

Abstract
Objectives
This research aimed to investigate the relative level of Runt‐related transcription factor 2 (RUNX2) in giant cell tumor of bone (GCTB). Through the histopathological similarities between osteoporosis and GCTB, the biological functions of exogenous RUNXS were demonstrated in GCTB cell lines. This generated awareness of the molecular mechanism of the biogenesis and metastasis of GCTB, as well as showing the pathways and processes involved in this study. This research also expected to provide hints for the clinical treatment of patients with GCTB, to release the tumor burden and reduce the recurrence rate and metastasis of patients with this condition.
Methods
The expression of RUNX2 in the tumors was verified by Western Blot, qRT‐PCR and immunohistochemistry, compared with the normal tissues’ adjacent tumors. Subsequently, the plasmids expressing RUNX2 were constructed, amplified and transfected into the 0404 cell line through transfection kits (0.4, 0.8, 1.6, 2.4 ng/μl). After that, the proliferation, migration, invasion, cellular viability and apoptosis of 0404 cell lines were examined by EDU assay, wound healing assay, transwell assay, annexin v staining, and CCK8 assay, respectively.
Results
The messenger RNA (mRNA) level of RUNX2 in tumors was over 100 folds more than the normal tissues. The protein level of tumors upregulated 8.32(±4.41) folds relatively. After the transfection of RUNX2 overexpressed plasmids into the 0404 cell line, the mRNA level of RUNX2 increased approximately 530.11(±24.87), 1117.96(±77.68), 2835.09(±45.22) and 4781.51(±79.37) folds respectively, and the protein level was upregulated about 4.12(±1.15), 16.73(±1.63), 21.53(±2.41) and 23.39(±0.85) folds respectively. The proliferation of 0404 cells was inhibited by 2.13(±1.02)% of 1.6 ng/μl group and 3.03(±1.76)% of 2.4 ng/μl group. And the migration was inhibited about 45.56(±6.13)%, 50.79(±5.27)%, 63.15(±8.62)% and 93.90(±3.65)% respectively. The invasion was decreased about 14.49(±5.4)%, 37.02(±6.52)%, 42.24(±2.59)% and 48.97(±10.61)% respectively. Meanwhile, FITC Annexin V/PI apoptosis assay demonstrated that RUNX2 plasmids could promote apoptosis rate around 4.15(±0.27)%, 5.07(±0.27)%, 7.61(±0.45)% and 11.32(±1.02)% respectively, and CCK8 proved these plasmids could weaken cellular viability in a concentration‐dependent manner with the time passing.
Conclusions
RUNX2 is highly expressed in giant cell tumors of bone. The RUNX2 overexpressed plasmids we constructed could be successfully transfected into 0404 cell line. Far more importantly, the exogenous RUNX2 can seriously block the biological functions of 0404 cell line in a concentration‐dependent manner, including proliferation, translocation, invasion, cellular viability, and apoptosis. Meanwhile, the mechanism was hypothesized and discussed in the article.
Keywords: Functions, GCTB, Inhibition, Overexpression, RUNX2
Introduction
Giant cell tumor of bone(GCTB) is a relatively rare primary borderline tumor, but it is easy to relapse and metastasize to local and distant tissues1. At present, the main therapies include surgery, radiotherapy and systemic treatment2. Denosumab is a molecular therapeutic drug targeting the receptor activator of nuclear factor‐κ B Ligand (RANKL). It was approved by the Food and Drug Administration (FDA) for the treatment of GCTB in June 2013. A large number of clinical studies have proven it effective3. It is also one of the treatments for osteoporosis. The pathology of giant cell tumor of bone is mainly characterized by giant multinucleated cells and stromal cells with osteolytic properties. These cells have obvious osteoclast characteristics and a high expression level of RANKL4.
Runt‐related transcription factor 2 (RUNX2), also known as Cbfa1, is a multifunctional transcription factor and considered to be one of the most important markers during the process of osteoblast differentiation and maturation5. Yuki Ikebuchi et al. found that RANKL bound to RANK on the surface of osteoblasts and transmitted signals to increase the expression of downstream RUNX2, thereby promoting the differentiation and maturation of osteoblasts6. Yang et al. found that circular RNA Circ‐VANGL1 can upregulate the expression of RUNX2 in bone tissue by adsorbing miR‐217 to alleviate the osteoporosis7. Ahmad et al. found that miR‐672‐5p can increase the level of RUNX2 by inhibiting the level of Smurf1, in order to reverse the bone loss and muscle reduction in mice after ovariectomy8. Yin et al. found that CircRunx2 regulates RUNX2 expression through miR‐203 to prevent osteoporosis9. It can be seen that RUNX2 is lowly expressed in patients with osteoporosis and increasing its level can prevent and treat osteoporosis. The histopathological features of osteoporosis have similarities with giant cell tumor of bone. Therefore, we speculate that RUNX2 can inhibit the differentiation of stromal cells to osteoclast in GCTB, so as to release the tumor burden. Meanwhile, there have been few studies on the use of RUNX2 in GCTB.
In this study, we compared the expression of RUNX2 in tumor tissues and adjacent normal tissues through the patients' specimens collected from our hospital within 3 years, and investigated whether the expression of RUNX2 affected the biological function of giant cell tumor of bone in vitro, to analyze the role of RUNX2 in GCTB and its molecular therapy.
Materials and Methods
Collection of Tumors and Adjacent Normal Tissues
Specimens were collected from patients with giant cell tumors of bone that were diagnosed and operated on from June 2016 to June 2019 in the Department of Orthopedics of the Jinling Hospital (Table 1). At the same time, the normal tissues surrounding the tumor were obtained during the operation and stored in a refrigerator at −80°C.
Table 1.
The general information of patients
| Number | Gender | Age(years) | Tumor location |
|---|---|---|---|
| Patient 1 | female | 17 | distal right femur |
| Patient 2 | male | 26 | distal right femur |
| Patient 3 | male | 49 | distal left femur |
| Patient 4 | female | 62 | distal right femur |
| Patient 5 | female | 35 | proximal left tibia |
| Patient 6 | female | 33 | proximal right tibia |
| Patient 7 | male | 22 | distal left humerus |
| Patient 8 | male | 25 | distal left femur |
| Patient 9 | female | 34 | distal right femur |
| Patient 10 | female | 31 | proximal left tibia |
| Patient 11 | male | 59 | distal right radius |
| Patient 12 | female | 46 | distal left radius |
| Patient 13 | female | 49 | distal left humerus |
| Patient 14 | female | 19 | distal right humerus |
| Patient 15 | male | 33 | distal left femur |
| Patient 16 | female | 25 | distal right femur |
| Patient 17 | male | 35 | proximal left tibia |
| Patient 18 | male | 41 | proximal right tibia |
| Patient 19 | male | 42 | proximal left tibia |
| Patient 20 | female | 60 | distal right femur |
The patients all provided their written consent for their tissues to be obtained for this study. This research has been approved by the Ethics Committee of Jinling Hospital.
Cell Culture
This study used the giant cell tumor 0404 cell line, which were cultured in the condition medium containing 10% fetal bovine serum (FBS, Gibco, USA) + 90% 1640 medium (Gibco, USA) + 1% penicillin–streptomycin (Gibco, USA) in a humidified atmosphere of 5% CO2 at 37°C. When cells occupied above 90% of the subsurface of the flask, the adherent cells were separated with 1/10 cell suspension volume of trypsin for 2 min, collected with three times trypsin volume of serum‐contained medium, and centrifuged for 3 min. The precipitate, mainly made up of cells, was resuspended with the culture medium, and 1/3 of the suspension averagely was spread to the well plate to continue the culture.
Plasmids Transfection
When cells occupied 70%–80% of the subsurface of the well plate (culturing for about 24 h, logarithmic growth phase), the cell transfection efficiency was the highest. Cell transfection was carried out by the transfection kit Lipofectamine® 3000 (Invitrogen, USA). Taking a six‐well plate as an example, transfection solutions were prepared, each of which contained 5 μl Lipo3000 or P3000 with 100 μl Opti‐MEM I reduced serum medium (Gibco, USA), respectively. Then, the plasmids solutions (prepared according to concentration) were added into the P3000 solution, mixed with Lipo3000 transfection, and reacted for 20 min. Finally, the mixture was averagely added to the well plate, followed with adding the 1640 culture solution containing 2% FBS. After that, the cells were cultured for 24–36 h before they were ready to be tested. Meanwhile, to reflect the efficiency of transfection, the plasmids containing eGFP sequence could emit green fluorescence under the blue excitation light, and the density and intensity of colour could represent the efficiency.
EdU Assay
This experiment was conducted with the Cell‐LightTM EdU Apollo567 kit (Ruibo, China). All the procedures were performed according to the instructions of manufacturers. After transfection, the cells were proliferated to about 80% subsurface of the well plate. According to the instructions, reagent A and conditioned medium were added in a certain ratio (1/1000–1/5000) for about 4–6 h, and then the culture supernatant was discarded and the cells adherent to the subsurface were washed with phosphate buffer (PBS) several times. Then, 500 ul triformol was added in each well can fix the cells. Then 500 μl of 5% Triton‐X PBS (Invitrogen, USA) added in each well was used to increase the permeability of cell membranes. After that, regent B, regent C, regent D and regent E were mixed to a certain ratio provided by the instructions. The mixture was cultured with cells for 30 min to stain the labeled uracil, followed by PBS washing three times. Then 5% Triton‐X PBS was used again. After that, the cells were cultured with regent F (prepared with distilled water in 1/100) to stain nucleus. After staining, observation and photograph were performed by a fluorescence microscope EVOS M7000, Thermo Fisher, USA). The uracil could emit red fluorescence under green laser and the nucleus could emit blue fluorescence under wathet laser. When focusing on one field, the numbers of red and blue were counted and calculated with the help of Image J (NIH, USA). The relative proliferation rate was represented by the ratio of red and blue.
Transwell Assay
In a sterilized and ventilated place, the Transwell nests (CORNING, USA) were conversely placed on the cell culture dish, and 200 μl matrix glue was evenly spread on the nested bottom porous membrane to prepare the transwell chambers. After 5 min, 160‐180 μl glue was discarded from the edge, then the chambers were illuminated overnight with UV light. After the cells were cultured to a certain amount (90%–100%), they were separated with trypsin and washed by PBS, followed by centrifugation. At the same time, 500 μl (24‐well plate exampled) condition medium containing 20% FBS was added to the bottom of the well plate and the prepared chambers were placed in the well plate. The serum‐free 1640 medium (about 100–150 μl) was used to resuspend the cell precipitate to make each chamber contain around 20,000 cells. After culturing for 24–48 h (depending on cellular characteristics), the supernatant medium was discarded and the cells adherent to the subsurface was washed by PBS several times. Then 500 μl paraformaldehyde in each well can fix the cells. Then 500 μl 5% Triton‐X PBS added in each well was used to increase the permeability of cell membranes. The cells were stained by 500 μl crystal violet in each well for 30 min. After that, they were stained with blue and photographed by the microscope (EVOS M7000, Thermo Fisher, USA). Focusing on one field, the blue spots represented the cells that traversed through the porous membrane and glue to reflect the relative invasion rate. The numbers of blue spots were counted and calculated by Image J (NIH, USA).
Wound Healing Assay
The scratching test, also known as the wound healing assay, is generally performed with 12‐well plates. When cells were increased to 100% subsurface of the well plate, a 1 mm‐wide scratch was drawn in each well and the medium was adjusted to free‐serum medium. The width of the scratches was measured by Image J (NIH, USA) and photographed by the microscope (EVOS M7000, Thermo Fisher, USA). After culturing for 24–48 h (depending on cellular characteristics), the width of the scratch was measured and photographed again. The ratio between the width at 0 h and 24 h (exampled) was calculated and represented the relative translocation rate.
Apoptosis Assay
In this experiment, the apoptosis rate was measured using the Annexin V FITC Apop Dtect kit (BD, USA). When the number of cells in the six‐well plate was increased to 90% or more, they were digested with trypsin and collected into a 1.5 ml EP tube, washed twice with PBS, and then made into a suspension of 1 × 106 cells/ml with 1 × Binding Buffer. After that, 5 μl FITC Annexin V and PI solution were added to each tube respectively to stain the cellular membranes with FITC or PI. After culturing for 20 min in dark, 400 μl Binding Buffer and the prepared cell solution were mixed to stop staining and added into the Falcon tubes, and then the flow cytometry (FACSCalibur, BD, USA) was used to examine the staining of the single cell. The cells only stained with FITC meant they were in early apoptosis phase, while cells stained with PI meant they were in late apoptosis phase. If cells were stained with both, it meant the cells have been dead and should be excluded. Finally, the total cells in early and late phases were recorded and represented as the relative apoptosis rates.
Western Blot
Firstly, 10% gel, 1 × Running Buffer, 1 × Trans Buffer and 1 × Tri‐HCL buffering solution and Tween (TBST) were prepared according to the instructions of manufacturer (Thermo Fisher, USA). The total protein in cells were extracted with RIPA, culturing for 30 min. The solution was then mixed with 5 × SDS solution and heated for 5 min to disrupt the superior structure of the protein. The total protein‐SDS solution was added to the gel according to the estimated content of the target protein, and constant voltage of 80 V was applied for electrophoresis with the help of the electrophoresis apparatus (EPS‐300‐IIV, CBS, USA). After the separation of the protein, the gel was taken out and placed in the transfer capsules with a sponge‐thick filter‐gel‐PVDF membrane‐filter paper structure. And constant current of 0.3 A was applied to transfer the film, for 90 min. After taking out of the PVDF membranes, according to the ladder, the corresponding bands containing the internal reference protein GAPDH and the target protein on the membranes were cut out. After blocking for 1 h with 5% milk, the bands were immersed in the respective first antibody (sc‐390715, Santa Cruze, USA) solution overnight. Then, the bands were washed by 1 × TBST several times. And the bands were incubated with the secondary antibody (sc‐47724, Santa Cruze, USA) for 1 h, followed by TBST washing several times. Then the bands were reacted with enhanced chemiluminescent (SuperSignal West Femto, Thermo Fisher, USA) and examined by chemiluminescence detector (Fluoroskan, Thermo Fisher, USA). The instrument can collect the signals from the chemiluminescent and the product of intensities and areas could be considered as the parameters of tested proteins. These were recorded and calculated by Image J (NIH, USA). The ratio of the parameters of targeted protein and GAPDH (control) represented the relative level of the targeted proteins.
qRT‐PCR
RNA was extracted from cells by TRIZOL (Invitrogen, USA) with 1 ml in each sample. And 500 μl chloroform in each sample was used to filter TRIZOL, culturing for 15 min. After that, 400 μl supernatant each sample was extracted and mixed with 400 μl isopropanol to precipitate the total RNAs. And 1 ml ethyl alcohol was used for each sample to wash out DNA, followed by centrifugation. The residue RNAs were dissolved in diethyl pyrocarbonate water. The reverse transcription solution (Takara, Japan) was prepared according to the RNA reverse transcription system and the cDNAs were obtained by the PCR instrument (MiniAmp, Thermo Fisher, USA) with a certain program. After that, real‐time quantitative PCR solution (Takara, Japan) was prepared according to the respective system. And then real‐time quantitative PCR was carried out by the SYBR Green method (Takara, Japan) with LightCycler 480 system (Roche, Penzberg, Germany). During the duplication of cDNAs, the SYBR green dye can stain the structures between the two strands and the instrument can collect the signals from the staining. After the reaction, the instrument can analyse the Ct values according to the signals. Through the threshold, targeted gene and GAPDH (internal reference) had their own Ct values. And the deviation between the two Ct values can be considered as △Ct. The relative mRNA levels of targeted gene were represented by 2‐△Ct. The RUNX2 primer was designed as F: CGGAATGCCTCTGCTGTTAT, R: AGCTTCTGTCTGTGCCTTCT.
CCK8 Assay
After transfection, the cells were cultured to 100% subsurface of six‐well plates. Then, cells were separated with trypsin and suspended with 1 ml condition medium per well. After that, 100 μl cell suspension each well was added into a tube containing 6 ml condition medium to dilute the concentration. Then the new suspension was averagely spread into 96‐well plates with 100 ul each well. After culturing for 24 h, 48 h, 72 h, 96 h, and 120 h, respectively, 10 μl CCK8 reaction mixture (APExBIO, USA) was added to each well. The enzyme‐labeled instrument (Multiskan GO, Thermo Fisher, USA) was used to detect the absorbance of OD450 in each well after culturing the mixture of CCK8 reaction and cells for 2 h. Every time point had an absorbance which could represent the cellular viability in that time and then comparison of the trend of the absorbance of each sample was the assay result.
Measured Parameter
Levels of Genes
The expression of genes includes the levels of mRNAs and proteins. The levels of mRNA could be defined as the total amount of the targeted mRNA in certain samples. Meanwhile, the levels of proteins are the total amount of the targeted proteins in certain samples. Thus, qRT‐PCR, Western Blot and immunohistochemistry were used to measure the relative level of genes. In qRT‐PCR, the SYBR green dye can stain the structures during the cDNA replication, which produces a signal within one replication. The instrument can collect the signals and make some analysis. Through the threshold, targeted gene and GAPDH (internal reference) had their own Ct values. And the deviation between the two Ct values can be considered as △Ct. The relative mRNA levels of targeted gene were represented by 2‐△Ct. In Western Blot, the targeted proteins can react with the primary antibody to form a complex, then the complex can react with the secondary antibody to form luminous enzyme. With the addition of luminous substrates and the help of the instruments, the targeted proteins can form gray signals; the signals’ intensities and areas reflect the level of proteins. In immunohistochemistry, as mentioned in the methods of “Western Blot”, the targeted proteins can form complexes with the primary and secondary antibodies, which are then stained with blue dye. The size and number of blue points can reflect the relative levels of proteins.
Proliferation Rate
The proliferation rate is the ratio of proliferous cells and the total cells during a certain amount of time. In our research, the cells proliferation rates were measured by EDU assay. The total nuclei could be stained with blue, while the nuclei of proliferous cells were also stained with red. Under the fluorescence microscope, the red fluorescence of new nuclei and the blue fluorescence of total nuclei were presented and taken as photos. The ratio of the red points and blue points could represent the relative proliferation rates.
Migration Rate
The migration rate is the number of translocated cells during a certain amount of time. With the wound healing assays, we can measure the relative migration rates. At 0 h, on the subsurface of each well of plates, which were full of cells, scratches were made, with around 1 cm width. After a certain time, the widths of the scratches were recorded under the microscope and measured by Image J. The ratio between the width in 0 h and 24 h (exampled) was calculated and represented the relative translocation rates.
Invasion Rate
The invasion rate is the number of invaded cells during a certain amount of time. The transwell assays were utilized to measure the relative invasion rates. The cells were cultured in a serum‐free medium in chambers, and their subsurface had the porous membranes. The chambers containing cells were hang on the serum‐rich medium. The cells in the chambers could be attracted to the serum‐rich medium through the micropore and stalled in the membranes. After a certain time, the cells in the membranes were stained with violet, which can be recorded under the microscope and counted by Image J. The ratio of the numbers of the two time points can represent the invasion rates.
Cellular Viability
The cellular viability can be considered as the total number of cells which have normal proliferous abilities. We used CCK8 assays to quantify the cellular viability. On the cellular membranes of live cells with high cellular viability, CCK8 reaction fluids can stain some structures. After a certain time, the reaction complex can produce signals under OD450, which can be collected by the instruments. The consecutive absorbance of OD450 could reflect the relative cellular viability.
Apoptosis Rate
Apoptosis rate is defined as the total number of cells which are at the phase of apoptosis, including early apoptosis and late apoptosis. The apoptosis rates were measured by the Annexin V FITC assays in our research. The cellular membranes of early apoptosis cells could be solely stained by PI (propidium lodide), while the cellular membranes of apoptosis cells could be solely stained by FITC. The flow cytometry can recognize the stained signals produced by PI and FITC. Then, the cells with certain staining could be classified and counted. Thus, the sum of cells which were stained by PI or FITC (not both) could reflect the apoptosis rate.
Statistical Analysis
All data statistics were performed with SPSS 20.0 (IBM, USA). Data are expressed as mean ± standard deviation. Student's t test was used to test the two groups of data with normality, homogeneity and independence of variance. ANOVA was used to test whether multiple sets of datawere statistically significant. Dunnett‐t test was used for that the data of treatment groups was compared to a control group. *, **, ***, **** that indicate P < 0.05, P < 0.01, P < 0.001, P < 0.0001 respectively.
Results
RUNX2 is Highly Expressed in Giant Cell Tumor of Bone
In order to investigate the expression of RUNX2 in human giant cell tumors (GCTB), we collected seven specimens of patients with GCTB (information shown in Table 1) admitted to the Jinling Hospital from June 2016 to June 2019, and performed qRT‐PCR. RUNX2 mRNA was highly expressed in GCTB compared to normal tissues adjacent to the tumors (Fig. 1A). The mRNA level in tumors was over 100 times that in normal tissues. Subsequently, we confirmed the results through Western Blot which showed that RUNX2 had a higher protein expression level in GCTB (Fig. 1B). Comparatively, relative protein level of RUNX2 in tumors was appropriately 8.32(±4.41) folds higher than that in normal tissues. Immunohistochemistry on the pathological section of 20 pairs of tumor tissues and normal tissues from our pathology department had been completed for deeper examination of the expression of RUNX2 (Fig. 1C).
Figure 1.
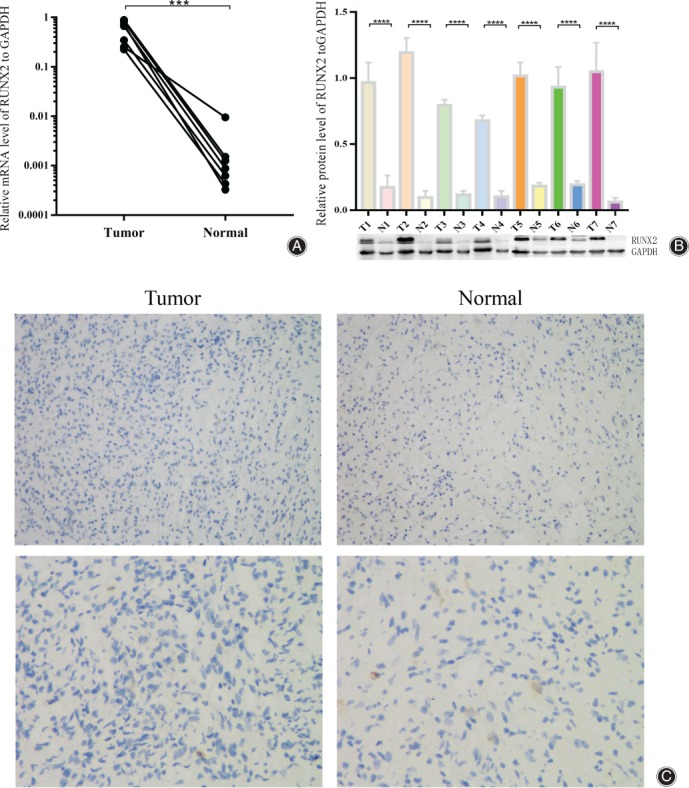
RUNX2 is highly expressed in giant cell tumor tissue. (A) Tumor tissues and normal tissues of seven patients with GCTB were examined by qRT‐PCR for RUNX2 mRNA level, and the results were normalized to tumor 1. (B) Western Blot results of RUNX2 in aforementioned tissues, standardized by tumor 1, where “T1” stands for “tumor 1”, “N1” stands for “normal tissue 1”, and so on. (C) Immunohistochemistry of RUNX2 in 20 pairs of tissues. The photos upside were taken at 200X, while the photos downside were at 400X. “***, ****” means P < 0.001 and P < 0.0001, respectively.
Construction of RUNX2 Plasmids
To induce RUNX2 to overexpress in 0404 cell line, we designed the RUNX2 plasmids (Fig. 2A, B, Appendix S1, S2) and transiently transfected them into the 0404 cell line with different concentration (0.4, 0.8, 1.6, 2.4 ng/μl). We also transfected control vector (2.4 ng/μl) into 0404 cell line. Results were compared with the group “MOCK”. Subsequently, qRT‐PCR (Fig. 2C) and Western Blot experiments (Fig. 2D) were performed, confirming that the designed RUNX2 plasmids were able to increase the mRNA and protein levels of RUNX2 in the cell line. To be more specific, The qRT‐PCR showed that these groups with progressively increased concentration could increase the mRNA level about 530.11(±24.87), 1117.96(±77.68), 2835.09(±45.22) and 4781.51(±79.37) folds respectively. And Western Blot showed that the protein level increased about 4.12(±1.15), 16.73(±1.63), 21.53(±2.41) and 23.39(±0.85) respectively. Meanwhile, we detected the fluorescence level of eGFP in MOCK, CV (0.4 ng/μl) and RUNX2 plasmids (0.4 ng/μl) after transfection for 12 h (Fig. 2E) to verify the transfection and expression of plasmids.
Figure 2.
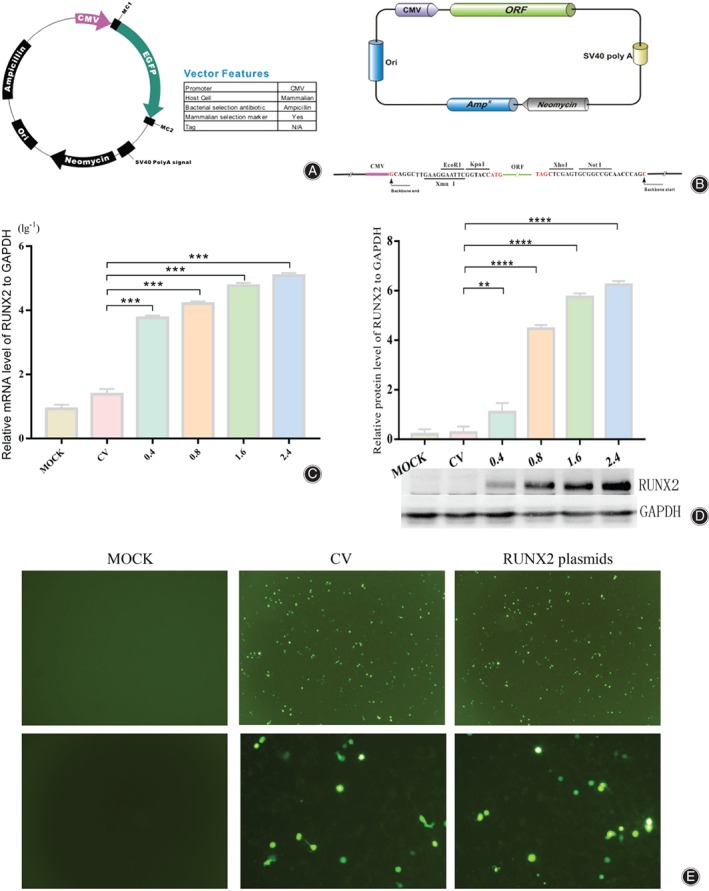
Construction of the RUNX2 overexpression plasmids. (A)Schematic diagram of control vector (CV); (B) Schematic diagram of RUNX2 plasmids structure, and RUNX2 sequence is between ORF and poly A; (C) After the transfection of CV and RUNX2 plasmids, mRNA of RUNX2 in different transfection concentrations of 0404 cells were examined by qRT‐ PCR. Each group of results were transformed by lg10, and standardized by mock group; (D) After transfecting CV and RUNX2 plasmids, Western Blot shows the level of RUNX2 protein in 0404 cells with different transfection concentrations, which were standardized by 0.4 ng/ul group. (E) The fluorescence level of eGFP in MOCK, CV and RUNX2 plasmids. Photos upside were taken at 40X, while the photos downside were taken at 200X. “***” means P < 0.001.
RUNX2 Plasmids Inhibit the Biological Functions of 0404 Cells
To examine the effect of RUNX2 plasmids on the biological functions of giant cell tumor 0404 cell line in vitro, the control vector (CV) and RUNX2 plasmids were transfected into the 0404 cell line. The EDU assay showed that the proliferation of the 0404 cell line was inhibited after overexpression of RUNX2 (Fig. 3A), presenting that the 1.6 ng/ul group decreased about 2.13(±1.02)% and 2.4 ng/μl group decreased about 3.03(±1.76)%. Subsequently, we performed the scratching assay on 0404 cell line in the same experimental group to test the migration. The results showed that the cell migration, after overexpression of RUNX2, was significantly weakened (Fig. 3B) in a concentration‐dependent way, decreasing about 45.56(±6.13)%, 50.79(±5.27)%, 63.15(±8.62)% and 93.90(±3.65)% respectively. The invasion was then tested by Transwell assay and similar results were found (Fig. 3C). The inhibition of invasion was around 14.49(±5.4)%, 37.02(±6.52)%, 42.24(±2.59)% and 48.97(±10.61)%, respectively. After that, we examined whether RUNX2‐overexpressed plasmids affected apoptosis in 0404 cells, and the results exhibited that high expression of RUNX2 can induce the apoptosis of 0404 cell line (Fig. 3D) with the apoptosis rate about 4.15(±0.27)%, 5.07(±0.27)%, 7.61(±0.45)% and 11.32(±1.02)% respectively. Finally, through the CCK8 assay, we detected that, with the increasing transfection level of RUNX2 plasmids, the viability of 0404 cell line had been seriously inhibited as time passed. In summary, the transfection of RUNX2 plasmids into the giant cell tumor 0404 cell line suppressed proliferation, migration, invasion and cellular viability of 0404 cell line, and promoted their apoptosis in a concentration‐dependent manner.
Figure 3.
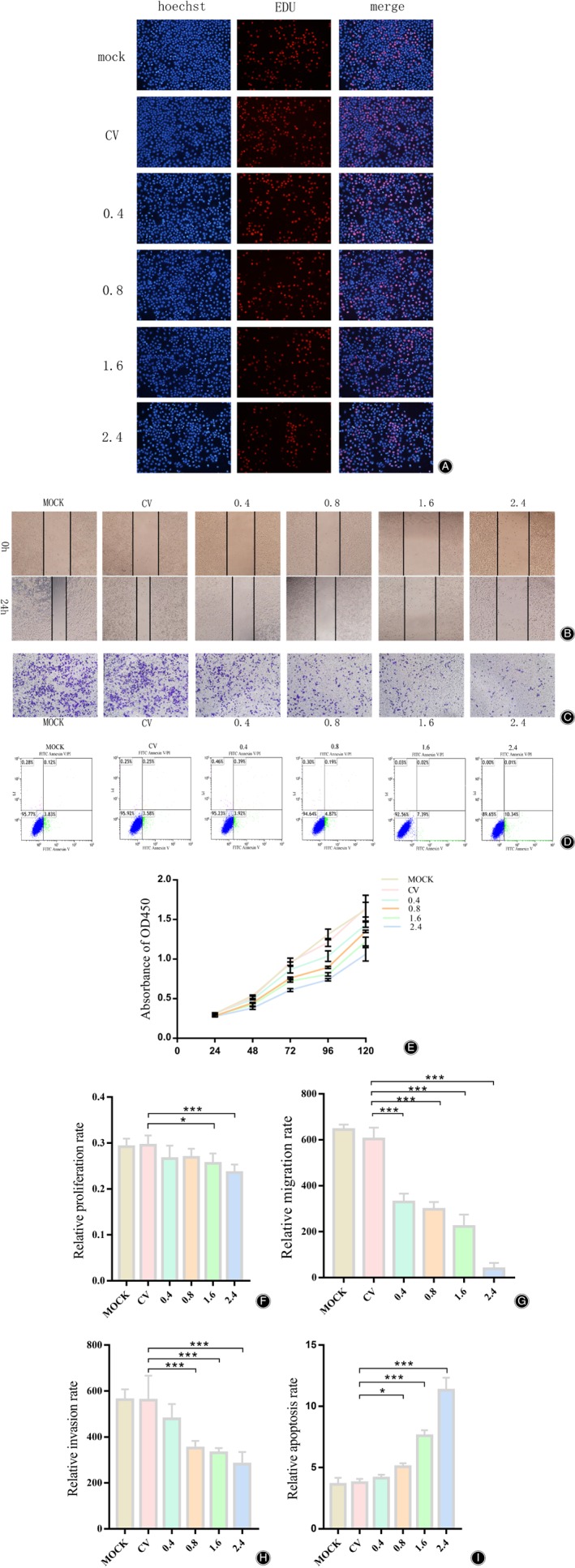
The biological function of the 0404 cell line was inhibited after transfection of the RUNX2 plasmids. (A, F) Experimental and statistical results of the EdU assay after the transfection of CV and RUNX2 plasmids; (B, G) Experimental and statistical results of the scratching assay after the transfection of CV and RUNX2 plasmids. The first row is the photo taken just after the scratching, and the second row is the photo taken after 24h; (C, H) Experimental and statistical results of the transwell invasion assay. The time to pass through Matrigel is about 36h; (D, I) Experimental and statistical results of the apoptosis assay. 0404 cells were stained by FITV Annexin V / PI approximately 24 h after transfection, and detected by flow cytometry. (E) Experimental and statistical results of CCK8 assay. “*” and “***” indicate P < 0.05 and P < 0.001, respectively.
Discussion
Main Pathological Features in Our Cell Line
Giant cell tumor of bone (GCTB) is a borderline tumor that has a tendency to recur and metastasize. According to Kito, GCTB consisted of three types of cells, including multinucleated giant cells, CD68+ mononuclear circle cells and spindle cells (stromal cells). He also found the mononuclear spindle cells could determine the main properties of GCTB, including proliferation, invasion and translocation10, 11. Abe thought that these spindle cells could obviously express PCNA, which is an indicator for proliferation12. Thus, we considered that only with interference of the spindle cells of GCTB can we finally influence the biological functions of GCTB. To confirm the ingredient of 0404 cell line, we conducted the immunofluorescence of CD68 and PCNA in 0404 cell line (Appendix S3, S4, S5).
Forced Expression of RUNX2 could Repress Functions of GCTB In Vitro because of the Hypothesis of SNP
RUNX2 is a transcription factor that promotes the differentiation and maturation of bone marrow mesenchymal stem cells into osteoblasts. Its expression is reduced in patients with osteoporosis, and can be regulated by various factors to affect the degree of osteoporosis. In recent years, a variety of microRNAs (miRNA) have been found to inhibit the development of giant cell tumors by regulating RUNX2 expression levels13, 14. In addition to miRNA, RUNX2 also interacts with MMP‐1315, TWIST16 in GCTB. In this study, we compared the expression of RUNX2 in some giant cell tumor tissues with the normal tissues, and found that RUNX2 was highly expressed in GCTB. We constructed plasmids which included the sequence of RUNX2 and transiently transfected them into 0404 cell line. The proliferation, migration, invasion and cellular viability of 0404 cells were inhibited, and the apoptosis was enhanced. Therefore, we speculated that the increase of exogenous RUNX2 expression in giant cell tumor of bone was associated with the single nucleotide polymorphism (SNP) in the RUNX2 gene. During the development of GCTB, RUNX2 was highly expressed to inhibit the differentiation of stroma cells in GCTB into osteoclasts. However, due to SNP, RUNX2 cannot be translated into the right proteins, where it cannot play the normal inhibitory role. The continuous expression of wrong RUNX2 was stimulated by the development of tumors. Nonetheless, the biological functions of RUNX2 were inhibited when the right RUNX2 plasmids were transfected into the cells. Last but not least, we supposed that RUNX2 plasmids mainly influenced the spindle cells (stroma cells) to inhibit their biological functions.
Prospects and Challenges
According to some researchers, GCTB has similar histopathological features as osteoporosis; this means that Denosumab can be used to treat patients with GCTB. Combining our findings with the studies in the past, we speculate that some other key factors functioning in osteoporosis can be studied further in GCTB. Meanwhile, there exists many significant molecules found in the process of bone metabolism which may also function in GCTB, such as RUNX2.
This study has some limitations. We have only studied the effect of RUNX2 in giant cell tumor 0404 cell line, whereas it lacks the study in vivo. However, at present, there have been few scholars who have completed the construction of in situ models of GCTB, while some scholars consider the GCTB animal model can be replaced by the model of osteoporosis. It is worthwhile examining this possibility in the future.
Conclusion
RUNX2 was highly expressed in GCTB. Through the transfection of RUNX2 plasmids, the GCTB 0404 cells showed their inhibited biological functions, including proliferation, translocation, invasion, and enhanced apoptosis rates. Although Denosumab had been approved by FDA for GCTB, there are a shortage of new therapies for this condition. This article aims to provide a new angle for deep exploration of GCTB and a novel target for the treatment of GCTB.
Supporting information
Appendix S1 & S2 The sequences of control vector (S1) and RUNX2 plasmids (S2). (presented with two attachment files).
Supporting Information
Appendix S3 The immunofluorescence level of CD68 in 0404 cell line. The photos upside were taken at 200X, while the photos downside were taken at 400X. The red fluorescence represents CD68 and the blue represents nucleus.
Appendix S4 The immunofluorescence level of PCNA in 0404 cell line. The photos upside were taken at 200X, while the photos downside were taken at 400X. The red fluorescence represents PCNA and the blue represents nucleus.
Appendix S5 The morphology of 0404 cell line is shown. The left photo was taken at 40X, while the right photo was taken at 200X. We can see that cells grew with colony, and mainly consist of spindle cells with a few of mononuclear circle cells and multinucleated giant cells.
Appendix S6 The information of patients with GCTB treated in out hospitals during 2016–2018.
Funding: NSFC (Natural Science Foundation of China). ID: 81472508. Name: Novel therapeutic approaches for osteosarcoma developed based on exosome‐mediated mircoRNA transport mechanisms in vivo.
References
- 1. Turcotte RE. Giant cell tumor of bone. Orthop Clin North Am, 2006, 37: 35–51. [DOI] [PubMed] [Google Scholar]
- 2. Wei G, Jianmin L, Jingnan S, Chongqi T. Guideline for clinical evidence⁃based diagnosis and treatment of giant cell tumor of bone. J Bone Joint Surg Ch, 2018, 11: 41–52. [Google Scholar]
- 3. Rutkowski P, Ferrari S, Grimer RJ, et al Surgical downstaging in an open‐label phase II trial of denosumab in patients with giant cell tumor of bone. Ann Surg Oncol, 2015, 22: 2860–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Orosz Z, Athanasou NA. Giant cell–containing tumors of bone. Surg Pathol Clin, 2017, 10: 553–573. [DOI] [PubMed] [Google Scholar]
- 5. Komori T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell, 1997, 89: 755–764. [DOI] [PubMed] [Google Scholar]
- 6. Ikebuchi Y, Aoki S, Honma M, et al Coupling of bone resorption and formation by RANKL reverse signaling. Nature, 2018, 561: 195–200. [DOI] [PubMed] [Google Scholar]
- 7. Yang L, Zeng Z, Kang N, Yang JC, Wei X, Hai Y. Circ‐VANGL1 promotes the progression of osteoporosis by absorbing miRNA‐217 to regulate RUNX2 expression. Eur Rev Med Pharmacol Sci, 2019, 23: 949–957. [DOI] [PubMed] [Google Scholar]
- 8. Ahmad N, Kushwaha P, Karvande A, et al MicroRNA‐672‐5p identified during weaning reverses osteopenia and sarcopenia in ovariectomized mice. Mol Ther Nucleic Acids, 2019, 14: 536–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin Q, Wang J, Fu Q, Gu S, Rui Y. CircRUNX2 through has‐miR‐203 regulates RUNX2 to prevent osteoporosis. J Cell Mol Med, 2018, 22: 6112–6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kito M. Identification of cytokines produced by cells cultured from human giant cell tumors of bone. Nihon Seikeigeka Gakkai Zasshi, 1991, 65: 918–930. [PubMed] [Google Scholar]
- 11. Kito M, Moriya H, Mikata A, et al Establishment of a cell line from a human giant cell tumor of bone. Clin Orthop Relat Res, 1993, 9: 353–360. [PubMed] [Google Scholar]
- 12. Abe Y, Yonemura K, Nishida K. Giant cell tumor of bone: analysis of proliferative cells by double‐labeling immunohistochemistry with anti‐proliferating cell nuclear antigen antibody and culture procedure. Nihon Seikeigeka Gakkai Zasshi, 1994, 68: 407–414. [PubMed] [Google Scholar]
- 13. Jiang ZY, Jiang JJ, Ma YS, et al Downregulation of miR‐223 and miR‐19a induces differentiation and promotes recruitment of osteoclast cells in giant‐cell tumor of the bone via the Runx2/TWIST‐RANK/RANKL pathway. Biochem Biophys Res Commun, 2018, 505: 1003–1009. [DOI] [PubMed] [Google Scholar]
- 14. Huang Q, Jiang Z, Meng T, et al MiR‐30a inhibits osteolysis by targeting RunX2 in giant cell tumor of bone. Biochem Biophys Res Commun, 2014, 453: 160–165. [DOI] [PubMed] [Google Scholar]
- 15. Mak IW, Cowan RW, Popovic S, Colterjohn N, Singh G, Ghert M. Upregulation of MMP‐13 via Runx2 in the stromal cell of giant cell tumor of bone. Bone, 2009, 45: 377–386. [DOI] [PubMed] [Google Scholar]
- 16. Singh S, Mak IW, Cowan RW, Turcotte R, Singh G, Ghert M. The role of TWIST as a regulator in giant cell tumor of bone. J Cell Biochem, 2011, 112: 2287–2295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 & S2 The sequences of control vector (S1) and RUNX2 plasmids (S2). (presented with two attachment files).
Supporting Information
Appendix S3 The immunofluorescence level of CD68 in 0404 cell line. The photos upside were taken at 200X, while the photos downside were taken at 400X. The red fluorescence represents CD68 and the blue represents nucleus.
Appendix S4 The immunofluorescence level of PCNA in 0404 cell line. The photos upside were taken at 200X, while the photos downside were taken at 400X. The red fluorescence represents PCNA and the blue represents nucleus.
Appendix S5 The morphology of 0404 cell line is shown. The left photo was taken at 40X, while the right photo was taken at 200X. We can see that cells grew with colony, and mainly consist of spindle cells with a few of mononuclear circle cells and multinucleated giant cells.
Appendix S6 The information of patients with GCTB treated in out hospitals during 2016–2018.