mice. A recent study suggests that the Cyp2c-cluster may encodes enzymes that mediate C-6 hydroxylation of bile acids in mice (19). The species-dependent difference in bile acid pool composition also exists in that mice and rats predominantly use taurine for bile acid conjugation while the bile acid pool in humans and some non-human primates contains both glycine-conjugates and taurine-conjugates in roughly 3:1 ratio (20,21). The BAAT substrate specificity and possibly the availability of taurine and glycine in the peroxisomes have been suggested to dictate the preferential use of glycine or taurine for bile acid conjugation in humans and rodents (2,22,23). Liver is a major organ for taurine synthesis from cysteine, which is catalyzed by cysteine dioxygenase 1 (CDO1) and cysteine sulfinic acid decarboxylase (CSAD) (24). Recent studies showed that both enzymes in taurine synthesis are repressed by bile acids and FXR, suggesting that taurine synthesis and bile acid synthesis are coordinately regulated by bile acids (25-27). Over-activation of the taurine synthesis pathway may deplete hepatic cysteine and glutathione to increase hepatic sensitivity to oxidative stress (26).
Enterohepatic circulation of bile acids
The enterohepatic circulation of bile acids refers to the process of bile acid secretion across the canalicular membrane of hepatocytes into bile, bile acid release into the small intestine, re-absorption at the terminal ileum, and basolateral uptake into hepatocytes for re-secretion into bile. The ATP-binding cassette (ABC) transporter bile salt export pump (BSEP, ABCB11) mediates the canalicular bile acid secretion into bile against the concentration gradient, which is the rate-limiting step in bile formation (28,29). BSEP shows a hepatocyte-specific expression and high substrate specificity for conjugated bile acids. In contrast, the multidrug resistance-associated protein-2 (MRP2, ABCC2) mediates the apical secretion of sulfated and conjugated bile acids, bilirubin glucuronide and glutathione. In hepatocytes, the apical ABCG5 and ABCG8 heterodimer mediates cholesterol secretion into bile (30) and the multi-drug resistant 3 (MDR3, ABCB4) mediates the apical secretion of phosphatidylcholine (31). Bile acids, cholesterol and phospholipids are three major constituents in bile. They form mixed micelles, which is necessary to prevent cholesterol precipitation in bile and the toxicity of free bile acids to biliary epithelial cells. Under fasting, the high tone of the sphincter of Oddi provides high pressure resistance that promotes bile to enter the gallbladder where bile acids are further concentrated in bile. Postprandial release of the gut hormone cholecystokinin by the neuroendocrine cells stimulates gallbladder contraction and sphincter of Oddi relaxation to release bile into small intestine (32), where bile acids form mixed micelles with dietary lipids to facilitate their digestion and absorption. The terminal ileum is the major site of active bile acid transport across the enterocyte into the portal circulation. The apical sodium-dependent bile salt transporter (ASBT, SLC10A2) mediates the apical uptake of conjugated bile acids into enterocytes (33-35). Bile acids bind to the bile acid binding protein (I-BABP) intracellularly and are secreted across the basolateral membrane by the organic solute transporters OSTα (SLC51A) and OSTβ (SLC51B) heterodimer into the portal circulation (36-38). About 95% of the bile acids are re-absorbed in the intestine and transported back to the liver. The Na+-taurocholate co-transporting polypeptide (NTCP, SCL10A1) is the major basolateral conjugated-bile acid uptake transporter in hepatocytes (39-41) and several isoforms of the organic anion transporters (OATPs) mediate Na+-independent basolateral uptake of conjugated and un-conjugated bile acids (18,42). The first pass bile acid extraction rate is about 90% and for this reason the systemic bile acid concentration is markedly lower than the portal bile acid concentration under normal physiology (43).
Regulation of bile acid homeostasis
Bile acids regulate various biological pathways via activation of intracellular and cell surface bile acid receptors. The well-studied bile acid receptors are farnesoid x receptor (FXR), Takeda G receptor 5 (TGR5) and sphingosine 1 phosphate receptor (S1PR), which mediate bile acid regulation of bile acid homeostasis, nutrient metabolism, immune response and cell proliferation. FXR belongs to the nuclear receptor superfamily of ligand-activated transcription factors (44). Conjugated and unconjugated CDCA and CA are endogenous ligands for FXR. Among all major bile acid species, CDCA is the strongest FXR agonist with an EC50 value of about 10–20 µM. Hydrophilic bile acids such as UDCA and MCAs do not activate FXR (45). TGR5 is a G protein-coupled receptor expressed in adipocytes, skeletal muscle, intestine and macrophages but not hepatocytes. Secondary bile acids LCA and DCA activate TGR5 (6,46). Liver and intestine are exposed to high levels of endogenous bile acids. However, bile acid concentration in the systemic circulation is usually well below the EC50 of FXR and TGR5. Potent FXR and TGR5 agonists have been developed and have shown great potential as therapeutics for cholestasis, NAFLD and diabetes. The molecular basis for the beneficial effects of bile acid receptor agonists are briefly discussed below.
As mentioned early, about 5% of bile acids is lost daily in feces. This amount roughly equals to the amount produced via de novo synthesis in liver, and a constant bile acid pool is maintained. There are several FXR-mediated bile acid sensing mechanisms in the liver and intestine that help regulate bile acid homeostasis. In general, when intracellular bile acid concentration increases, FXR inhibits bile acid uptake and promotes bile acid secretion to prevent intracellular bile acid accumulation. In response to increased intracellular bile acid concentration, FXR also inhibits the transcription of genes in bile acid synthesis. When bile acid concentration decreases, hepatic bile acid synthesis is induced and intestine bile acid preservation is maintained.
The mechanisms by which bile acids feedback inhibit bile acid synthesis have been extensively investigated and are relatively well understood. CYP7A1, the rate limiting enzyme in the classic bile acid synthesis pathway is mainly controlled at the transcriptional level. The human, mouse and rat CYP7A1 gene proximal promoter contains a bile acid response element-I (BARE-I) that binds two nuclear receptors hepatocyte nuclear factor 4α (HNF4α) and liver related homolog-1 (LRH-1). The endogenous ligands for HNF4α and LRH-1 are unknown. It is believed that HNF4α and LRH-1 are constitutively active and maintains CYP7A1 expression in hepatocytes. Mutations of the BARE-I sequence significantly reduced CYP7A1 promoter activity. Hepatocyte FXR induces a nuclear receptor small heterodimer partner (SHP), which acts as a co-repressor of HNF4α and LRH-1 to inhibit CYP7A1 gene transcription (47,48). Bile acid re-uptake mainly occurs at the terminal ileum where ASBT is highly expressed. Here FXR induces the transcription of an endocrine hormone fibroblast growth factor 15 (FGF15), which inhibits CYP7A1 gene transcription in hepatocytes by binding to the cell surface FGF receptor 4 (FGFR4) (49). FGF15 repression of CYP7A1 partially depends on intracellular activation of the ERK1/2 signaling, but the downstream events are still incompletely understood (50). FGF19 is the human orthologue of FGF15. Similar to FGF15, FXR induces FGF19 transcription (50,51) and FGF19 binds FGFR4 to repress CYP7A1 gene transcription (50,51). However, human primary hepatocytes express FGF19 but mouse hepatocytes do not express FGF15 (50,52). Intrahepatic bile acid accumulation induces FGF19 to repress CYP7A1 via autocrine manner in humans, but this mechanism is absent in mice (49,52). It has been reported that CYP7A1 mRNA has a relatively short half-life of about 30 min (53), which renders CYP7A1 sensitive to post-transcriptional regulation at the level of mRNA stability. Studies have shown that FGF19 treatment rapidly repressed CYP7A1 mRNA within 2 h of treatment initiation (54-56). A new study reported that FXR induced a RNA-binding protein ZFP36L1 to promote CYP7A1 mRNA degradation (57). These findings suggest that there are redundant pathways mediating the bile acid feedback inhibition of bile acid synthesis. Abolishing a single pathway will less likely prevent bile acid feedback inhibition of bile acid synthesis. To date, reports of regulation of CYP7A1 function at the enzymatic activity level are still scarce.
FXR regulation of bile acid transport in the enterocyte is an important mechanism to maintain a relatively constant bile acid pool size. In enterocytes, FXR induces I-BABP, OSTα and OSTβ and inhibits ASBT (58-60), leading to reduced uptake and increased secretion. This mechanism does not promote trans-enterocyte bile acid transport but instead prevents intracellular bile acid accumulation. Intra-enterocyte accumulation of bile acids has been shown to promote cellular injury and gut junction disruption (61-65). In hepatocytes, FXR induces BSEP (66) and inhibits NTCP and prevent intrahepatic bile acid accumulation (67). During cholestasis, the hepatic basolateral transporters OSTα/β and MRP isoforms are induced to efflux bile acids into the systemic circulation (37,68,69). FXR regulation of bile acid synthesis and transport serve as important rationale for using FXR agonist to cholestasis treatment. Recently, the potent FXR agonist obeticholic acid (OCA) has been approved for treating primary biliary cholangitis in patients with insufficient response to UDCA treatment.
Gut microbiota, bile acids and energy metabolism
The fetal intestine is sterile. After birth, bacteria rapidly colonize the intestine. In humans, it is estimated that the terminal ileum contains about 108 bacteria while the colon contains about 1013 to 1014 bacteria (70,71). Bacteria-mediated bile acid transformation therefore mainly occurs in the terminal ileum and colon. On the other hand, bile acids are known to have antimicrobial activity and partially responsible for the relatively low abundance of bacteria in the small intestine (72,73). Therefore, bile acids help shape the gut microbiota, while gut microbiota modulates bile acid biochemical and biological properties. Here we will discuss how the complex interactions between bile acids and gut microbiota regulate host metabolism and immunity in health and metabolic diseases.
Obesity is associated with altered gut microbiota (74). Intestinal microbes utilize polysaccharides, starch and bile acids to generate short chain fatty acids including acetate, butyrate and propionate, and amino acids, which increases host energy harvest (75-77). In this way, altered gut microbiota may have significantly impacts on lipid, glucose and energy metabolism in the host. Consistently, germ free mice showed resistance to diet-induced obesity when compared to the conventionally raised mice (75,78). When germ-free mice receive caecal microbiota transplant from obese mice, they were more prone to weight gain than germ free mice receiving caecal microbiota from lean mice (75,78). Gut microbiota can also alter host metabolism by regulating gut hormones including peptide YY and glucagon-like peptide-1 (GLP1) (79-82). In humans, Firmicutes and Bacteroidetes account for about 90% of the gut microbiome and their relative abundance is altered in obesity (83). Increased Firmicutes to Bacteroidetes ratio is associated with increased energy harvest and obesity (84). Several mechanisms mediate bile acid effects in shaping the gut microbiota. TCA has been shown to increase gut Bacteroidetes and Bilophila wadsworthia (85). The secondary bile acid DCA has the most potent antimicrobial activity in the gut, and increased DCA abundance in the colon causes selective inhibition of gut bacteria growth, resulting in altered gut microbiota (86). Consistently, biliary obstruction and reduced bile flow to the small intestine were associated with gut bacterial overgrowth, gut epithelial injury and bacterial translocation in humans and animal models (87-89). Mice subjected to bile duct ligation showed increased bacterial overgrowth in the ileum and cecum, which was reduced by administration of non-steroid FXR agonist GW4064 (90). It is interesting to note that GW4064 treatment did not reduce gut bacterial overgrowth in mice lacking FXR, suggesting that the antimicrobial effect of GW4064 was due to FXR signaling activation but not its biochemical property. FXR induction of genes including Ang1, iNos and IL18 may play a role in mediating the antimicrobial activity of FXR (91-93). Recent studies showed that probiotics administration significantly reduced hepatic steatosis and inflammation in patients with fatty liver diseases, suggesting that modulating gut microbiota represents a promising treatment for fatty liver disease (94,95).
Understanding of how microbiota in turn regulate bile acid metabolism has also been an area of focus in recent years. As mentioned earlier, bacterial BSH and 7α-hydroxysteroid dehydrogenase activities mediate the secondary bile acid synthesis and modulate bile acid composition in the enterohepatic circulation. In ileum and colon, BSH activity has been detected in many gram-positive bacteria genera including Clostridium, Enterococcus, Bifidobacterium and Lactobacillus, and gram-negative genus Bacteroides (15). These anaerobic bacteria also express 7α-hydroxysteroid dehydrogenase activities. Mice raised in germ-free environment and antibiotic-treated mice showed increased hepatic bile acid synthesis and enlarged bile acid pool size compared to conventionally raised mice (45). Germ-free mice and antibiotic-treated mice showed increased abundance of T-βMCA (96). Because T-βMCA acts as an FXR antagonist, such change in bile acid pool composition was associated with decreased FXR activation, reduced intestine FGF15 expression, which may account for increased hepatic bile acid synthesis and bile acid pool expansion (45,97). Furthermore, reduced gut bile acid deconjugation also significantly decreased fecal unconjugated bile acid excretion and increased bile acid preservation in the gut (98).
Bile acid therapeutics in NASH
NAFLD is a spectrum of chronic liver abnormalities ranging from simple steatosis to NASH (99). Simple steatosis, defined as more than 5% or more hepatic fat accumulation, is a benign condition that does not required treatment. In contrast, NASH is a more debilitating condition with the presence of hepatocyte cell death and inflammatory infiltration. It is estimated that ~30% of NAFLD patients may progress to develop NASH, and ~10–30% of NASH patients may progress to cirrhosis with increased risk of hepatocellular carcinoma. NAFLD is closely associated with obesity and type-2 diabetes (100,101). Currently, there is no clinically approved drugs for NASH treatment. Better understanding of the drivers of NAFLD progression to NASH holds key to developing effective therapeutics for NASH treatment. The pathogenesis of NASH is multifactorial and its risk is determined by complex interactions between many genetic and environmental factors. Insulin resistance is a major risk factor for NASH. Excessive hepatic accumulation of free fatty acids, free cholesterol and other lipid intermediates including diacylglycerols, acyl-carnitines and ceramides are thought to cause organelle dysfunction, lipotoxicity and hepatocyte apoptosis (102). Altered gut microbiota and permeability also contribute to NASH development via gut-liver axis. Several bile acid-based therapies have been investigated in clinical trials for NASH treatment and will be reviewed here (103-115). The physiological effects of these therapies and their potential clinical applications are summarized in Table 1.
Table 1. Summary of the bile acid-based therapeutics.
| Therapeutics | Physiological effects | Potential clinical applications |
|---|---|---|
| FXR agonists | Bile acid synthesis ↓ | Cholestasis |
| Lipogenesis ↓ | NASH | |
| Gluconeogenesis ↓ | ||
| Inflammation ↓ | ||
| LDL cholesterol ↑ | ||
| Pruritus ↑ | ||
| TGR5 agonist | Energy expenditure ↑ | Obesity |
| GLP-1 secretion ↑ | Type-II diabetes | |
| Inflammation ↓ | NASH | |
| Atherosclerosis | ||
| FGF19 mimetic | Bile acid synthesis ↓ | Cholestasis |
| Lipogenesis ↓ | NASH | |
| Gluconeogenesis ↓ | Type-II diabetes | |
| LDL cholesterol ↑ | ||
| ASBT inhibitor | Bile acid synthesis ↑ | Cholestasis |
| Insulin sensitivity ↑ | Hypercholesterolemia | |
| LDL cholesterol ↓ | NASH | |
| Pruritus ↓ | Type-II diabetes | |
| Diarrhea ↑ | Pruritus | |
| Bile acid sequestrants | Bile acid synthesis ↑ | Cholestasis |
| Insulin sensitivity ↑ | Hypercholesterolemia | |
| LDL cholesterol ↓ | Type-II diabetes | |
| Pruritus ↓ | Pruritus |
NASH, non-alcoholic steatohepatitis; FXR, farnesoid X receptor; TGR5, Takeda G receptor 5; ASBT, apical sodium-bile acid transporter; LDL, low-density lipoprotein.
FXR agonists for NASH treatment
Extensive studies in experimental NAFLD models have shown that pharmacological activation of FXR improves metabolic homeostasis and decreases inflammatory response in the liver. OCA is a 6α-ethyl derivative of CDCA and a selective FXR agonist with ~100-fold higher potency than CDCA in stimulating FXR activity (116). In animal experiments, OCA attenuated hepatic steatosis, improved insulin sensitivity and decreased hepatic inflammation and fibrosis (106,117). The roles of FXR in regulating metabolic homeostasis and proinflammatory response have been investigated in pharmacological models and genetic KO mice and will only be briefly summarized here. FXR activation inhibits de novo lipogenesis. In diabetes and NAFLD, hyperinsulinemia induces sterol regulatory element-binding protein-1 (SREBP-1)-mediated de novo lipogenesis in the liver. In addition, chronic hyperglycemia also promotes hepatic lipogenesis by increasing substrate flux and carbohydrate-response element-binding protein (CREBP) activity. FXR activation has been shown to repress both SREBP-1 (118) and ChREBP (119). Studies have shown that FXR activation can induce the transcription of several target genes that promote fatty acid oxidation. FXR induces peroxisomal proliferator-activated receptor α (PPARα) (120) and fibroblast growth factor 21 (FGF21) (121), which is a fasting-induced hormone that promotes lipid oxidation and ketogenesis (122-124). In addition, FXR induces liver carboxylesterase 1 (CES1) (125). Hepatic CES1 mediates the hydrolysis of triglycerides that fuels the mitochondrial fatty acid oxidation. FXR activation also antagonizes NF-κB-stimulated inflammation in liver (126) and preserves the intestinal barrier function in inflammatory bowel disease (127). These findings laid the molecular basis for using FXR agonist to treat human NASH patients. To date, OCA treatment significantly improved liver histology in NASH patients in phase-2 clinical trials (108,128). On the other hand, findings from clinical trials also showed that OCA inhibited hepatic bile acid synthesis and their use was associated with hypercholesterolemia (128). Its long-term use raises concerns of higher risk of cardiovascular disease, which is the leading cause of mortality in NASH (129-132). Since OCA is a bile acid derivative, OCA caused pruritus in a significant portion of the NASH patients, especially in patients receiving high dose of OCA (108,128). Finally, due to its induction of FGF19, concerns of tumorigenesis upon its chronic use has been raised (133). Phase-3 trial is currently ongoing to evaluate the efficacy of OCA in NASH resolution and fibrosis. The effects of OCA in patients with NAFLD/NASH has been recently summarized (134). FXR-induced FGF15/19 has been shown to inhibit hepatic steatosis and improved insulin sensitivity and thus partially mediates the metabolic benefits of FXR activation (56,135-137). Intestine-restricted FXR agonist fexaramine activated the gut FXR-FGF15 without causing systemic FXR activation and improved metabolic homeostasis (137,138). Studies in animal models have shown that FGF19 promote liver cancer (139-141), which raised concerns for its use in treating chronic diseases. A non-tumorigenic FGF19 analogue NGM282 has been developed that retained the signaling property in regulating bile acid synthesis and improving hepatic steatosis without exhibiting tumorigenic effect (141-143). New results showed that NGM282 significantly decreased hepatic steatosis and improved fibrosis in human NASH (143,144). NGM282 inhibited hepatic bile acid synthesis and caused hypercholesterolemia. Recent study showed that hypercholesterolemia could be sufficiently addressed with addition of statin (145).
Intestinal FXR antagonism as a therapeutic approach for NASH treatment
Blocking intestine bile acid re-uptake by bile acid sequestrants stimulates hepatic bile acid synthesis and decreases plasma cholesterol concentration. Colesevelam, a second generation of bile acid sequestrant, also improved glycemic control in type-2 diabetes mellitus (146-148). It has been reported that bile acid sequestrants increased gut incretin GLP-1 secretion to improve insulin sensitivity (149). GLP-1 is secreted from the ileal and colonic L-cells in response to macronutrient stimulation. GLP-1 acts on endocrine pancreas to promote insulin secretion from the pancreatic β cells and inhibits glucagon production from the α cells (150). Activation of TGR5 by bile acid or TGR5 agonist has been shown to stimulate GLP-1 production (151,152). It was suggested that by preventing bile acid re-uptake in the ileum bile acid sequestrants increased bile acid concentration in the distal ileum and colon where TGR5 was highly expressed in intestine L cells (153,154). Another possible mechanism mediating the insulin sensitizing effect of bile acid sequestrant is that bile acid sequestrants may delay dietary fat absorption, which causes a higher concentration of dietary fatty acids to reach the distal ileum to induce GLP-1 secretion (155). Various studies have also shown that bile acid sequestrants prevented diet-induced obesity and hepatic steatosis in experimental models (156,157). In addition to promoting GLP1 secretion, bile acid sequestrants may also improve hepatic mitochondrial function, autophagy flux. However, human studies have shown inconsistent findings. Colestimide treatment for 24-week was reported to promote weight loss and improve hepatic steatosis and liver enzymes in NASH patients (158). In contrast, another study showed that 24-week treatment of colesevelam increased hepatic fat content measured by magnetic resonance in biopsy-proven NASH patients (159). Bile acid sequestrant treatment is known to cause transient elevation of plasma triglycerides with unclear underlying causes (160).
More recently, intestine-specific ASBT inhibitors have been developed as potential therapeutics for cholestasis, type-2 diabetes and NASH. This therapeutic approach brought beneficial effects including increased GLP-1 secretion and reduced plasma glucose in experimental animal models and human diabetic patients (161-164). In experimental NAFLD models, ASBT inhibitor also decreased hepatic steatosis and inflammation (162). These findings suggest that ASBT inhibitors may simultaneously offer multiple benefits for diabetes and NASH-associated disorders and are being tested for treating type-II diabetes and NASH in multiple clinical trials. Blocking bile acid re-absorption in the intestine increases bile acid spillover into the colon, which enhances gut incretin secretion and alters gut microbiota (45,152,165-167). Currently, how ASBT inhibitor decreases hepatic fat accumulation is still largely unknown. Its effects in NASH patients remain to be determined. Opposing to FXR agonist treatment, ASBT inhibitors increase hepatic bile acid synthesis and decrease plasma cholesterol. In addition, ASBT inhibitors decrease bile acid re-circulating to the liver to reduce hepatic bile acid load, which may be desirable in conditions of impaired liver function. Although ASBT inhibitor use is obviously not associated with pruritus, currently clinical findings showed that diarrhea is a common side effect of ASBT inhibitor due to bile acid malabsorption, and no available therapeutic approach has been reported to address this issue.
As mentioned earlier, germ-free mice showed increased bile acid synthesis and enlarged bile acid pool, and were resistant to diet-induced weight gain (168). Germ-free mice showed increased tauro-β-muricholic acid (T-βMCA) (96). It is possible that T-βMCA is preserved as a result of lack of bacteria-mediated de-conjugation. Increased T-MCAs acted as FXR antagonists and reduced FGF15 expression, leading to increased hepatic bile acid synthesis and enlarged bile acid pool (45). Treating mice with an antioxidant tempol caused a shift of the microbial community from Firmicutes towards Bacteroidetes, which is consistent with reduced short chain fatty acid production and weight loss (169,170). Tempol also decreased the abundance of BSH-harboring bacteria, which resulted in T-βMCA accumulation and intestinal FXR antagonism. Decreased intestinal FXR activation as a result of genetic FXR knockout, modulation of gut microbiota or treatment with glycine-β-MCA has been shown to reduce ceramide synthesis and circulating ceramide and improve metabolic homeostasis in mice (166,167).
In summary, new evidence from various experimental models suggest that decreasing intestinal bile acid/FXR signaling also results in improved metabolic homeostasis. In addition to increased hepatic bile acid synthesis, increased gut GLP-1 secretion and reduced gut ceramide production, the metabolic impact of decreasing intestine bile acid uptake and FXR activation requires further investigation.
Bile acid signaling in ALD
ALD is a major cause of liver cirrhosis in the US (171). For over 40 years, standard treatments for ALD remain to be abstinence, corticosteroids and nutritional support. There is clearly an unmet need for effective therapies for ALD. Better understanding of the pathogenic factors of ALD is key to developing therapeutic strategies to treat ALD. In the liver, ethanol is metabolized by alcohol dehydrogenase and CYP2E1 to acetaldehyde, which is further metabolized to acetate by aldehyde dehydrogenase. Ethanol metabolism increases cellular NADH/NAD+ ratio and promotes hepatic fat accumulation via modulating hepatic fatty acid oxidation and lipogenic pathways (172). Ethanol metabolism generates oxidative stress and reactive intermediates that form DNA and protein adducts to cause hepatocellular organelle dysfunction, injury and innate and adaptive immune responses (171). In the intestine, alcohol consumption induces gut permeability and alters gut microbiome to promote endotoxin translocation to the liver and systemic circulation (173). Increased endotoxins promote hepatic inflammation and synergize with ethanol to cause liver injury in ALD. The gut-liver axis is increasingly recognized as a major cause of ALD (173). In both humans and experimental models, acute and chronic alcohol consumption was associated with significantly elevated serum endotoxins of gut microbial origin (174,175). Ethanol and its toxic metabolite acetaldehyde produced in the intestine have been shown to inhibit junction proteins and disrupt tight junction integrity (176,177).
The relationship between bile acid metabolism and ALD is incompletely understood. However, recent studies revealed decreased hepatic bile acid synthesis and elevated serum bile acids in ALD patients, suggesting that ALD is associated with disrupted bile acid homeostasis (178,179). Furthermore, serum bile acid levels positively correlated with ALD severity (178,179). In experimental mouse models, bile acid composition was altered and bile acid pool size was increased in chronic 8-week Lieber DeCarli diet-fed mice (180). In mice fed 10-day ethanol diet followed by a single dose of ethanol binge (chronic plus binge model), hepatic CYP7A1 expression was repressed and bile acid pool size was increased (181). In addition, CYP7A1-deficient mice were more sensitive to ethanol-induced hepatic steatosis and inflammation, while hepatic CYP7A1 overexpression in mice were protective against ethanol-induced liver injury (181). Expansion of bile acid pool appeared to be a unique pathological change in ALD because diet-induced NAFLD was not associated with a smaller bile acid pool (182). Interestingly, new studies showed that whole body FXR KO mice but not hepatocyte-specific FXR KO mice were more susceptible to alcohol-induced liver injury (183-185), suggesting a protective role of extrahepatic FXR in ALD. Recent studies showed that both systemic FXR activation and intestine-restricted activation of the FXR signaling improved hepatic steatosis and inflammation in ethanol-fed mice (180,186). Overexpression of FGF19 also ameliorated steatosis and liver injury in ethanol-fed mice, suggesting that FGF15 mediates the beneficial effects of intestine-restricted FXR activation. These studies provided new evidence that targeting FXR may be a potential treatment for ALD. Further studies are needed to evaluate the potential benefits of FXR agonist in human ALD patients. Despite distinct etiology, ALD and NASH share overlapping pathological and histological abnormalities. It is therefore reasonable to expect that the therapeutic benefits of FXR activation in ALD may likely be mediated by improved lipid homeostasis and reduced inflammatory response. On the other hand, the close association between elevated serum bile acid concentration and ALD severity may imply the presence of cholestasis in advanced ALD (178,179). The effect of FXR activation on bile acid homeostasis in ALD remains to be determined. In addition, pruritus and hypercholesterolemia may also be potential undesirable effects in the chronic use of FXR agonist in ALD. The potential benefits of blocking intestine bile acid re-uptake and intestinal FXR agonism in ALD have not been studied.
Conclusion and future perspective
Bile acids are both physiological detergents and metabolic regulators. Bile acids plays a key role in mediating the gut-liver signaling communication to regulate metabolic homeostasis and inflammation. Basic research in last 20 years has unveiled important functions of bile acids in physiology and pathophysiology, which laid the molecular basis for developing bile acid-based therapies for treating liver and metabolic diseases. In this regard, significant progress has been made in translating basic research findings in bile acid biology to clinical applications. Colesevelam has been approved as second- and third-line drugs to control blood glucose in type-2 diabetes. FXR agonist OCA has recently been approved for treating primary biliary cholangitis and is being evaluated in phase-3 clinical trials for NASH treatment. Bile acids and their derivatives have the unwanted side effect of pruritus that may cause low patient compliance. Non-bile acid-based agonists specific for FXR may be developed for treating NASH. Findings from experimental models suggest that targeting intestine bile acid transport and FXR antagonism represent new therapeutic strategies to treat NASH. The molecular mechanisms require further investigation and the beneficial effects in human NASH patients remain to be determined. Alcoholic cirrhosis is a leading cause of liver transplantation in the United States (171). There is still unmet need for developing novel and effective treatments for ALD. New findings demonstrated disrupted bile acid homeostasis in ALD patients and beneficial effects of bile acid-based therapies in experimental ALD models. These findings suggest that bile acids may serve as a potential biomarker for ALD, and targeting bile acid metabolism and signaling may be a potential strategy to treat ALD.
Acknowledgments
Funding: This work was supported in part by NIH grants 1R01DK102487-01 and R01 DK117965-01A1 to T Li, and DK44442 and DK58379 to JYL Chiang.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the noncommercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-ncnd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the Guest Editor (Yu-Jui Yvonne Wan) for the series “Gut Microbiome and Liver Disease” published in Hepatobiliary Surgery and Nutrition. The article was sent for external peer review organized by the Guest Editor and the editorial office.
Conflicts of Interest: The series “Gut Microbiome and Liver Disease” was commissioned by the editorial office without any funding or sponsorship. The authors have no other conflicts of interest to declare.
References
- 1.Russell DW, Setchell KD. Bile acid biosynthesis. Biochemistry 1992;31:4737-49. 10.1021/bi00135a001 [DOI] [PubMed] [Google Scholar]
- 2.Falany CN, Johnson MR, Barnes S, et al. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N-acyltransferase. J Biol Chem 1994;269:19375-9. [PubMed] [Google Scholar]
- 3.Jones RS, Meyers WC. Regulation of hepatic biliary secretion. Annu Rev Physiol 1979;41:67-82. 10.1146/annurev.ph.41.030179.000435 [DOI] [PubMed] [Google Scholar]
- 4.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science 1999;284:1362-5. 10.1126/science.284.5418.1362 [DOI] [PubMed] [Google Scholar]
- 5.Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 2001;98:3369-74. 10.1073/pnas.051551698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 2002;298:714-9. 10.1016/S0006-291X(02)02550-0 [DOI] [PubMed] [Google Scholar]
- 7.Li T, Chiang JY. Bile Acid Signaling in Metabolic Disease and Drug Therapy. Pharmacol Rev 2014;66:948-83. 10.1124/pr.113.008201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell DW. The Enzymes, Regulation, and Genetics of Bile Acid Synthesis. Annu Rev Biochem 2003;72:137- 74. 10.1146/annurev.biochem.72.121801.161712 [DOI] [PubMed] [Google Scholar]
- 9.Myant NB, Mitropoulos KA. Cholesterol 7 alpha-hydroxylase. J Lipid Res 1977;18:135-53. [PubMed] [Google Scholar]
- 10.Axelson M, Mork B, Sjovall J. Occurrence of 3 beta-hydroxy-5-cholestenoic acid, 3 beta,7 alpha-dihydroxy-5-cholestenoic acid, and 7 alpha-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J Lipid Res 1988;29:629-41. [PubMed] [Google Scholar]
- 11.Matsuzawa N, Takamura T, Kurita S, et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007;46:1392-403. 10.1002/hep.21874 [DOI] [PubMed] [Google Scholar]
- 12.Mihalik SJ, Steinberg SJ, Pei Z, et al. Participation of two members of the very long-chain acyl-CoA synthetase family in bile acid synthesis and recycling. J Biol Chem 2002;277:24771-9. 10.1074/jbc.M203295200 [DOI] [PubMed] [Google Scholar]
- 13.Pellicoro A, van den Heuvel FA, Geuken M, et al. Human and rat bile acid-CoA:amino acid N-acyltransferase are liver-specific peroxisomal enzymes: implications for intracellular bile salt transport. Hepatology 2007;45:340-8. 10.1002/hep.21528 [DOI] [PubMed] [Google Scholar]
- 14.Ferrell JM, Boehme S, Li F, et al. Cholesterol 7{alpha}-hydroxylase-deficient mice are protected from high fat/high cholesterol diet-induced metabolic disorders. J Lipid Res 2016;57:1144-54. 10.1194/jlr.M064709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006;47:241-59. 10.1194/jlr.R500013-JLR200 [DOI] [PubMed] [Google Scholar]
- 16.Bjorkhem I, Einarsson K, Melone P, et al. Mechanism of intestinal formation of deoxycholic acid from cholic acid in humans: evidence for a 3-oxo-delta 4-steroid intermediate. J Lipid Res 1989;30:1033-9. [PubMed] [Google Scholar]
- 17.Hylemon PB, Melone PD, Franklund CV, et al. Mechanism of intestinal 7 alpha-dehydroxylation of cholic acid: evidence that allo-deoxycholic acid is an inducible side-product. J Lipid Res 1991;32:89-96. [PubMed] [Google Scholar]
- 18.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 2003;83:633-71. 10.1152/physrev.00027.2002 [DOI] [PubMed] [Google Scholar]
- 19.Takahashi S, Fukami T, Masuo Y, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 2016;57:2130-7. 10.1194/jlr.M071183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hofmann AF, Hagey LR, Krasowski MD. Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res 2010;51:226-46. 10.1194/jlr.R000042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moschetta A, Xu F, Hagey LR, et al. A phylogenetic survey of biliary lipids in vertebrates. J Lipid Res 2005;46:2221-32. 10.1194/jlr.M500178-JLR200 [DOI] [PubMed] [Google Scholar]
- 22.Falany CN, Fortinberry H, Leiter EH, et al. Cloning, expression, and chromosomal localization of mouse liver bile acid CoA:amino acid N-acyltransferase. J Lipid Res 1997;38:1139-48. [PubMed] [Google Scholar]
- 23.Hardison WG. Hepatic taurine concentration and dietary taurine as regulators of bile acid conjugation with taurine. Gastroenterology 1978;75:71-5. 10.1016/0016-5085(78)93767-8 [DOI] [PubMed] [Google Scholar]
- 24.Stipanuk MH, Dominy JE, Jr, Lee JI, et al. Mammalian cysteine metabolism: new insights into regulation of cysteine metabolism. J Nutr 2006;136:1652S-9S. 10.1093/jn/136.6.1652S [DOI] [PubMed] [Google Scholar]
- 25.Kerr TA, Matsumoto Y, Matsumoto H, et al. Cysteine sulfinic acid decarboxylase regulation: A role for farnesoid X receptor and small heterodimer partner in murine hepatic taurine metabolism. Hepatol Res 2014;44:E218-28. 10.1111/hepr.12230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Li J, Matye D, et al. Bile acids regulate cysteine catabolism and glutathione regeneration to modulate hepatic sensitivity to oxidative injury. JCI Insight 2018. doi: . 10.1172/jci.insight.99676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Matye D, Nguyen N, et al. HNF4alpha Regulates CSAD to Couple Hepatic Taurine Production to Bile Acid Synthesis in Mice. Gene Expr 2018;18:187-96. 10.3727/105221618X15277685544442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boyer JL. Bile formation and secretion. Compr Physiol 2013;3:1035-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Childs S, Yeh RL, Georges E, et al. Identification of a sister gene to P-glycoprotein. Cancer Res 1995;55:2029-34. [PubMed] [Google Scholar]
- 30.Berge KE, Tian H, Graf GA, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 2000;290:1771-5. 10.1126/science.290.5497.1771 [DOI] [PubMed] [Google Scholar]
- 31.Smit JJ, Schinkel AH, Oude Elferink RP, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993;75:451-62. 10.1016/0092-8674(93)90380-9 [DOI] [PubMed] [Google Scholar]
- 32.Otsuki M. Pathophysiological role of cholecystokinin in humans. J Gastroenterol Hepatol 2000;15 Suppl:D71-83. 10.1046/j.1440-1746.2000.02178.x [DOI] [PubMed] [Google Scholar]
- 33.Shneider BL, Dawson PA, Christie DM, et al. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J Clin Invest 1995;95:745-54. 10.1172/JCI117722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dawson PA, Haywood J, Craddock AL, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem 2003;278:33920-7. 10.1074/jbc.M306370200 [DOI] [PubMed] [Google Scholar]
- 35.Oelkers P, Kirby LC, Heubi JE, et al. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J Clin Invest 1997;99:1880-7. 10.1172/JCI119355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gong YZ, Everett ET, Schwartz DA, et al. Molecular cloning, tissue distribution, and expression of a 14-kDa bile acid-binding protein from rat ileal cytosol. Proc Natl Acad Sci U S A 1994;91:4741-5. 10.1073/pnas.91.11.4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ballatori N, Christian WV, Lee JY, et al. OSTalpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005;42:1270-9. 10.1002/hep.20961 [DOI] [PubMed] [Google Scholar]
- 38.Rao A, Haywood J, Craddock AL, et al. The organic solute transporter alpha-beta, Ostalpha-Ostbeta, is essential for intestinal bile acid transport and homeostasis. Proc Natl Acad Sci U S A 2008;105:3891-6. 10.1073/pnas.0712328105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hagenbuch B, Stieger B, Foguet M, et al. Functional expression cloning and characterization of the hepatocyte Na+/bile acid cotransport system. Proc Natl Acad Sci U S A 1991;88:10629-33. 10.1073/pnas.88.23.10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest 1994;93:1326-31. 10.1172/JCI117091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meier PJ, Stieger B. Bile salt transporters. Annu Rev Physiol 2002;64:635-61. 10.1146/annurev.physiol.64.082201.100300 [DOI] [PubMed] [Google Scholar]
- 42.Suga T, Yamaguchi H, Sato T, et al. Preference of Conjugated Bile Acids over Unconjugated Bile Acids as Substrates for OATP1B1 and OATP1B3. PLoS One 2017;12:e0169719. 10.1371/journal.pone.0169719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meier PJ. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. Am J Physiol 1995;269:G801-12. [DOI] [PubMed] [Google Scholar]
- 44.Mangelsdorf DJ, Thummel C, Beato M, et al. The nuclear receptor superfamily: the second decade. Cell 1995;83:835-9. 10.1016/0092-8674(95)90199-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sayin SI, Wahlstrom A, Felin J, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 2013;17:225-35. 10.1016/j.cmet.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 46.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem 2003;278:9435-40. 10.1074/jbc.M209706200 [DOI] [PubMed] [Google Scholar]
- 47.Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 2000;6:517-26. 10.1016/S1097-2765(00)00051-4 [DOI] [PubMed] [Google Scholar]
- 48.Lu TT, Makishima M, Repa JJ, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 2000;6:507-15. 10.1016/S1097-2765(00)00050-2 [DOI] [PubMed] [Google Scholar]
- 49.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2005;2:217-25. 10.1016/j.cmet.2005.09.001 [DOI] [PubMed] [Google Scholar]
- 50.Song KH, Li T, Owsley E, et al. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 2009;49:297-305. 10.1002/hep.22627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holt JA, Luo G, Billin AN, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev 2003;17:1581-91. 10.1101/gad.1083503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schaap FG, van der Gaag NA, Gouma DJ, et al. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 2009;49:1228-35. 10.1002/hep.22771 [DOI] [PubMed] [Google Scholar]
- 53.Baker DM, Wang SL, Bell DJ, et al. One or more labile proteins regulate the stability of chimeric mRNAs containing the 3'-untranslated region of cholesterol-7alpha -hydroxylase mRNA. J Biol Chem 2000;275:19985-91. 10.1074/jbc.M002351200 [DOI] [PubMed] [Google Scholar]
- 54.Kong B, Wang L, Chiang JY, et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012;56:1034-43. 10.1002/hep.25740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong B, Guo GL. Soluble expression of disulfide bond containing proteins FGF15 and FGF19 in the cytoplasm of Escherichia coli. PLoS One 2014;9:e85890. 10.1371/journal.pone.0085890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Potthoff MJ, Boney-Montoya J, Choi M, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab 2011;13:729-38. 10.1016/j.cmet.2011.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tarling EJ, Clifford BL, Cheng J, et al. RNA-binding protein ZFP36L1 maintains posttranscriptional regulation of bile acid metabolism. J Clin Invest 2017;127:3741-54. 10.1172/JCI94029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee H, Zhang Y, Lee FY, et al. FXR regulates organic solute transporters alpha and beta in the adrenal gland, kidney, and intestine. J Lipid Res 2006;47:201-14. 10.1194/jlr.M500417-JLR200 [DOI] [PubMed] [Google Scholar]
- 59.Grober J, Zaghini I, Fujii H, et al. Identification of a bile acid-responsive element in the human ileal bile acid-binding protein gene. Involvement Of the farnesoid x receptor/9-cis-retinoic acid receptor heterodimer. J Biol Chem 1999;274:29749-54. 10.1074/jbc.274.42.29749 [DOI] [PubMed] [Google Scholar]
- 60.Neimark E, Chen F, Li X, et al. Bile acid-induced negative feedback regulation of the human ileal bile acid transporter. Hepatology 2004;40:149-56. 10.1002/hep.20295 [DOI] [PubMed] [Google Scholar]
- 61.Ferrebee CB, Li J, Haywood J, et al. Organic Solute Transporter alpha-beta Protects Ileal Enterocytes From Bile Acid-Induced Injury. Cell Mol Gastroenterol Hepatol 2018;5:499-522. 10.1016/j.jcmgh.2018.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li T, Francl JM, Boehme S, et al. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem 2012;287:1861-73. 10.1074/jbc.M111.305789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ocvirk S, O'Keefe SJ. Influence of Bile Acids on Colorectal Cancer Risk: Potential Mechanisms Mediated by Diet - Gut Microbiota Interactions. Curr Nutr Rep 2017;6:315-22. 10.1007/s13668-017-0219-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu T, Coulter S, Yoshihara E, et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019;176:1098-112.e18. 10.1016/j.cell.2019.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gadaleta RM, Garcia-Irigoyen O, Moschetta A. Bile acids and colon cancer: Is FXR the solution of the conundrum? Mol Aspects Med 2017;56:66-74. 10.1016/j.mam.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 66.Ananthanarayanan M, Balasubramanian N, Makishima M, et al. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem 2001;276:28857-65. 10.1074/jbc.M011610200 [DOI] [PubMed] [Google Scholar]
- 67.Denson LA, Sturm E, Echevarria W, et al. The Orphan Nuclear Receptor, shp, Mediates Bile Acid-Induced Inhibition of the Rat Bile Acid Transporter, ntcp. Gastroenterology 2001;121:140-7. 10.1053/gast.2001.25503 [DOI] [PubMed] [Google Scholar]
- 68.Boyer JL, Trauner M, Mennone A, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol 2006;290:G1124-30. 10.1152/ajpgi.00539.2005 [DOI] [PubMed] [Google Scholar]
- 69.Cui YJ, Aleksunes LM, Tanaka Y, et al. Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol Sci 2009;110:47-60. 10.1093/toxsci/kfp094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu Q, Gao R, Wu W, et al. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumour Biol 2013;34:1285-300. 10.1007/s13277-013-0684-4 [DOI] [PubMed] [Google Scholar]
- 71.Joyce SA, Gahan CG. The gut microbiota and the metabolic health of the host. Curr Opin Gastroenterol 2014;30:120-7. 10.1097/MOG.0000000000000039 [DOI] [PubMed] [Google Scholar]
- 72.Stacey M, Webb M. Studies on the antibacterial properties of the bile acids and some compounds derived from cholanic acid. Proc R Soc Med 1947;134:523-37. [DOI] [PubMed] [Google Scholar]
- 73.Kurdi P, Kawanishi K, Mizutani K, et al. Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J Bacteriol 2006;188:1979-86. 10.1128/JB.188.5.1979-1986.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aron-Wisnewsky J, Gaborit B, Dutour A, et al. Gut microbiota and non-alcoholic fatty liver disease: new insights. Clin Microbiol Infect 2013;19:338-48. 10.1111/1469-0691.12140 [DOI] [PubMed] [Google Scholar]
- 75.Turnbaugh PJ, Ley RE, Mahowald MA, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027-31. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- 76.Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science 2006;312:1355-9. 10.1126/science.1124234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 2001;81:1031-64. 10.1152/physrev.2001.81.3.1031 [DOI] [PubMed] [Google Scholar]
- 78.Backhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718-23. 10.1073/pnas.0407076101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reimer RA, McBurney MI. Dietary fiber modulates intestinal proglucagon messenger ribonucleic acid and postprandial secretion of glucagon-like peptide-1 and insulin in rats. Endocrinology 1996;137:3948-56. 10.1210/endo.137.9.8756571 [DOI] [PubMed] [Google Scholar]
- 80.Kok NN, Morgan LM, Williams CM, et al. Insulin, glucagon-like peptide 1, glucose-dependent insulinotropic polypeptide and insulin-like growth factor I as putative mediators of the hypolipidemic effect of oligofructose in rats. J Nutr 1998;128:1099-103. 10.1093/jn/128.7.1099 [DOI] [PubMed] [Google Scholar]
- 81.Cani PD, Dewever C, Delzenne NM. Inulin-type fructans modulate gastrointestinal peptides involved in appetite regulation (glucagon-like peptide-1 and ghrelin) in rats. Br J Nutr 2004;92:521-6. 10.1079/BJN20041225 [DOI] [PubMed] [Google Scholar]
- 82.Delzenne NM, Cani PD, Daubioul C, et al. Impact of inulin and oligofructose on gastrointestinal peptides. Br J Nutr 2005;93 Suppl 1:S157-61. 10.1079/BJN20041342 [DOI] [PubMed] [Google Scholar]
- 83.Ley RE, Turnbaugh PJ, Klein S, et al. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022-3. 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- 84.Clarke SF, Murphy EF, Nilaweera K, et al. The gut microbiota and its relationship to diet and obesity: new insights. Gut Microbes 2012;3:186-202. 10.4161/gmic.20168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Devkota S, Wang Y, Musch MW, et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 2012;487:104-8. 10.1038/nature11225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Islam KB, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011;141:1773-81. 10.1053/j.gastro.2011.07.046 [DOI] [PubMed] [Google Scholar]
- 87.Berg RD. Bacterial translocation from the gastrointestinal tract. Trends Microbiol 1995;3:149-54. 10.1016/S0966-842X(00)88906-4 [DOI] [PubMed] [Google Scholar]
- 88.Ding JW, Andersson R, Soltesz V, et al. The role of bile and bile acids in bacterial translocation in obstructive jaundice in rats. Eur Surg Res 1993;25:11-9. 10.1159/000129252 [DOI] [PubMed] [Google Scholar]
- 89.Lorenzo-Zuniga V, Bartoli R, Planas R, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003;37:551-7. 10.1053/jhep.2003.50116 [DOI] [PubMed] [Google Scholar]
- 90.Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A 2006;103:3920-5. 10.1073/pnas.0509592103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wallace JL, Miller MJ. Nitric oxide in mucosal defense: a little goes a long way. Gastroenterology 2000;119:512-20. 10.1053/gast.2000.9304 [DOI] [PubMed] [Google Scholar]
- 92.Biet F, Locht C, Kremer L. Immunoregulatory functions of interleukin 18 and its role in defense against bacterial pathogens. J Mol Med (Berl) 2002;80:147-62. 10.1007/s00109-001-0307-1 [DOI] [PubMed] [Google Scholar]
- 93.Hooper LV, Stappenbeck TS, Hong CV, et al. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol 2003;4:269-73. 10.1038/ni888 [DOI] [PubMed] [Google Scholar]
- 94.Kobyliak N, Abenavoli L, Mykhalchyshyn G, et al. A Multi-strain Probiotic Reduces the Fatty Liver Index, Cytokines and Aminotransferase levels in NAFLD Patients: Evidence from a Randomized Clinical Trial. J Gastrointestin Liver Dis 2018;27:41-9. [DOI] [PubMed] [Google Scholar]
- 95.Porras D, Nistal E, Martinez-Florez S, et al. Intestinal Microbiota Modulation in Obesity-Related Non-alcoholic Fatty Liver Disease. Front Physiol 2018;9:1813. 10.3389/fphys.2018.01813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Swann JR, Want EJ, Geier FM, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A 2011;108 Suppl 1:4523-30. 10.1073/pnas.1006734107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Miyata M, Takamatsu Y, Kuribayashi H, et al. Administration of ampicillin elevates hepatic primary bile acid synthesis through suppression of ileal fibroblast growth factor 15 expression. J Pharmacol Exp Ther 2009;331:1079-85. 10.1124/jpet.109.160093 [DOI] [PubMed] [Google Scholar]
- 98.Miyata M, Yamakawa H, Hamatsu M, et al. Enterobacteria modulate intestinal bile acid transport and homeostasis through apical sodium-dependent bile acid transporter (SLC10A2) expression. J Pharmacol Exp Ther 2011;336:188-96. 10.1124/jpet.110.171736 [DOI] [PubMed] [Google Scholar]
- 99.Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol 2007;128:837-47. 10.1309/RTPM1PY6YGBL2G2R [DOI] [PubMed] [Google Scholar]
- 100.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 2006;43:S99-112. 10.1002/hep.20973 [DOI] [PubMed] [Google Scholar]
- 101.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol 2010;5:145-71. 10.1146/annurev-pathol-121808-102132 [DOI] [PubMed] [Google Scholar]
- 102.Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta 2010;1801:299-310. [DOI] [PubMed]
- 103.Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678-93. 10.1038/nrd2619 [DOI] [PubMed] [Google Scholar]
- 104.Zollner G, Trauner M. Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br J Pharmacol 2009;156:7-27. 10.1111/j.1476-5381.2008.00030.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fiorucci S, Baldelli F. Farnesoid X receptor agonists in biliary tract disease. Curr Opin Gastroenterol 2009;25:252-9. 10.1097/MOG.0b013e328324f87e [DOI] [PubMed] [Google Scholar]
- 106.Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today 2012;17:988-97. 10.1016/j.drudis.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 107.McMahan RH, Wang XX, Cheng LL, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem 2013;288:11761-70. 10.1074/jbc.M112.446575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mudaliar S, Henry RR, Sanyal AJ, et al. Efficacy and safety of the farnesoid x receptor agonist obeticholic Acid in patients with type 2 diabetes and nonalcoholic Fatty liver disease. Gastroenterology 2013;145:574-82.e1. 10.1053/j.gastro.2013.05.042 [DOI] [PubMed] [Google Scholar]
- 109.Lian F, Xing X, Yuan G, et al. Farnesoid X receptor protects human and murine gastric epithelial cells against inflammation-induced damage. Biochem J 2011;438:315-23. 10.1042/BJ20102096 [DOI] [PubMed] [Google Scholar]
- 110.Hollman DA, Milona A, van Erpecum KJ, et al. Anti-inflammatory and metabolic actions of FXR: insights into molecular mechanisms. Biochim Biophys Acta 2012;1821:1443-52. [DOI] [PubMed]
- 111.Stojancevic M, Stankov K, Mikov M. The impact of farnesoid X receptor activation on intestinal permeability in inflammatory bowel disease. Can J Gastroenterol 2012;26:631-7. 10.1155/2012/538452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pols TW, Noriega LG, Nomura M, et al. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. Journal of hepatology 2011;54:1263-72. 10.1016/j.jhep.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pols TW, Nomura M, Harach T, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell metabolism 2011;14:747-57. 10.1016/j.cmet.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stepanov V, Stankov K, Mikov M. The bile acid membrane receptor TGR5: a novel pharmacological target in metabolic, inflammatory and neoplastic disorders. J Recept Signal Transduct Res 2013;33:213-23. 10.3109/10799893.2013.802805 [DOI] [PubMed] [Google Scholar]
- 115.Duboc H, Tache Y, Hofmann AF. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig Liver Dis 2014;46:302-12. 10.1016/j.dld.2013.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pellicciari R, Costantino G, Camaioni E, et al. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem 2004;47:4559-69. 10.1021/jm049904b [DOI] [PubMed] [Google Scholar]
- 117.Cipriani S, Mencarelli A, Palladino G, et al. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res 2010;51:771-84. 10.1194/jlr.M001602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 2004;113:1408-18. 10.1172/JCI21025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caron S, Huaman Samanez C, Dehondt H, et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol Cell Biol 2013;33:2202-11. 10.1128/MCB.01004-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pineda Torra I, Claudel T, Duval C, et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 2003;17:259-72. 10.1210/me.2002-0120 [DOI] [PubMed] [Google Scholar]
- 121.Cyphert HA, Ge X, Kohan AB, et al. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem 2012;287:25123-38. 10.1074/jbc.M112.375907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Y, Wong K, Walsh K, et al. Retinoic acid receptor beta stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J Biol Chem 2013;288:10490-504. 10.1074/jbc.M112.429852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab 2007;5:415-25. 10.1016/j.cmet.2007.05.003 [DOI] [PubMed] [Google Scholar]
- 124.Badman MK, Pissios P, Kennedy AR, et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 2007;5:426-37. 10.1016/j.cmet.2007.05.002 [DOI] [PubMed] [Google Scholar]
- 125.Xu J, Li Y, Chen WD, et al. Hepatic carboxylesterase 1 Is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology 2014;59:1761-71. 10.1002/hep.26714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang YD, Chen WD, Wang M, et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008;48:1632-43. 10.1002/hep.22519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011;60:463-72. 10.1136/gut.2010.212159 [DOI] [PubMed] [Google Scholar]
- 128.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 2015;385:956-65. 10.1016/S0140-6736(14)61933-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Reaven GM. Multiple CHD risk factors in type 2 diabetes: beyond hyperglycaemia. Diabetes Obes Metab 2002;4 Suppl 1:S13-8. 10.1046/j.1462-8902.2001.00037.x [DOI] [PubMed] [Google Scholar]
- 130.Taskinen MR. Strategies for the management of diabetic dyslipidaemia. Drugs 1999;58 Suppl 1:47-51; discussion 75-82. 10.2165/00003495-199958001-00011 [DOI] [PubMed] [Google Scholar]
- 131.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol 2013;10:330-44. 10.1038/nrgastro.2013.41 [DOI] [PubMed] [Google Scholar]
- 132.Lonardo A, Sookoian S, Chonchol M, et al. Cardiovascular and systemic risk in nonalcoholic fatty liver disease - atherosclerosis as a major player in the natural course of NAFLD. Curr Pharm Des 2013;19:5177-92. 10.2174/1381612811319290003 [DOI] [PubMed] [Google Scholar]
- 133.Schaap FG, Jansen PLM, Olde Damink SWM. Chronic elevation of plasma fibroblast growth factor 19 in long-term farnesoid X receptor agonist therapy, a happy marriage or cause for oncological concern? Hepatology 2018;67:782-4. 10.1002/hep.29511 [DOI] [PubMed] [Google Scholar]
- 134.Abenavoli L, Falalyeyeva T, Boccuto L, et al. Obeticholic Acid: A New Era in the Treatment of Nonalcoholic Fatty Liver Disease. Pharmaceuticals (Basel) 2018. doi: . 10.3390/ph11040104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tomlinson E, Fu L, John L, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002;143:1741-7. 10.1210/endo.143.5.8850 [DOI] [PubMed] [Google Scholar]
- 136.Fu L, John LM, Adams SH, et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004;145:2594-603. 10.1210/en.2003-1671 [DOI] [PubMed] [Google Scholar]
- 137.Fang S, Suh JM, Reilly SM, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 2015;21:159-65. 10.1038/nm.3760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pathak P, Cen X, Nichols RG, et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018;68:1574-88. 10.1002/hep.29857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 2002;160:2295-307. 10.1016/S0002-9440(10)61177-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by Oncogenomic screening. Cancer Cell 2011;19:347-58. 10.1016/j.ccr.2011.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhou M, Wang X, Phung V, et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res 2014;74:3306-16. 10.1158/0008-5472.CAN-14-0208 [DOI] [PubMed] [Google Scholar]
- 142.Wu X, Ge H, Lemon B, et al. Separating mitogenic and metabolic activities of fibroblast growth factor 19 (FGF19). Proc Natl Acad Sci U S A 2010;107:14158-63. 10.1073/pnas.1009427107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Harrison SA, Rinella ME, Abdelmalek MF, et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018;391:1174-85. 10.1016/S0140-6736(18)30474-4 [DOI] [PubMed] [Google Scholar]
- 144.Harrison SA, Rossi SJ, Paredes AH, et al. NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients with Nonalcoholic Steatohepatitis. Hepatology 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 145.Rinella ME, Trotter JF, Abdelmalek MF, et al. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol 2019;70:735-44. 10.1016/j.jhep.2018.11.032 [DOI] [PubMed] [Google Scholar]
- 146.Goldberg RB, Fonseca VA, Truitt KE, et al. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med 2008;168:1531-40. 10.1001/archinte.168.14.1531 [DOI] [PubMed] [Google Scholar]
- 147.Staels B, Kuipers F. Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs 2007;67:1383-92. 10.2165/00003495-200767100-00001 [DOI] [PubMed] [Google Scholar]
- 148.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med 1994;121:416-22. 10.7326/0003-4819-121-6-199409150-00004 [DOI] [PubMed] [Google Scholar]
- 149.Sonne DP, Hansen M, Knop FK. Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. Eur J Endocrinol 2014;171:R47-65. 10.1530/EJE-14-0154 [DOI] [PubMed] [Google Scholar]
- 150.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev 2007;87:1409-39. 10.1152/physrev.00034.2006 [DOI] [PubMed] [Google Scholar]
- 151.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun 2005;329:386-90. 10.1016/j.bbrc.2005.01.139 [DOI] [PubMed] [Google Scholar]
- 152.Thomas C, Gioiello A, Noriega L, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 2009;10:167-77. 10.1016/j.cmet.2009.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Harach T, Pols TW, Nomura M, et al. TGR5 potentiates GLP-1 secretion in response to anionic exchange resins. Sci Rep 2012;2:430. 10.1038/srep00430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Potthoff MJ, Potts A, He T, et al. Colesevelam suppresses hepatic glycogenolysis by TGR5-mediated induction of GLP-1 action in DIO mice. Am J Physiol Gastrointest Liver Physiol 2013;304:G371-80. 10.1152/ajpgi.00400.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hofmann AF. Bile acid sequestrants improve glycemic control in type 2 diabetes: a proposed mechanism implicating glucagon-like peptide 1 release. Hepatology 2011;53:1784. 10.1002/hep.24100 [DOI] [PubMed] [Google Scholar]
- 156.Zhu A, Chen J, Wu P, et al. Cationic Polystyrene Resolves Nonalcoholic Steatohepatitis, Obesity, and Metabolic Disorders by Promoting Eubiosis of Gut Microbiota and Decreasing Endotoxemia. Diabetes 2017;66:2137-43. 10.2337/db17-0070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang Y, Ding Y, Li J, et al. Targeting the Enterohepatic Bile Acid Signaling Induces Hepatic Autophagy via a CYP7A1-AKT-mTOR Axis in Mice. Cell Mol Gastroenterol Hepatol 2016;3:245-60. 10.1016/j.jcmgh.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Taniai M, Hashimoto E, Tobari M, et al. Treatment of nonalcoholic steatohepatitis with colestimide. Hepatol Res 2009;39:685-93. 10.1111/j.1872-034X.2009.00507.x [DOI] [PubMed] [Google Scholar]
- 159.Le TA, Chen J, Changchien C, et al. Effect of colesevelam on liver fat quantified by magnetic resonance in nonalcoholic steatohepatitis: a randomized controlled trial. Hepatology 2012;56:922-32. 10.1002/hep.25731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sjoberg BG, Straniero S, Angelin B, et al. Cholestyramine treatment of healthy humans rapidly induces transient hypertriglyceridemia when treatment is initiated. Am J Physiol Endocrinol Metab 2017;313:E167-74. 10.1152/ajpendo.00416.2016 [DOI] [PubMed] [Google Scholar]
- 161.Wu Y, Aquino CJ, Cowan DJ, et al. Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J Med Chem 2013;56:5094-114. 10.1021/jm400459m [DOI] [PubMed] [Google Scholar]
- 162.Rao A, Kosters A, Mells JE, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Sci Transl Med 2016;8:357ra122. 10.1126/scitranslmed.aaf4823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nunez DJ, Yao X, Lin J, et al. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes Obes Metab 2016;18:654-62. 10.1111/dom.12656 [DOI] [PubMed] [Google Scholar]
- 164.Chen L, Yao X, Young A, et al. Inhibition of apical sodium-dependent bile acid transporter as a novel treatment for diabetes. Am J Physiol Endocrinol Metab 2012;302:E68-76. 10.1152/ajpendo.00323.2011 [DOI] [PubMed] [Google Scholar]
- 165.Gonzalez FJ, Jiang C, Xie C, et al. Intestinal Farnesoid X Receptor Signaling Modulates Metabolic Disease. Dig Dis 2017;35:178-84. 10.1159/000450908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jiang C, Xie C, Lv Y, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 2015;6:10166. 10.1038/ncomms10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jiang C, Xie C, Li F, et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 2015;125:386-402. 10.1172/JCI76738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wostmann BS. Intestinal bile acids and cholesterol absorption in the germfree rat. J Nutr 1973;103:982-90. 10.1093/jn/103.7.982 [DOI] [PubMed] [Google Scholar]
- 169.Mitchell JB, Xavier S, DeLuca AM, et al. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radic Biol Med 2003;34:93-102. 10.1016/S0891-5849(02)01193-0 [DOI] [PubMed] [Google Scholar]
- 170.Li F, Jiang C, Krausz KW, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 2013;4:2384. 10.1038/ncomms3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572-85. 10.1053/j.gastro.2011.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol 2019;70:237-48. 10.1016/j.jhep.2018.10.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015;148:30-6. 10.1053/j.gastro.2014.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Parlesak A, Schafer C, Schutz T, et al. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol 2000;32:742-7. 10.1016/S0168-8278(00)80242-1 [DOI] [PubMed] [Google Scholar]
- 175.Bala S, Marcos M, Gattu A, et al. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS One 2014;9:e96864. 10.1371/journal.pone.0096864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dunagan M, Chaudhry K, Samak G, et al. Acetaldehyde disrupts tight junctions in Caco-2 cell monolayers by a protein phosphatase 2A-dependent mechanism. Am J Physiol Gastrointest Liver Physiol 2012;303:G1356-64. 10.1152/ajpgi.00526.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang Y, Tong J, Chang B, et al. Effects of alcohol on intestinal epithelial barrier permeability and expression of tight junction-associated proteins. Mol Med Rep 2014;9:2352-6. 10.3892/mmr.2014.2126 [DOI] [PubMed] [Google Scholar]
- 178.Brandl K, Hartmann P, Jih LJ, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396-405. 10.1016/j.jhep.2018.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yang Z, Kusumanchi P, Ross RA, et al. Serum Metabolomic Profiling Identifies Key Metabolic Signatures Associated With Pathogenesis of Alcoholic Liver Disease in Humans. Hepatol Commun 2019;3:542-57. 10.1002/hep4.1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hartmann P, Hochrath K, Horvath A, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018;67:2150-66. 10.1002/hep.29676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Donepudi AC, Ferrell JM, Boehme S, et al. Deficiency of cholesterol 7alpha-hydroxylase in bile acid synthesis exacerbates alcohol-induced liver injury in mice. Hepatol Commun 2017;2:99-112. 10.1002/hep4.1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen C, Li J, Matye DJ, et al. Hepatocyte Sortilin 1 knockout and treatment with a Sortilin 1 inhibitor reduced plasma cholesterol in Western diet-fed mice. J Lipid Res 2019;60:539-49. 10.1194/jlr.M089789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kong B, Zhang M, Huang M, et al. FXR deficiency alters bile acid pool composition and exacerbates chronic alcohol induced liver injury. Dig Liver Dis 2019;51:570-6. 10.1016/j.dld.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang M, Kong B, Huang M, et al. FXR deletion in hepatocytes does not affect the severity of alcoholic liver disease in mice. Dig Liver Dis 2018;50:1068-75. 10.1016/j.dld.2018.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Manley S, Ni HM, Williams JA, et al. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol 2014;2:991-1002. 10.1016/j.redox.2014.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Iracheta-Vellve A, Calenda CD, Petrasek J, et al. FXR and TGR5 Agonists Ameliorate Liver Injury, Steatosis, and Inflammation After Binge or Prolonged Alcohol Feeding in Mice. Hepatol Commun 2018;2:1379-91. 10.1002/hep4.1256 [DOI] [PMC free article] [PubMed] [Google Scholar]