

Abstract
Purpose
This work is aimed at identifying several molecular markers correlated with the diagnosis and development of basal cell carcinoma (BCC).
Methods
The available microarray datasets for BCC were obtained from the Gene Expression Omnibus (GEO) database, and differentially expressed genes (DEGs) were identified between BCC and healthy controls. Afterward, the functional enrichment analysis and protein-protein interaction (PPI) network analysis of these screened DEGs were performed. An external validation for the DEG expression level was also carried out, and receiver operating characteristic curve analysis was used to evaluate the diagnostic values of DEGs.
Result
In total, five microarray datasets for BCC were downloaded and 804 DEGs (414 upregulated and 390 downregulated genes) were identified. Functional enrichment analysis showed that these genes including CYFIP2, HOXB5, EGFR, FOXN3, PTPN3, CDC20, MARCKSL1, FAS, and PTCH1 were closely correlated with the cell process and PTCH1 played central roles in the BCC signaling pathway. Moreover, EGFR was a hub gene in the PPI network. The expression changes of six genes (CYFIP2, HOXB5, FOXN3, PTPN3, MARCKSL1, and FAS) were validated by an external GSE74858 dataset analysis. Finally, ROC analysis revealed that CYFIP2, HOXB5, PTPN3, MARCKSL1, PTCH1, and CDC20 could distinguish BCC and healthy individuals.
Conclusion
Nine gene signatures (CYFIP2, HOXB5, EGFR, FOXN3, PTPN3, CDC20, MARCKSL1, FAS, and PTCH1) may serve as promising targets for BCC detection and development.
1. Introduction
Basal cell carcinoma (BCC) is one of common malignant epithelial neoplasms that derive from basal cells and accounts for nearly 75% of skin cancers [1]. It was reported that the BCC incidence rates have been increasing and roughly reached 2.75 million cases around the world [2]. BCC was generally categorized into several types such as superficial, nodular, and nevoid BCC on the basis of its histological and clinical characteristics. Notably, the majority of lesions for BCC occurred on the head and neck [3]. The convincing evidence has suggested that gender, age, sunlight exposure (especially ultraviolet radiation), smoking, chemicals, and fair skin were all BCC risk factors [4, 5]. Although BCC recurrence was extremely frequent due to local tissue destruction, it rarely metastasizes or leads to death [6]. The imiquimod application, surgical excision, and radiation therapy have been unsatisfactory for BCC treatment. Therefore, it is imperative to develop promising strategies for early diagnosis and intervention of BCC.
Fortunately, the next-generation high-throughput sequencing provides fascinating opportunities for bioinformatics analysis. A growing number of studies have demonstrated that gene expression profiling could effectively screen molecular makers and predict pathogenesis for tumorigenesis in the last decades, thereby offering novel therapeutic strategies for cancer treatment [7]. Fang et al. previously found that the expression level of EGR1 (early growth response 1) was decreased in BCC based on a microarray analysis. Furthermore, they also noted that EGR1 might suppress epidermal proliferation via modulating CDC25A that is critical for cell cycle progression from G1 to S phase [8]. Heller et al. conducted a global gene expression analysis and identified BCC-associated differentially expressed gene (DEG) targets, providing several new diagnostic signatures for the susceptibility of BCC [9]. Later, Yunoki et al. identified three BCC-correlated genes including BCL2 (B-cell lymphoma 2), PTCH1 (Patched 1), and SOX9 (SRY-box 9) and unique gene networks involved with tumorigenesis through a microarray analysis [10]. Additionally, Jee et al. classified BCC into squamous cell carcinoma- (SCC-) like BCC, normal-like BCC, and classical BCC subtypes by a comparative analysis of gene expression profiles from BCC, SCC, and normal skin tissues. Moreover, functional analysis revealed that hedgehog signaling pathways were possibly related to classical BCC development [11]. Although numerous investigations have extracted several BCC-related gene makers, a deeper insight into the underlying molecular mechanisms of BCC has not been completely achieved.
In this study, the eligible mRNA microarray datasets from BCC tissues were retrieved and downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database. Then, the DEGs between BCC and normal tissues were screened followed by functional enrichment analyses. The protein-protein (PPI) network was also constructed to explore underlying interactions among key genes. Finally, the expression levels of DEGs were further examined by an external dataset, and diagnostic performance of these genes were also evaluated. This work provided novel gene signatures for BCC diagnosis and promoted a deeper understanding for BCC progression.
2. Materials and Methods
2.1. Data Acquisition
The publicly accessible microarray datasets in BCC were searched and downloaded from the NCBI-GEO [12] database (https://www.ncbi.nlm.nih.gov/geo/). The keyword terms of carcinoma, basal cell (MeSH Terms) OR basal cell carcinoma (All Fields) AND “Homo sapiens” (porgn) AND “gse” (Filter) were used for precise searching. The inclusive criteria for datasets were as follows: (i) the datasets were gene expression profiles, (ii) the data was obtained from tumor tissues of patients with BCC or normal skin tissues from healthy individuals (normal controls), and (iii) the patients did not receive medication or other treatments. Finally, five datasets (GSE103439, GSE53462, GSE42109, GSE39612, and GSE7553) were chosen according to the abovementioned screening criteria as shown in Table 1. Of these, four datasets (GSE103439, GSE42109, GSE39612, and GSE7553) were generated by the GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array platform, and the platform for GSE53462 was GPL10558 Illumina Human HT-12 V4.0 expression beadchip. These datasets would be used for the following integrated analyses, which consisted of 48 BCC tissues and 85 normal tissue samples.
Table 1.
The microarray datasets for basal cell carcinoma.
| GEO accession | Control | Case | Platform | Year | Country | Author |
|---|---|---|---|---|---|---|
| GSE103439 | 2 | 4 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 2017 | Japan | Yoshiaki Tabuchi |
| GSE53462 | 5 | 16 | GPL10558 Illumina Human HT-12 V4.0 expression beadchip | 2014 | South Korea | Hyun Goo Woo |
| GSE42109 | 10 | 11 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array; GPL571 [HG-U133A_2] Affymetrix Human Genome U133A 2.0 Array | 2013 | USA | Mayte Suarez-Farinas |
| GSE39612 | 64 | 2 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 2012 | USA | Paul Harms |
| GSE7553 | 4 | 15 | GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array | 2008 | USA | Sean Yoder |
GEO: Gene Expression Omnibus.
2.2. Identification of Differentially Expressed Genes (DEGs)
A meta-analysis of five microarray datasets from different platforms was carried out according to the guidelines provided by Ramasamy et al. [13]. The preprocessing of raw data across different platforms, mainly including background correction, normalization, log2 transformation, and gene expression calculation, was performed. The overlapped genes in five datasets were selected. Then, the R metaMA package was used to calculate effect sizes from unpaired data by either classical or moderated t-tests (Limma) for each study and combine these effect sizes [14]. The DEGs between BCC and normal controls were screened according to the threshold of the multiple comparison correction false discovery rate (FDR) < 0.05. The whole R sentences for data processing and identification of DEGs in this study were shown in Supplementary Materials (available here). Finally, the hierarchical clustering analysis of identified DEGs was performed by the pheatmap package (https://cran.r-project.org/package=pheatmap) in R language.
2.3. Gene Ontology (GO) and Pathway Enrichment Analyses
To explore the possible biological roles of the DEGs, the functional analyses were conducted. Firstly, the GO analysis involving three categories of molecular function (MF), cellular component (CC), and biological process (BP) was conducted with BiNGO plugin in cytoscape software [15]. The parameters were set as follows: (i) hypergeometric test was used as the selected statistical test; (ii) Benjamini and Hochberg FDR correction was set as selected correction; (iii) selected significance level was 0.05; and (iv) for testing option, we used whole annotation as the reference set. In addition, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was undertaken using KOBAS (http://kobas.cbi.pku.edu.cn/index.php), which is a web-accessible tool and could identify enriched pathways for an input set of genes by mapping to genes using known pathways in the KEGG database [16]. Moreover, the statistically significantly enriched pathways were selected with the cutoff of P value < 0.05.
2.4. Protein-Protein Interaction (PPI) Network
The Biological General Repository for Interaction Datasets (BioGRID) database is available online and provides 1,728,498 biological PPI interactions by August 2019. Here, we performed a PPI analysis for screened DEGs based on this database to identify significant protein pairs. The cytoscape software (http://apps.cytoscape.org/apps/cytonca) was utilized to construct the PPI network. Moreover, the CytoNCA [17] (http://apps.cytoscape.org/apps/cytonca), a cytoscape plugin, was used to analyze topological characteristics of PPI nodes using the without weight parameter. Several metrics such as Degree Centrality (DC), Betweenness centrality (BC), and Closeness centrality (CC) were estimated. In this study, hub protein was determined on the basis of high scores. In addition, the MCODE plugin (http://apps.cytoscape.org/apps/mcode) in cytoscape software was employed to further identify several PPI submodules using the default parameters of degree cutoff = 2, node score cutoff = 0.2, K‐core = 2, and max.depth = 100 [18]. Notably, the PPI score > 3 was considered as the cutoff for screening significantly enriched functional modules from the PPI network.
2.5. Validation of DEG Expression Levels by a Noncoding RNA Expression Profile
The GSE74858 dataset, including 3 BCC patients and 3 healthy individuals, was obtained from the GEO database and used as a verification dataset to evaluate the expression levels of DEGs. Furthermore, the receiver operating characteristic (ROC) analysis was also carried out to assess the performance of DEGs in this research using the “pROC” package (https://cran.r-project.org/web/packages/pROC/index.html) in R language. The area under the ROC curve (AUC) was then computed. Herein, if AUC values of the genes were greater than 0.9, these genes were considered to discriminate patients undergoing BCC from healthy controls with high specificity and sensitivity.
3. Results
3.1. DEG Screening
The meta-analysis of five gene expression profiles was conducted, and the DEGs were identified. Consequently, a total of 804 DEGs were extracted between BCC and normal controls according to screening criteria described in Materials and Methods. Of these, there were 414 upregulated genes and 390 downregulated genes in patients with BCC. Moreover, the clustering analysis of these DEGs indicated that they could clearly distinguish BCC and healthy controls as exhibited in Figure 1, suggesting that the differential gene expression was possibly responsible for BCC occurrence.
Figure 1.

The heatmap of differentially expressed genes in basal cell carcinoma. The “label” represents sample type (basal cell carcinoma and normal skin samples). The light red color shows the basal cell carcinoma samples, and light blue color shows normal skin samples.
3.2. Functional Enrichment Analyses of DEGs
As shown in Table 2, the GO functional annotation analysis showed that identified DEGs were enriched in 53 GO-BP terms including cellular component organization, cell cycle, cell proliferation, and ectoderm development. Meanwhile, they were focused on 54 GO-CC terms such as intracellular part, intracellular, and cytoplasm. It was also observed that there were three GO-MF terms (protein binding, binding, and ribonuclease H activity). Notably, numerous genes were closely associated with the cellular process, including CYFIP2 (cytoplasmic FMR1 interacting protein 2; upregulated), HOXB5 (homeobox B5; downregulated), EGFR (epidermal growth factor receptor; downregulated), FOXN3 (forkhead box N3; upregulated), PTPN3 (protein tyrosine phosphatase nonreceptor type 3; downregulated), CDC20 (cell division cycle 20; upregulated), MARCKSL1 (myristoylated alanine rich protein kinase C substrate like 1; upregulated), FAS (fas cell surface death receptor; downregulated), and upregulated PTCH1. More specifically, FOXN3, CDC20, and EGFR played essential roles in the cell cycle process, and PTCH1 and EGFR were involved in cell proliferation. Additionally, KEGG pathway enrichment analysis revealed that these genes were primarily enriched in pathways in cancer, p53 signaling pathway and BCC pathway. We also noted that PTCH1 exerted critical biological roles in the BCC pathway (Figure 2).
Table 2.
The functional analyses of differentially expressed genes in BCC.
| Category | Term | Count | P value | Genes |
|---|---|---|---|---|
| GOTERM_BP (top 5) | Cellular component organization | 183 | 3.92E − 10 | RNH1, KDM1A, ZWILCH, DSCC1, HDAC11, INPPL1, TESK2, BUB1B, UBE3A, FNBP1L,… |
| Cellular process | 507 | 1.22E − 09 | CYFIP2, RPL7, HOXA5, HOXB5, EGFR, FOXN3, PTPN3, CDC20, FAS, PTCH1, IDH2,… | |
| Cell cycle phase | 49 | 8.98E − 09 | ZWILCH, CDC20, EGFR, FOXN3, HSPA2, NDC80, ZWINT, TPX2, CENPF, KIF18A, MC1R,… | |
| M phase | 42 | 1.86E − 08 | CDCA3, ZWILCH, DSCC1, CDCA8, BUB1B, TTK, MKI67, NCAPH, CDC20, PSMC3IP,… | |
| Cell proliferation | 48 | 2.47E − 08 | ACHE, CSE1L, EGFR, PTCH1, WNT5A, EMP1, SOX11, CBFA2T3, EREG, TPX2,… | |
|
| ||||
| GOTERM_CC (top 5) | Intracellular part | 552 | 1.83E − 12 | CYFIP2, PTPN3, EGFR, CDC20, FAS, FOXN3, NUP107, RNH1, TESK2, NOC2L,… |
| Intracellular | 560 | 5.23E − 11 | CYFIP2, PTPN3, EGFR, CDC20, FAS, FOXN3, SPI1, NUP107, RNH1, TESK2,… | |
| Cytoplasm | 403 | 4.48E − 09 | CYFIP2, PTPN3, EGFR, CDC20, FAS, RNH1, TESK2, FNBP1L, RPL7, AKT1,… | |
| Intracellular organelle | 473 | 4.91E − 09 | PTPN3, EGFR, CDC20, FAS, FOXN3, SPI1, NUP107, RNH1, TESK2, NOC2L,… | |
| Spindle | 26 | 5.26E − 09 | CDCA8, BUB1B, TTK, CDC14A, CDC20, CCNB1, RACGAP1, HAUS7, MYC, AKT1,… | |
|
| ||||
| GOTERM_MF | Protein binding | 448 | 2.09E − 12 | CYFIP2, EGFR, FOXN3, PTPN3, CDC20,FAS, PTCH1, MARCKSL1, CNTFR, SPI1,… |
| Binding | 603 | 7.51E − 09 | CYFIP2, EGFR, FOXN3, PTPN3, CDC20, FAS, PTCH1, MARCKSL1, HOXB5, CNTFR,… | |
| Ribonuclease H activity | 4 | 1.21E − 04 | FEN1, RNASEH2A, RNH1, EXO1 | |
GO: Gene Ontology; BP: biological process; MF: molecular function; CC: cellular component; BCC: basal cell carcinoma.
Figure 2.
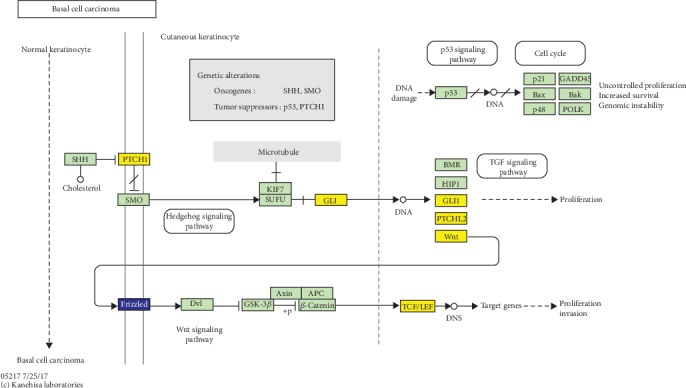
Basal cell carcinoma pathway from the Kyoto Encyclopedia of Genes and Genomes enrichment analysis. The yellow color shows the upregulated genes while blue color represents the downregulated genes.
3.3. PPI Analysis
PPI network analysis was conducted to explore the underlying interactions of DEGs. In total, 549 gene nodes (296 upregulated genes and 254 downregulated genes) and 1462 protein pairs were obtained in the PPI network (Figure 3(a)). The top 5 gene nodes with a high degree were EGLN3 (Egl-9 family hypoxia inducible factor 3; degree = 82), XPO1 (exportin 1; degree = 81), EGFR (degree = 77), CFTR (CF transmembrane conductance regulator; degree = 48), and MCM2 (minichromosome maintenance complex component 2; degree = 47), and they were regarded as hub genes. Furthermore, the PPI submodule analysis suggested that 3 significantly enriched submodules were identified based on the PPI score > 3 (Figures 3(b)–3(d)). Interestingly, we found that the genes in submodule 1 were all upregulated.
Figure 3.
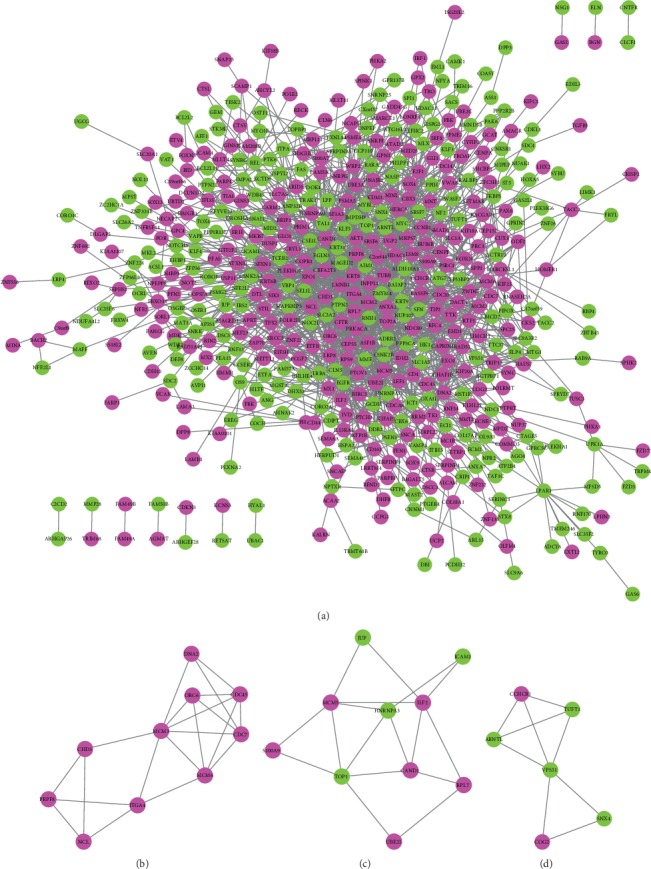
The protein-protein interaction (PPI) network analysis: (a) PPI network of differentially expressed genes: (b) submodule 1; (c) submodule 2; (d) submodule 3. The submodules in the PPI network were extracted by MCODE based on PPI score > 3. The rosy red circular nodes denote upregulated genes. The green circular nodes represent significantly downregulated genes. The node size represents degree.
3.4. Verification of DEG Expression Levels and ROC Analysis
A noncoding RNA profile GSE74858 was downloaded from the GEO repository, and there were 145 overlapped genes between this dataset and the integrated datasets. We found that 25 overlapped genes exhibited significant differential expression, and their expression trends were also consistent with our integration analysis. More notably, six genes (CYFIP2, HOXB5, FOXN3, PTPN3, MARCKSL1, and FAS) were predominately correlated with the cellular biological process, and their expression patterns were validated by an external GSE74858 dataset (Figure 4). Besides, the ROC curve analysis was conducted to assess the diagnostic values of DEGs in BCC by five microarray datasets. There were 439 genes according to AUC > 0.9, and 129 genes were overlapped with DEGs by integrated analysis and then extracted. Our findings revealed that CYFIP2 (AUC = 0.949), HOXB5 (AUC = 0.908), PTPN3 (AUC = 0.952), MARCKSL1 (AUC = 0.962), PTCH1 (AUC = 0.981), and CDC20 (AUC = 0.956) could significantly distinguish BCC samples and healthy controls, implying that these genes might serve as diagnostic makers for BCC detection (Figure 5).
Figure 4.

Validation of the expression levels of six differentially expressed genes in T basal cell carcinoma based on GSE74858 dataset. The x-axis represents basal cell carcinoma and normal controls. The y-axis represents the expression read counts.
Figure 5.

The receiver operating characteristic (ROC) curve analyzed the diagnostic value of six differentially expressed genes in basal cell carcinoma. The ROC curves were used to evaluate the diagnostic value of these six differentially expressed genes in basal cell carcinoma with sensitivity and 1-specificity. The x-axis shows 1-specificity, and the y-axis shows sensitivity. AUC: area under the ROC curve.
4. Discussion
In the present study, we performed an integrated bioinformatics analysis using five microarray datasets about BCC. In total, 804 DEGs (414 upregulated and 390 downregulated genes) were screened between BCC and normal control tissues. The GO functional analysis showed that several genes, including CYFIP2, HOXB5, EGFR, FOXN3, PTPN3, CDC20, MARCKSL1, FAS, and PTCH1, primarily played pivotal roles in cellular processes. Moreover, the expression patterns of six genes (CYFIP2, HOXB5, FOXN3, PTPN3, MARCKSL1, and FAS) were validated in an external noncoding RNA dataset. Additionally, CYFIP2, HOXB5, PTPN3, MARCKSL, CDC20, and PTCH1 had superior diagnostic values for BCC prediction.
Overwhelming evidence has suggested that the patched gene (PTCH) family played vital roles in BCC induction and progression [19, 20]. A previous study highlighted that PTCH could encode a receptor for the hedgehog signaling pathway which was important for vertebrate development and tumorigenesis [21]. Undén et al. found that the expression level of PTCH mRNA was overexpressed in BCC cells compared with nontumor epidermal cells, which was similar to our finding that PTCH1 was upregulated in BCC patients [22]. Interestingly, numerous researchers pointed out that approximately 90% of function mutations in PTCH1 were strongly related to BCC initiation and progression [23]. Gianferante et al. evaluated 18 nevoid BCC syndrome National Cancer Institute families by whole exome sequencing and found that 89% of families exhibited a pathogenic PTCH1 mutation [24]. A recent investigation has reported that PTCH1 had two mutational statuses (germinal and somatic mutation). Moreover, there was a higher expression level of PTCH1 in BCC with germinal and somatic PTCH1 mutations than that only with germinal PTCH1 mutation [25]. Additionally, our functional analyses showed that PTCH1 played significant roles in cell proliferation and BCC pathway. However, few reports investigated the detailed molecular mechanisms of the biological roles of PTCH1 on BCC cell proliferation and growth. Herein, we performed a ROC curve analysis of PTCH1, and the result indicated that this gene was capable of discriminating BCC patients and healthy controls with a relatively high AUC value (AUC = 0.981). Taken together, our findings suggested that PTCH1 might be involved in the pathogenesis of BCC and has a great diagnostic value for BCC detection.
We found that four genes (upregulated CYFIP2 and MARCKSL1 and downregulated HOXB5 and PTPN3) were also key players in cell biological processes. Furthermore, their expression patterns were validated by the differential expression analysis based on an external dataset. Besides, these four genes could clearly differentiate BCC patients from normal controls. CYFIP2 was reported to be a direct p53 target gene, and its overexpression was closely linked with the development of various cancers [26, 27]. Ling et al. analyzed p53 mutations in sporadic and hereditary BCC tumors, and genetic alterations of p53 were responsible for BCC progression [28]. Later on, Huang et al. stated that p53 activation by imiquimod contributed to cell apoptosis of a skin BCC/KMC1 cell line [29]. These findings provided indirect evidence for the hypothesis that CYFIP2 participated in the pathogenic mechanism of BCC. HOXB5 is a member of the homeobox (HOX) gene B cluster and plays key roles in several cancers [30, 31]. Lee et al. suggested that HOXB5 increased cell proliferation and invasiveness in estrogen receptor- (ER-) positive breast cancer [32]. More interestingly, this research group also found that HOXB5 could upregulate EGFR expression and thereby promote HOXB5-driven invasion in ER-positive breast cancer [33]. In this work, we noted that EGFR was involved in cell cycle and proliferation and served as a pivotal hub gene in PPI analysis. Whether the HOXB5/EGFR axis participated in BCC development remains to be illuminated in the following analysis. PTPN3 (also known as PTPH1) was also reported to be strongly correlated with diverse cancers including colorectal cancer and gastric adenocarcinoma [34–36]. Furthermore, Li et al. unraveled that the PTPN3 depletion could block the degradation of EGFR, which accelerated cell proliferation and tumorigenicity in lung cancer cells [37]. The potential roles of the PTPN3/EGFR axis on BCC occurrence also need to be clarified in the future. MARCKSL1 is a cytoskeletal regulator and implicated with the initiation of multiple cancers [38]. Our results implied that MARCKSL1 might be a promising diagnostic maker for BCC prediction. However, the underlying pathogenic mechanisms of the four-gene signature (CYFIP2, MARCKSL1, HOXB5, and PTPN3) for BCC progression have not been expatiated.
The FAS gene contains a highly conserved cytoplasmic death domain and encodes a transmembrane protein of the tumor necrosis factor receptor superfamily. Existing evidence has shown that FAS could bind to a death ligand, FAS ligand (FASL), to induce the cell apoptotic pathway [39]. The early research indicated that FAS normally was underexpressed and even undetectable in BCC while there was a high FASL level, possibly inducing cell death and contributing to cancerization [40]. A later study examined the expression level of FAS/FASL in BCCs using the immunohistochemical method. The results implied that FASL was located on the cell membrane of keratinocytes at the basal cell layers and FAS/FASL was markedly decreased in BCC [41]. Wang et al. revealed that FAS/FASL mRNA expression and protein levels were reduced in the BCC compared to the normal skin samples and FASL immunostaining levels were strongly related to the ability of tumor invasiveness and metastasis [42]. Similarly, we found that there was a lower FAS expression in BCC tissues than healthy controls, which was also confirmed by a bioinformatics analysis with another BCC dataset. Therefore, we speculated that FAS might be a key gene driver in BCC development. The other two genes' (CDC20 and FOXN3) levels were increased in BCC tissues compared to normal tissues, and functional analysis showed that they were mainly involved in the cell cycle process. We also observed that CDC20 exhibited a good performance for BCC diagnosis based on a ROC analysis. Therefore, we inferred that the aberrant expression of CDC20 and FOXN3 was probably associated with BCC development. However, the influence of these two genes on BCC pathogenesis has not been fully investigated.
There are still limitations in our analysis. Firstly, only one external noncoding RNA dataset GSE74858 was obtained and used to evaluate expression levels of identified DEGs due to lack of gene expression profile of BCC. Notably, the expression patterns of six genes (CYFIP2, HOXB5, FOXN3, PTPN3, MARCKSL1, and FAS) were consistent with our initial differential expression analysis in the training dataset. However, the expression levels of other key genes still need to be further evaluated by using a larger sample size. Secondly, the corresponding experimental assays such as cell and animal experiments were not performed to validate our conclusion primarily due to lack of enough patients' samples and limited research funding. Thirdly, the underlying molecular mechanism between several signaling pathways involving key genes and BCC development remains to be deciphered. Fourthly, the relevant clinical characteristics are also required to be collected to assess BCC prognosis.
In summary, nine gene signatures (CYFIP2, HOXB5, EGFR, FOXN3, PTPN3, CDC20, MARCKSL1, FAS, and PTCH1) may play central roles in the initiation and progression of BCC, which provides deeper insights into BCC management. However, experimental verification and integrated bioinformatics analyses still need to be carried out in the future.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Supplementary Materials
The whole R sentences for data processing and identification of differentially expressed genes in this study.
References
- 1.Ballester-Sánchez R., Pons-Llanas O., Candela-Juan C., et al. Efficacy and safety of electronic brachytherapy for superficial and nodular basal cell carcinoma. Journal of Contemporary Brachytherapy. 2015;3(3):231–238. doi: 10.5114/jcb.2015.52140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verkouteren J. A. C., Ramdas K. H. R., Wakkee M., Nijsten T. Epidemiology of basal cell carcinoma: scholarly review. The British Journal of Dermatology. 2017;177(2):359–372. doi: 10.1111/bjd.15321. [DOI] [PubMed] [Google Scholar]
- 3.Rubin A. I., Chen E. H., Ratner D. Basal-cell carcinoma. The New England Journal of Medicine. 2005;353(21):2262–2269. doi: 10.1056/NEJMra044151. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y., Liu J. The prognostic roles of cyclooxygenase-2 for patients with basal cell carcinoma. Artificial Cells, Nanomedicine, and Biotechnology. 2019;47(1):3053–3057. doi: 10.1080/21691401.2019.1643731. [DOI] [PubMed] [Google Scholar]
- 5.Flohil S. C., Seubring I., van Rossum M. M., Coebergh J. W. W., de Vries E., Nijsten T. Trends in basal cell carcinoma incidence rates: a 37-year Dutch observational study. The Journal of Investigative Dermatology. 2013;133(4):913–918. doi: 10.1038/jid.2012.431. [DOI] [PubMed] [Google Scholar]
- 6.Tanese K. Diagnosis and management of basal cell carcinoma. Current Treatment Options in Oncology. 2019;20(2):p. 13. doi: 10.1007/s11864-019-0610-0. [DOI] [PubMed] [Google Scholar]
- 7.Wadlow R., Ramaswamy S. DNA microarrays in clinical Cancer Research. Current Molecular Medicine. 2005;5(1):111–120. doi: 10.2174/1566524053152834. [DOI] [PubMed] [Google Scholar]
- 8.Fang M., Wee S. A., Ronski K., Fan H., Tao S., Lin Q. Evidence of EGR1 as a differentially expressed gene among proliferative skin diseases. Genomic Medicine. 2007;1(1-2):75–85. doi: 10.1007/s11568-007-9010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heller E. R., Gor A., Wang D., et al. Molecular signatures of basal cell carcinoma susceptibility and pathogenesis: a genomic approach. International Journal of Oncology. 2013;42(2):583–596. doi: 10.3892/ijo.2012.1725. [DOI] [PubMed] [Google Scholar]
- 10.Yunoki T., Tabuchi Y., Hirano T., Miwa S., Imura J., Hayashi A. Gene networks in basal cell carcinoma of the eyelid, analyzed using gene expression profiling. Oncology Letters. 2018;16(5):6729–6734. doi: 10.3892/ol.2018.9484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jee B. A., Lim H., Kwon S. M., et al. Molecular classification of basal cell carcinoma of skin by gene expression profiling. Molecular Carcinogenesis. 2015;54(12):1605–1612. doi: 10.1002/mc.22233. [DOI] [PubMed] [Google Scholar]
- 12.Barrett T., Troup D. B., Wilhite S. E., et al. NCBI GEO: mining tens of millions of expression profiles--database and tools update. Nucleic Acids Research. 2007;35(Database):D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramasamy A., Mondry A., Holmes C. C., Altman D. G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Medicine. 2008;5(9, article e184) doi: 10.1371/journal.pmed.0050184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marot G., Foulley J. L., Mayer C. D., Jaffrezic F. Moderated effect size and P-value combinations for microarray meta-analyses. Bioinformatics. 2009;25(20):2692–2699. doi: 10.1093/bioinformatics/btp444. [DOI] [PubMed] [Google Scholar]
- 15.Shannon P., Markiel A., Ozier O., et al. cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xie C., Mao X., Huang J., et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Research. 2011;39(Supplement_2):W316–W322. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang Y., Li M., Wang J., Pan Y., Wu F. X. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Bio Systems. 2015;127:67–72. doi: 10.1016/j.biosystems.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z. K., Zhang R. Y., Yong Y. L., et al. Identification of crucial genes based on expression profiles of hepatocellular carcinomas by bioinformatics analysis. PeerJ. 2019;7, article e7436 doi: 10.7717/peerj.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan H., Oro A. E., Scott M. P., Khavari P. A. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nature Medicine. 1997;3(7):788–792. doi: 10.1038/nm0797-788. [DOI] [PubMed] [Google Scholar]
- 20.Lacour J. P. Carcinogenesis of basal cell carcinomas: genetics and molecular mechanisms. The British Journal of Dermatology. 2002;146(Supplement 61):17–19. doi: 10.1046/j.1365-2133.146.s61.5.x. [DOI] [PubMed] [Google Scholar]
- 21.Stone D. M., Hynes M., Armanini M., et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384(6605):129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 22.Undén A. B., Zaphiropoulos P. G., Bruce K., Toftgård R., Ståhle-Bäckdahl M. Human patched (PTCH) mRNA is overexpressed consistently in tumor cells of both familial and sporadic basal cell carcinoma. Cancer Research. 1997;57(12):2336–2340. [PubMed] [Google Scholar]
- 23.Bresler S. C., Padwa B. L., Granter S. R. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Head and Neck Pathology. 2016;10(2):119–124. doi: 10.1007/s12105-016-0706-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gianferante D. M., Rotunno M., Dean M., et al. Whole-exome sequencing of nevoid basal cell carcinoma syndrome families and review of Human Gene Mutation Database PTCH1 mutation data. Molecular Genetics & Genomic Medicine. 2018;6(6):1168–1180. doi: 10.1002/mgg3.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez M. F., Romano M., Martinez A., et al. Nevoid basal cell carcinoma syndrome: PTCH1 mutation profile and expression of genes involved in the hedgehog pathway in Argentinian patients. Cells. 2019;8(2):p. 144. doi: 10.3390/cells8020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saller E., Tom E., Brunori M., et al. Increased apoptosis induction by 121F mutant p53. The EMBO Journal. 1999;18(16):4424–4437. doi: 10.1093/emboj/18.16.4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jackson R. S., II, Cho Y.-J., Stein S., Liang P. CYFIP2, a direct p53 target, is leptomycin-B sensitive. Cell Cycle. 2007;6(1):95–103. doi: 10.4161/cc.6.1.3665. [DOI] [PubMed] [Google Scholar]
- 28.Ling G., Ahmadian A., Persson Å., et al. PATCHED and p53 gene alterations in sporadic and hereditary basal cell cancer. Oncogene. 2001;20(53):7770–7778. doi: 10.1038/sj.onc.1204946. [DOI] [PubMed] [Google Scholar]
- 29.Huang S. W., Chang S. H., Mu S. W., et al. Imiquimod activates p53-dependent apoptosis in a human basal cell carcinoma cell line. Journal of Dermatological Science. 2016;81(3):182–191. doi: 10.1016/j.jdermsci.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Zhang B., Li N., Zhang H. Knockdown of homeobox B5 (HOXB5) inhibits cell proliferation, migration, and invasion in non-small cell lung cancer cells through inactivation of the Wnt/β-catenin pathway. Oncology Research. 2018;26(1):37–44. doi: 10.3727/096504017X14900530835262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu N., Wu Y. P., Yin H. B., Xue X. Y., Gou X. Molecular network-based identification of competing endogenous RNAs and mRNA signatures that predict survival in prostate cancer. Journal of Translational Medicine. 2018;16(1):p. 274. doi: 10.1186/s12967-018-1637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee J. Y., Hur H., Yun H. J., et al. HOXB5 promotes the proliferation and invasion of breast cancer cells. International Journal of Biological Sciences. 2015;11(6):701–711. doi: 10.7150/ijbs.11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee J. Y., Kim J. M., Jeong D. S., Kim M. H. Transcriptional activation of EGFR by HOXB5 and its role in breast cancer cell invasion. Biochemical and Biophysical Research Communications. 2018;503(4):2924–2930. doi: 10.1016/j.bbrc.2018.08.071. [DOI] [PubMed] [Google Scholar]
- 34.Han S., Williams S., Mustelin T. Cytoskeletal protein tyrosine phosphatase PTPH1 reduces T cell antigen receptor signaling. European Journal of Immunology. 2000;30(5):1318–1325. doi: 10.1002/(SICI)1521-4141(200005)30:5<1318::AID-IMMU1318>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 35.Wu C. W., Chen J. H., Kao H. L., et al. PTPN3 and PTPN4 tyrosine phosphatase expression in human gastric adenocarcinoma. Anticancer Research. 2006;26(2B):1643–1649. [PubMed] [Google Scholar]
- 36.Wang Z., Shen D., Parsons D. W., et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304(5674):1164–1166. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 37.Li M. Y., Lai P. L., Chou Y. T., et al. Protein tyrosine phosphatase PTPN3 inhibits lung cancer cell proliferation and migration by promoting EGFR endocytic degradation. Oncogene. 2015;34(29):3791–3803. doi: 10.1038/onc.2014.312. [DOI] [PubMed] [Google Scholar]
- 38.Chen Z., Liu Y., Yao L., Guo S., Gao Y., Zhu P. The long noncoding RNA lncZic2 drives the self-renewal of liver tumor-initiating cells via the protein kinase C substrates MARCKS and MARCKSL1. The Journal of Biological Chemistry. 2018;293(21):7982–7992. doi: 10.1074/jbc.RA117.001321. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Dicker T., Siller G., Saunders N. Molecular and cellular biology of basal cell carcinoma. The Australasian Journal of Dermatology. 2002;43(4):241–246. doi: 10.1046/j.1440-0960.2002.00609.x. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez-Steil C., Wrone-Smith T., Sun X., Krueger J. G., Coven T., Nickoloff B. J. Sunlight-induced basal cell carcinoma tumor cells and ultraviolet-B-irradiated psoriatic plaques express Fas ligand (CD95L) The Journal of Clinical Investigation. 1998;101(1):33–39. doi: 10.1172/JCI1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jang T. J. Expression of CD40 and Fas ligand in Bowen's disease, squamous cell carcinoma and basal cell carcinoma. Yonsei Medical Journal. 2002;43(3):304–308. doi: 10.3349/ymj.2002.43.3.304. [DOI] [PubMed] [Google Scholar]
- 42.Wang X. Y., Zhang R., Lian S. Aberrant expression of Fas and FasL pro-apoptotic proteins in basal cell and squamous cell carcinomas. Clinical and Experimental Dermatology. 2011;36(1):69–76. doi: 10.1111/j.1365-2230.2010.03860.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The whole R sentences for data processing and identification of differentially expressed genes in this study.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.