

Abstract
Objective:
Genetic variants in the region of TNFAIP3 interacting protein 1 (TNIP1) are associated with autoimmune disease and reduced TNIP1 gene expression. This study aimed to define the functional genetic mechanisms driving TNIP1 hypomorphic expression imparted by the SLE-associated TNIP1 H1 risk haplotype.
Methods:
Dual-luciferase expression and EMSA assays were used to evaluate the allelic effects 11 risk variants on enhancer function and nuclear protein binding in immune cell line models (EBV B, Jurkat, THP1 cells) stimulated with or without PMA/Ionomycin (P/I). HiChIP and qRT-PCR were used to analyze long-range regulatory effects of the TNIP1 haplotype.
Results:
Bioinformatic analyses of 50 SNPs on the TNIP1 H1 risk haplotype identified 11 non-protein-coding variants with high likelihood of influencing TNIP1 gene expression. Eight variants in EBV B, 5 in THP-1, and 2 in Jurkat cells exhibited various allelic effects on enhancer activation resulting in a cumulative suppressive effect on TNIP1 expression (−7.14, −6.80, −2.44 fold, respectively; n>3). Specifically, in EBV B cells, only 2 variants (rs10057690, rs13180950) exhibited both allele-specific loss of enhancer activity (p<0.01) and nuclear protein binding (p<0.01). In contrast, the rs10036748 risk allele reduced binding affinities of transcriptional repressors, Bhlhe40/DEC-1 (p<0.05) and CREB-1, in EBV B cells, resulting in a gain of enhancer activity (p<0.05). HiChIP and qRT-PCR revealed that overall transcriptional repression of the TNIP1 haplotype extends to neighboring genes, DCTN4 (p<0.001) and GMA2 (p<0.01), that share a 3D chromatin network.
Conclusion:
Hypomorphic TNIP1 expression results from the combined concordant and opposing effects of multiple risk variants carried on the TNIP1 risk haplotype, with the strongest regulatory effect in B lymphoid lineage cells. Further, the TNIP1 risk haplotype effect extends to neighboring genes within a shared chromatin network.
Introduction
Systemic lupus erythematosus (SLE) is a challenging autoimmune disease characterized by a loss of immunologic self-tolerance and widespread innate and adaptive immune dysfunctions that cause systemic end-organ damage, severe morbidity, and early mortality. Genome-wide association studies (GWAS) and targeted genetic scans of SLE case-control populations have identified more than 100 genetic loci associated with increased SLE susceptibility (1,2). TNFAIP3 interacting protein 1 (TNIP1), located on 5q32-33.1, has been identified by multiple GWAS, large-scale replication, and genetic fine-mapping studies as a strong SLE susceptibly locus shared across multiple racial groups including European American, African American, Gullah, Hispanic, Hispanic Dominican Republic, Chinese and Japanese (3–9). Multiple independent and race-specific SLE risk haplotypes spanning the TNIP1 locus have been identified, including independent H1 and H2 haplotypes that we previously identified in European Americans (3,6). Genetic variability within the TNIP1 locus has also been associated with several other autoimmune diseases, including Sjögren’s syndrome (10), systemic sclerosis (11,12), psoriatic arthritis (13), and psoriasis (13–16), as well as asthma (17), and some cancers (18–20).
TNIP1 encodes the polyubiquitin binding protein, A20 binding inhibitor of NF-κB 1 (ABIN1). TNIP1/ABIN1 is a critical negative regulator of the proinflammatory NF-κB signaling pathway (21), and has been implicated in regulation of Toll-like receptor (TLR) (22), peroxisome proliferator-activated receptor (PPAR) (23), retinoic acid receptor (24), and C/EBPβ (25) signaling pathways. Loss of TNIP1/ABIN1 in mice exacerbates NF-κB and C/EBPβ signaling, resulting in chronic inflammation and the progressive development of lupus-like inflammatory phenotypes including immune cell expansion and activation, circulating autoantibodies, and renal dysfunction (21,25,26). These findings strongly implicate the hypomorphic TNIP1 expression associated with the SLE TNIP1 risk haplotypes as an important contributing factor in the pathogenesis of SLE (3,27).
Despite strong genetic association and the identification of several risk variants, the mechanisms that modulate the cell type- and state-specific transcriptional regulation of TNIP1 in the context of SLE is not well understood. Using a combination of in vitro assays across different immune cell line lineages, with and without stimulation, we functionally characterized SLE risk variants located in enhancers spanning the TNIP1 H1 risk haplotype. Our findings suggest that multiple variants with concordant and opposing allelic effects in vitro collectively function to suppress TNIP1 gene expression, most potently in B lymphoid lineage cells. Moreover, we also demonstrate that the hypomorphic expression, induced by the TNIP1 risk haplotype, extends beyond TNIP1 to other genes that share 3D chromatin contact domains with the risk haplotype.
Methods
Antibodies and cell lines
Jurkat and THP-1 cells were purchased from ATCC. Epstein-Barr virus transformed human B (EBV B) cell lines were obtained from the Lupus Family Registry and Repository (LFRR) at the Oklahoma Medical Research Foundation (OMRF) with IRB approval (28). EBV B cell lines were selected using genotype data corresponding to the different TNIP1 variants. Genotypes were verified by Sanger sequencing. Cell lines were maintained in RPMI 1640 medium supplemented with 10% FBS, 1X penicillin-streptomycin antibiotic mixture (Atlanta Biologicals Inc., GA), and 2 mM L-glutamine (Lonza, Basel, Switzerland). THP-1 cell medium was also supplemented with 50 μM ß-mercaptoethanol. Cells were stimulated without or with phorbol 12-myristate 13-acetate and ionomycin (P/I; 50 ng/mL, 500 ng/mL) for 2 h where indicated. The following antibodies were used in the study: anti-Early growth response 1 (Egr1; #44D5), anti-cAMP Responsive Element Binding Protein 1 (CREB-1; #48H2) (Cell Signaling Technology, Beverly, MA); anti-basic helix-loop-helix family member 40 (Bhlhe40/DEC1; #NB100-1800; Novus Biologicals, Littleton, CO); anti-Histone H3 (acetyl K27; #ab4279; Abcam, Cambridge, MA); anti-β-Actin (beta-Actin; #ab8226; Abcam, Cambridge, MA), anti-Flag (F1804; Sigma-Aldrich, St. Louis, MO), and Normal Rabbit IgG (Millipore, Billerica, CA). All stock laboratory chemicals were from Sigma Aldrich (St. Louis, MO).
Dual-Luciferase Reporter Assay
DNA sequences, approximately 350 bp in length, surrounding selected non-risk or risk TNIP1 variants were PCR amplified (Supplementary Table 1) and cloned into a minimal promoter luciferase plasmid, pGL4.23 (Promega, Madison, WI). CREB1 (GenScript; #OHu22955D) and BHLHE40/DEC1 (GenScript; #OHu17520) cloned into flag-tag overexpression cloning vector, pcDNA3.1/C-(K)DYK were purchased from GenScript (Piscataway, NJ). Indicated plasmids was transiently co-transfected with the pRL-TK plasmid into Jurkat (Nucleofector SE kit, #V4XC-1032), THP-1 (Nucleofector SG kit, #V4XC-3032), or EBV B (Nucleofector SF kit, #V4XC-2032) cells using Amaxa Nucleofector (Lonza, Portsmouth, NH; SF kit). The pRL-TK plasmid was used for normalization and to calculate transfection efficiency. Twenty-four hours post transfection, cells were treated with P/I (50 ng/ml, 500 ng/ml) for 2 h, then the enhancer activity was measured using the Dual-Luciferase reporter assay (Promega, Madison, WI) as previously described (29).
Electrophoretic Mobility Shift Assay (EMSA)
Complementary pairs of 40 bp non-risk and risk probes, chemically synthesized and 5’ end biotinylated by IDT (Coralville, IA), were annealed by heating at 95 °C for 5 min, then cooled to room temperature (Supplementary Table 1). Ten micrograms of nuclear protein from quiescent or P/I-stimulated (50 ng/mL, 500 ng/mL) Jurkat, EBV B, or THP-1 cells were incubated with 20 fmol biotin-end-labeled probes in binding buffer (1 μg poly dI-dC, 20 mM HEPES, 10% glycerol, 100 mM KCl, and 0.2 mM EDTA, pH 7.9) for 20 min at room temperature. DNA-protein complexes were resolved on a non-denaturing 5% acrylamide gel for approximately 60 min at 100V in 0.5X Tris borate/EDTA before being transferred onto a positively charged nylon membrane (#AM10104; Thermo Fisher Scientific, Waltham, MA) in 0.5X Tris borate/EDTA at 300 mA for 30 min. Membrane-bound DNA-protein complexes were UV cross-linked at 120 mJ/cm2 using a UV Stratalinker 1800 (Stratagene, La Jolla, CA), then detected by horseradish peroxidase-conjugated streptavidin chemiluminescence (LightShift™ chemiluminescent EMSA kit, #89880, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Chemiluminescence was captured on x-ray films exposed for 1-5 min and developed using a Mini-Med 90 X-ray Film Processor (AFP Manufacturing, Peachtree City, GA). For competition assays, 10, 50, and 200-fold excess of unlabeled non-risk or risk probes were added to the EMSA binding reactions (Supplementary Figure 5). Semiquantitative densitometry was performed using NIH Image J Software.
DNA-affinity pulldown of nuclear proteins
Streptavidin magnetic beads (200 μg; Dynabeads M-280 Streptavidin (#112-06D); Invitrogen, Carlsbad, CA) were subjected to two rounds of blocking with 1% bovine serum albumin (BSA) in phosphate buffered saline (PBS) for 15 min followed by washing with PBS containing 1M NaCl and TE buffer. Biotinylated non-risk or risk oligonucleotides were bound to 100 μL of the BSA-blocked streptavidin beads by incubating for 30 min at room temperature in TE buffer, followed by washing with TE buffer. A biotinylated scrambled oligonucleotide served as a negative control (Supplementary Table 1). Fifty micrograms of nuclear extract were pre-cleared by incubating with 100 μL of the BSA-blocked beads in binding buffer (250 mM NaCl, 50 mM Tris Cl, 50% glycerol, 2.5 mM DTT, 2.5 mM EDTA, pH 7.6) containing 15 ng/μL poly dI:dC (#81349-500UG; Sigma-Aldrich, St. Louis, MO), 0.5 μg/mL BSA, and 0.1% NP40 for 30 min on ice. Pre-cleared nuclear extracts were incubated with the oligonucleotide-linked Streptavidin beads for 30 min in a 37 °C water bath. Samples were gently shaken every 5 min. Beads were subsequently washed three times with binding buffer containing 0.1% NP40. The proteins were eluted in 50 μL of 0.2% SDS sample buffer by boiling for 5 min then resolved on a Criterion XT 4%–12% Bis-Tris gel (#3450124; BioRad, Hercules, CA) followed by western blotting.
Western blotting
Cells were pelleted and washed in cold PBS and lysed with RIPA lysis buffer (25 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, pH 8, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) containing a protease inhibitor cocktail (#539132; EMD Millipore, Burlington, MA) and Halt phosphatase inhibitors (#1862495; Thermo Fisher Scientific, Waltham, MA). Cells were lysed for 15 min on ice followed by syringe lysis with a 27g needle, then cleared by centrifugation at max speed for 20 min.
Protein concentrations were determined by Qubit Protein Assay Kit (#Q33212, Thermo Fisher Scientific, Waltham, MA). Proteins were denatured in 2X SDS loading buffer by heating to 95 °C for 5 min, then separated by SDS-PAGE, transferred to PVDF membrane (#1620177, BioRad, Hercules, CA), blocked with 5% non-fat dairy milk, and analyzed by western blotting using the indicated antibodies. Proteins were detected using Pierce ECL Western Blotting Substrate (#32106; Thermo Fisher Scientific, Waltham, MA) and visualized using a ChemiDocMP Imaging System (Bio-Rad, Hercules, CA).
Chromatin immunoprecipitation and Real-Time Quantitative Reverse Transcription-PCR (ChIP-qPCR)
ChIP assays were performed using Covaris truChIP chromatin shearing kit and Magna ChIP protein A/G beads (MilliporeSigma, Burlington, MA) according to the manufacturer’s recommendations. In brief, 1x107 EBV B cells carrying the TNIP1 rs10036748 non-risk or risk alleles were treated with P/I (50ng/ml, 500 ng/ml) in growth medium for 2 h, then cross-linked with 1% formaldehyde. Nuclei were isolated and sonicated in 1 mL of lysis buffer with a Covaris S1 sonicator (Woburn, MA). Five hundred microliters of chromatin-protein complexes were immunoprecipitated overnight at 4°C by mild agitation with antibodies specific for CREB-1, Bhlhe40/DEC1, or normal rabbit IgG (negative control). DNA was eluted from the immunoprecipitated chromatin complexes, reverse-crosslinked, purified by Agencourt AMPure XP beads (Beckman Coulter, Brea, CA), and subjected to qRT-PCR analysis using RT2 SYBR Green (Qiagen, Germantown, MD) and primers neighboring the rs10036748 variant (Supplementary Table 1).
HiChIP
H3K27ac-mediated chromatin interactions were measured for the whole genome of EBV B cells as part of a previously published study (30). HiChIP raw reads (fastq files) were aligned to the hg19 human reference genome using HiC-Pro (31). Aligned data were processed and analyzed through the hichipper pipeline (32). Loops were derived from the linked paired-end reads that overlap with anchors, then analyzed and visualized using the R package diffloop (33). Data are available at the Gene Expression Omnibus, accession #GSE116193.
RNA extraction and Real-time Quantitative Reverse Transcription-PCR (qRT-PCR)
Total RNA from EBV B cells carrying the non-risk or risk TNIP1 H1 haplotype was isolated using Direct-zol RNA MiniPrep Plus kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol (3). cDNA synthesis was performed using QuantiTect reverse transcriptase kit (Qiagen, Germantown, MD) as per the manufacturer’s recommendations. Gene expression was measured by real-time PCR analysis using RT2 SYBR Green (Qiagen). Gene expression primers for human ANXA6 (QT00066941), human DCTN4 (QT00038766), human GM2A (QT00071967), human SMIM3 (QT01028090), human TNIP1 (QT00044072), and human GAPDH (PPH00150F) were purchased from Qiagen.
Results
SLE risk variants function collectively to suppress TNIP1 expression
We used RegulomeDB, which collates bioinformatic data from ENCODE and other sources, to prioritize variants on the SLE-associated risk haplotypes that are most likely to be functional regulators of TNIP1 expression (RegulomeDB score ≤ 3a) (34). Of the 50 variants on the risk haplotype (3), RegulomeDB identified 11 that were located in non-protein-coding regions enriched for H3K4me1 (mark of a poised enhancer) and H3K27ac (mark of an active enhancer) and ChIP-seq transcription factor binding sites in several cell lines, as well as peripheral blood mononuclear cells and primary CD19+ B cells (Figure 1; Supplementary Figure 1; Supplementary Table 2). To begin functionally characterizing the allelic and cell type-specific regulatory potential of these variants, we created a series of luciferase expression vectors by PCR-amplifying an approximate 350 bp sequence containing the respective non-risk or risk alleles from EBV B cells carrying the non-risk or risk haplotype, and then cloning it into a minimal promoter luciferase vector designed to measure the activity of upstream DNA elements using a dual-luciferase assay (29). We transiently transfected the luciferase constructs into B lymphoid (EBV B), monocytoid (THP-1), or T lymphoid (Jurkat) lineage cells for 24 h and then measured luciferase activity at rest or after 2 h of stimulation with P/I. After normalization to the vector-only control, an increase in luciferase activity over that of the vector-only control was interpreted as increased enhancer activity. Cell type-specific increases in enhancer activity were observed for ten variants in EBV B cells (Figure 2A), nine variants in THP-1 cells (Figure 2B), and five variants in Jurkat cells (Figure 2C).
Figure 1. Bioinformatic analysis of 11 SLE risk variants located in regulatory elements of the TNIP1 locus.
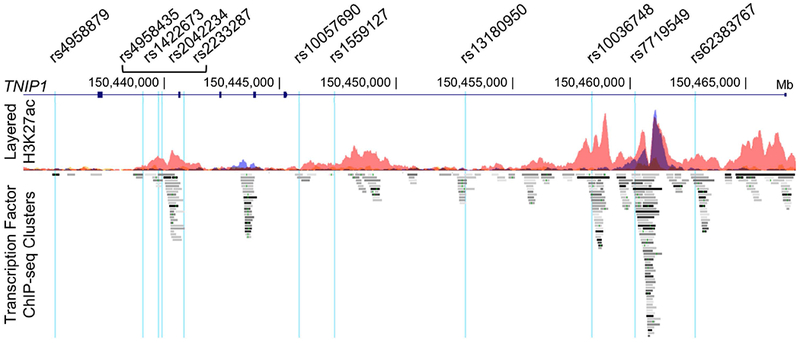
Eleven non-protein-coding SLE risk variants on the TNIP1 locus were predicted by RegulomeDB to have regulatory functions, and are positioned in enhancer regions identified by enrichment of H3K27ac marks and ChIP-seq transcription factor binding. Variant positions are indicated on the UCSC Genome Browser tracks: Gene Symbol, custom track of the TNIP1 SLE risk SNPs, ENCODE H3K27Ac chromatin marks for GM12878, H1-hESC, and K562 cell lines, and ENCODE ChIP-seq transcription factor enrichment.
Figure 2. The TNIP1 locus has a complex regulatory mechanism with cell type and stimuli-dependent regulatory elements.
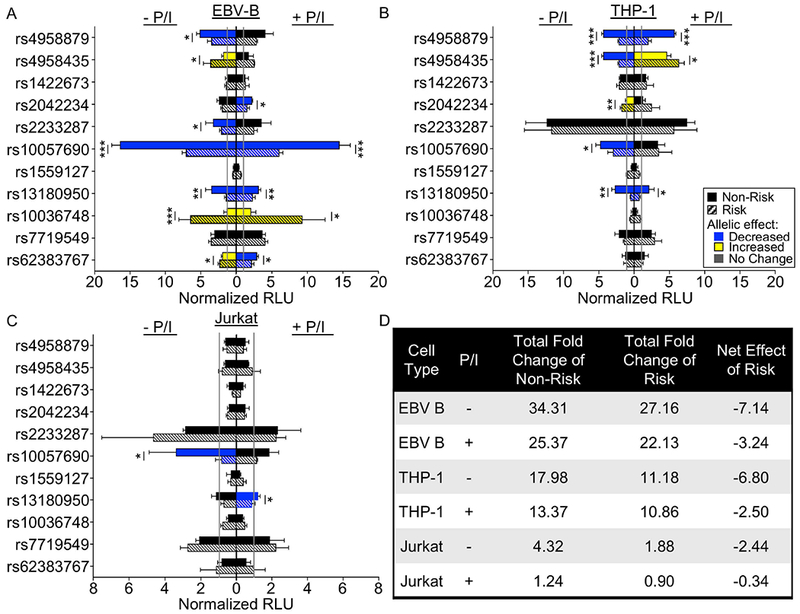
(A-C) Sequences carrying the non-risk (solid bar) or risk (striped bar) alleles of the indicated variants were cloned into a luciferase vector with a minimal promoter. Luciferase activity was measured after transient transfection in EBV B (A), THP-1 (B), or Jurkat (C) cells at rest (left) or after P/I stimulation (right). Luciferase activity was normalized to the vector-only (gray line), and presented as Normalized Relative Luciferase Units (Normalized RLU). Statistical comparisons were performed using Student’s t-test, n>3. Blue bars indicate a significant decrease in luciferase activity in the risk relative to non-risk; yellow bars indicate an increase in the risk relative to non-risk; black bars indicate a lack of allelic effect; * indicates p<0.05; ** indicates p<0.01; ***indicates p<0.001. (D) Summary analysis of the TNIP1 haplotype effect on luciferase activity in EBV B, THP-1, and Jurkat cells. Total fold change of non-risk alleles or risk alleles over vector-only control were calculated for all variants exhibiting significant allelic effects, then the cumulative effect was estimated by subtracting the total non-risk effect from the total risk effect.
Of the ten variants demonstrating enhancer activity in EBV B cells, eight showed significant allele-specific effects (Figure 2A). Three variants retained the allelic effect irrespective of stimulation (rs10057690, rs13180950, rs10036748). Only one variant, rs2042234, produced a significant allelic effect with stimulation, while three others (rs4958879, rs4958435, rs2233287) lost evidence for allele-specific activity with stimulation. The rs62383767 variant flipped from increased to decreased luciferase activity from the risk allele with stimulation. Interestingly, the two variants with the strongest enhancer effect exhibited opposing allelic effects where the risk allele of rs10057690 significantly reduced enhancer activity and the risk allele of rs10036748 significantly increased enhancer activity.
Of the nine variants demonstrating enhancer activity in THP-1 cells (Figure 2B), five demonstrated significant allelic effects on luciferase activity. Risk alleles of rs4958879 and rs13180950 significantly impaired enhancer activity irrespective of stimulation. The increased enhancer activity from the rs2042234 risk allele and the decreased enhancer activity of the rs10057690 risk allele were only observed in the unstimulated condition. Stimulation flipped the allelic effect of rs4958435, increasing luciferase activity for the risk allele over the non-risk allele. Only five variants in Jurkat cells exhibited enhancer effects, with two variants, rs10057690 and rs13180950, showing reduced luciferase activity for the risk alleles; however, with opposing stimulation dependence (Figure 2C).
To summarize the cumulative effect of these variants in each cell line at rest and following stimulation, we calculated the difference in the sum of the fold change between risk and non-risk variants for each condition (Figure 2D). EBV B and THP-1 cells demonstrated the strongest net effect of the risk alleles in attenuating luciferase activity (−7.14 and −6.80, respectively), compared with Jurkat cells (−2.44), suggesting that the risk haplotype is most potent in suppressing TNIP1 expression in B lymphoid and monocytoid cell types. Stimulation with P/I augmented luciferase expression from the risk allele to some degree in each cell type, but not enough to reverse the cumulative suppression of luciferase activity. Overall, these results are consistent with the hypomorphic TNIP1 gene expression observed in vivo, in carriers of the TNIP1 risk haplotype.
SLE risk variants alter nuclear protein complex binding at the TNIP1 locus
We performed EMSAs using extracts from Jurkat, THP-1, or EBV B cells stimulated with or without P/I to determine if the risk alleles of the 11 variants altered nuclear protein complex binding affinities. In all three cell types, nearly all variants that exhibited binding demonstrated a qualitative loss of nuclear protein complex binding to the risk allele (Supplementary Figures 2–4). Densitometry was used to obtain semiquantitative estimates of nuclear protein complex binding for the cell types in resting and stimulated conditions. Variants that did not demonstrate detectable binding were omitted from the semiquantitative analysis.
Only two variants (rs4958879 and rs10036748) exhibited a significant loss of binding to the risk allele in unstimulated Jurkat cells (Figure 3A; Supplementary Figure 2). The allelic effect of rs4958879 was lost with stimulation (Figure 3B; Supplementary Figure 2). In THP-1 cells, no significant allelic effects were observed (Figure 3C,D; Supplementary Figure 3). The nuclear extracts from EBV B cells demonstrated the most robust allele-specific effects with three risk alleles (rs10057690, rs13180950; rs10036748) significantly reducing binding of nuclear proteins from unstimulated cells (Figure 3E; Supplementary Figures 4 and 5). Stimulation resulted in a loss of allele-specific significance for the rs10057690 risk variant, but produced a gain of allele-specific significance for the risk allele of rs7719549 (Figure 3F; Supplementary Figure 4). Together, the combined luciferase and EMSA results suggest that the functional potency of the TNIP1 risk haplotype is most active in B lymphoid lineage cells.
Figure 3. TNIP1 SLE risk alleles differentially affect nuclear complex binding in immune cells.
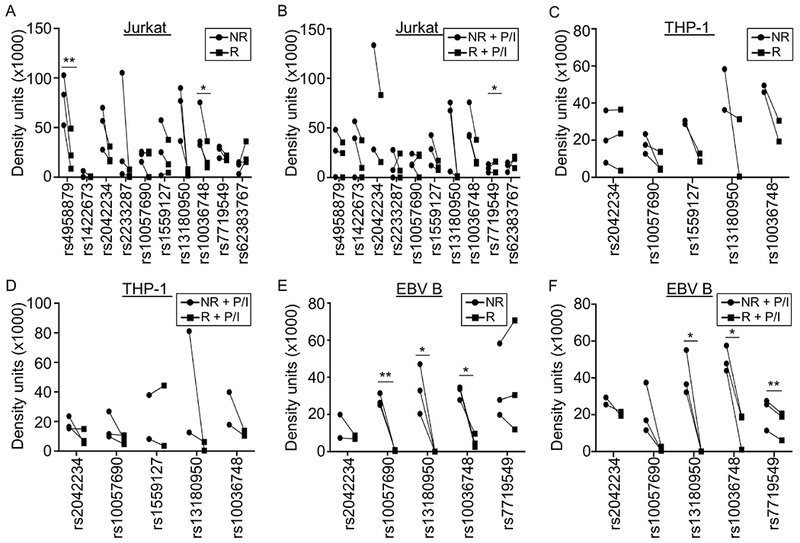
EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts were from Jurkat (A,B), THP-1 (C,D), or EBV B (E,F) cells at rest (A,C,E) or after P/I stimulation (B,D,F). Statistical comparisons were performed using paired t-test; * indicates p<0.05; ** indicates p<0.01. Circles indicate non-risk allele; Squares indicate risk allele. Representative images are in Supplementary Figures 2–4.
SLE risk allele rs10036748 impairs nuclear factor binding
In EBV B cells, two variants, rs10057690 and rs13180950, showed concordant loss of nuclear protein binding and reduced luciferase activity for the risk, relative to non-risk alleles (Figure 2A; Figure 3E,F). In contrast, rs10036748 showed discordant effects with the risk allele showing loss of binding in the EMSAs, but increased luciferase activity (Figure 2A; Figure 3E,F), suggesting that this allele may oppose the overall reduction in TNIP1 gene expression imparted by the risk haplotype in vivo. We hypothesized that the loss of binding of a transcription inhibitor complex was responsible for enhanced allele-specific luciferase activity by the rs10036748 risk (A) allele. To test this, we used bioinformatic analyses to identify nuclear proteins predicted to bind the rs10036748 variant. ENCODE and Genomatix databases identified early growth response-1 (Egr1), basic helix-loop-helix family member 40 (Bhlhe40/DEC1), and cAMP-responsive element binding protein 1 (CREB-1). Egr1, CREB-1, and Bhlhe40/DEC1 are all transcription factors reportedly involved in the activation, proliferation, differentiation and/or function of different immune cell subtypes (35–40). In addition, Bhlhe40/DEC1 and CREB-1 have both been reported to function as transcriptional repressors in specific cell types and states. While Bhlhe40/DEC1 is an essential transcriptional repressor of IL-10 production by Th1 cells and in mice infected with Mycobacterium tuberculosis (41–43), CREB-1 reportedly competes against CBP/p300 to impair NF-κB signaling in germinal B cells (40,44).
We performed DNA-affinity pulldowns using nuclear extracts from Jurkat, EBV B, and THP-1 cells to determine the allele-specific binding of Egr1, Bhlhe40/DEC1, or CREB-1 to the rs10036748 variant. Egr1 from Jurkat cell nuclear extracts exhibited reduced binding to the risk (A) allele, relative to the non-risk (G) allele of rs10036748 (Figure 4A). We did not observe allele-specific binding by Egr1 in EBV B cells (Figure 4A), and THP-1 nuclear extracts did not pulldown detectable levels of Egr1 (Figure 4A). In contrast, Bhlhe40/DEC1 and CREB-1 exhibited reduced binding to the risk (A) allele in all three cell types (Figure 4A). To confirm the allele-specific binding of Bhlhe40/DEC1 and CREB-1 in situ, we performed ChIP-qPCR in EBV B cells carrying the non-risk or risk TNIP1 haplotype. Bhlhe40/DEC1 and CREB-1 both showed decreased binding to the risk (A) allele (Figure 4B,C), but only reduced Bhlhe40/DEC1 binding reached significance (Figure 4B). Collectively, these results suggest that the risk (A) allele of rs10036748 impairs nuclear protein binding, particularly Bhlhe40/DEC1, in immune-relevant cell lines.
Figure 4. Rs10036748 SLE risk allele reduces binding of Egr1, Bhlhe40/DEC1, and CREB-1.
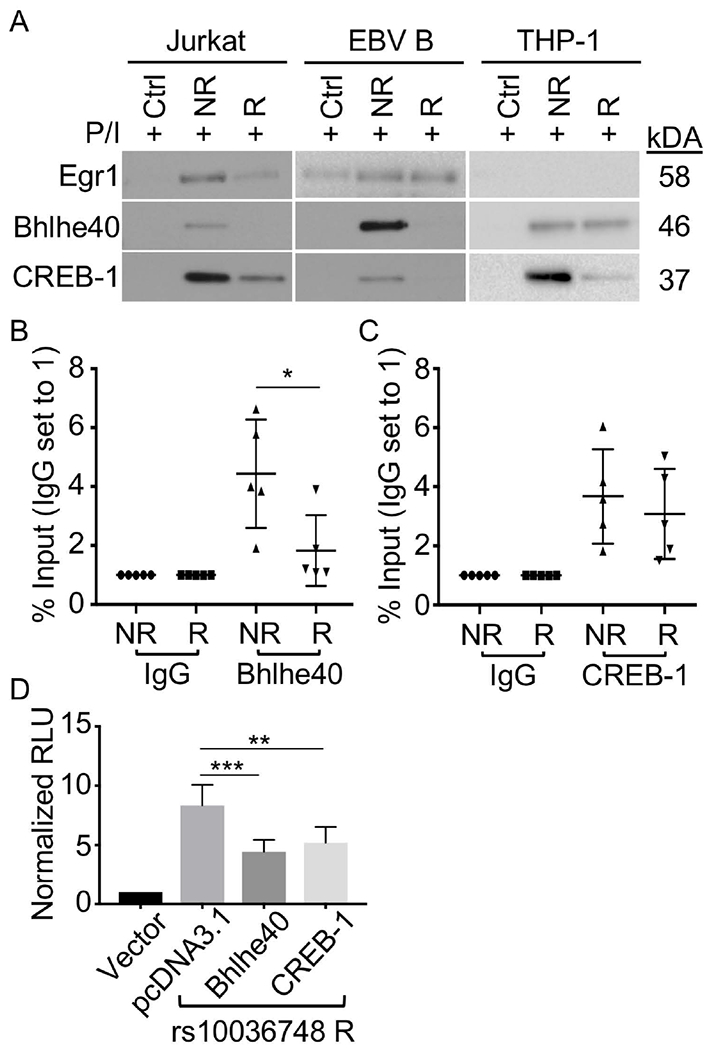
(A) DNA affinity pulldown performed using annealed biotinylated oligonucleotides containing the non-risk (NR) or risk (R) allele of rs10036748 and nuclear extracts from Jurkat, EBV B, or THP-1 cells stimulated with P/I for 2 h. Eluted proteins were separated by SDS-PAGE and analyzed by Western blot using antibodies specific against Egr1, Bhlhe40/DEC1, or CREB-1 as indicated. Images are representative of n=3. (B-C) ChIP-qPCR performed using homozygous EBV B cell lines (non-risk (NR) or risk (R); n=5 cell lines per genotype) stimulated with P/I for 2 h, and antibodies specific against Bhlhe40/DEC1 (B), CREB-1 (C), or Rabbit IgG isotype control (B,C). (D) Luciferase activity was measured in EBV B cells transiently transfected with Firefly luciferase vector-only (vector), or co-transfected with luciferase vector carrying the rs10036748 risk allele and pcDNA3.1, pcDNA3.1-Bhlhe40/DEC1, or pcDNA3.1-CREB-1. Luciferase activity was normalized to the vector control, and presented as Normalized Relative Luciferase Units (RLU). Statistical comparisons were made using Student’s t-test, * indicates p<0.05; ** indicates p<0.01 *** indicates p<0.001.
Bhlhe40/DEC1 and CREB-1 demonstrated reduced binding to the risk allele in affinity pull-down and ChIP-qPCR assays, and are reported to function as transcription repressors. Therefore, we reasoned that the increased luciferase activity we observed for the rs10036478 risk allele may result from an allele-dependent loss of these transcriptional inhibitors. Further, we hypothesized that the overexpression of either Bhlhe40/DEC1 or CREB-1 could overcome the lower affinity binding to the rs10036748 risk allele, thereby suppressing the allelic increase in luciferase activity. Indeed, Bhlhe40/DEC1 or CREB-1 transient overexpression in EBV B cells expressing the luciferase vector carrying the rs10036748 risk allele significantly reduced the allele-specific increase in luciferase activity (Figure 4D; Supplementary Figure 6).
The TNIP1 locus influences gene expression locally and at a distance.
Enhancers can regulate gene expression locally or at a distance through long-range DNA looping (45), therefore we hypothesized that the hypomorphic effect of the TNIP1 risk haplotype may also manifest an effect on other genes within its 3D chromatin network. To define the 3D chromatin network of the TNIP1 locus, we performed HiChIP in EBV B cells with chromatin precipitated using antibodies for H3K27ac (30). Several long-range interactions were observed between the enhancer spanning the TNIP1 risk haplotype and flanking genes known to be expressed in immune cell types (www.gtexportal.org)(46), including SMIM3, ANXA6, DCTN4, and GM2A (Figure 5A). Similar chromatin looping events were also observed in publicly available Hi-C data from the GM12878 EBV B cell line (Supplementary Figure 7) (47,48). To determine if the TNIP1 risk haplotype influenced the expression of these distant genes, we performed qRT-PCR using RNA isolated from resting EBV B cells carrying the TNIP1 non-risk or risk H1 haplotype. As expected, the TNIP1 SLE risk haplotype significantly reduced TNIP1 expression (Figure 5B). EBV B cells carrying the TNIP1 risk haplotype also exhibited a significant loss of DCTN4 and GM2A expression. Although showing a similar trend, no significant differences in the expression of SMIM3 and ANXA6 were observed. Collectively, these data suggest that the effect of the TNIP1 risk haplotype extends beyond TNIP1 to include other genes whose promoters are contacted by the TNIP1 haplotype through long-range DNA interactions.
Figure 5. The TNIP1 locus has a complex regulatory structure that influences gene expression locally and at a distance.
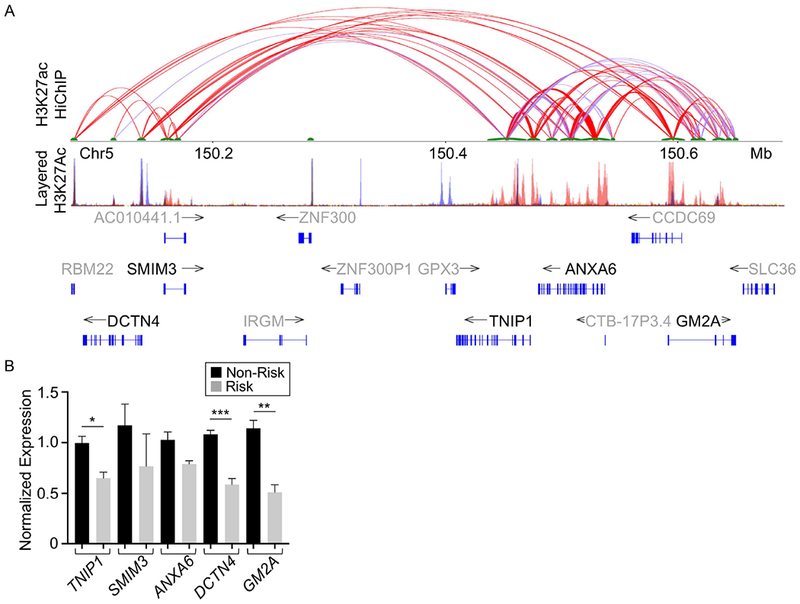
(A) H3K27ac HiChIP looping interactions within the TNIP1 region were visualized as a two-dimensional looping diagram. Arc thickness is proportional to the frequency of observed paired-end tags (6 PET threshold). The enhancer with a strong H3K27ac ChIP peak spanning the TNIP1 gene locus forms long-range interactions with multiple H3K27ac anchors proximal to distant genes. H3K27Ac Peak Track was adapted from the UCSC Genome Browser ENCODE H3K27Ac chromatin marks for GM12878, H1-hESC, and K562 cell lines (B). Expression of distant genes in the H3K27ac regulatory network with TNIP1 was measured in resting EBV B cell lines carrying the non-risk (NR) or risk (R) H1 TNIP1 haplotype using qRT-PCR. Expression was normalized to GAPDH, then TNIP1 expression in non-risk EBV B controls. Statistical comparisons were performed using Student’s t-test, n>3, * indicates p<0.05; ** indicates p<0.01; *** indicates p<0.001.
Discussion
Despite reported associations between the TNIP1 SLE risk haplotype, hypomorphic TNIP1 expression, and dysregulation of key inflammatory signaling pathways, few studies have investigated the regulatory mechanisms controlling TNIP1 expression in the context of the genetic risk haplotype. Using a combination of in vitro assays aimed at characterizing TNIP1 risk variants that are predicted to have functional effects, we discovered a complex regulatory mechanism whereby the cumulative loss of nuclear protein complex binding and loss of enhancer activity at specific risk alleles drives the hypomorphic expression of TNIP1. Moreover, both EMSA and luciferase assays revealed the most significant and reproducible effects in the EBV B cells, suggesting that the TNIP1 risk haplotype may be most potent in cells of B lymphoid lineage.
With respect to the effect of stimulation on enhancer activity and allele-specific effects, the luciferase assays provided the most definitive information. Our data demonstrated that stimulation with P/I alleviated the suppressive effects of the risk alleles on luciferase activity across all cell types (Figure 2D), but not enough to completely reverse the suppressive effect of the TNIP1 haplotype. This suggests that the magnitude of suppression of TNIP1 expression by the TNIP1 risk haplotype can be modulated by the overall activation of cellular pathways. Since we used a cell surface receptor-independent activation method, we cannot conclude how the activation of pathways downstream of cell surface receptors, such as the tumor necrosis factor receptor (TNFR) and TLR, might alter the suppressive effect of the TNIP1 risk alleles.
We chose to further study the TNIP1 risk variant, rs10036748, which is an index SNP for this locus in multiple SLE GWAS studies and is shared among several risk haplotypes and across multiple racial groups, including the H1 and H2 haplotypes in European Americans and the Block 2 haplotype in the Han Chinese (3–9), because it exhibited an allele-specific increase in enhancer activity, but a discordant reduction in nuclear protein complex binding. This illustrates how genetic variants not only modulate gene expression through loss of binding of transcription-activating proteins, but also through the loss of binding of transcriptional repressor proteins. Our affinity pulldown and ChIP-qPCR data suggest that the rs10036748 risk allele promotes enhancer activity by reducing the binding affinity of Bhlhe40/DEC1 and CREB-1. Forced expression of Bhlhe40/DEC1 and CREB-1 can overcome the low affinity binding of the risk allele, returning the luciferase activity to the level of the non-risk allele. Together these results suggest that the risk allele of rs10036748 opposes the hypomorphic expression of the TNIP1 risk haplotype by lowering the binding affinity of Bhlhe40/DEC1 and CREB-1 transcriptional repressors. Further studies using genetic engineering methods that swap the risk and non-risk alleles at rs10036748 will be necessary to confirm our observations.
Another interesting observation from our study is that the transcription suppression induced by the TNIP1 risk haplotype is not limited to TNIP1. Within the 3D chromatin network defined by H3K27ac HiChIP data, we observed significant reductions in the transcription of DCTN4 and GM2A. Similar trends were also observed for SMIM3 and ANXA6, but did not reach statistical significance. These results suggest that the functional impact of the TNIP1 risk haplotype likely extends beyond its effect of suppressing TNIP1 expression towards pathways involving DCTN4 and GMA2 expression. For example, DCTN4 missense variants have recently been shown to be associated with shorter time to developing chronic Pseudomonas aeruginosa infection, a condition associated with worse long-term pulmonary disease and survival in patients with cystic fibrosis (CF) (49). CF patients biallelic for DCTN4 missense variants and the TNIP1 risk haplotype could be at risk of even earlier infection due to reduced expression of wild type DCTN4 by the TNIP1 risk haplotype.
Our study has some limitations that must be considered in the interpretation of our data. First, the EMSA and luciferase assays do not assess function of the risk alleles in their natural genomic context. Second, binding of proteins to the probes used in both assays is dependent, to some extent, on the length of the probe and the sequences contained therein. Third, we measured the function of risk variants in immortalized cell lines. Both the Jurkat and THP-1 cell lines are derived from spontaneous malignant transformations while EBV B cells are virally transformed; the molecular composition of these cell lines is not expected to fully reflect the precise molecular composition of the primary cells from which they were originally derived. Despite these limitations, our data demonstrated that the majority of the risk alleles reduced nuclear protein complex binding with variable allelic effects on enhancer activity, resulting in an overall affect (Figure 2D) that is consistent with hypomorphic TNIP1 expression; therefore, we are confident that our data provides a reasonable approximation for how the TNIP1 risk haplotype operates in primary immune cells.
Supplementary Material
Supplementary Figure 1. Bioinformatic analysis of 11 SLE risk variants located in regulatory elements of the TNIP1 locus. Eleven of fifty non-protein-coding SLE risk variants on the TNIP1 locus were predicted by RegulomeDB to have regulatory functions, and are positioned in enhancer regions identified by enrichment of H3K4me1 and H3K27ac chromatin marks and ChIP-seq transcription factor binding. Variant positions are indicated on the UCSC Genome Browser tracks: Gene Symbol, custom track of the TNIP1 SLE risk SNPs, ENCODE H3K4me1 and H3K27Ac chromatin marks for GM12878, H1-hESC, and K562 cell lines, NIH Roadmap Epigenomics H3K4me1 and H3K27Ac chromatin marks for peripheral mononuclear blood cells and primary CD19+ B cells, and ENCODE ChIP-seq transcription factor enrichment.
Supplementary Figure 2. TNIP1 SLE risk alleles differentially effect nuclear factor binding in Jurkat cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were from Jurkat cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 3. TNIP1 SLE risk alleles differentially effect nuclear factor binding in THP-1 cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were from THP-1 cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 4. TNIP1 SLE risk alleles differentially effect nuclear factor binding in EBV B cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were derived from EBV B cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 5. Competition EMSA assay demonstrates nuclear factor binding specificity to rs10036748. Competition EMSA assay was performed using increasing concentrations of unlabeled oligonucleotides containing the non-risk (G) or risk (A) allele of rs10036748 to compete against biotinylated oligonucleotides containing the non-risk (G) or risk (A) allele of rs10036748. Nuclear extracts (N.E.) were derived from EBV B cells at rest. Representative image of n=3.
Supplementary Figure 6. Bhlhe40/DEC1 and CREB-1 overexpression in EBV B cells. EBV B cells were transiently transfected with empty pcDNA3.1 (A,B), pcDNA3.1-Bhlhe40/DEC1 (A), or pcDNA3.1-CREB-1 (B). Whole cell lysates were separated by SDS-PAGE. Overexpressed flag-tagged Bhlhe40/DEC1 (A) or CREB-1 (B) was detected by Western blot using an antibody specific against Flag. ß-actin was used as a loading control. Images are representative of n=3.
Supplementary Figure 7. The TNIP1 locus has a complex chromatin architecture. Hi-C chromatin looping events at the TNIP1 locus in the EBV B cell line, GM12878, were visualized as a heatmap. The heatmap was adapted from the Penn State Computational and Functional Genomics/Epigenomics Browser.
Supplementary Table 1. Oligonucleotide Sequences.
Supplementary Table 2. RegulomeDB Scores of Screened TNIP1 SNPs
Acknowledgements
We want to thank Kiely Grundahl for her technical assistance and helpful discussions. Research reported in this publication was supported by the National Institutes of Health (NIH) grants: AR063124, AR073606, AR056360, GM110766, and AI082714; as well as the Presbyterian Health Foundation. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Financial Support: Research was supported by National Institutes of Health (NIH) grants: AR063124, AR073606, AR056360, GM110766, and AI082714; as well as the Presbyterian Health Foundation.
Footnotes
COI Disclosures: None
References
- 1.Chen L, Morris DL, Vyse TJ. Genetic advances in systemic lupus erythematosus. Curr Opin Rheumatol 2017;29:423–33. [DOI] [PubMed] [Google Scholar]
- 2.Deng Y, Tsao BP. Updates in Lupus Genetics. Curr Rheumatol Rep 2017;19:68. [DOI] [PubMed] [Google Scholar]
- 3.Adrianto I, Wang S, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. Association of two independent functional risk haplotypes in TNIP1 with systemic lupus erythematosus. Arthritis Rheum 2012;64:3695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han J-W, Zheng H-F, Cui Y, Sun L-D, Ye D-Q, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet 2009;41:1234–7. [DOI] [PubMed] [Google Scholar]
- 5.He C-F, Liu Y-S, Cheng Y-L, Gao J-P, Pan T-M, Han J-W, et al. TNIP1, SLC15A4, ETS1, RasGRP3 and IKZF1 are associated with clinical features of systemic lupus erythematosus in a Chinese Han population. Lupus 2010;19:1181–6. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D-M, Cheng L-Q, Zhai Z-F, Feng L, Zhong B-Y, You Y, et al. Single-nucleotide Polymorphism and Haplotypes of TNIP1 Associated with Systemic Lupus Erythematosus in a Chinese Han Population. J Rheumatol 2013;40:1535–44. [DOI] [PubMed] [Google Scholar]
- 7.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet 2009;41:1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawasaki A, Ito S, Furukawa H, Hayashi T, Goto D, Matsumoto I, et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic lupus erythematosus in a Japanese population: a case-control association study. Arthritis Res Ther 2010;12:R174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Z, Yu Y, Yue Y, Hearth-Holmes M, Lopez PD, Tineo C, et al. Genetic Alleles Associated with SLE Susceptibility and Clinical Manifestations in Hispanic Patients from the Dominican Republic. Curr Mol Med 2019;19:164–71. [DOI] [PubMed] [Google Scholar]
- 10.Lessard CJ, Li H, Adrianto I, Ice JA, Rasmussen A, Grundahl KM, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 2013;45:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allanore Y, Saad M, Dieudé P, Avouac J, Distler JHW, Amouyel P, et al. Genome-Wide Scan Identifies TNIP1, PSORS1C1, and RHOB as Novel Risk Loci for Systemic Sclerosis. PLoS Genet 2011;7:e1002091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bossini-Castillo L, Martin JE, Broen J, Simeon CP, Beretta L, Gorlova OY, et al. Confirmation of TNIP1 but not RHOB and PSORS1C1 as systemic sclerosis risk factors in a large independent replication study. Ann Rheum Dis 2013;72:602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowes J, Orozco G, Flynn E, Ho P, Brier R, Marzo-Ortega H, et al. Confirmation of TNIP1 and IL23A as susceptibility loci for psoriatic arthritis. Ann Rheum Dis 2011;70:1641–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Yan H, Song Z, Chen F, Wang H, Niu J, et al. Downregulation of TNIP1 Expression Leads to Increased Proliferation of Human Keratinocytes and Severer Psoriasis-Like Conditions in an Imiquimod-Induced Mouse Model of Dermatitis. PLoS One 2015;10:e0127957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet 2009;41:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan KX, Zhang YJ, Han L, Huang Q, Zhang ZH, Fang X, et al. TT genotype of rs10036748 in TNIP 1 shows better response to methotrexate in a Chinese population: a prospective cohort study. Br J Dermatol 2019;doi: 10.1111/bjd.17704:bjd.17704. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Ampleford EJ, Howard TD, Moore WC, Torgerson DG, Li H, et al. Genome-wide association studies of asthma indicate opposite immunopathogenesis direction from autoimmune diseases. J Allergy Clin Immunol 2012;130:861–868.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Zhao Z, Zhou J, Liu Y, Wang H, Zhao X. Relationship between the TERT, TNIP1 and OBFC1 genetic polymorphisms and susceptibility to colorectal cancer in Chinese Han population. Oncotarget 2017;8:56932–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng Y, Jiang X, Jin J, Luo X, Chen W, Li Q, et al. TNIP1 Polymorphisms with the Risk of Hepatocellular Carcinoma Based on Chronic Hepatitis B Infection in Chinese Han Population. Biochem Genet 2019;57:117–28. [DOI] [PubMed] [Google Scholar]
- 20.Liu Z, Shi Y, Na Y, Zhang Q, Cao S, Duan X, et al. Genetic polymorphisms in TNIP1 increase the risk of gastric carcinoma. Oncotarget 2016;7:40500–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.G’Sell RT, Gaffney PM, Powell DW. A20-Binding Inhibitor of NF-κB Activation 1 is a Physiologic Inhibitor of NF-κB: A Molecular Switch for Inflammation and Autoimmunity. Arthritis Rheumatol (Hoboken, NJ) 2015;67:2292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shamilov R, Aneskievich BJ. TNIP1 in Autoimmune Diseases: Regulation of Toll-like Receptor Signaling. J Immunol Res 2018;2018:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flores AM, Gurevich I, Zhang C, Ramirez VP, Devens TR, Aneskievich BJ. TNIP1 is a corepressor of agonist-bound PPARs. Arch Biochem Biophys 2011;516:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurevich I, Zhang C, Francis N, Struzynsky CP, Livings SE, Aneskievich BJ. Human TNFα-induced protein 3-interacting protein 1 (TNIP1) promoter activation is regulated by retinoic acid receptors. Gene 2013;515:42–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian T, Chen F, Shi X, Li J, Li M, Chen Y, et al. Upregulation of the C/EBP β LAP isoform could be due to decreased TNFAIP3/TNIP1 expression in the peripheral blood mononuclear cells of patients with systemic lupus erythematosus. Mod Rheumatol 2017;27:657–63. [DOI] [PubMed] [Google Scholar]
- 26.Kuriakose J, Redecke V, Guy C, Zhou J, Wu R, Ippagunta SK, et al. Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. J Clin Invest 2019;129:2251–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caster DJ, Korte EA, Nanda SK, McLeish KR, Oliver RK, G’Sell RT, et al. ABIN1 Dysfunction as a Genetic Basis for Lupus Nephritis. J Am Soc Nephrol 2013;24:1743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rasmussen A, Sevier S, Kelly JA, Glenn SB, Aberle T, Cooney CM, et al. The Lupus Family Registry and Repository. Rheumatology (Oxford) 2011;50:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An Enhancer Element Harboring Variants Associated with Systemic Lupus Erythematosus Engages the TNFAIP3 Promoter to Influence A20 Expression. PLoS Genet 2013;9:e1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelikan RC, Kelly JA, Fu Y, Lareau CA, Tessneer KL, Wiley GB, et al. Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks. Nat Commun 2018;9:2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 2015;16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lareau CA, Aryee MJ. hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods 2018;15:155–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lareau CA, Aryee MJ. diffloop: a computational framework for identifying and analyzing differential DNA loops from sequencing data. Bioinformatics 2018;34:672–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bencheikh L, Diop MK, Rivière J, Imanci A, Pierron G, Souquere S, et al. Dynamic gene regulation by nuclear colony-stimulating factor 1 receptor in human monocytes and macrophages. Nat Commun 2019;10:1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kharbanda S, Nakamura T, Stone R, Hass R, Bernstein S, Datta R, et al. Expression of the early growth response 1 and 2 zinc finger genes during induction of monocytic differentiation. J Clin Invest 1991;88:571–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y, Liu M, Tang L, Du F, Liu Y, Hao P, et al. Iguratimod represses B cell terminal differentiation linked with the inhibition of PKC/EGR1 axis. Arthritis Res Ther 2019;21:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mora-López F, Pedreño-Horrillo N, Delgado-Pérez L, Brieva JA, Campos-Caro A. Transcription of PRDM1 , the master regulator for plasma cell differentiation, depends on an SP1/SP3/EGR-1 GC-box. Eur J Immunol 2008;38:2316–24. [DOI] [PubMed] [Google Scholar]
- 39.Dinkel A, Warnatz K, Ledermann B, Rolink A, Zipfel PF, Bürki K, et al. The transcription factor early growth response 1 (Egr-1) advances differentiation of pre-B and immature B cells. J Exp Med 1998;188:2215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol 2010;185:6413–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu F, Sharma S, Jankovic D, Gurram RK, Su P, Hu G, et al. The transcription factor Bhlhe40 is a switch of inflammatory versus antiinflammatory Th1 cell fate determination. J Exp Med 2018;215:1813–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huynh JP, Lin C-C, Kimmey JM, Jarjour NN, Schwarzkopf EA, Bradstreet TR, et al. Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. J Exp Med 2018;215:1823–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao M, Xiaofei L, Gang C, Wei L, Jing X, Gang H, et al. DEC1 binding to the proximal promoter of CYP3A4 ascribes to the downregulation of CYP3A4 expression by IL-6 in primary human hepatocytes. Biochem Pharmacol 2012;84:701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer SN, Scuoppo C, Vlasevska S, Bal E, Holmes AB, Holloman M, et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity 2019;51:535–547.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014;159:1665–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol 2018;doi: 10.1186/s13059-018-1519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet 2012;44:886–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Bioinformatic analysis of 11 SLE risk variants located in regulatory elements of the TNIP1 locus. Eleven of fifty non-protein-coding SLE risk variants on the TNIP1 locus were predicted by RegulomeDB to have regulatory functions, and are positioned in enhancer regions identified by enrichment of H3K4me1 and H3K27ac chromatin marks and ChIP-seq transcription factor binding. Variant positions are indicated on the UCSC Genome Browser tracks: Gene Symbol, custom track of the TNIP1 SLE risk SNPs, ENCODE H3K4me1 and H3K27Ac chromatin marks for GM12878, H1-hESC, and K562 cell lines, NIH Roadmap Epigenomics H3K4me1 and H3K27Ac chromatin marks for peripheral mononuclear blood cells and primary CD19+ B cells, and ENCODE ChIP-seq transcription factor enrichment.
Supplementary Figure 2. TNIP1 SLE risk alleles differentially effect nuclear factor binding in Jurkat cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were from Jurkat cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 3. TNIP1 SLE risk alleles differentially effect nuclear factor binding in THP-1 cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were from THP-1 cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 4. TNIP1 SLE risk alleles differentially effect nuclear factor binding in EBV B cells. EMSAs were performed using biotinylated oligonucleotides containing the non-risk or risk alleles of the indicated variants. Nuclear extracts (N.E.) were derived from EBV B cells at rest or stimulated with P/I for 2 h. Representative image of n>3.
Supplementary Figure 5. Competition EMSA assay demonstrates nuclear factor binding specificity to rs10036748. Competition EMSA assay was performed using increasing concentrations of unlabeled oligonucleotides containing the non-risk (G) or risk (A) allele of rs10036748 to compete against biotinylated oligonucleotides containing the non-risk (G) or risk (A) allele of rs10036748. Nuclear extracts (N.E.) were derived from EBV B cells at rest. Representative image of n=3.
Supplementary Figure 6. Bhlhe40/DEC1 and CREB-1 overexpression in EBV B cells. EBV B cells were transiently transfected with empty pcDNA3.1 (A,B), pcDNA3.1-Bhlhe40/DEC1 (A), or pcDNA3.1-CREB-1 (B). Whole cell lysates were separated by SDS-PAGE. Overexpressed flag-tagged Bhlhe40/DEC1 (A) or CREB-1 (B) was detected by Western blot using an antibody specific against Flag. ß-actin was used as a loading control. Images are representative of n=3.
Supplementary Figure 7. The TNIP1 locus has a complex chromatin architecture. Hi-C chromatin looping events at the TNIP1 locus in the EBV B cell line, GM12878, were visualized as a heatmap. The heatmap was adapted from the Penn State Computational and Functional Genomics/Epigenomics Browser.
Supplementary Table 1. Oligonucleotide Sequences.
Supplementary Table 2. RegulomeDB Scores of Screened TNIP1 SNPs