

Abstract
Background:
The treatment of high-risk neuroblastoma continues to present a formidable challenge to pediatric oncology. Previous studies have shown that BET (Bromodomain and extra-terminal) inhibitors can inhibit MYCN expression and suppress MYCN-amplified neuroblastoma in vivo. Furthermore, alterations within RAS-MAPK (mitogen-activated protein kinase) signaling play significant roles in neuroblastoma initiation, maintenance, and relapse, and MEK (mitogen-activated extracellular signal-regulated kinase) inhibitors demonstrate efficacy in subsets of neuroblastoma preclinical models. Finally, hyperactivation of RAS-MAPK signaling has been shown to promote resistance to BET inhibitors. Therefore, we examined the anti-tumor efficacy of combined BET/MEK inhibition utilizing I-BET726 or I-BET762 and trametinib in high-risk neuroblastoma.
Procedure:
Utilizing a panel of genomically annotated neuroblastoma cell line models, we investigated the in vitro effects of combined BET/MEK inhibition on cell proliferation and apoptosis. Furthermore, we evaluated the effects of combined inhibition in neuroblastoma xenograft models.
Results:
Combined BET and MEK inhibition demonstrated synergistic effects on the growth and survival of a large panel of neuroblastoma cell lines through augmentation of apoptosis. Combination therapy slowed tumor growth in a MYCN-non-amplified, NRAS mutated neuroblastoma xenograft model, but had no efficacy in a MYCN-amplified model harboring a loss-of-function mutation in NF1.
Conclusions:
Combinatorial BET and MEK inhibition was synergistic in the vast majority of neuroblastoma cell lines in the in vitro setting but showed limited anti-tumor activity in vivo. Collectively, these data do not support clinical development of this combination in high-risk neuroblastoma.
Keywords: neuroblastoma, BET, MAPK, MEK, MYCN, MYC
INTRODUCTION
High-risk neuroblastoma, an aggressive tumor that arises from the developing peripheral sympathetic nervous system, results in considerable morbidity and mortality and remains a substantial challenge within pediatric oncology.1 MYCN amplification occurs in approximately 40% of patients with high-risk neuroblastoma and is significantly correlated with poor prognosis, even in patients who have otherwise favorable disease features.1,2 Genetically engineered mouse and zebrafish models in which MYCN is overexpressed in the peripheral sympathetic nervous system develop tumors that closely recapitulate human neuroblastomas3,4. Previous investigations have demonstrated that high MYC/MYCN gene signatures predict poor prognosis in patients with neuroblastoma, even in the absence of MYCN amplification.5,6 In support of these studies, a significant subset of non-MYCN-amplified tumors express high levels of MYCN or MYC protein, demonstrating a potent role for both MYCN and MYC in neuroblastoma pathogenesis.7,8. Given the key role of both MYCN and MYC in neuroblastoma aggression, therapeutically inhibiting the MYC family has been the focus of years of investigation.
Targeting the MYC family, however, presents a formidable challenge, as these transcription factors orchestrate the expression of thousands of genes.9,10 The discovery that Bromodomain and extraterminal (BET) proteins mediate the transcription of MYC, coupled with the development of BET inhibitors, provided a foundation for beginning to surmount this therapeutic challenge.11,12,13,14 BET proteins are “chromatin readers” that bind to acetylated lysines in chromatin, recruiting additional chromatin modifying complexes to dynamically influence both gene activation and repression.9,15 Several groups showed that BET inhibitors target the transcription of MYC, disrupt the orchestration of its transcriptional program, and display therapeutic efficacy in multiple MYC-driven cancers11,12,13,14, including neuroblastoma.16,17
In addition to the MYC family, various components of the RAS-MAPK (mitogen-activated protein kinase) network are deregulated in neuroblastoma. Activation of this signalling network occurs through mutations and amplifications in upstream receptor tyrosine kinases (ALK)18,19,20,21, mutations in signal transduction proteins (NRAS, KRAS),22 and mutations or copy number loss of negative regulators (NF1, PTPN11).22 Lesions in the RAS-MAPK signaling pathway occur in 3–5% of newly diagnosed neuroblastomas23 and RAS-MAPK mutations are significantly enriched in relapsed neuroblastomas.24 Intriguingly, in ovarian cancer, chronic BET inhibition leads to the activation of multiple Receptor Tyrosine Kinases (RTK) and downstream RAS/PI3K networks, ultimately conferring resistance to BET inhibitors.25 In these models, ovarian cells became exquisitely dependent on RTK-driven networks, and combination therapies targeting RTK/MAPK networks, with BET inhibition, results in increased inhibition of cell growth.
Given the deregulation of MYCN/MYC and RAS-MAPK networks in primary and relapsed neuroblastomas, as well as the molecular crosstalk between these two networks, we hypothesized that combined BET and MEK (mitogen-activated extracellular signal-regulated kinase) inhibition would be more effective than single agent therapy.
METHODS
Cell Lines
Cell lines were obtained from the Children’s Hospital of Philadelphia cell line bank and maintained in RPMI-1640 media containing 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin at 37°C and 5% CO2. Annual genotyping (AmpFISTR Identifier Kit) and a single-nucleotide polymorphism (SNP) array analysis (Illumina H550) of cell lines were performed to ensure maintenance of cell identity, as previously described.26
Cell Growth and Viability Assays
I-BET726 and I-BET762 were provided by GSK and trametinib was purchased from Cellagen. To assess the combination efficacy of I-BET726 and trametinib, neuroblastoma cell lines were seeded in 96-well plates and treated with the combination of I-BET726 and trametinib. Cells were plated at a density of 1,000–4,000 cells/well of a 96-well plate and treated in triplicate 24 hours later with the same concentration range of either compound or the combination. Concentrations used were based upon single agent IC50 values determined by Cell Titer-Glo. For combination studies, concentration selection included a range from 1/4x to 4x (with x indicating single agent IC50) and cells were plated in duplicate wells in a matrix formation for treatment with both compounds. Viability was measured after 6 days with Cell Titer-Glo, and IC50 values were calculated as previously described.26 Drug synergy was analyzed by isobologram and combination index methods and synergy scores were determined by Chalice.27,28
Western Blotting
To extract protein, we utilized lysis buffer containing 150 mM NaCl, 25 mM Tris, 1 mM EDTA, 1mM EGTA, 1mM DTT, and 1% Triton X-100 with 1% Halt protease/phosphatase inhibitor (Thermo Scientific, #78440). Protein concentration was determined via the BCA protein assay (Thermo Scientific). Approximately 20–40 μg of protein were resolved by SDS-PAGE and were blotted as previously described.26 The following antibodies were utilized (Cell Signaling, unless otherwise noted): p-ERK (1:4000, #4370), ERK (1:4000, #4695), MYCN (1:1000, #9405), MYC (1:1000, #13987), PARP (1:1000, #9542), c-PARP (1:1000, #5625), Ku80 (1:2000, #2753), and β-Actin (1:5000, Santa Cruz #sc-47778). All immunoblots were stained with Ponceau S prior to immunoblotting to confirm equal protein loading among lanes.
Flow Cytometry
Following treatment at the indicated time points, cells were trypsinized, washed with PBS supplemented with 1% FBS, and fixed overnight at −20°C with 70% ethanol. Cells were then washed twice, stained with Fx-Cycle Violet (Invitrogen #F10347) as per the manufacturer’s protocol, and analyzed on an Attune flow cytometer (Life Technologies). Analysis was performed using FlowJo v10, with cells gated to exclude non-cell debris and doublets. Proportions of cells in sub-G1, G1, S, and G2/M phases of the cell cycle were then determined using FlowJo’s Cell Cycle Univariate Analysis feature and normalized to 100% prior to graphing.
Xenograft Studies
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed. Female, CB17 SCID−/− mice aged 5–7 weeks were obtained from Taconic Biosciences. To evaluate the synergy of I-BET762 and trametinib, neuroblastoma cell line-derived xenografts were implanted subcutaneously into the right flank of each mouse. Animals bearing engrafted tumors of 0.2 – 0.5 cm3 were then randomized (n ≥ 9 mice/group) into the following groups for oral treatment: 1) 25 mg/kg I-BET762, once daily, 2) 1 mg/kg trametinib, once daily, 3) 25 mg/kg I-BET762 and 1 mg/kg trametinib, or 4) vehicle control. Tumor volume was measured throughout treatment using calipers and was calculated as: volume = (π/6)(diameter3), as previously described.26 Mice were euthanized when tumor volume reached 3 cm3, in accordance with the Children’s Hospital of Philadelphia IACUC guidelines. A linear mixed effects model was used to measure differences in tumor growth rates between the vehicle, single agent, and combinations.
Statistics
Linear mixed effects modeling was used to quantitate differences in the rate of tumor volume changing over time among different groups. The model included group, day, and group-by-day interaction as fixed effects, and included a random intercept and slope for each mouse. A significant group-by-day interaction suggests that the tumor volume changes at different rates for the two comparison groups. The model used the control group as reference and created separate group indicators and interaction terms for the remaining treatment groups. Appropriate contrast statements were created to compare treatment groups against the control group.
Data Availability
Data available on request from the authors.
RESULTS
Combined BET and MEK inhibition demonstrates synergy across multiple neuroblastoma cell line models
We sought to examine the anti-tumor efficacy of combined BET and MEK inhibition utilizing I-BET726, a prototype BET inhibitor that possesses good in vitro solubility, and trametinib. To maximize the pre-clinical relevance of these studies, we examined a large panel of genomically annotated human neuroblastoma cell line models.29 The table summarizes MYCN/MYC status, alterations in the RAS-MAPK network (including ALK mutations and NF1 status), and alterations in the TP53 network (including TP53 and MDM2 status). As depicted in Table 1, there was a wide range in sensitivity to both agents, with a median IC50 value for I-BET726 of 186 nM (range 25–1142 nM) and median IC50 value for trametinib of 91 nM (range 8–312400 nM), consistent with previous studies.24 There was no apparent correlation of I-BET726 cytotoxicity with MYCN status, whereas canonical MAPK mutations were enriched in the lines most sensitive to trametinib. Combination treatments were performed in a matrix design across a range of doses for each compound guided by single agent IC50 values, as previously described.26 As shown in Table 1, BET/MEK inhibition demonstrated synergy in 21/22 cases, with very strong or strong synergy in 10/22 neuroblastoma cell lines and moderate synergy or synergy in 11/22 cases. Notably, there was no clear biomarker identified that predicted synergy.
Table 1. Effects of IBET-726 and trametinib alone and in combination across neuroblastoma cell line models annotated with clinically relevant mutations.
Genomic analysis was performed utilizing a focused gene panel (Foundation Medicine platform). Cell lines are ranked according to synergy scores (most to least sensitive). Synergy values were determined by Chalice analysis of cell survival data obtained from CellTiter-Glo assays using a matrix treatment schedule from 1/4x-4x IC50 (+++++ very strong synergy; ++++ strong synergy; +++ synergy; ++ moderate synergy; + slight synergy; +/− additive, - antagonistic).
| Cell Line | MYCN Status | MYC Amplification | MYC Expression (FPKM) | RAS-MAPK Pathway Mutations | ALK Mutations | TP53 Mutations | MDM2 Amplification | NF1 Deletion | IC50 GSK726 (nM) | IC50 Trametinib (nM) | Combination Index | Synergy |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NB1691 | MYCN amplified | Non-amp | 0.12 | Amplified | 1142 | 508 | 0.03 | +++++ | ||||
| SK-N-FI | Non-amp | 1.75 | M246R | Deletion (homozygous) |
1023 | 40 | 0.2 | ++++ | ||||
| NLF | MYCN amplified | Non-amp | 0.13 | V203M | 875 | 248 | 0.03 | +++++ | ||||
| NB-SD | MYCN amplified | Non-amp | 0.01 | FGFR1 P770S | F1174L | C176F | 820 | 66 | 0.35 | +++ | ||
| SMS-SAN | MYCN amplified | Non-amp | 0.03 | F1174L | 797 | 1275 | 0.28 | ++++ | ||||
| SK-N-BE(2)-C | MYCN amplified | Non-amp | 0.06 | NF1 N664fs | C135F | Deletion (hemizygous) | 706 | 64 | 0.18 | ++++ | ||
| NGP | MYCN amplified | Non-amp | 0.12 | A159D C141W |
541 | 91 | 0.92 | ± | ||||
| NB-1643 | MYCN amplified | Non-amp | 0.02 | R1275Q | 515 | 341 | 0.35 | +++ | ||||
| NB69 | Non-amp | 165.14 | Deletion (hemizygous) | 279 | 3920 | 0.59 | +++ | |||||
| NB-Ebc1 | Non-amp | 0.12 | KRAS G12D | 271 | 15 | 0.44 | +++ | |||||
| LA-N-5 | MYCN amplified | Non-amp | 0.07 | R1275Q | 186 | 426 | 0.49 | +++ | ||||
| SK-N-AS | Non-amp | 10.24 | NRAS Q61K | 166 | 8 | 0.69 | +++ | |||||
| SK-N-BE(2) | MYCN amplified | Non-amp | 0.24 | NF1 N664fs | C135F | 157 | 61 | 0.03 | +++++ | |||
| IMR-05 | MYCN amplified | Non-amp | 0.00 | 109 | 262 | 0.29 | ++++ | |||||
| SK-N-DZ | MYCN amplified | Non-amp | 0.02 | R110L | 91 | 13,470 | 0.53 | +++ | ||||
| Kelly | MYCN amplified | Non-amp | 0.02 | F1174L | P177T | 86 | 174 | 0.2 | ++++ | |||
| NBL-S | Non-amp | 0.19 | 50 | 19 | 0.22 | ++++ | ||||||
| SY-5Y | Non-amp | 15.63 | PTPN11 T507K | F1174L | 30 | 43 | 0.35 | +++ | ||||
| CHP-212 | MYCN amplified | Non-amp | 0.05 | NRAS Q61K | 30 | 31 | 0.15 | ++++ | ||||
| SK-N-SH | Non-amp | 25.72 | F1174L | 28 | 72 | 0.68 | +++ | |||||
| CHP-134 | MYCN amplified | Non-amp | 0.18 | 25 | 312,400 | 0.83 | ++ |
| CI Interval | Symbol | Observed synergy |
|---|---|---|
| <0.1 | +++++ | Very strong |
| 0.1 – 0.3 | ++++ | Strong |
| 0.3 – 0.7 | +++ | Synergy |
| 0.7 – 0.85 | ++ | Moderate synergism |
| 0.85 – 0.9 | + | Slight synergism |
| 0.9 – 1.1 | ± | Additive |
| >0.1 | - | Antagonistic |
Single agent treatment decreases cell cycle progression
As BET and MEK inhibition demonstrated synergy in the majority of neuroblastoma cell line models examined, we first tested the effect of combined inhibition on cell cycle progression. We studied 2 MYCN-amplified neuroblastoma models, NLF and SK-N-BE(2)C, that are NF1 wild-type and NF1 mutated (loss–of- function), respectively, as well as 2 non-MYCN-amplified models, NBL-S and SK-N-AS, NF1 wild-type and NRAS mutated, respectively (Table 1). Cells were treated with control, single agent, or combined treatment, and flow cytometry performed to determine cell cycle distribution (Figure 1). Both single agent and combined BET/MEK inhibition, to varying degrees, diminished progression through the cell cycle, increasing the percentage of cells in G0/G1 phase and reducing the percentage in S phase. This influence on cell cycle progression was most prominent in NLF, NBL-S, and SK-N-AS; it was less striking in SK-N-BE(2)C, which may be due in part to the fact that the concentrations of I-BET726 and trametinib were below the individual IC50s in this model. To confirm that BET and MEK inhibitors inhibited MYCN/MYC and MAPK signaling, respectively, we assessed MYCN/MYC protein levels and phosphorylated ERK protein levels in the SK-N-BE(2)C (Supplementary Figure S1A and S1B) and SK-N-AS models (Supplementary Figure S1C and S1D). In these cell lines, BET inhibition led to decreased levels of MYCN/MYC and MEK inhibition led to decreased phosphorylated ERK. Combined treatment, however, did not lead to further decreases in the levels of MYCN/MYC and phosphorylated ERK.
Figure 1. Effects of IBET-726 and trametinib alone and in combination on cell cycle distribution.
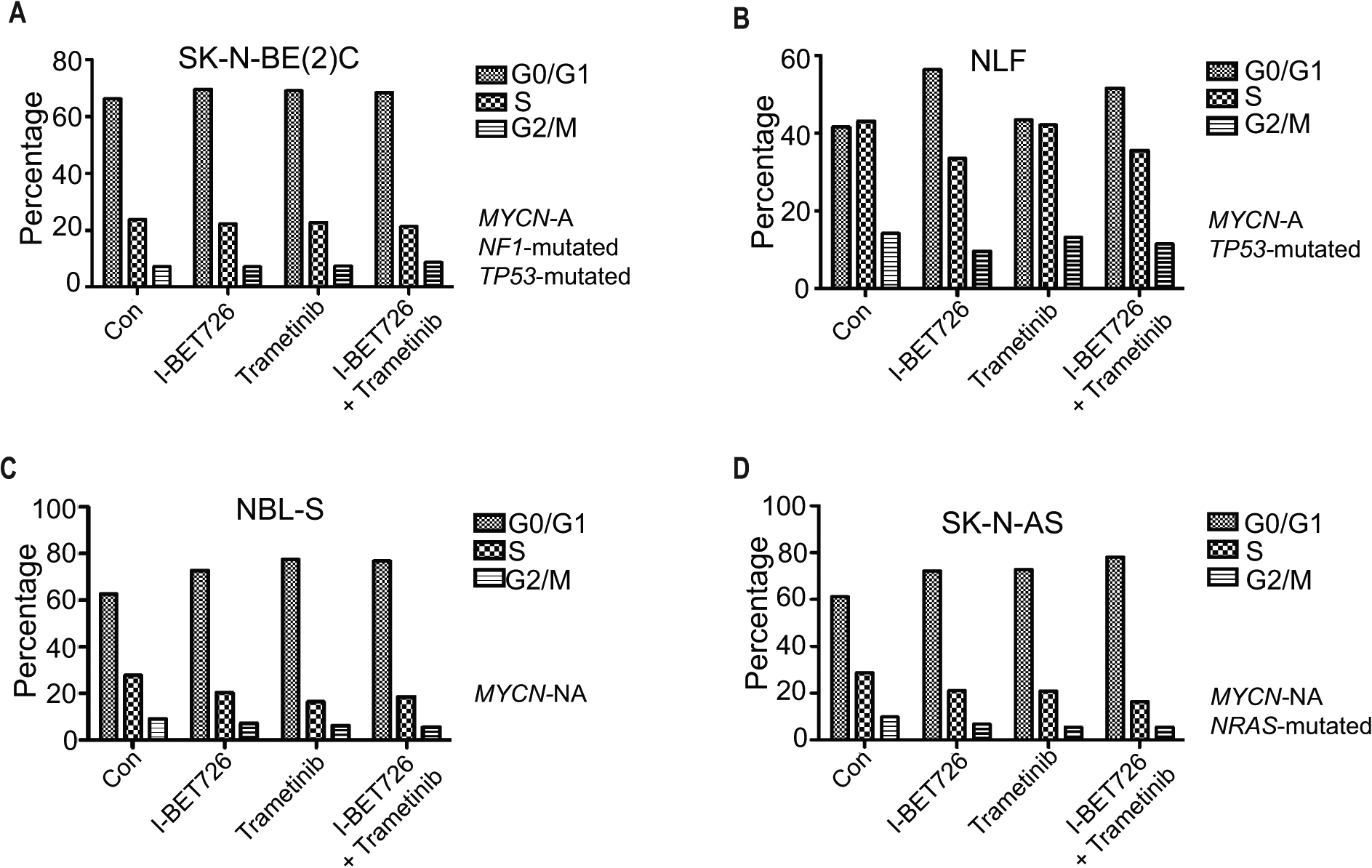
(A-D) Neuroblastoma cell lines SK-N-BE(2C) (A), NLF (B), NBL-S (C), and SK-N-AS (D) were treated with control, I-BET726 (500 nM), trametinib (25 nM), or I-BET726/trametinib for 72 hours and stained with PI. Flow cytometry was performed to detect the proportions of cells present in G0/G1 phase, S phase, and G2/M phase.
Combined BET and MEK inhibition augments apoptosis
We next determined the effect of single agent versus combined BET/MEK inhibition on apoptosis. Cells were treated with control, single agent, or combined treatment for up to 96 hours and apoptosis assessed by quantitating PARP cleavage. Treatment with I-BET726 alone resulted in apoptosis in neuroblastoma cell line models, while treatment with single agent trametinib did not (Figure 2). In SK-N-BE (2)C (Figure 2A), NBL-S (Figure 2C) and SK-N-AS (Figure 2D), we observed increased levels of cleaved PARP with combined BET/MEK inhibition compared to single agent alone. In NLF, combined treatment did not increase the levels of cleaved PARP, suggesting that the predominant effect may be on the cell cycle.
Figure 2. Effects of IBET-726 and trametinib alone and in combination on apoptosis.
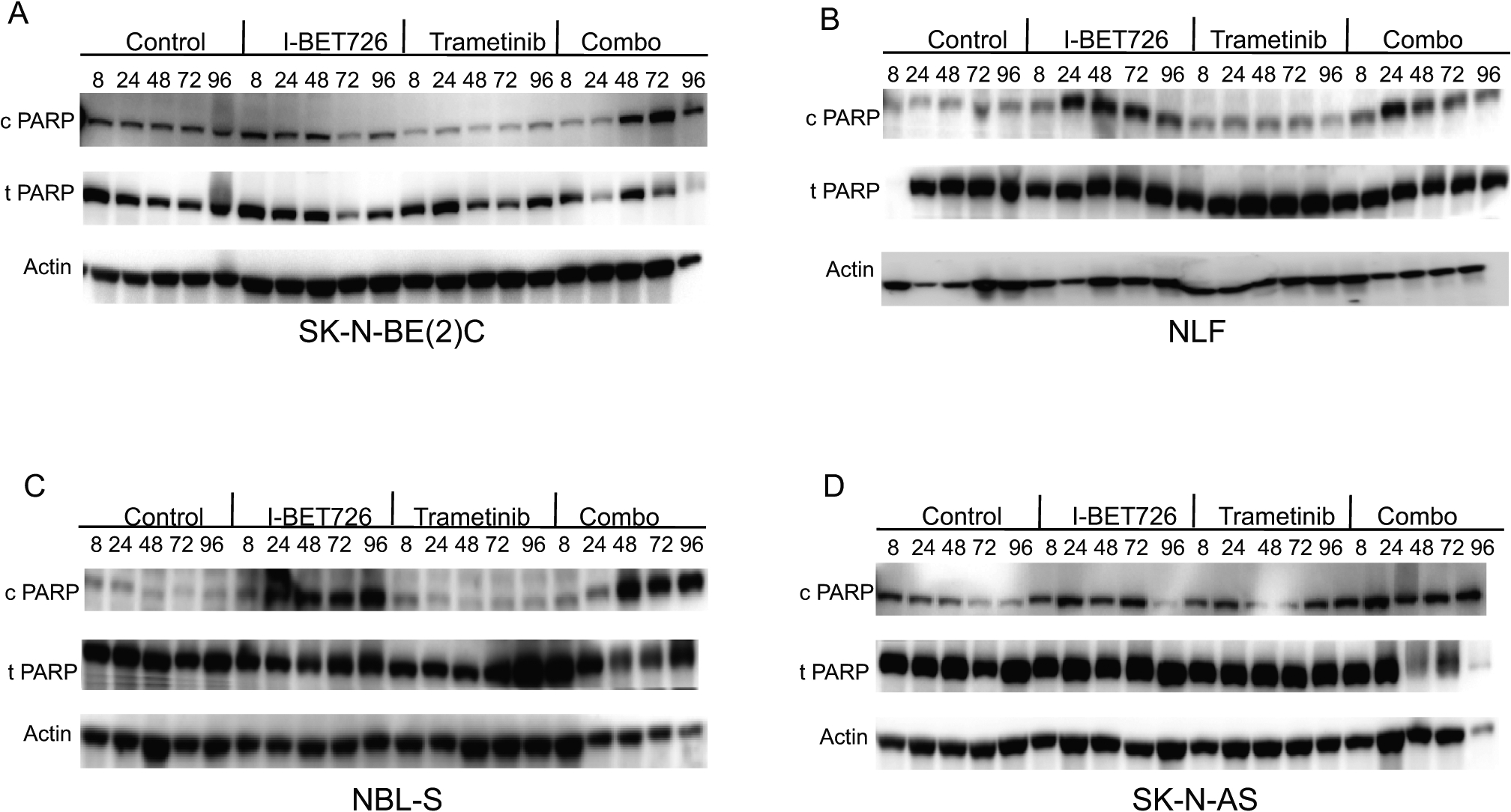
(A-D) Neuroblastoma cell lines SK-N-BE(2C) (A), NLF (B), NBL-S (C), and SK-N-AS (D) were treated with control, I-BET726 (500 nM), trametinib (25 nM), or I-BET726/trametinib for varying amounts of time (8–96 hours). Immunoblotting for cleaved PARP, total PARP, and actin was performed.
Combined BET and MEK inhibition shows limited activity against neuroblastoma cell line xenografts
Two previous studies have demonstrated the potential efficacy of BET inhibition in MYCN- amplified neuroblastoma xenograft models.16,30 To assess the impact of BET inhibition in additional neuroblastoma models, we used I-BET762, which displays poor in vitro solubility, but possesses superior in vivo pharmacokinetic properties compared to I-BET726. We treated two MYCN-amplified xenograft models (NGP and NB-1691) with I-BET762 (20 mg/kg once/day), but did not observe significant responses (Supplementary Figure S2A and S2B).31 We then escalated the dose and administered I-BET762 at 25 mg/kg 1x/day or 12.5 mg/kg 2x/day, but again did not observe any effect on tumor growth in SK-N-BE(2) C, a MYCN-amplified xenograft model (Supplementary Figure S2C). Finally, we further increased the dose and treated another MYCN-amplified xenograft model, IMR5, with a higher dose of I-BET762 (30 mg/kg daily) and observed minimal effects on tumor growth (Supplementary Figure S2D).
Given these results, we next assessed the impact of combined BET and MEK inhibition on the SK-N-BE(2)C xenograft, which is MYCN-amplified and also has a loss-of -function mutation within NF1. Neuroblastoma xenografts were implanted subcutaneously and allowed to grow to approximately 200 mm3 at which point they were randomized to one of four treatment arms: 1) vehicle (daily), 2) I-BET762 (daily, 25 mg/kg), 3) trametinib (daily, 1 mg/kg), or 4) I-BET762-trametinib.32 In this model, we did not observe tumor growth delay with either single agent or combined treatment (Figure 3A; p= 0.197, control versus I-BET762; p= 0.840, control versus trametininb; p= 0.809, control versus combination). Neither single agent nor combined inhibition prolonged survival (Figure 3B), although the regimen was well-tolerated, without significant weight loss (Figure 3C).
Figure 3. Effects of IBET-762 and trametinib in two neuroblastoma xenograft models.
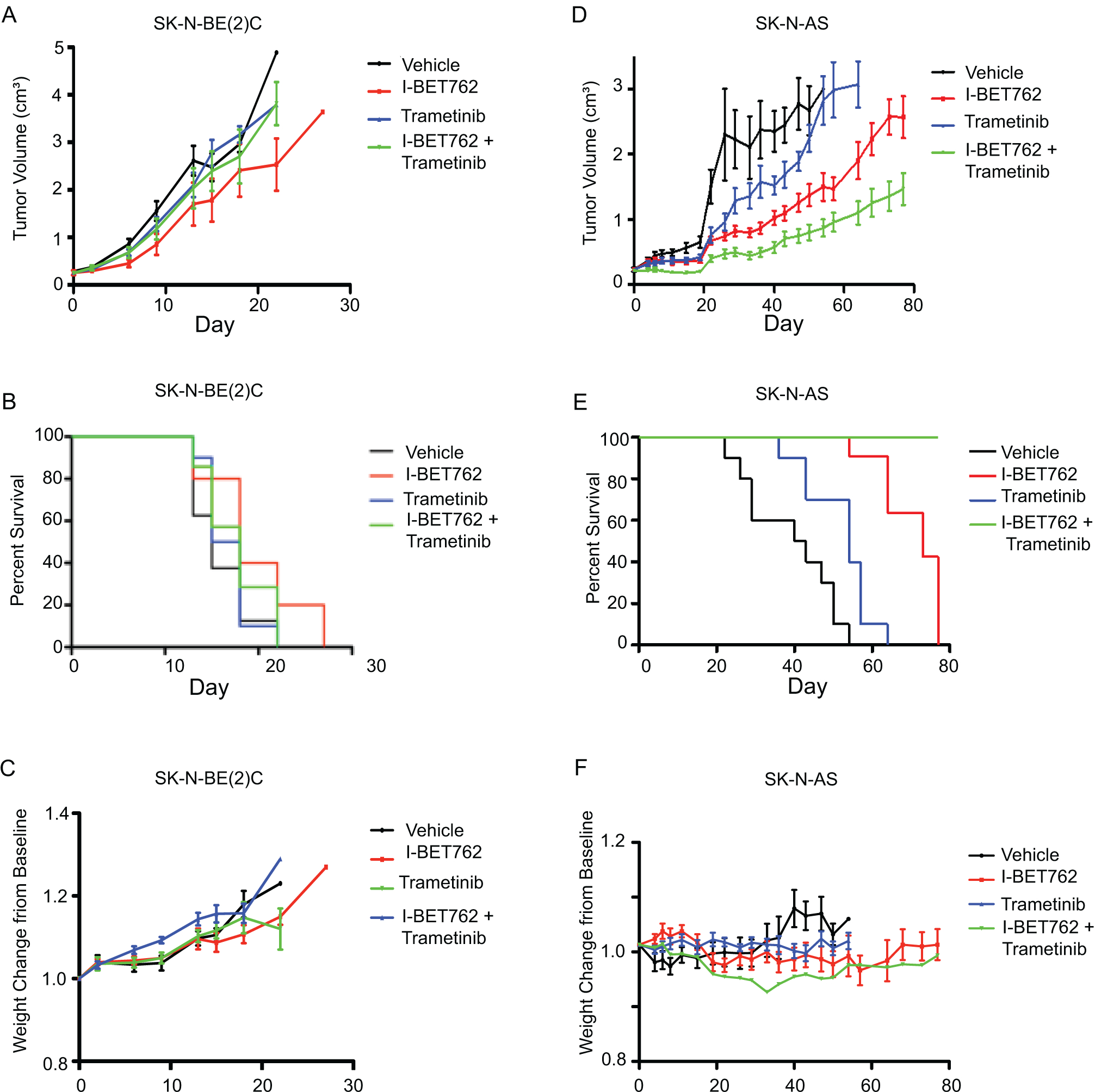
Xenografts were treated with: 1) vehicle (daily), 2) I-BET762 (daily, 25 mg/kg), 3) trametinib (daily, 1 mg/kg), or 4) I-BET762-trametinib. (A) Response of tumor volumes to study arms for SK-N-BE(2)C xenografts. (B) Kaplan-Meier analyses for SK-N-BE(2)C xenografts. (C) Weight change from baseline on study arms for SK-N-BE(2)C xenografts. (D-F) Response of tumor volumes (D), Kaplan-Meier analyses (E), and weight changes (F), respectively, for SK-N-AS xenografts.
As recent investigations have demonstrated there are subsets of aggressive neuroblastomas that do not express MYCN, but rather, MYC, we examined the role of combined BET/MEK inhibition in SK-N-AS, a non-MYCN-amplified xenograft model that expresses high levels of MYC and harbors an NRAS mutation. We observed tumor growth delay with single agent I-BET762 (p<0.0001) and single agent trametinib (p=0.0011) treatments compared to vehicle (Figure 3D). In comparison to vehicle and single agent trametinib, combined I-BET762 and trametinib treatment further delayed tumor growth (p<0.0001 and p=0.0002, respectively; Figure 3D). Moreover, combined BET and MEK inhibition prolonged survival compared to vehicle, single agent trametinib, or single agent IBET-762 (p<0.0001 for all comparisons; Figure 3E). This dosing strategy was well tolerated, without evidence of weight loss or other concerns for toxicity (Figure 3F).
DISCUSSION
In this study we evaluated the efficacy of combined BET and MEK inhibition. We demonstrated that in vitro treatment with I-BET726 alone significantly inhibits cell proliferation in both MYCN-amplified and non-MYCN- amplified neuroblastoma cell line models, consistent with previous reports.17 Combined BET/MEK inhibition demonstrated synergistic effects on cell proliferation across a panel of neuroblastoma cell lines in vitro, augmenting apoptosis, as compared to single agent treatment. These findings are consistent with studies that suggest a prominent role for the MYC family, broadly considered, in large subsets of neuroblastoma.7,8,5
In contrast to recent studies in which the combination of the clinical BET inhibitor GSK525762 (I-BET762; 15 mg/kg daily) and trametinib (1 mg/kg daily) demonstrated synergy across aggressive adult malignancies31, we did not demonstrate substantive efficacy of combined inhibition in neuroblastoma xenografts. Notably, in our studies, we used a higher dose of I-BET762 (25 mg/kg daily) and the same dose of trametinib, and yet did not observe significant effects in neuroblastoma xenografts.
In the SK-N-BE(2)C xenograft model, neither single agent treatment nor combined inhibition delayed tumor growth. In contrast, in SK-N-AS, a non-MYCN-amplified xenograft model with a canonical NRAS mutation, we observed sensitivity to both single agent treatment and a greater response to combined inhibition. These results further show that BET inhibition may demonstrate efficacy in some non-MYCN-amplified tumors and provide further support for the role of MYC in neuroblastoma tumorigenesis. Indeed, in a zebrafish model of neuroblastoma, MYC overexpression led to the development of neuroblastoma tumors, with similar kinetics to what is observed in this model with MYCN overexpression.33 While our results in the SK-N-AS xenograft model are statistically significant, they represent progressive disease and are unlikely to be clinically impactful. In aggregate, our findings do not provide support for the clinical development of this combination in neuroblastoma.
In addition to the MAPK family, multiple kinases, including ALK (Anaplastic lymphoma kinase)18,19,20,21, AURKA (Aurora kinase A),34,35,36 CDK4 (Cyclin -dependent kinase 4)37,38, and CHK1 (Checkpoint kinase 1)39,40 play prominent roles in neuroblastoma tumorigenesis. Inhibitors directed against these kinases are being studied for incorporation into the treatment of subsets of patients with neuroblastoma, with ALK inhibitors currently being studied with frontline standard of care therapy.1,41 While targeted kinase inhibition is effective in some tumors, resistance to such agents, which is driven in part by epigenetic alterations,42 remains a significant challenge in oncology. In breast cancer cells exhibiting amplification of the RTK ERBB2, treatment with the ERBB2 inhibitor lapatinib leads to the upregulation of multiple RTK networks and this kinome reprogramming is inhibited through BET inhibition.43 While our preclinical studies do not show efficacy of combined BET and MEK inhibition, it will be of interest to test whether combining BET inhibitors, or other inhibitors directed against the malignant epigenome, with agents directed against ALK, AURKA, CDK4, or CHK1, might constitute rational combinations in defined subsets of high-risk neuroblastoma patients.
Supplementary Material
Supplementary Figure S1, related to Figure 1. Effects of IBET-726 and trametinib alone and in combination on molecular targets. (A-B). SK-N-BE(2C) was treated with control or varying combinations of inhibitors as indicated (I-BET726 of 1 corresponds to 500 nM and trametinib of 1 corresponds to 25 nM). Lysates were harvested and blotted for MYCN, p-ERK (phosphorylated ERK), t-ERK (total ERK), and Ku80. (C-D). SK-N-AS was treated with control or varying combinations of inhibitors. Lysates were harvested and blotted for MYC, p-ERK, t-ERK, and Ku80.
Supplementary Figure S2, related to Figure 3. Effects of IBET-762 on multiple neuroblastoma xenograft models. (A-B) NGP and NB-1691 xenografts were treated with vehicle or I-BET762 (daily, 20 mg/kg). Response of tumor volumes is shown. (C) SK-N-BE(2)C xenografts were treated with vehicle or I-BET762 (25 mg/kg/day or 12.5 mg/kg 2x/day). Response of tumor volumes is shown. (D) IMR5 xenografts were treated with vehicle or I-BET762 (daily, 30 mg/kg). Response of tumor volumes is shown.
ACKNOWLEDGMENTS
This work was supported in part by a research grant from GlaxoSmithKline (J.M.M.), NIH Grants R35 CA220500 (J.M.M.), K12 HD04324 (R.W.S), K08-7K08CA194162-02 (R.W.S.), T32GM008562 (J.R.H.), the Aflac Cancer and Blood Disorders Center Trust (R.W.S.), and the William G. Woods, MD, Aflac Clinical Investigator Chair (R.W.S).
Abbreviations Key:
- BET
Bromodomain and extraterminal
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated extracellular signal-regulated kinase
- RTK
receptor tyrosine kinase
- ALK
anaplastic lymphoma kinase
- AURKA
Aurora kinase A
- CDK4
cyclin–dependent kinase 4
Footnotes
CONFLICT OF INTEREST STATEMENT
Anastasia Wyce and Olena Barbash are full-time employees and shareholders at GlaxoSmithKline.
REFERENCES
- 1.Matthay KK, Maris JM, Schleiermacher G, et al. Neuroblastoma. Nat Rev Dis Primers. 2016;2:16078. [DOI] [PubMed] [Google Scholar]
- 2.Bagatell R, Beck-Popovic M, London WB, et al. Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol. 2009;27(3):365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu S, Lee JS, Guo F, et al. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 2012;21(3):362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredlund E, Ringner M, Maris JM, Pahlman S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc Natl Acad Sci U S A. 2008;105(37):14094–14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valentijn LJ, Koster J, Haneveld F, et al. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci U S A. 2012;109(47):19190–19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang LL, Teshiba R, Ikegaki N, et al. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children’s Oncology Group study. Br J Cancer. 2015;113(1):57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niemas-Teshiba R, Matsuno R, Wang LL, et al. MYC-family protein overexpression and prominent nucleolar formation represent prognostic indicators and potential therapeutic targets for aggressive high-MKI neuroblastomas: a report from the children’s oncology group. Oncotarget. 2018;9(5):6416–6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schnepp RW, Maris JM. Targeting MYCN: a good BET for improving neuroblastoma therapy? Cancer Discov. 2013;3(3):255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang CV, Reddy EP, Shokat KM, Soucek L. Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer. 2017;17(8):502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc Natl Acad Sci U S A. 2011;108(40):16669–16674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018;8(1):24–36. [DOI] [PubMed] [Google Scholar]
- 16.Puissant A, Frumm SM, Alexe G, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wyce A, Ganji G, Smitheman KN, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One. 2013;8(8):e72967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosse YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455(7215):975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janoueix-Lerosey I, Lequin D, Brugieres L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455(7215):967–970. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455(7215):971–974. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Takita J, Hiwatari M, et al. Mutations of the PTPN11 and RAS genes in rhabdomyosarcoma and pediatric hematological malignancies. Genes Chromosomes Cancer. 2006;45(6):583–591. [DOI] [PubMed] [Google Scholar]
- 23.Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45(3):279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eleveld TF, Oldridge DA, Bernard V, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet. 2015;47(8):864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016;16(5):1273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart LS, Rader J, Raman P, et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin Cancer Res. 2017;23(7):1785–1796. [DOI] [PubMed] [Google Scholar]
- 27.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 28.Lehar J, Krueger AS, Avery W, et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27(7):659–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harenza JL, Diamond MA, Adams RN, et al. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci Data. 2017;4:170033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henssen A, Althoff K, Odersky A, et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin Cancer Res. 2016;22(10):2470–2481. [DOI] [PubMed] [Google Scholar]
- 31.Wyce A, Matteo JJ, Foley SW, et al. MEK inhibitors overcome resistance to BET inhibition across a number of solid and hematologic cancers. Oncogenesis. 2018;7(4):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyce A, Degenhardt Y, Bai Y, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. 2013;4(12):2419–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimmerman MW, Liu Y, He S, et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov. 2018;8(3):320–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20(2):189–193. [DOI] [PubMed] [Google Scholar]
- 35.Otto T, Horn S, Brockmann M, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15(1):67–78. [DOI] [PubMed] [Google Scholar]
- 36.Mills CC, Kolb EA, Sampson VB. Recent Advances of Cell-Cycle Inhibitor Therapies for Pediatric Cancer. Cancer Res. 2017;77(23):6489–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rader J, Russell MR, Hart LS, et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin Cancer Res. 2013;19(22):6173–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moreno L, Caron H, Geoerger B, et al. Accelerating drug development for neuroblastoma - New Drug Development Strategy: an Innovative Therapies for Children with Cancer, European Network for Cancer Research in Children and Adolescents and International Society of Paediatric Oncology Europe Neuroblastoma project. Expert Opin Drug Discov. 2017;12(8):801–811. [DOI] [PubMed] [Google Scholar]
- 39.Cole KA, Huggins J, Laquaglia M, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci U S A. 2011;108(8):3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowery CD, VanWye AB, Dowless M, et al. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin Cancer Res. 2017;23(15):4354–4363. [DOI] [PubMed] [Google Scholar]
- 41.Mosse YP. Anaplastic Lymphoma Kinase as a Cancer Target in Pediatric Malignancies. Clin Cancer Res. 2016;22(3):546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angus SP, Zawistowski JS, Johnson GL. Epigenetic Mechanisms Regulating Adaptive Responses to Targeted Kinase Inhibitors in Cancer. Annu Rev Pharmacol Toxicol. 2018;58:209–229. [DOI] [PubMed] [Google Scholar]
- 43.Stuhlmiller TJ, Miller SM, Zawistowski JS, et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015;11(3):390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1, related to Figure 1. Effects of IBET-726 and trametinib alone and in combination on molecular targets. (A-B). SK-N-BE(2C) was treated with control or varying combinations of inhibitors as indicated (I-BET726 of 1 corresponds to 500 nM and trametinib of 1 corresponds to 25 nM). Lysates were harvested and blotted for MYCN, p-ERK (phosphorylated ERK), t-ERK (total ERK), and Ku80. (C-D). SK-N-AS was treated with control or varying combinations of inhibitors. Lysates were harvested and blotted for MYC, p-ERK, t-ERK, and Ku80.
Supplementary Figure S2, related to Figure 3. Effects of IBET-762 on multiple neuroblastoma xenograft models. (A-B) NGP and NB-1691 xenografts were treated with vehicle or I-BET762 (daily, 20 mg/kg). Response of tumor volumes is shown. (C) SK-N-BE(2)C xenografts were treated with vehicle or I-BET762 (25 mg/kg/day or 12.5 mg/kg 2x/day). Response of tumor volumes is shown. (D) IMR5 xenografts were treated with vehicle or I-BET762 (daily, 30 mg/kg). Response of tumor volumes is shown.
Data Availability Statement
Data available on request from the authors.