

Microtubule-associated serine/threonine kinase like (MASTL), also known as Greatwall (Gwl) kinase, has an important role in the regulation of mitosis. By inhibiting protein phosphatase 2A (PP2A), it plays a crucial role in activating one of the most important mitotic kinases, known as cyclin-dependent kinase 1 (CDK1). MASTL has been seen to be upregulated in various types of cancers and is also involved in tumor recurrence. It is activated by CDK1 through phosphorylations in the activation/T-loop, but the complete mechanism of its activation is still unclear.
KEYWORDS: Akt, cancer, Greatwall kinase, MASTL, mitosis, PP2A
ABSTRACT
Microtubule-associated serine/threonine kinase like (MASTL), also known as Greatwall (Gwl) kinase, has an important role in the regulation of mitosis. By inhibiting protein phosphatase 2A (PP2A), it plays a crucial role in activating one of the most important mitotic kinases, known as cyclin-dependent kinase 1 (CDK1). MASTL has been seen to be upregulated in various types of cancers and is also involved in tumor recurrence. It is activated by CDK1 through phosphorylations in the activation/T-loop, but the complete mechanism of its activation is still unclear. Here, we report that AKT phosphorylates MASTL at residue T299, which plays a critical role in its activation. Our results suggest that AKT increases CDK1-mediated phosphorylation and hence the activity of MASTL, which, in turn, promotes mitotic progression through PP2A inhibition. We also show that the oncogenic potential of AKT is augmented by MASTL activation, since AKT-mediated proliferation in colorectal cell lines can be attenuated by inhibiting and/or silencing MASTL. In brief, we report that AKT plays an important role in the progression of mitosis in mammalian cells and that it does so through the phosphorylation and activation of MASTL.
INTRODUCTION
Microtubule-associated serine/threonine kinase like (MASTL), also known as Greatwall (Gwl) kinase, is one of the main proteins that are essential for mitotic entry in mammalian cells (1) and is currently regarded as the master regulator of mitosis (2–4). It is required for timely activation of mitosis by the activation of the cyclin-dependent kinase 1 (CDK1)–cyclin B complex via inhibition of protein phosphatase 2A (PP2A) (5). PP2A is one of the major cellular serine/threonine phosphatases that regulate a number of signaling pathways as well as cell cycle progression (6). It is a very complex enzyme consisting of three subunits: the structural A subunit, the catalytic C subunit, and the regulatory B subunit (the most complex of the three), forming a trimeric holoenzyme ABC complex. Each of the three subunits is, in turn, composed of many isoforms (7). It is believed that the particular combination of the three subunit isoforms imparts a specific function to a given PP2A complex within the cell. The B subunit is the most diverse subunit in PP2A and has a critical role in giving a specific localization and function to the enzyme. Among its isoforms, the PP2A-B55δ isoform is known to be responsible for the dephosphorylation of downstream targets of CDK1 to prevent premature mitotic entry (8). It has been reported that Greatwall kinase, an orthologue of human MASTL (hMASTL), phosphorylates ARPP19 (19 kDa) and α-endosulfine (15 kDa; a ligand for the sulfonyl urea receptor) at serine 62 and serine 67, respectively (9, 10). After phosphorylation, these proteins strongly associate with PP2A-B55δ, resulting in the inhibition of this phosphatase. Inhibition of PP2A is essential for the mitotic entry of mammalian cells, since it promotes the phosphorylation-mediated activation of numerous proteins, such as CDK1-cyclin B, Plk1, and AurK, and their substrates, which play critical roles in mitotic entry and progression (11). Further evidence has shown that MASTL is necessary for the recruitment of cyclin B to anaphase-promoting complex (APC) at late metaphase, independently of its Cdc20 subunit. The efficient degradation of cyclin B at this step is necessary for the transition of cells from metaphase to anaphase (12).
In addition to its role in mitotic entry, MASTL plays an important role in mitotic exit, where its deactivation has been seen to be an essential requirement for the cells to exit from mitosis (13, 14). This inactivation of MASTL results in the reactivation of PP2A, which is also essential for the mitotic exit (15–17). Since it is so critical for mitosis, MASTL has been investigated in various types of carcinomas and has been seen to be upregulated in a number of malignancies (18–20). In addition, higher expression of MASTL in cancer patients increases chromosomal instability (CIN) and also correlates well with poor survival (19, 21, 22). It has been shown that knockdown/knockout of MASTL in various breast cancer cell lines substantially decreased their proliferation, migration/invasion, and colony formation properties, as well as tumor formation potential, in xenograft models (19).
The role of MASTL in tumor progression has been attributed to the increased rate of cell proliferation driven by activation/upregulation of this kinase in tumor cells. Some recent evidence has also shown that MASTL promotes tumor formation and invasiveness via AKT activation. MASTL has been observed to stabilize S473 phosphorylation of AKT by activating glycogen synthase kinase 3 (GSK3), which results in the ubiquitination of PHLPP, a phosphatase responsible for the dephosphorylation of AKT on S473 (23). Moreover, some high-throughput studies have shown that in comparison to many other kinases, MASTL can be a better therapeutic target for the prevention of tumor progression and the development of resistance against anticancer drugs (18, 24, 25). Taking these findings together, MASTL regulation seems to be critical for cells both in terms of correct timing of mitotic entry and in the regulation of normal cell proliferation to avoid tumor progression. Although it has been shown that CDK1-cyclin B and possibly Plx1 kinase (the Xenopus homolog of mammalian Plk1) are responsible for phosphorylating and activating Greatwall kinase (26), the complete mechanism of its regulation is still not fully understood, particularly in mammalian cells.
Here, we report that in addition to its phosphorylation and partial activation by CDK1-cyclin B, MASTL is also phosphorylated by AKT at residue T299. This phosphorylation leads to a further increase in, and/or stabilization of, CDK1-cyclin B-mediated phosphorylation and, hence, full activation of MASTL. Moreover, coexpression of MASTL and AKT in 293T cells, as well as in cancer cell lines such as SW480, HeLa, and U2OS, strongly promotes the mitotic entry of these cells. We also show that the cell proliferation-promoting potential of MASTL increases substantially in the presence of AKT. Together, these results show that MASTL is a bona fide substrate of AKT and that the role of AKT in tumorigenesis may depend largely on the activation of MASTL. These results also delineate the unknown mechanism by which AKT promotes the mitotic progression of mammalian cells through the activation of MASTL and the consequent inactivation of PP2A.
RESULTS
MASTL is a potential phosphorylation target for AKT.
Since the mechanism of the regulation of MASTL/Gwl is not fully understood, we wanted to obtain some further leads by using various bioinformatics tools. We used Scansite (https://scansite4.mit.edu/4.0), an online computational tool, to predict the kinases and/or binding partners that could potentially target MASTL to regulate its activity. We used the protein sequence of human MASTL for this analysis. The results indicated that human MASTL could be a possible substrate for AKT, since it has a perfect consensus site for AKT phosphorylation [RXRXX(S/T), where R represents arginine, X represents any amino acid, S represents serine, and T represents threonine] at residue T299 (Fig. 1A). A comparison of the sequences from different mammalian species using Clustal Omega software (https://www.ebi.ac.uk/Tools/msa/clustalo) showed that this site is highly conserved in many mammalian species (Fig. 1B) but not in Xenopus laevis or Drosophila melanogaster (Fig. 1C). To directly test whether MASTL is a bona fide substrate for AKT, we cotransfected pCMV-HA-MASTL, encoding human MASTL protein, with increasing amounts of pCDNA3.1-HA-AKT into HEK 293T cells. The results showed that at higher expression levels of AKT (>1.0 μg), MASTL protein levels were markedly reduced (Fig. 1D and E). This was probably because AKT phosphorylates MASTL at its consensus motif, leading to proteasomal degradation. To test whether that is so, we mutated the human MASTL construct at threonine 299 to alanine (T299A) and cotransfected this mutated construct (mutMASTL) with increasing amounts of AKT. Our results showed that the mutant MASTL (T299A) protein was not degraded even after the addition of 1.5 μg of AKT DNA to 293T cells (Fig. 1F and G). To confirm that this effect was specifically due to AKT, we cotransfected hemagglutinin-tagged MASTL (HA-MASTL) with an HA-AKT, FLAG-ERK1, or FLAG-p38α construct into HEK 293T cells and compared the protein levels of MASTL among these samples. The results showed that MASTL protein was specifically degraded by AKT, while ERK1 and p38α had no such effect on MASTL protein levels (Fig. 1H and I).
FIG 1.

MASTL is a potential target for AKT phosphorylation. (A) Scansite analysis (https://scansite4.mit.edu/4.0) of the human MASTL protein sequence (NCBI Protein accession no. NP_001165774.1) showing the presence of a conserved AKT phosphorylation site (RKRLAT) on the hMASTL sequence at residue T299. The analysis was carried out at a medium level of stringency on the Scansite portal. (B) Alignment of MASTL protein sequences from various mammalian species (as shown) using the Clustal Omega alignment tool. (C) Alignment of protein sequences of Drosophila melanogaster and Xenopus laevis in comparison with those of the mammalian species (human and mouse). (D) MASTL was overexpressed along with increasing expression of AKT by use of the pCMV-HA-MASTL and pCDNA3.1-HA-AKT constructs in 293T cells. The blot shows HA-MASTL protein levels after the addition of increasing amounts of HA-AKT DNA (amounts given in panel E). (F) The HA-MASTL mutant (T299A) showed no change in expression after the addition of HA-AKT DNA (amounts given in panel G). (H) Western blot showing that HA-MASTL was specifically degraded by HA-AKT and not by the FLAG-ERK1 or FLAG-p38α construct. (E, G, and I) Quantification analyses of the blots in panels D, F, and H, respectively. Immunoblotting for various proteins was carried out with the indicated antibodies. The experiments were repeated three times. Data are expressed as means ± standard errors of the means of triplicates.
To check whether MASTL protein degradation was due to AKT-mediated phosphorylation followed by proteasomal degradation, we used bortezomib, a known proteasomal inhibitor (27). The results showed that in the presence of bortezomib (1 μM for 24 h), MASTL protein levels were stabilized back to normal levels (Fig. 2A and B). Furthermore, to confirm that AKT is responsible for the degradation of MASTL protein, LY294002, a well-known inhibitor of the phosphatidylinositol 3-kinase (PI3K) pathway, was used to inhibit AKT activation. Treatment of 293T cells overexpressing HA-MASTL and HA-AKT with 20 μM LY294002 also prevented the degradation of MASTL protein by AKT (Fig. 2C and D). Moreover, a kinase-dead (KD) mutant of AKT (pCDNA3-T7-AKT1-K179M-T308A-S473A) had hardly any effect on MASTL protein levels when transiently expressed in 293T cells (Fig. 2E and F). In support of these results, coexpression of a hyperactive catalytic subunit of a PI3K mutant (pBABE-puro-HA-PIK3CA [H1047R]) (28), which is known to have oncogenic properties, with wild-type HA-MASTL in 293T cells degraded MASTL protein levels to an extent similar to that with wild-type HA-AKT (Fig. 2G and H). To test whether these constructs and LY294002 were working properly, we cotransfected 293T cells with wild-type HA-AKT or its kinase-dead mutant HA-AKT-KD and FLAG-FOXO1, one of the downstream targets of AKT. Western blotting results showed increased phosphorylation levels of FLAG-FOXO1 in the presence of wild-type HA-AKT, but not in the presence of the kinase-dead mutant, over those for the control (Fig. 2I and J).
FIG 2.

MASTL degradation is caused by AKT activation. (A) Western blot showing HA-MASTL protein levels in the presence of HA-AKT after the addition of 1 μM bortezomib for 24 h. Two micrograms of pCMV-HA-MASTL and 1 μg of pCDNA3.1-HA-AKT were cotransfected into 293T cells. The pEGFP-F construct (1 μg) was used as a transfection control. (C) Blot showing that the proteasomal degradation of HA-MASTL caused by HA-AKT (second lane from left) was rescued by inhibiting AKT activity by the addition of 20 μM LY294002 for 18 h (fourth lane). (E) Western blot showing that the degradation of HA-MASTL protein in the presence of wild-type HA-AKT was not observed with the kinase-dead mutant (HA-AKT-KD). (G) The hyperactive mutant of the catalytic subunit of PI3K (the pBABE-puro-HA-H1047R construct) (1 μg), when cotransfected with pCMV-HA-MASTL (2 μg), showed an effect similar to that of wild-type HA-AKT. (I) Phosphorylation of FLAG-FOXO1 showing the activity of wild-type HA-AKT compared to that of mutant HA-AKT-KD. (B, D, F, H, and J) Quantification of the protein amounts from the blots in panels A, C, E, G, and I, respectively. Immunoblotting was carried out using the indicated antibodies against the proteins as shown. The experiments were repeated three times, and data are expressed as means ± standard errors of the means of triplicates.
We also wanted to confirm that the AKT pathway was functional in the cells in the presence of the different constructs and reagents used in these experiments. For that purpose, we checked the AKT substrate phosphorylation levels in cell lysates in the presence and absence of HA-AKT, HA-AKT-KD, PI3K-HA-H1047R, and LY294002 (a PI3K kinase inhibitor). The results clearly showed an increase in AKT substrate phosphorylation in the presence of HA-AKT and PI3K-HA-H1047R but not in the presence of HA-AKT-KD. Also, treatment of these cells with LY294002 resulted in a decrease in these phosphorylations from those for untreated cells (Fig. 3A and B). In view of these results, we next asked whether the half-life of MASTL was decreased in the presence of AKT. We cotransfected HA-MASTL and HA-AKT constructs into HEK 293T cells and added cycloheximide to inhibit protein translation in a time-dependent manner. The results showed that the total protein levels of HA-MASTL were decreased much faster in the presence of HA-AKT than in its absence (Fig. 3C and D). Together, these results confirmed that activation of the PI3K-AKT pathway promotes the degradation of MASTL in cells.
FIG 3.

MASTL is phosphorylated by AKT on its substrate site. (A and B) AKT substrate phosphorylations showing the activity of wild-type HA-AKT (1 μg), the kinase-dead HA-AKT-KD construct (1 μg), PI3K-H1047R (1 μg), and LY294002 (20 μM for 18 h). An anti-AKT substrate phosphorylation antibody was used to detect total cellular AKT substrate phosphorylation levels in the different cell extracts as shown. (C and D) Cycloheximide assay showing the decreased half-life of HA-MASTL in the presence of overexpressed HA-AKT. MASTL and AKT expression was detected with an anti-HA antibody. GAPDH was used as a loading control and was detected with an anti-GAPDH antibody. In panel C, lanes show results without HA-AKT (–) or with HA-AKT (+) at 0, 1, 2, 3, and 4 h after cycloheximide treatment from left to right, respectively. (E) Lysates from pCMV-HA-MASTL- and pCDNA3.1-HA-AKT-transfected 293T cells were subjected to immunoprecipitation (IP) by an anti-HA antibody, followed by Western blotting (immunoblotting [IB]) and detection with an anti-phospho-AKT substrate antibody. (F) An anti-HA antibody was used on the same blot to detect HA-MASTL and HA-AKT expression (after reprobing). (G) The same experiment was repeated with wild-type HA-MASTL and mutant HA-MASTL T299A (mMASTL). (H) Detection of all the transfected proteins in the blot shown in panel G after probing with an anti-HA antibody. IgG was used as a loading control in immunoprecipitation experiments.
MASTL is phosphorylated by AKT on its substrate site.
The proteasomal degradation of MASTL in the presence of AKT implied that MASTL might be phosphorylated by AKT. Therefore, our next aim was to determine directly whether AKT phosphorylates human MASTL protein on its potential substrate site at residue T299. pCMV-HA-MASTL and pCDNA3.1-HA-AKT were cotransfected into 293T cells, and untransfected cells were used as a control. This was followed by immunoprecipitation of exogenously expressed HA-MASTL using an anti-HA antibody. The immunoprecipitated proteins were blotted with an anti-phospho-AKT substrate antibody. This antibody recognizes the potential substrates that are phosphorylated by AKT on its consensus site [RXRXX(S/T)] and is popularly used to identify AKT substrates. The results clearly showed the detection of a phosphorylation signal for an AKT substrate in the lane for MASTL- and AKT-cotransfected cells, while it was absent in the MASTL lane (Fig. 3E). The same blot was reprobed using an anti-HA antibody to confirm the presence of HA-MASTL and HA-AKT (Fig. 3F). To further validate that the phosphorylation was done by AKT specifically at its substrate site on MASTL, the same experiment was repeated using both the wild-type (HA-MASTL) and mutant (HA-mutMASTL [T299A]) constructs along with HA-AKT. Our results clearly showed that the anti-phospho-AKT antibody specifically detected wild-type MASTL, but not mutant MASTL (T299A), when cotransfected with AKT (Fig. 3G and H). This blot was also reprobed with an anti-HA antibody to detect the expression of HA-MASTL, HA-mutMASTL (T299A), and HA-AKT in these samples (Fig. 3H). These results thus established that AKT phosphorylates MASTL specifically at residue T299.
AKT phosphorylates MASTL and increases CDK1 substrate phosphorylation through PP2A inhibition, leading to mitotic progression.
From the results presented above, it was confirmed that AKT phosphorylates hMASTL at residue T299. Since MASTL is a mitotic protein that is required in its active form primarily during mitosis, we asked whether MASTL would be phosphorylated by AKT specifically during this stage. To check that, we cotransfected HA-MASTL and HA-AKT into HEK 293T cells and arrested them in mitosis by using nocodazole (at 50 nM for 18 h). Western blotting results indeed confirmed that AKT phosphorylates MASTL during the mitotic phase, as seen by a distinct band shift on HA-MASTL (Fig. 4A).
FIG 4.

AKT-mediated phosphorylation on MASTL increases CDK1 substrate phosphorylation. (A) pCMV-HA-MASTL (2 μg) was cotransfected with pCDNA3.1-HA-AKT (1 μg) into HEK 293T cells, and pEGFP-F (1 μg) was used as a transfection control. At 24 h posttransfection, cells were arrested in mitosis by use of 50 nM nocodazole for 18 h. Western blotting showed a band shift of HA-MASTL in the presence of HA-AKT (indicated by the arrow). (B) Increasing amounts of pCDNA3.1-HA-AKT were cotransfected with pCMV-HA-MASTL (2 μg) into 293T cells. The blot shows increasing phosphorylation of MASTL as well as other CDK substrates in the presence of increasing amounts of HA-AKT. Phosphorylation levels of Aurora kinase (pAur) were used as mitotic markers. For blots below the dashed line, the same lysates as those above the line were loaded again and run on a separate gel. (D) Western blot showing CDK-mediated phosphorylation of mutant MASTL (mutMASTL) or other CDK substrates in the presence of increasing amounts of HA-AKT. (F) An antibody against phospho-CDK1-T14 (pThr14) was used to determine PP2A activity in the presence of the cotransfected constructs HA-MASTL (2 μg) and HA-AKT1 (in increasing amounts [0.25, 0.5. 0.75, 1.0, and 1.5 μg in the second to sixth lanes, respectively]). The amounts of transfected HA-AKT were detected using an anti-AKT antibody, while MASTL was detected with an anti-MASTL antibody. (G) Blot showing CDK1 substrate phosphorylation levels in the presence of wild-type HA-MASTL (2 μg) and increasing amounts of pKH3-RSK1. (C, E, and H) Quantification of the anti-pSer-CDK1 substrate signal intensities in panels B, D, and G, respectively. The amounts of transfected plasmids used in panels B, D, and G are given in panels C, E, and H, respectively. The experiments were repeated three times. Data are expressed as means ± standard errors of the means of triplicates.
Next, we were interested in determining the effect of AKT phosphorylation on MASTL activation. If AKT-mediated phosphorylation at T299 truly activates MASTL, then cotransfection of HA-MASTL and HA-AKT into cells should promote mitotic entry in the host cells. Since MASTL activates CDK1 and its substrates by inhibiting PP2A (10, 16), we used the level of CDK substrate phosphorylation as a mitotic marker in MASTL- and AKT-cotransfected cells. This blotting method has been used by several groups as an assay to detect CDK-MASTL activity and the consequent inhibition of PP2A (9, 12, 16, 26, 29). For that purpose, increasing amounts of HA-AKT, from 0.25 μg to 1.5 μg, were cotransfected with HA-MASTL into 293T cells, and the extracted lysates were subjected to Western blotting using an anti-phospho-CDK substrate antibody. The results showed that increasing amounts of HA-AKT, when cotransfected with HA-MASTL, led to increased CDK1-cyclin B-mediated phosphorylation of MASTL as well as of other substrates of CDK1-cyclin B. Moreover, enhanced phospho-Aurora kinase protein levels also confirmed higher percentages of mitotic cells when cells were transfected with increasing amounts of HA-AKT (Fig. 4B and C). While HA-AKT increased the CDK1-cyclin B-mediated phosphorylation levels in the presence of wild-type HA-MASTL, it was unable to do so when mutant HA-MASTL was cotransfected into the cells (Fig. 4D and E). These results thus provided strong evidence that AKT promotes mitotic entry through the activation of MASTL by phosphorylating it at residue T299, leading to PP2A inhibition.
Since MASTL regulates mitosis through the inhibition of PP2A (5, 6, 9, 10, 16), we asked whether AKT also promotes mitosis through MASTL-induced PP2A inhibition. Therefore, we evaluated PP2A activity by Western blotting using a phospho-CDK1-threonine 14 antibody (pThr14). CDK1 is phosphorylated by Wee1/Myt1 at threonine 14 or tyrosine 15 (T14 or Y15), leading to the inactivation of the CDK1-cyclin B complex. Wee1/Myt1 are themselves phosphorylated and inactivated by CDK1-cyclin B, while they are dephosphorylated and activated by PP2A. Therefore, phosphorylation of T14 or Y15 on CDK1 is used as a readout of PP2A activity (16, 30, 31). Western blotting results indeed showed that when cotransfected with HA-MASTL, increasing amounts of HA-AKT (0.25 to 1.0 μg) led to reduced phospho-CDK1-T14 signals, thus indicating enhanced mitotic entry of these cells (Fig. 4F). Surprisingly, at larger amounts of AKT (1.5 to 2.0 μg), phospho-CDK1-T14 levels sharply increased, with concomitant decreases in MASTL levels. This was, however, consistent with our results in Fig. 1 and 2, which showed drastic reductions in MASTL protein levels at higher AKT levels due to proteasomal degradation following phosphorylation. These results thus strongly support our argument that activation of AKT and MASTL leads to PP2A inhibition and hence promotes mitotic entry of cells.
Furthermore, a previous study on the regulation of MASTL has shown that besides phosphorylation by CDK1, MASTL is probably also phosphorylated and activated by one of the AGC kinase family members, ribosomal S6 kinase (RSK2) (26). This family of kinases includes AKT as well as RSK, and all members have the same substrate motif for phosphorylation [(R/K)X(R/K)XX(S/T)], although they may have slightly different preferences for the basic residues at positions −3 and −5 of the S/T phosphorylation residue (where X is any amino acid) (32, 33). We wanted to compare the activities of RSK and AKT toward the phosphorylation of MASTL. Our results showed that cotransfection of RSK1 with HA-MASTL in 293T cells had a very modest effect, if any, on CDK substrate levels, even with larger amounts of HA-RSK1 transfected into these cells (Fig. 4G and H). Hence, these results confirmed that AKT, rather than RSK, is the bona fide and potent regulator of MASTL.
Next, we wanted to determine whether the decrease in the phosphorylation levels of CDK substrates, including that of MASTL, in the presence of large amounts of AKT (Fig. 4B) was due to proteasomal degradation. We cotransfected HA-MASTL and HA-AKT into 293T cells and added 1 μM bortezomib for 24 h to inhibit proteasomal degradation. The results confirmed that bortezomib treatment could prevent the degradation of MASTL and other CDK substrates in the presence of overexpressed HA-AKT (Fig. 5A and B). To gain further support for the role of AKT in the phosphorylation of CDK substrates, we cotransfected a KD mutant of HA-AKT along with HA-MASTL into 293T cells. Western blotting results clearly indicated that, unlike wild-type HA-AKT, the kinase-dead mutant of HA-AKT had no impact on CDK substrate phosphorylation, even at larger amounts (Fig. 5C and D). In support of these results, cotransfection of a constitutively active mutant of PI3K (HA-H1047R) with HA-MASTL into 293T cells showed an effect similar to that of HA-AKT (Fig. 5E and F).
FIG 5.

The proteasomal degradation of MASTL and other CDK1 substrates is mediated by AKT. (A) HA-MASTL (2 μg) and HA-AKT (1 μg) were overexpressed in HEK 293T cells as indicated. At 24 h posttransfection, cells were treated with 1 μM bortezomib for 24 h more and were blotted with an anti-CDK1 substrate antibody. The pEGFP-F vector was used as a transfection control and was detected with an anti-GFP antibody. (C) CDK substrate phosphorylation in 293T cells after cotransfection of HA-MASTL (2 μg) with wild-type HA-AKT (1 μg) or the kinase-dead mutant HA-AKT-KD (1 μg). (E) Effect of cotransfection of a hyperactive mutant of the catalytic subunit of PI3K (HA-H1047R) (1 μg) with HA-MASTL (2 μg) on CDK substrate phosphorylation levels in 293T cells. (B, D, and F) Quantification analyses for the phospho-CDK1 substrate levels in the blots shown in panels A, C, and E, respectively. The experiments were conducted in triplicate, and the data are expressed as means ± standard errors of the means of triplicates.
AKT promotes cell growth and proliferation through MASTL.
AKT is well known to stimulate the survival, growth, protein translation, and proliferation of mammalian cells through its numerous downstream substrates (34–36). To see whether it promotes the growth and proliferation of cells through MASTL, we first examined endogenous MASTL expression in various cell lines (Fig. 6A). Among these, the SW480 cell line showed highly upregulated expression of MASTL protein levels. We selected the SW480 cell line for our further studies, since it showed higher expression of MASTL than other cell lines. We obtained cell lines stably expressing myristoylated FLAG-AKT1, an inducible construct containing short hairpin RNA (shRNA) against MASTL (shMASTL), and two different inducible shAKT constructs (shAKT1 and shAKT2) in order to target AKT knockdown. The expression of these proteins in the corresponding cell lines was confirmed by Western blotting (Fig. 6B to H). We next wanted to study the effect of AKT (overexpression and knockdown) and MASTL (knockdown) on the growth rate of the SW480 cell line. For that purpose, we seeded 6-well plates in duplicate with equal numbers of these cells, keeping one of the plates as a control (without doxycycline [DOX]). The cells were allowed to grow for 2 days until they reached approximately 60% confluence. At this stage, the control plate (Fig. 6I, top) was stained with crystal violet, and this was taken as the 0-h time point. At the same time point, the shMASTL- and shAKT-expressing cell lines were induced by doxycycline addition and were allowed to grow for a further 24 h, followed by crystal violet staining (Fig. 6I, bottom). As can be seen from the results, the AKT-overexpressing SW480 cell line showed a higher intensity of crystal violet staining than the control, indicating faster growth (Fig. 6I and J). In contrast, silencing of MASTL using shMASTL substantially reduced the growth rate from that of the control SW480 cell line. Similarly, in the AKT-overexpressing SW480 cell line, silencing of MASTL by shMASTL and silencing of AKT by shAKT1 or shAKT2 resulted in the killing of cancerous SW480 cells to almost comparable levels. In addition to the crystal violet staining, a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was performed to compare the proliferation of the cells under similar experimental conditions. The results obtained were comparable to those obtained with crystal violet staining, thus further strengthening our results showing that AKT promotes cell growth and proliferation through MASTL (Fig. 6K).
FIG 6.

Silencing of MASTL is sufficient to overcome the growth of the AKT-overexpressing SW480 cell line. (A) Cancerous cell lines showing endogenous expression of MASTL. SW480 cells had the highest MASTL expression among these cell lines. (B) Western blot of SW480 cell extracts showing expression of the stably transfected pWZL-Neo-MF-AKT construct. Expression of exogenous AKT was detected by an anti-FLAG antibody. (C to E) Western blots of extracts of the SW480 colorectal cell line stably expressing the inducible PLKO-0.1-shMASTL, shAKT1, and shAKT2 constructs, respectively, showing the silencing of each protein after induction by doxycycline (DOX) addition (2 μg/ml). Expression of MASTL and AKT was detected by using anti-MASTL and anti-AKT antibodies, respectively. (F to H) Quantification of the relative expression of MASTL and AKT in panels C, D, and E, respectively. (I) Growth and proliferation assays of SW480 cells stably expressing the HA-AKT, shMASTL, shAKT1, and shAKT2 constructs. Equal numbers of cells from these cell lines (as indicated) were plated in 6-well plates in duplicate. The cells were allowed to grow for 2 days. One plate was used as a control and was stained with crystal violet (shown as –DOX, 0 h) (top). In the other plate, knockdown of MASTL (shMASTL) or AKT (shAKT1 and -2) was induced by the addition of doxycycline (2 μg/ml), and cells were grown for a further 24 h and were stained with crystal violet (shown as +DOX, 24 h) (bottom). The plates were dried and were used for imaging to compare the cell densities. (J) Quantification of the cell densities in different wells after destaining. The absorbances of destained solutions were measured at 590 nm. (K) Quantification of cell proliferation by an MTT assay. Experiments were carried out as for panel I in 12-well plates. Data are expressed as means ± standard errors of the means of triplicates.
Given the above results showing that AKT and MASTL promote CDK1 activity, cell growth, and cell proliferation, we checked whether the increase in the proliferation of SW480-AKT cells was accompanied by an increase in mitosis. We subjected cells of the SW480, SW480-AKT, SW480-shMASTL, and SW480-AKT-shMASTL lines to cell cycle analysis by flow cytometry. The results indicated an approximately 10% increase in the mitotic population (at the G2/M peak as seen in fluorescence-activated cell sorter [FACS] analysis) of SW480-AKT cells over that of SW480 control cells. However, this increase in the G2/M peak was almost fully reversed by silencing MASTL expression in the SW480-AKT cell line (Fig. 7A to D).
FIG 7.

Cell cycle analysis using FACS. (A to H) The SW480 (A and E), SW480-AKT (B and F), SW480 shMASTL (C and G), and SW480-AKT-shMASTL (D and H) cell lines were used to check cell cycle distribution using FACS analysis. For panels E to H, cells were treated with nocodazole (NOC) at 50 nM for 16 to 20 h in order to arrest cells in mitosis. (I and J) Graphical representations of FACS data obtained from panels A to D and E to H, respectively.
To further strengthen our findings that both MASTL and AKT play important roles in mitotic entry and regulation, we repeated these experiments in the presence of nocodazole, which arrests cells in mitosis. As expected, most of the SW480 and SW480-AKT cells showed high G2/M population peaks. These peaks showed sharp decreases in the SW480-shMASTL and SW480-AKT-shMASTL cell lines, with concomitant increases in the G1 peaks (Fig. 7E to H). These results thus clearly showed that MASTL expression is critical for the entry of cells into the mitotic phase and that overexpression of AKT is not able to promote mitosis in the absence of MASTL expression. The results of FACS analysis are summarized graphically in Fig. 7I and J.
AKT promotes mitosis in mammalian cells through the activation of MASTL and the inhibition of PP2A.
Since MASTL plays a critical role in mitotic entry and progression, we next wanted to confirm independently whether AKT would further stimulate the ability of MASTL to promote cells into mitosis. Despite our repeated efforts with different approaches, we did not succeed in obtaining stable cell lines overexpressing MASTL. This is most likely due to several mitotic abnormalities resulting in slower growth/division and death, as has been seen for MCF10A cells stably expressing MASTL (2, 21). We therefore chose HeLa cell lines for these experiments, since MASTL has been reported to be overexpressed in these cell lines, which was also observed in our results (Fig. 6A). Also, studies with HeLa cells have been useful in delineating the role of MASTL in mitosis in mammalian cells (29). Moreover, due to their morphology and sticky nature, these cells are quite useful for immunofluorescence experiments. In addition, we used U2OS cells for these experiments for the same reasons, although they have lower expression of MASTL than HeLa cells (data not shown).
To study the roles of AKT and MASTL in mitotic regulation, we obtained HeLa and U2OS cell lines stably expressing FLAG-AKT (Fig. 8A). Since AKT and MASTL supposedly inhibit PP2A, thus promoting cells into mitosis, we asked whether treatment with okadaic acid (a PP2A inhibitor) would make AKT-expressing cells more sensitive to PP2A inhibition and hence promote mitotic entry. As can be seen in Fig. 8B and C, the percentages of cells in mitosis (as seen by rounded morphology) were substantially higher in HeLa-AKT and U2OS-AKT cells than in the parental HeLa and U2OS (control) cells in the presence of a low okadaic acid concentration (25 nm). It is well known that cells in mitosis undergo extensive cytoskeletal rearrangements and spindle formation, resulting in a rounded morphology (37, 38).
FIG 8.

AKT enhances the mitotic index of MASTL-expressing cells. (A) U2OS and HeLa cell lines stably overexpressing FLAG-AKT were derived by retroviral infection. AKT expression was confirmed by Western blotting using an anti-AKT antibody. Upper bands in the second and fourth lanes represent exogenous FLAG-AKT in the U2OS-AKT and HeLa-AKT cell lines, respectively, while the endogenous AKT (lower band) is seen in all four lanes. (B and C) AKT overexpression in HeLa-AKT and U2OS-AKT cells increased the percentages of mitotic cells (as seen by rounded morphology) over those in control HeLa and U2OS cells, respectively, in the presence of okadaic acid (OA) (at 25 nM for 24 h). Images were taken at ×10 magnification. (D to G) Mitosis was confirmed by immunofluorescence using an anti-phospho-histone 3-Ser10 (pH3-Ser10) antibody (red) in HeLa and HeLa-AKT cells (D) and in U2OS and U2OS-AKT cells (F). DAPI (blue) was used to stain the nuclei. Images were taken at ×20 magnification. (E and G) Graphical representations of the mitotic indices for panels D and F, respectively. Data are expressed as means ± standard errors of the means of triplicates (n, 962 HeLa cells, 1,408 HeLa-AKT cells, 1,697 U2OS cells, and 1,675 U2OS-AKT cells).
To be certain that the rounded morphology represented mitotic cells, we compared the mitotic index of HeLa-AKT with that of the control HeLa cell line by using an anti-phospho-histone 3-Ser10 (pH3-S10) antibody, which is popularly used as a marker for mitotic cells. The cells were subjected to a double thymidine block for 18 h so as to synchronize them at the G1/S phase. At about 10 h after thymidine block release, the cells were used for immunofluorescence studies. At this stage, they were expected to be predominantly in the mitotic phase. The anti-pH3-S10 staining results clearly showed that AKT-overexpressing HeLa cells (HeLa-AKT cells) had a mitotic index about 4-fold higher than that of control (HeLa) cells (Fig. 8D and E). These results were consistent with the FACS data we obtained with SW480 cells (Fig. 7). Similar results were also obtained in U2OS and U2OS-AKT cells (Fig. 8F and G). These results clearly established that AKT promotes mitotic progression in mammalian cells.
It is noteworthy that the overall mitotic indices of HeLa and HeLa-AKT cells (Fig. 8D and E) were higher than those of U2OS and U2OS-AKT cells (Fig. 8F and G). This is probably related to the higher expression of MASTL in HeLa cells than in U2OS cells (data not shown). In addition, HeLa cells are known to be LKB1 null (LKB−/−). In some other studies, we have shown that LKB1 expression negatively regulates AKT activation (Z. Sarwar, S. Bhat, Q. Gillani, I. Reshi, M. U. Nisa, G. Adelmant, J. Marto, and S. Andrabi, submitted for publication [https://www.biorxiv.org/content/10.1101/691402v1]). This indicates that HeLa cells would have higher AKT activity than U2OS cells, which have wild-type LKB1 expression. This feature would result in higher MASTL activation and concomitant PP2A inhibition in HeLa cells, promoting mitotic entry and progression in these cells.
Since HeLa-AKT cells showed a higher proportion of cells in mitosis than the control HeLa cells, they provided a good resource for further investigations. To gain additional evidence that AKT plays a role in enhancing the mitotic index in mammalian cells, HeLa-AKT cells were synchronized at the G1/S checkpoint by a double thymidine block and were then released. After 3 h postrelease, when they were expected to be predominantly in S phase and prior to mitotic entry, they were treated individually either with the PP2A inhibitor okadaic acid, with LY294002 to inhibit AKT activity, or with both simultaneously. In the presence of okadaic acid, cells quickly assumed a more-rounded morphology, showing increased mitosis, as expected. LY294002 clearly reduced the rounded/mitotic phenotype both on its own and in the presence of okadaic acid (Fig. 9A). To confirm these results, we performed immunofluorescence experiments using phospho-histone 3 staining. The results obtained (Fig. 9B) were in complete conformity with the cellular morphology results mentioned above. While okadaic acid treatment enhanced the mitotic index of HeLa-AKT cells by >20-fold, LY294002 reduced it to a level even lower than that of control HeLa-AKT cells. More importantly, when HeLa-AKT cells were treated with both the inhibitors simultaneously, LY294002 treatment substantially reduced the okadaic acid-induced mitotic index of cells (Fig. 9B and C). We also performed Western blotting on the cell lysates obtained from similar experiments, using the anti-pH3-Ser10 antibody. In agreement with the immunofluorescence results, phospho-histone 3 protein levels were higher in HeLa-AKT cells treated with okadaic acid, and lower in the presence of LY294002, than in control HeLa-AKT cells (Fig. 9D). Similarly, LY294002 treatment also reduced the phospho-histone 3 band intensity in lysates obtained from HeLa-AKT cells treated with okadaic acid.
FIG 9.

AKT activation promotes the mitotic index through PP2A inhibition. (A) HeLa-AKT cells were treated with okadaic acid (OA) (100 nM), LY294002 (40 μM), or both for 6 h. Enhanced cell rounding morphology indicates increased mitosis (magnification, ×10). (B) Immunofluorescence staining for phospho-histone 3 (Ser10) was carried out to confirm the mitotic phase in these cells (red), while DAPI (blue) was used to stain the nuclei. A total of 191 cells were used for HeLa-AKT-Con (Ctrl), 278 for HeLa-AKT-OA (OA), 259 for HeLa-AKT-LY (LY), and 383 for HeLa-AKT-OA-LY (OA + LY). (C) Graphical analysis of the mitotic indices obtained in the experiment for which results are shown in panel B. (D) Western blot showing phospho-histone 3 protein levels in HeLa-AKT cells under different conditions. OA (100 nM), LY294002 (40 μM), or both together were added for 6 h. Immunoblotting was carried out with anti-phospho-histone 3 (pH3) and β-actin antibodies as shown. (E) Western blot of lysates from stable HeLa, HeLa-SVST, HeLa-AKT, and HeLa-AKT-SVST cell lines. Cell extracts were blotted with antibodies against AKT substrates phospho-AKT (S473), total AKT, HA-SVST, and β-actin, as indicated. The anti-AKT antibody detected both endogenous AKT (bottom double bands) and exogenously expressed AKT (FLAG-AKT) (top band). (F) Immunofluorescence results for HeLa, HeLa-SVST, HeLa-AKT, and HeLa-AKT-SVST cells with anti-phospho-histone 3 antibodies (red). DAPI was used to stain the nuclei (blue). A total of 602 cells were used for HeLa-Con (Ctrl), 948 for HeLa-SVST (SVST), 746 for HeLa-AKT (AKT), and 957 for HeLa-AKT-SVST (SVST+AKT). All immunofluorescence images were taken at ×20 magnification. (G) Graphical representation of the mitotic indices of the cell lines shown in panel F. The experiments were repeated two or three times.
The results presented above clearly showed the role of AKT activation and PP2A inhibition in promoting the mitotic progression of cells. Next, we wanted to use an independent assay to further substantiate these findings. Simian virus 40 small-t antigen (SV40 ST) is a well-known inhibitor of PP2A and is more specific than okadaic acid, since it displaces the regulatory B56γ subunit(s) of PP2A (39). By virtue of this inhibition, it also activates AKT by enhancing its phosphorylation levels and hence its activity, thus promoting the transformation of mammalian cells (40, 41). Given the results obtained with okadaic acid, we presumed that SV40 ST expression would also promote mitotic progression through PP2A inhibition and AKT activation, which would, in turn, lead to MASTL activation. To test this idea, we made HeLa and HeLa-AKT cell lines stably overexpressing SV40 ST (HeLa-SVST and HeLa-AKT-SVST, respectively). The expression of SV40 ST in these cell lines was confirmed by Western blotting (Fig. 9E). Inhibition of PP2A and activation of AKT in cells expressing SV40 ST was confirmed by enhanced AKT substrate phosphorylation and also by increased phosphorylation of AKT at the serine 473 residue. As expected, HeLa-SV40ST and HeLa-AKT-SV40ST cells clearly had higher mitotic indices than HeLa and HeLa-AKT cells, respectively, as evidenced by increased phospho-histone 3 levels shown by immunofluorescence (Fig. 9F and G).
Taken together, these results provide strong evidence that AKT expression increases cell proliferation through MASTL activation and PP2A inhibition, which consequently promote the entry of cells into mitosis. And the increase in the growth/division of cells expressing AKT can be significantly reduced by silencing MASTL.
DISCUSSION
Greatwall (Gwl) kinase is one of the important regulators of mitosis that was originally discovered several years ago in Drosophila (42, 43). It was also later found in Xenopus and mammals, where it is known as microtubule-associated serine/threonine kinase like (MASTL) (1, 29). Although the role of MASTL in mitotic regulation has been extensively studied, the mechanism of its action is still not fully clear. A few studies have reported that Greatwall kinase/MASTL is regulated mainly by CDK1-mediated phosphorylation at residues T193 and T206 (Xenopus numbering; equivalent to T194 and T207 in hMASTL, respectively). This is followed by another critical phosphorylation at residue S883 (S875 in hMASTL) in the tail region, which further activates Gwl/MASTL. The identity of the kinase responsible for this tail residue phosphorylation is still controversial; it has been proposed to be Plx1 (Xenopus Polo kinase 1) or the Greatwall kinase itself, through autophosphorylation (26, 29, 44). Importantly, the same studies have suggested that besides CDK1, there might be some other regulator(s) of MASTL, potentially from its own AGC family members, which would enhance its kinase activity. This is because unlike other AGC kinases, MASTL does not have a hydrophobic motif in its tail that could bind its own hydrophobic pocket, which is needed for its activation. It is therefore believed that like phosphoinositide-dependent kinase (PDK), MASTL would require another AGC family member that would provide the hydrophobic tail needed to bind to the hydrophobic pocket. This would result in the conformational change in the MASTL catalytic region, thus enhancing its kinase activity.
In this study, we attempted to elucidate the mechanism of regulation of MASTL activity during mitosis. We employed the Scansite 4 analysis tool to search for potential regulators of MASTL and found that AKT was one of the kinases that could potentially phosphorylate MASTL. To confirm this, we overexpressed MASTL and AKT together in HEK 293T cells and found out that MASTL protein is degraded after coexpression with AKT. Using various biochemical assays, we showed that AKT indeed phosphorylates hMASTL at residue T299 during mitosis. This phosphorylation leads to increases in the phosphorylation of CDK1 downstream substrates, with a significant increase in the phosphorylation of MASTL itself. It has been observed that various phosphorylations at the N-terminal and C-terminal domains of MASTL lead these two lobes to interact with each other, resulting in the stabilization of phosphorylations of this kinase (26). Our results add to the complexity of this mechanism, suggesting that AKT-mediated phosphorylation on MASTL results in the stabilization of the CDK1-mediated phosphorylation of this kinase, leading to its further activation. We believe that earlier groups (26, 29, 42, 44) have missed this AKT phosphorylation site on MASTL because they have predominantly used Xenopus and/or Drosophila Gwl proteins that do not have this motif, which is highly conserved in mammalian species (Fig. 1B and C).
Our results showed that stable expression of AKT in the SW480 colorectal cell line, in which endogenous MASTL is highly upregulated, increased mitotic proliferation of these cells, as confirmed by cell growth assays and FACS analysis. This increase in SW480-AKT cell growth was substantially reduced by silencing of MASTL as well as AKT in these cells. Using immunofluorescence experiments, we also showed that AKT overexpression enhanced the mitotic indices of HeLa and U2OS cells (Fig. 8). This increase in the mitotic index was further enhanced by treatment of these AKT-overexpressing cells with okadaic acid or SV40 ST, both of which are known inhibitors of PP2A, while it was reversed in the presence of LY294002 (Fig. 9). Overall, these findings suggest that AKT overexpression leads to the activation of MASTL, which promotes the entry of cells into mitosis, and that AKT, MASTL, and PP2A are connected in a pathway that plays an important role in mitotic entry and regulation.
A previous study suggested that MASTL could be activated by a phosphorylated hydrophobic peptide motif of RSK2 (PIFtide) (26). In light of these results, we had already carried out an experiment to check the phosphorylation and activation of MASTL with RSK1 and the resulting CDK1-cyclin B substrate phosphorylations, using an anti-phospho-CDK substrate antibody. Our results clearly showed that AKT has a much stronger impact than RSK1 on MASTL phosphorylation/activation (Fig. 4G). We believe that our results could be more relevant, since we used the full RSK protein rather than the hydrophobic peptide (PIFtide) used by the other group. Furthermore, while this article was in revision, it was reported that mouse MASTL is phosphorylated at the T297 site (equivalent to the human T299 site) (45), which we have shown to be phosphorylated by AKT1 in our study. The fact that this site was also detected by an AKT substrate antibody as reported in the above-mentioned paper (45) lends further support to our data showing that MASTL is a genuine AKT substrate. Although Soeda et al. (45) have shown evidence that MASTL is a target for RSK1 phosphorylation, these studies were carried out mostly in mouse oocytes undergoing meiosis during fertilization, where the extracellular signal-related kinase (ERK)/RSK pathway plays a major role in the delayed pronuclear (PN) formation. Therefore, it is quite possible that while ERK1-RSK1-MASTL may play a role during meiosis in germ cells, in contrast, AKT-MASTL could be significant in mitotic regulation in somatic cells. Alternatively, this site may be phosphorylated by both these AGC kinases, though under different conditions or cellular processes. It is also pertinent to mention here that the MASTL sequence in humans or mice (RXRXXT or RKRLGT) is a more suitable substrate for AKT than other AGC kinases, such as RSK and S6K1, which have similar substrate motifs but prefer K residues to R residues at the −5 and −3 sites at the N terminus of phosphoacceptor S/T residues (33, 46, 47). Nevertheless, the studies by Soeda et al. (45) and our group together establish that mammalian MASTL is phosphorylated at T297 (mouse) or T299 (human) and is regulated by a member(s) of the AGC family of kinases.
The fact that AKT regulates the mitotic entry and progression of cells through MASTL is not entirely surprising. AKT has been shown to be highly activated during the G2/M transition in epithelial cells and to promote mitotic entry and progression (48). Another study showed that activated AKT enabled cells to overcome G2/M arrest in a p53-independent manner in the presence of various inhibitors (49). In both those studies, the authors proposed that AKT must activate a potential mitotic kinase to promote G2/M transition. Surprisingly, however, the exact mechanism by which AKT promotes mitotic progression has not been investigated further. We propose that activated AKT phosphorylates and activates MASTL, which, in turn, phosphorylates endogenous inhibitors such as ENSA/ARPP19, thus leading to the inactivation of PP2A and the subsequent progression of cells from G2 to the mitotic (M) phase, as reported previously (9, 10). Although AKT is well known to promote the G1/S transition through the phosphorylation and inactivation of several proteins enforcing this checkpoint, such as p21, p27, FOXO1/3, and GSK3 (33, 50, 51), to our knowledge, this is the first mechanistic study explaining how AKT promotes the G2/M transition and hence mitosis. Based on our findings and published reports, we thus propose a model explaining the role of AKT in mitotic regulation (Fig. 10).
FIG 10.
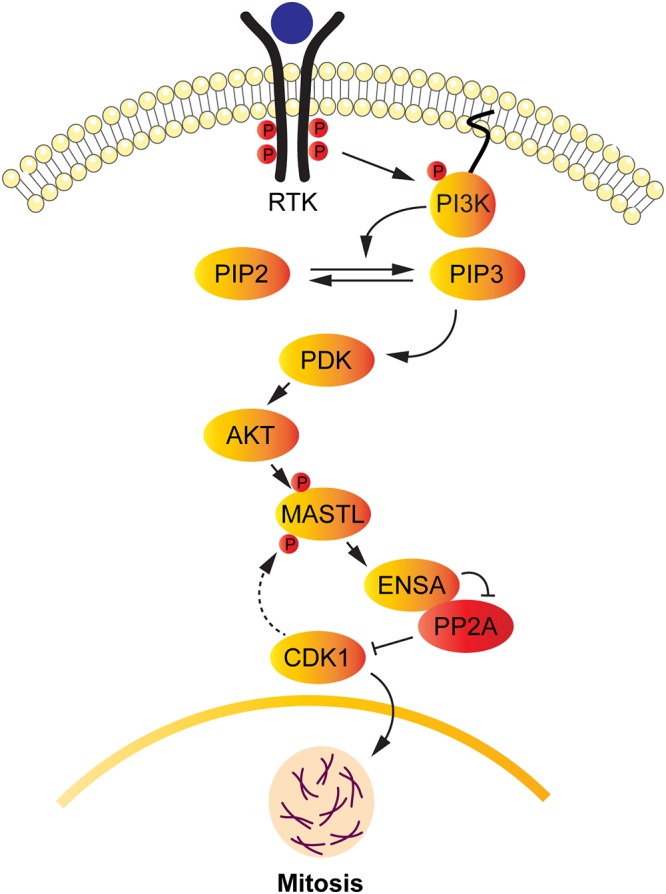
Proposed model showing the connecting link between AKT and MASTL signaling during mitosis. Activation of the PI3K pathway leads to the phosphorylation and activation of AKT. Activated AKT, in turn, leads to the phosphorylation of MASTL at residue T299, thereby leading to MASTL activation. Once activated, MASTL leads to the phosphorylation of ENSA, resulting in the inactivation of PP2A-B55δ, a regulatory B subunit of PP2A. As a result, the CDK1-cyclin B complex remains activated, leading to the phosphorylation of numerous substrates, including MASTL, which plays an important role in the entry and progression of the cells into mitosis through PP2A inhibition.
Our findings are also complementary to another report mentioning that Gwl leads to the phosphorylation of AKT at S473 and consequently its activation (23). Our studies have shown the reciprocal mechanism. It is quite possible that AKT and MASTL phosphorylate and activate each other by feedback mechanisms.
In summary, as determined by various genetic and biochemical assays, our data strongly support the argument that AKT phosphorylates hMASTL at residue T299, leading to its activation and PP2A inhibition, thereby promoting mitosis. These results assume significance because both AKT and MASTL are reported to be activated and/or overexpressed in many types of cancers and are popular targets for anticancer therapy. Since AKT regulates numerous cell survival pathways, the use of anti-AKT drugs usually has drastic effects on patient survival. On the other hand, identifying and inhibiting specific downstream candidates, such as MASTL, that regulate mitotic progression may prove to more beneficial for the development of specific anticancer therapeutic agents.
MATERIALS AND METHODS
Materials and reagents.
Doxycycline, rabbit monoclonal anti-HA, protease and phosphatase inhibitors, Polybrene, agarose A/G beads, propidium iodide, RNase A, MTT, and crystal violet were obtained from Sigma-Aldrich. The primers used for PCR were purchased from Integrated DNA Technologies (IDT). Anti-AKT substrate, anti-CDK substrate, anti-phospho-histone 3 (Ser 10), anti-pCDK1-T14, anti-MASTL, anti-Myc tag, anti-green fluorescent protein (anti-GFP), anti-phospho-Aurora, anti-cyclin B, anti-HA, anti-FLAG, anti-FOXO1, anti-phospho-FOXO1/FOXO3A (T24/T32), anti-pAKT 473, and secondary antibodies (DyLight 680 and DyLight 800) for the Li-Cor Odyssey-CLx imaging system were purchased from Cell Signaling Technology, USA. Secondary antibodies (Alexa Fluor 488 and Alexa Fluor 594) for immunofluorescence were purchased from Jackson Immunochemicals, USA. Glass slides and rounded coverslips were purchased from Thermo Fisher Scientific. Antifade mounting solution and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Thermo Fisher Scientific. The nitrocellulose membrane, protein ladder, acrylamide solutions, and Bradford reagent were purchased from Bio-Rad Laboratories, USA. Drugs (nocodazole, okadaic acid, LY294002, bortezomib) were purchased from Calbiochem/EMD Biosciences. Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin, G418, puromycin, and trypsin were obtained from Invitrogen/Life Technologies. Q5 high-fidelity DNA polymerase, deoxynucleoside triphosphates (dNTPs), T4 DNA ligase, DpnI, and all restriction enzymes that were used in our experiments were purchased from New England Biolabs, USA. The transfection reagent polyethyleneimine (PEI) was obtained from Polysciences, USA. The MASTL construct was a kind gift from Anna Castro, France (29). It was amplified by PCR using adaptors containing XhoI and NotI restriction sites and was later cloned into pCMV-HA-3.7, using the primer sequences for MASTL (5′-GGCCTCGAGTAGATCCCACCGCGGGAAGCAA-3′ and 3′-GGCCGCGGCCGCCTACAGACTAAATCCAGATAC-5′) by following standard cloning procedures. Other plasmids used were pCDNA3.1 HA-AKT1 (Addgene plasmid 9008) (50), human PKH3-RSK1 (Addgene plasmid 13841) (52), the AKT kinase-dead construct pCDNA3 T7 AKT1 K179M T308A S473A (Addgene plasmid 9031) (50), pCMV-FLAG-hErk1 (Addgene plasmid 49328), pCDNA3-FLAG-p38α (Addgene plasmid 20351), pCDNA3/FLAG-FOXO1, and pWZL-Neo-Myr-FLAG-AKT (Addgene plasmid 20422). pEGFP-F (Clontech) was used as a transfection control. pCDNA3-FLAG-FOXO1 (also known as FKHR) was a kind gift from Kunliang Guan (51). pBABE-puro-HA-PIK3CA (H1047R) (28) and the 293T, Phoenix, and SW480 cell lines were kindly given as gifts by Thomas Roberts, Dana-Farber Cancer Institute, Harvard University, USA. shAKT1 and shAKT2 were cloned into pLKO.1 puro using the CGCGTGACCATGAACGAGTTT sequence for shAKT1 and the CGAGTTTGAGTACCTGAAGCT sequence for shAKT2 (53). Similarly, for the shMASTL construct, CCCATTCATTGTCCATTTGTA was cloned into pLKO.1. The source of the sequence that was used for cloning was from Sigma-Aldrich (TRCN0000002278).
Site-directed mutagenesis.
For site-directed mutagenesis, pCMV-HA-MASTL, cloned in our lab (as described above), was used as the template to generate the mutant MASTL. Overlapping primers 5′-AGGAAAAGGCTGGCCGCATCCAGTGCCAGTAGT-3′ and 5′-ACTACTGGCACTGGATGCGGCCAGCCTTTTCCT-3′ containing a point mutation (A to G at the 895th nucleotide of MASTL) were used to amplify the template. This mutation was used to alter the amino acid serine to alanine at the 299th position on MASTL, corresponding to the AKT phosphorylation site. The protocol from the Agilent QuikChange site-directed mutagenesis kit was followed to create the mutant. The amplified clones were sent to SciGenom Labs, India, for sequencing to confirm the incorporation of the desired mutation.
Cell culture.
Cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin using standard procedures. Protein expression was induced by adding doxycycline at a 2-μg/ml concentration.
Transfection.
For transient transfections, pCMV-HA-MASTL, pCDNA3.1-HA-AKT, enhanced GFP (EGFP), pBABE-puro-HA-PIK3CA (H1047R), pCDNA3T7-AKT1-K179M-T308A-S473A (AKT-KD), pKH3-RSK, pCMV-HA-mutant MASTL, pFLAG-CMV-hErk1, and pCDNA3-FLAG-p38α were transfected into HEK 293T cells using PEI as the transfection reagent. The cells were harvested after 48 h using NP-40 lysis buffer (1% NP-40, 50 mM Tris-Cl, 150 mM NaCl, 10% glycerol, and 2 mM EDTA) containing protease and phosphatase inhibitors and were incubated on ice for about 30 min. Denaturation was done by adding 5× loading dye and boiling for about 3 min.
Retroviral transductions.
SW480, HeLa, and U2OS cell lines stably overexpressing AKT were obtained by a standard retroviral transduction procedure as follows. pWZL-Neo-MF-AKT (2.5 μg; Addgene USA) along with 2.0 μg of vectors expressing gag/pol and vsvg at a 2:1 ratio was transfected into Phoenix cells that were about 80% confluent at the time of transfection. On the next day, the medium of the transfected cells was changed, and the cells were allowed to grow further for about 24 h. After incubation, the viral supernatants of these transfected Phoenix cells, along with 8 μg/ml Polybrene, were added to the target cells (SW480, HeLa, and U2OS) for at least 6 h. At the end of the day, cells were washed with 1× phosphate-buffered saline (PBS), fresh medium was added for overnight incubation, and the same procedure was repeated on the next day. On the third day, the cells were washed, and the selection of stably transfected cells was started by the addition of 500 μg/ml G418 to the medium. After selection of the cells for about 2 weeks, each cell line was amplified and used for further experiments. The HeLa-SV40ST and HeLa-AKT-SV40ST cell lines were also obtained by retroviral infections as mentioned above, using the pWZL-blast-SV40ST construct that was available in our laboratory.
Western blotting.
The denatured cell lysates were run on SDS-PAGE gels, and the proteins were transferred to nitrocellulose membranes. After transfer, the blots were blocked with 5% nonfat dry milk for about 1 h. Then corresponding primary antibodies were added to each blot, and the blots were incubated overnight at 4°C. Subsequently, blots were washed three times with 1× Tris-buffered saline with Tween 20 (TBS-T, comprising 13.7 mM NaCl, 20 mM Tris [pH 7.4], and 0.05% Tween 20) for 10 min each time. This was followed by incubation of the blots in a secondary antibody for about 1 h. Finally, the blots were again washed three times with 1× TBS-T and were subjected to an infrared detection system using a Li-Cor Odyssey-CLx machine.
Immunoprecipitation.
An anti-HA antibody was premixed with protein G-agarose beads at a ratio of 1:20. The antibody-bead mixture was kept on a rotator for about 1 h at 4°C. The preloaded beads were then washed three times with 1× PBS at 3,000 rpm for 1 min each time. Equal amounts of each protein extract (as measured by the Bradford assay) were added to the protein G-agarose beads preloaded with the primary antibody. The samples were incubated/mixed overnight on a rotator at 4°C. After incubation, the beads were washed three times with 1× PBS at 3,000 rpm for 1 min each time. Finally, 2× loading buffer was added to the beads, and the mixtures were boiled at 100°C for 3 to 5 min. The samples were then subjected to Western blotting.
Cycloheximide chase assay.
293T cells were transfected with 1 μg of pCMV-HA-MASTL alone or with pCDNA-HA-AKT. The cells were cultured for 42 h, after which they were treated with cycloheximide (100 μg/ml) for 1, 2, 3, 4, or 5 h. The cells were then harvested and cell lysates prepared using 100 μl of lysis buffer (50 mM Tris chloride, 150 mM NaCl, 10% glycerol, 1% NP-40, and 2 mM EDTA) containing protease and phosphatase inhibitors. The lysates were then run on 8% SDS-PAGE gels and were subjected to Western blot analysis using an anti-HA antibody to detect HA-MASTL and HA-AKT bands.
Crystal violet staining.
Equal numbers of cells (1 × 105) were seeded in 6-well plates in duplicate. After 2 days, when the cell density reached 60%, shMASTL, shAKT1, and shAKT2 were induced with 2 μg/ml doxycycline for 24 h in one of the plates, while the other was kept as the 0-h control. After induction, the plates were taken and washed once with 1× PBS. Then the cells were fixed with methanol for 10 min and were washed again with 1× PBS. One milliliter of 0.1% crystal violet (0.1% crystal violet in 25% ethanol) was added per well and was left in the wells for about 10 min at room temperature. The dye was then aspirated from the plates, followed by washing with distilled water. The stained cells were allowed to dry at room temperature. For quantification, the dye attached to the cells was extracted with 2 ml of 10% acetic acid, and its absorbance was measured at 590 nm. The experiments were repeated three times.
MTT assay.
Equal numbers of cells (10,000) were plated in 12-well plates in duplicate. After a cell density of 60% was reached, shMASTL, shAKT1, and shAKT2 were induced with 2 μg/ml doxycycline for 24 h. Next, the medium of the cells was replaced with fresh medium, and 100 μl of 12 mM MTT stock (5 mg MTT in 1 ml of 1× PBS) was added to each well. After incubation of the cells with MTT for about 2 h in a CO2 incubator at 37°C, 1 ml of an SDS-HCl solution (5 g of SDS in 50 ml of 0.01 M HCl) was added to each well. The solutions were mixed thoroughly by pipetting and were incubated again at 37°C for about 4 h. Next, the plates were removed from the incubator, the solutions were again mixed by pipetting, and the absorbance was measured at 570 nm. The experiments were repeated three times.
FACS analysis.
For cell cycle analysis, equal numbers of cells were plated and were grown to 60 to 70% density. The cells were washed with 1× PBS (without Ca2+ and Mg2+). For the disaggregation of cells, 3 ml of 1× PBS supplemented with 0.1% EDTA was added to the plates, and cells were allowed to incubate for 5 min in a CO2 incubator. The cells were then dislodged from the plates by pipetting to obtain single-cell suspensions. Cells were collected in 15-ml tubes and were centrifuged at 1,000 × g for 5 min. Cell pellets were washed with 1× PBS containing 1% serum at 1,000 × g for 5 min. Finally, the cell pellets were resuspended in 0.5 ml of 1× PBS and were fixed by the dropwise addition of about 5 ml chilled ethanol with vortexing to prevent cell clumping. At this stage, the cells were stored at 4°C at least overnight in the dark. On the next day, centrifugation at 1,000 × g for 5 min was carried out, and the pellets were again washed once with 1× PBS containing 1% serum and were resuspended in 500 μl of a propidium iodide-RNase solution (50 μg/ml propidium iodide, 10 mM Tris [pH 7.5], 5 mM MgCl2, and 20 μg/ml RNase A). The resuspended mixtures were incubated at 37°C for 30 min to remove RNA molecules from the solution. The samples were then subjected to flow cytometric analysis using a FACSVerse flow cytometer and ModFit software.
Immunofluorescence.
HeLa, HeLa-AKT, U2OS, and U2OS-AKT cells were split and grown on coverslips overnight in regular culture medium (DMEM–10% fetal calf serum [FCS]–penicillin-streptomycin). Cells were subjected to a double thymidine block (using 2 mM thymidine) in order to obtain a synchronous population of cells. Then the cells were released from the thymidine block, and after 10 h, the medium was aspirated and the cells were washed with 1× PBS for 5 min. Next, the cells were fixed with 4% paraformaldehyde and then permeabilized with 0.2% Triton X-100. After a washing with 1× PBS, cells were treated with blocking buffer (2% bovine serum albumin [BSA] in PBS) for 1 h. After that, the cells were incubated with a phospho-histone 3 (Ser 10) antibody (1:100 in blocking buffer) overnight at 4°C. On the next day, the coverslips were washed twice with 1× PBS with Tween 20 (PBS-T) and once with 1× PBS for 5 min each time at room temperature. The cells were then incubated with a secondary antibody (Alexa Fluor 594) (dilution, 1:5,000) for 1 h at room temperature. Then the cells were washed twice with 1× PBS-T and once with 1× PBS for 5 min each time. The coverslips were fixed on glass slides over a DAPI-containing antifade mounting solution and were visualized using an inverted fluorescence microscope (Axio Observer 7) at ×20 magnification. Phospho-histone 3 staining was visualized at the red wavelength (594 nm), while nuclei were visualized at the blue wavelength (420 nm), at ×20 magnification. The mitotic index was calculated by counting the phospho-histone 3-stained cells (mitotic cells) and DAPI-stained nuclei (total number of cells) in a given field. The experiments were repeated three times.
ACKNOWLEDGMENTS
This study was supported by the Department of Biotechnology, Government of India, through grant BT/PR5743/BRB/10/1100/2012, a Ramalingaswami Fellowship, grant BT/PR2283/AGR/36/692/2011, and grant BT/PR13605/MED/30/1525/2015 to Shaida Andrabi, as well as by a DST-FIST grant to the Department of Biochemistry, University of Kashmir. A Junior Research Fellowship for Irfana Reshi from CSIR-UGC, India, is also gratefully acknowledged.
We declare that there is no conflict of interest.
REFERENCES
- 1.Voets E, Wolthuis R. 2010. MASTL is the human ortholog of Greatwall kinase that facilitates mitotic entry, anaphase and cytokinesis. Cell Cycle 9:3591–3601. doi: 10.4161/cc.9.17.12832. [DOI] [PubMed] [Google Scholar]
- 2.Marzec K, Burgess A. 2018. The oncogenic functions of MASTL kinase. Front Cell Dev Biol 6:162. doi: 10.3389/fcell.2018.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vigneron S, Robert P, Hached K, Sundermann L, Charrasse S, Labbe JC, Castro A, Lorca T. 2016. The master Greatwall kinase, a critical regulator of mitosis and meiosis. Int J Dev Biol 60:245–254. doi: 10.1387/ijdb.160155tl. [DOI] [PubMed] [Google Scholar]
- 4.Castro A, Lorca T. 2018. Greatwall kinase at a glance. J Cell Sci 131:jcs222364. doi: 10.1242/jcs.222364. [DOI] [PubMed] [Google Scholar]
- 5.Yu J, Zhao Y, Li Z, Galas S, Goldberg ML. 2006. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol Cell 22:83–91. doi: 10.1016/j.molcel.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 6.Mochida S, Hunt T. 2012. Protein phosphatases and their regulation in the control of mitosis. EMBO Rep 13:197–203. doi: 10.1038/embor.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seshacharyulu P, Pandey P, Datta K, Batra SK. 2013. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mochida S, Ikeo S, Gannon J, Hunt T. 2009. Regulated activity of PP2A-B55δ is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J 28:2777–2785. doi: 10.1038/emboj.2009.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gharbi-Ayachi A, Labbe JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, Lorca T. 2010. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330:1673–1677. doi: 10.1126/science.1197048. [DOI] [PubMed] [Google Scholar]
- 10.Mochida S, Maslen SL, Skehel M, Hunt T. 2010. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330:1670–1673. doi: 10.1126/science.1195689. [DOI] [PubMed] [Google Scholar]
- 11.Barr FA, Elliott PR, Gruneberg U. 2011. Protein phosphatases and the regulation of mitosis. J Cell Sci 124:2323–2334. doi: 10.1242/jcs.087106. [DOI] [PubMed] [Google Scholar]
- 12.Voets E, Wolthuis R. 2015. MASTL promotes cyclin B1 destruction by enforcing Cdc20-independent binding of cyclin B1 to the APC/C. Biol Open 4:484–495. doi: 10.1242/bio.201410793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manchado E, Guillamot M, de Cárcer G, Eguren M, Trickey M, García-Higuera I, Moreno S, Yamano H, Cañamero M, Malumbres M. 2010. Targeting mitotic exit leads to tumor regression in vivo: modulation by Cdk1, Mastl, and the PP2A/B55α, δ phosphatase. Cancer Cell 18:641–654. doi: 10.1016/j.ccr.2010.10.028. [DOI] [PubMed] [Google Scholar]
- 14.Rogers S, Fey D, McCloy RA, Parker BL, Mitchell NJ, Payne RJ, Daly RJ, James DE, Caldon CE, Watkins DN, Croucher DR, Burgess A. 2016. PP1 initiates the dephosphorylation of MASTL, triggering mitotic exit and bistability in human cells. J Cell Sci 129:1340–1354. doi: 10.1242/jcs.179754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hunt T. 2013. On the regulation of protein phosphatase 2A and its role in controlling entry into and exit from mitosis. Adv Biol Regul 53:173–178. doi: 10.1016/j.jbior.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Vigneron S, Brioudes E, Burgess A, Labbe JC, Lorca T, Castro A. 2009. Greatwall maintains mitosis through regulation of PP2A. EMBO J 28:2786–2793. doi: 10.1038/emboj.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Della Monica R, Visconti R, Cervone N, Serpico AF, Grieco D. 2015. Fcp1 phosphatase controls Greatwall kinase to promote PP2A-B55 activation and mitotic progression. Elife 4:e10399. doi: 10.7554/eLife.10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anania M, Gasparri F, Cetti E, Fraietta I, Todoerti K, Miranda C, Mazzoni M, Re C, Colombo R, Ukmar G, Camisasca S, Pagliardini S, Pierotti M, Neri A, Galvani A, Greco A. 2015. Identification of thyroid tumor cell vulnerabilities through a siRNA-based functional screening. Oncotarget 6:34629–34648. doi: 10.18632/oncotarget.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez-Fernandez M, Sanz-Flores M, Sanz-Castillo B, Salazar-Roa M, Partida D, Zapatero-Solana E, Ali HR, Manchado E, Lowe S, VanArsdale T, Shields D, Caldas C, Quintela-Fandino M, Malumbres M. 2018. Therapeutic relevance of the PP2A-B55 inhibitory kinase MASTL/Greatwall in breast cancer. Cell Death Differ 25:828–840. doi: 10.1038/s41418-017-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhuge BZ, Du BR, Meng XL, Zhang YQ. 2017. MASTL is a potential poor prognostic indicator in ER+ breast cancer. Eur Rev Med Pharmacol Sci 21:2413–2420. [PubMed] [Google Scholar]
- 21.Rogers S, McCloy RA, Parker BL, Gallego-Ortega D, Law AMK, Chin VT, Conway JRW, Fey D, Millar EKA, O’Toole S, Deng N, Swarbrick A, Chastain PD, Cesare AJ, Timpson P, Caldon CE, Croucher DR, James DE, Watkins DN, Burgess A. 2018. MASTL overexpression promotes chromosome instability and metastasis in breast cancer. Oncogene 37:4518–4533. doi: 10.1038/s41388-018-0295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon YN, Choe MH, Jung KY, Hwang SG, Oh JS, Kim JS. 2018. MASTL inhibition promotes mitotic catastrophe through PP2A activation to inhibit cancer growth and radioresistance in breast cancer cells. BMC Cancer 18:716. doi: 10.1186/s12885-018-4600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vera J, Lartigue L, Vigneron S, Gadea G, Gire V, Del Rio M, Soubeyran I, Chibon F, Lorca T, Castro A. 2015. Greatwall promotes cell transformation by hyperactivating AKT in human malignancies. Elife 4:e10115. doi: 10.7554/eLife.10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quaynor SD, Bosley ME, Duckworth CG, Porter KR, Kim SH, Kim HG, Chorich LP, Sullivan ME, Choi JH, Cameron RS, Layman LC. 2016. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Mol Cell Endocrinol 437:86–96. doi: 10.1016/j.mce.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Wong PY, Ma HT, Lee HJ, Poon RY. 2016. MASTL (Greatwall) regulates DNA damage responses by coordinating mitotic entry after checkpoint recovery and APC/C activation. Sci Rep 6:22230. doi: 10.1038/srep22230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vigneron S, Gharbi-Ayachi A, Raymond AA, Burgess A, Labbe JC, Labesse G, Monsarrat B, Lorca T, Castro A. 2011. Characterization of the mechanisms controlling Greatwall activity. Mol Cell Biol 31:2262–2275. doi: 10.1128/MCB.00753-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. 2011. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. 2005. The oncogenic properties of mutant p110α and p110β phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A 102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burgess A, Vigneron S, Brioudes E, Labbe JC, Lorca T, Castro A. 2010. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci U S A 107:12564–12569. doi: 10.1073/pnas.0914191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuck C, Zhang T, Potapova T, Malumbres M, Novak B. 2013. Robust mitotic entry is ensured by a latching switch. Biol Open 2:924–931. doi: 10.1242/bio.20135199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeong AL, Yang Y. 2013. PP2A function toward mitotic kinases and substrates during the cell cycle. BMB Rep 46:289–294. doi: 10.5483/bmbrep.2013.46.6.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, Cantley LC, Gygi SP, Rush J, Comb MJ. 2010. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal 3:ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manning BD, Cantley LC. 2007. AKT/PKB signaling: navigating downstream. Cell 129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chin YR, Toker A. 2009. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal 21:470–476. doi: 10.1016/j.cellsig.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo J, Manning BD, Cantley LC. 2003. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4:257–262. doi: 10.1016/s1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 37.Hoijman E, Rubbini D, Colombelli J, Alsina B. 2015. Mitotic cell rounding and epithelial thinning regulate lumen growth and shape. Nat Commun 6:7355. doi: 10.1038/ncomms8355. [DOI] [PubMed] [Google Scholar]
- 38.Stewart MP, Helenius J, Toyoda Y, Ramanathan SP, Muller DJ, Hyman AA. 2011. Hydrostatic pressure and the actomyosin cortex drive mitotic cell rounding. Nature 469:226–230. doi: 10.1038/nature09642. [DOI] [PubMed] [Google Scholar]
- 39.Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. 2004. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5:127–136. doi: 10.1016/s1535-6108(04)00026-1. [DOI] [PubMed] [Google Scholar]
- 40.Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC. 2003. Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3:483–495. doi: 10.1016/s1535-6108(03)00088-6. [DOI] [PubMed] [Google Scholar]
- 41.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. 2005. Cancer-associated PP2A Aα subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res 65:8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 42.Yu J, Fleming SL, Williams B, Williams EV, Li Z, Somma P, Rieder CL, Goldberg ML. 2004. Greatwall kinase: a nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. J Cell Biol 164:487–492. doi: 10.1083/jcb.200310059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, Zafiropoulos PJ, Yamaguchi S, Winter S, Carthew RW, Cooper M, Jones D, Frenz L, Glover DM. 2004. Genome-wide survey of protein kinases required for cell cycle progression. Nature 432:980–987. doi: 10.1038/nature03160. [DOI] [PubMed] [Google Scholar]
- 44.Blake-Hodek KA, Williams BC, Zhao Y, Castilho PV, Chen W, Mao Y, Yamamoto TM, Goldberg ML. 2012. Determinants for activation of the atypical AGC kinase Greatwall during M phase entry. Mol Cell Biol 32:1337–1353. doi: 10.1128/MCB.06525-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soeda S, Yamada-Nomoto K, Michiue T, Ohsugi M. 2018. RSK-MASTL pathway delays meiotic exit in mouse zygotes to ensure paternal chromosome stability. Dev Cell 47:363–376.e5. doi: 10.1016/j.devcel.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 46.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. 1996. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. 2002. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- 48.Shtivelman E, Sussman J, Stokoe D. 2002. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol 12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- 49.Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N. 2002. Activation of Akt/protein kinase B overcomes a G2/M cell cycle checkpoint induced by DNA damage. Mol Cell Biol 22:7831–7841. doi: 10.1128/mcb.22.22.7831-7841.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, Sellers WR. 1999. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang ED, Nunez G, Barr FG, Guan KL. 1999. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem 274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 52.Richards SA, Dreisbach VC, Murphy LO, Blenis J. 2001. Characterization of regulatory events associated with membrane targeting of p90 ribosomal S6 kinase 1. Mol Cell Biol 21:7470–7480. doi: 10.1128/MCB.21.21.7470-7480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P, Kim SY, Dunn IF, Schinzel AC, Sandy P, Hoersch S, Sheng Q, Gupta PB, Boehm JS, Reiling JH, Silver S, Lu Y, Stemke-Hale K, Dutta B, Joy C, Sahin AA, Gonzalez-Angulo AM, Lluch A, Rameh LE, Jacks T, Root DE, Lander ES, Mills GB, Hahn WC, Sellers WR, Garraway LA. 2009. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]