

Abstract
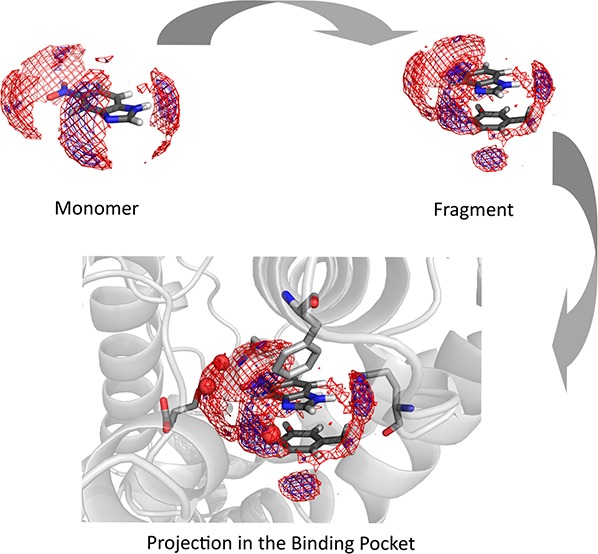
The use of fragments to biophysically characterize a protein binding pocket and determine the strengths of certain interactions is a computationally and experimentally commonly applied approach. Almost all drug like molecules contain at least one aromatic moiety forming stacking interactions in the binding pocket. In computational drug design, the strength of stacking and the resulting optimization of the aromatic core or moiety is usually calculated using high level quantum mechanical approaches. However, as these calculations are performed in a vacuum, solvation properties are neglected. We close this gap by using Grid Inhomogeneous Solvation Theory (GIST) to describe the properties of individual heteroaromatics and complexes and thereby estimate the desolvation penalty. In our study, we investigated the solvation free energies of heteroaromatics frequently occurring in drug design projects in complex with truncated side chains of phenylalanine, tyrosine, and tryptophan. Furthermore, we investigated the properties of drug-fragments crystallized in a fragment-based lead optimization approach investigating PDE-10-A. We do not only find good correlation for the estimated desolvation penalty and the experimental binding free energy, but our calculations also allow us to predict prominent interaction sites. We highlight the importance of including the desolvation penalty of the respective heteroaromatics in stacked complexes to explain the gain or loss in affinity of potential lead compounds.
Introduction
Molecular recognition in biological systems strongly depends on specific interactions between two molecules. In structure-based drug design these structure–activity relationships between ligands and their target molecules are rationalized and optimized and provide additional valuable information for the drug discovery process.1,2 However, the number of possible and favorable interaction types, which have to be considered in the drug design process, increased significantly over the past decades.3 Various options of possible interactions exist in a protein–ligand binding site. Crystal structures of protein–ligand complexes, such as the PDB, can elucidate which of those various interaction types are actually relevant in a given protein–ligand binding site.
Lead structures are starting points in medicinal chemistry, which sometimes lack the affinity which is required to function as drugs, and affinity optimization steps increase lipophilicity, molecular size, and molecular complexity.4,5 An attempt to characterize this druglikeness was proposed with the Lipinsky rule of 5, describing molecular size and hydrophilicity as primary risk factors in drug design.6
π-stacking interactions between aromatic rings play a central role in medicinal chemistry as an important contribution to ligand binding.1,7 Aromatic rings are frequently used in medicinal chemistry.10,8,9 Their characteristic features, such as planarity and the distinct π-electron cloud on top of and below the aromatic rings allow for addressing specific challenges in target recognition as they offer multiple interaction possibilities.7,10 These molecular interactions include π–π interactions,11 cation−π,12,13 amid−π,14 halogen−π,15 and hydrogen-bond interactions via heteroatoms.16 Before a drug can interact with the protein of interest, it has to be, at least partly, stripped of its solvation shell. However, in the field of computer aided molecular design of small molecules, usually high-level quantum mechanics calculations are performed to assess the strength of stacking interactions of aromatic heterocycles.17,18 Nevertheless, recent work has revealed a direct correlation of vacuum stacking interactions and the solvation free energy of heteroaromatics.19 Furthermore, the substitutions on a heteroaromatic core substantially influence the electrostatic properties and alter not only the strength and favored stacking geometry but also the solvation properties.20 Including hydration properties in computational approaches has been a hurdle in structure-based drug design.21 Programs like GIST,22,23 SZMAP,24 and WaterMap25−27 are well established to mainly characterize the solvation properties of the protein binding sites. Water molecules within the active site of a protein play a crucial role and therefore have to be considered in structure-based drug design.28,29 Displacement of water molecules from a protein binding site is considered a major contributor to protein–ligand binding.30,31 The shape and flexibility of a ligand-binding site in a protein is strongly influenced by water molecules and improves the complementarity between the protein and the ligand. The protein–ligand interaction is stabilized by a network of hydrogen bonds induced by water molecules.32 However, also the solvation of the ligand has to be taken into account, because the solvation changes by forming the protein–ligand complex, as the ligand has to strip off parts of its solvation shell.27,33 Different desolvation and solvation properties of ligands have shown to strongly influence receptor–ligand complex stabilities and highlight the critical role of ligand desolvation in determining binding affinity.34 Additionally, studies revealed that the desolvation effects represent a critical barrier in the binding event. The desolvation of a hydrophobic ligand and the active site of the β2-adrenergic receptor was shown to be the rate-limiting step in the ligand–receptor binding process.35−37 Introducing polar substituents contributed to an increase of the ligand desolvation barrier reflected in a substantial kon increase.38 Furthermore, it was shown that the energetic penalty resulting from desolvation of the polar groups compromises the observed binding affinity.38,39 Various studies confirmed that the inclusion of polar substituents results in a large penalty of desolvation upon binding and reveals a less favorable enthalpy of binding.33,40−42 Predicting and explaining the hydration properties of proteins as well as ligands is an ongoing challenge in the field of drug design.43,44
More general computational approaches to investigate the protein binding site make use of small molecules, acting as so-called probes, in short molecular dynamics simulations like SILCS45,46 or MDMix.47−49 Analyzing the positions of all fragments during these short simulations allows characterization of the binding site and generation of pharmacophore models. Besides these molecular dynamics based fragment methods, the idea of fragment docking was already established almost 20 years ago.50 This approach already included estimations of the desolvation penalty by using the generalized Born equation with numerical calculation of the Born radii.51 Newer scoring functions include already an implementation of the desolvation penalty and focus on characterization of the hydration and interaction hotspots of protein–ligand binding sites.52
Experimentally, a similar technique is used to assess the hydration properties and at the same time to explore the chemical space of possible ligands, namely, the fragment-based lead discovery (FBLD) approaches.53 When using FBLD, different cores with a variety of substituents are added to the protein of interest to act as probes. By measuring the affinities of these fragments, lead finding is facilitated. Additionally, the fragments are crystallized with the protein to give additional information on the binding pocket to advance the lead optimization process. In this study, we investigated an FBLD study of phosphodiesterase (PDE)-10-A.54 PDE-10 is one of the known 11 families of PDEs and reveals a unique primary amino acid sequence and distinct enzymatic activity. Chemical modulators for PDE-10 were revealed to be valuable, both in investigating enzyme properties and developing therapeutics, as shown by PDE-10 inhibitors. Inhibiting PDE-10-A might also be a novel treatment for psychosis.55
Here, we investigate how the solvation properties of aromatic complexes correlate with stacking interactions and highlight the importance of including solvation of ligands and the respective desolvation penalty to enhance the understanding of protein–ligand binding.
Methods
Data Set
The set of molecules investigated in this study has been already studied in terms of aromatic stacking interactions17,56 and is part of a fragment based lead optimization study on PDE-10-A (see Figure 1).54
Figure 1.

Heteroaromatics and amino acid mimics investigated in this study. The top row shows the 5 membered rings; rows two and three display the six membered rings with varying heteroatom content. Line four illustrates the amino acid mimics used for the QM and the GIST calculations. The colors of toluene, 4-methylphenol, and 3-methylindole reoccur in all following figures to allow a distinction between the respective data points.
Quantum Mechanical Calculations
Monomers were optimized using Gaussian 0957 at the ωB97XD58/cc-pVTZ59 level as proposed by Huber et al.60 in their investigation of stacking interactions of monocycles. To obtain the stacked complexes, we placed the truncated amino acid side chains in the center of a Cartesian coordinate system. For toluene and 4-methylphenol, we kept the center of the aromatic ring in the origin of the coordinate system. In the case of 3-methylindole, the center of the bond bridging the two aromatic cycles was kept at 0.0, 0.0, 0.0 while the aromatic system was kept flat in the x–y plane. In the z-direction we placed the heteroaromatics 3.9 Å on top using a MOE-SVL script.61 For the heteroaromatics in our test set, we kept the center of mass at x = 0.0 and y = 0.0 (Figure 2).
Figure 2.

Starting structure for the QM calculation of benzene, representing monocycles, stacking on a truncated Phe-side chain (toluene). The center of the phenyl ring of phenylalanine is at 0.0, 0.0, 0.0 and the center of the benzene ring is placed at 0.0, 0.0, 3.9.
These geometries were then optimized using the same setup employed for the monomers. For the structures of the FBLD we superposed the optimized structure with the fragments obtained from the PDB (electron density see Figure S2). Due to the fact that we deal with rigid molecules, the superposition did not cause a large deviation from the crystal coordinates. To mimic amino acids, we chose toluene (phenylalanine), 4-methylphenol (tyrosine), and 3-methylindole (tryptophan; Figure 2).
The interaction energies were calculated by subtracting the energies of the two geometry optimized monomers from the respective complex energy as proposed by the supermolecular approach.62 We did not use basis set superposition correction to allow for better comparability with previously published data.63
| 1 |
Simulation Details
The aromatic compounds were generated from SMILES64 using RdKIT. The structures of the stacked complexes were taken from the high-level QM calculations performed in the previous step. From the resulting files, we performed parametrizations using antechamber.65 The van der Waals, bond length, angle, and torsion parameters were taken from the General AMBER Force Field (GAFF).66 After performing a minimization of the molecules, using Gaussian 09,57 the partial charges were derived using the restrained electrostatic potential (RESP) method based on the QM calculation at the HF/6-31G* theory level. Topology and coordinate files for the molecular dynamics simulations were generated using AmberTools.67 The simulations were performed in a cubic water box of TIP3P68 water molecules with a minimal wall distance of 10 Å. To ensure proper equilibration, we followed the protocol outlined by Wallnoefer et al.69 In the production runs, we performed 10 ns NpT simulations with a time step of 2 fs. We applied restraints on the small molecules of 1000 kcal/(mol Å2). Spatial geometries were saved every 1000 steps, resulting in a final trajectory of 5000 frames. To allow for NpT conditions, we used the Berendsen barostat with a pressure relaxation time of 2.0 ps and Langevin dynamics with a friction coefficient of 2.0 ps–1.
Grid Inhomogeneous Theory
GIST allows a thermodynamic analysis of water molecules based on a molecular dynamics (MD) trajectory using a grid-based approach. GIST uses a grid to replace the spatial integrals from IST with discrete sums over the voxels on the constructed grid.22,23 One shortcoming of this method is that the solute has to be restrained to one conformation for each simulation. However, our systems are rigid rings, and therefore restraining the solute to only one conformation is no limitation. Recently published studies highlighted that GIST is a valid approach to study biomolecular systems and small molecules in combination with MD simulations, allowing to differentiate between a reference state, i.e., pure water, and an investigated state. A detailed description on the calculation of entropic and enthalpic terms is given by Nguyen et al. and in our previous and further recent publications.19,22,23,41,70−72
The free energy of solvation of a solute ΔGSolv can be approximated according to eq 2:
| 2 |
where kB is the Boltzmann constant, T is the temperature, GSolv(q) the solvation free energy of a solute in a single conformation q, and p(q) the probability to find the solute in this conformation q. GSolv(q), for a particular conformation q, is calculated by restraining its coordinates, making the solute retain its initial conformation. As mentioned, the compounds investigated in this work can be considered as rigid. Therefore, the error introduced by a single conformation approach is negligible. However, if our ligands would be more flexible, we could account for that by using multiple GIST calculations and combining them using eq 2.
Results
We performed calculations on 18 monocycles frequently occurring in computational drug design, which have been recently investigated in a study focusing on π-stacking of heteroaromatics17 (Figure 1). All these aromatic compounds show a local minimum in a parallel displaced stacked geometry. Using this optimized geometry, we further performed GIST analysis of the respective monomers and the resulting complex. We compared our vacuum stacking interaction energies with a published study, by the group of Wheeler,17 and found a good overall agreement with a Pearson correlation of 0.82. Due to the different approaches, global optimization in contrast to a grid based approach,17 the divergence can be explained by the very shallow energy surface of stacking heterocycles.60 The strongest differences are found in the stacking of 1,3,5-triacin (−8.18 vs −4.15 kcal/mol) and 1,2,5-triacin (−8.75 vs −5.30 kcal/mol) interacting with 3-methylindole. Comparing the different amino acid mimics individually, we identified a correlation of 0.86 for stacked compounds with toluene, a correlation of 0.88 for stacking on 4-methylphenol, and a correlation of 0.64 for 3-methylindol (Figure S1). By excluding the two mentioned triacin derivatives, the Pearson correlation of the latter improves significantly to 0.92. To investigate the influence of geometry, we performed GIST calculations using the structures published by Bootsma et al.17 for heteroaromatics stacking on toluene. We find a Pearson correlation of 0.98 for the desolvation penalty of the different geometries, indicating that the desolvation penalty of these small complexes is not that dependent on the conformation.
As shown in a recent publication on stacking interactions of benzene,19 we observe a good correlation for vacuum stacking interactions and the solvation free energy of the monomers for stacking on each amino acid mimic individually (Pearson coefficients of 0.87 for toluene, 0.80 for 4-methylphenol, and 0.77 for 3-methylindol). However, when comparing all three amino acid mimics, we find that the correlation vanishes (Figure 3). This is naturally the case as the solvation of the monomers remains constant, while the stacking interactions differ by more than 3 kcal/mol. As expected, we found that the vacuum stacking interactions are always stronger for 3-methylindole compared to toluene. To overcome this difference and to allow better comparability, we calculated the desolvation penalty as the difference between the solvation free energy of the complex and the solvation free energy of the respective monomers. For our data set of parallel displaced stacked geometries, we obtained a Pearson correlation of −0.74 for vacuum stacking interactions and the desolvation penalty of the optimized geometry.
Figure 3.

Correlation of the solvation free energies calculated using GIST and the vacuum stacking correlations (left) and the correlation of the desolvation penalty of the respective complexes with the corresponding vacuum stacking interactions (Pearson correlation: −0.74). Complexes with toluene are colored in black, with 4-methylphenol are colored in red, and with 3-methylindole are colored in green.
Besides the solvation free energies of the heteroaromatics and the respective complexes, we obtained maps containing hydration hot spots, i.e., positions where either enthalpically or entropically favored water positions can be identified.19 This allows us to compare which part of the hydration shell has to be broken to allow interactions between the two heteroaromatics.
For benzene and toluene, we can identify only the hydration hot-spot of the respective π clouds (Figure 4.A). For the complex, we find that the hydration site of each π cloud has to be replaced to form the complex. We calculate a desolvation penalty of 2.46 kcal/mol and a vacuum stacking interaction of −4.55 kcal/mol for the complex. With higher grades of substitutions, we observe a decrease of the solvation free energy of heteroaromatics, e.g., 1,3,5-triacin (−7.46 kcal/mol). However, in complex the nitrogen atoms can still form a major part of the monomer’s hydration shell (Figure 4B). Consequently, we obtained a resulting desolvation penalty of only 2.18 kcal/mol for 1,3,5-triacin, while vacuum stacking interactions revealed −5.77 kcal/mol for the complex. The lowest solvation free energy in our data set and the strongest vacuum stacking interactions are calculated for pyrimidone with −14.18 kcal/mol solvation free energy and −8.34 kcal/mol of stacking with toluene. This compound does also show the highest desolvation penalty with 3.50 kcal/mol (Figure 4C).
Figure 4.

GIST maps of benzene, 1,3,5-triacin, pyrimidone, and toluene as monomers and the maps of the respective complexes. The vacuum stacking interaction is obtained from quantum mechanical optimizations using DFT at the ωB97XD/cc-pVDZ level using Gaussian 09. The desolvation penalty was calculated as the difference of the sum of solvation free energy of the complex and the sum of the respective monomers. Red mesh: favorable solute–water enthalpy (cutoff −0.1 kcal/(mol·Å3)). Blue mesh: low water entropy (cutoff −0.08 kcal/mol·Å3)).
We applied our approach on a fragment-based lead discovery campaign on PDE-10-A, Table 1. We deleted everything from the crystal structure but the heteroaromatic ligand and Phe729, the stacking amino acid. We calculated the hydration free energy of this small fragment alone, because we assumed that the pocket is equal for all ligands and that the error introduced by the double sided π-cloud of the phenylalanine is compensated throughout the data set.
Table 1. Heteroaromatic Compounds Used in a FBLD Approach on PDE-10-A54 Including the Experimentally Obtained Binding Free Energies, Calculated from the Experimental Ki Values, the Desolvation Penalty Calculated from GIST and the QM Based Vacuum Stacking Interactions, All Given in kcal/mol.

As shown in previous studies for monocyclic heteroaromatics, stacking interaction correlates well with the dipole moment;60 however, we can also identify a correlation for the solvation free energy of these aromatic molecules and vacuum stacking energy as well as dipole moment.19 Due to the high number of substitutions on these fragments, and the deviation from ideal stacking geometries due to the binding pocket, we could not identify a correlation for the binding free energy either with vacuum stacking interaction (Pearson correlation −0.18) or with the solvation of the respective fragments (Pearson correlation −0.26). However, the Pearson correlation of these 10 fragments for experimental binding free energy and the calculated desolvation penalty resulted in 0.79, Figure 5 and Table S2.
Figure 5.

Correlation of desolvation penalty with experimentally determined binding affinity. The points are color coded by the calculated vacuum stacking interaction energy for the geometries found in crystal structures (left). On the right, we plot the stacking interactions against the experimental binding free energies. The points are color coded by the calculated desolvation penalty. The correlation for the experimental binding free energy and the desolvation penalty results in 0.79, while the vacuum stacking interaction shows a Pearson correlation of −0.18. The sum of the vacuum stacking interaction energy and the desolvation penalty revealed a Pearson correlation of merely 0.13. This is however due to 4-amino-2-methylphenol, which shows such a low vacuum stacking interaction that it cannot be compensated by the low desolvation penalty, SI Figure 4. After performing a geometry optimization, using the previously mentioned quantum mechanical setup, of the respective fragments, we obtained a Pearson correlation of −0.40 for the experimental binding energy and the energy for the optimized complex.
In the FBLD approach on PDE-10-A, the compound with the lowest binding affinity was identified as 4-amino-2-methylphenol (PDB accession code: 4LLP). While the vacuum stacking interactions would predict this specific fragment with the worst stacking interaction energy of this data set, we clearly see that it has the second lowest desolvation penalty. In the study by Recht et al.,54 two fragments are chosen for further development: 4-chloro-1,3-benzothiazol-2-amine (PDB accession code: 4MSH) and 8-nitroquinoline (4MSN). Judging solely from the desolvation penalty, these two compounds were ranked second and third in our desolvation penalty. While they also show substantially higher vacuum stacking interactions than 4-amino-2-methylphenol, at 6.66 and 6.90 kcal/mol, they still stack worse than the top stacking compounds, i.e., 5-nitroquinoline (PDB accession code: 4LM1) and 4-amino-1,7-dihydro-6H-pyrazolpyrimidine-6-thione (PDB accession code: 4LKQ) at −7.30 kcal/mol and −7.18 kcal/mol, respectively. Furthermore, we performed thermodynamic integration and MMPBSA calculations on the data set. Neither approach resulted in a correlation close the one obtained for the desolvation penalty, SI Figure 3.
Discussion
This study reveals that the desolvation penalty of complexes plays a crucial role in predicting binding properties of several aromatic cores and lead compounds.
The optimized positions of the unsubstituted fragments revealed a tendency to be displaced toward the methyl group of toluene and 4-methylphenol, equally (Figure 6A). This trend was also observed in a recent study on stacking interactions by Bootsma et al.17 This is most likely a result of the best possible alignment of the dipoles while not violating parallel displaced geometries and the rule of heteroatoms being positioned outside of the amino acid mimic π-cloud.60 However, when looking at the structures of the FBLD approach, we clearly see that the heteroaromatics are usually away from the protein backbone, mainly due to steric hindrance. Nevertheless, the change of environment from vacuum to water might also influence the optimal geometry. To test this hypothesis, we performed optimizations for pyridazine and 4-methylphenol in implicit solvent and were able to identify geometries with lower energies when the heteroaromatic compound is displaced from the methyl group of 4-methylphenol, Figure 6.
Figure 6.
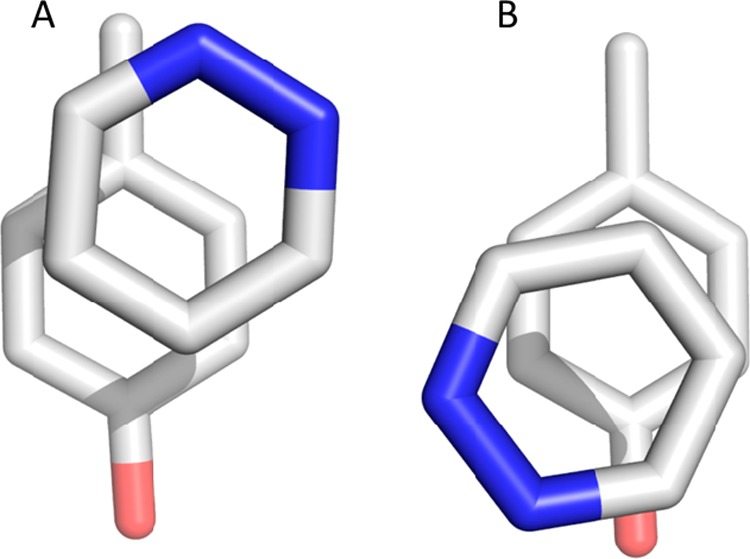
Comparison of local minima of the geometries optimized in a vacuum and in implicit solvent. Left: the geometry obtained in vacuum optimization. Right: the local minimum found when using implicit solvent in the QM calculations. In both cases, we used Gaussian 0957 at the ωB97XD58/cc-pVTZ59 level of theory. For the implicit solvation, we employed the polarizable continuum model (PCM), a reaction field calculation using the integral equation formalism model, implemented in Gaussian.73
It is known that the free energy surface of stacked heteroaromatics is rather shallow,60 therefore the two obtained local energy minima show very similar stacking interaction energies, −5.28 kcal/mol vs −5.25 kcal/mol. However, when we optimized the additional minimum, Figure 6B, in a vacuum, the geometry dissociated.
When projecting the enthalpy and entropy grids from the GIST calculation into the respective crystal structures, we can clearly identify where the water molecules of the solvation shell are or should be replaced by hydrophilic amino acids (Figure 7). Not only can we identify the amino acids mainly involved in the binding process, but we can also correctly predict the positions of water molecules modeled in the crystal structure (SI Video 1).
Figure 7.

GIST resulting from the calculations of the fragment inhibitors with toluene, projected into the respective crystal structures. Red mesh: favorable solute–water enthalpy (cutoff −0.1 kcal/(mol·Å3)). Blue mesh: low water entropy (cutoff −0.08 kcal/mol·Å3)). We find that the major hydration hot spots from the GIST calculations are replaced by interactions with Phe729 and GLN726.
Gln726 shows H bonds with all fragments presented in the study by Recht et al., regardless of their different binding geometries.54 We can see that the best binding fragment (PDB accession code: 4LLP) interacts not only in terms of π-stacking with Phe729 and H bonds with Gln726, but also the substituted hydroxyl group of the fragment points toward the solvent exposed region of the binding site. The hydration shell of the fragment is not completely broken in forming the complex, and the already small desolvation penalty can be mostly compensated. When comparing the desolvation penalties of the fragments in 4MSH and 4MSA, they are almost identical (Figure 7). However, in this case, we can at least partly attribute the higher affinity of 4MSH to the more favorable stacking geometry compared to 4MSA. The most unfavorable fragment in terms of experimental ΔGBind as well as in desolvation penalty results from 4,6-dimethylpyrimidin-2-amine with the PDB accession code 4LLX (Figure 7). We do not only observe a high desolvation penalty, but we can also see from the crystal structure despite finding H-bond interactions with GLN726 that the fragment solely offers hydrophobic interactions toward the solvent accessible part of the binding pocket.
Conclusion
In our study, we confirm the recently published correlation of solvation free energy of monomers with vacuum stacking interaction for amino acid mimics of phenylalanine, tyrosine, and tryptophan. Furthermore, we show that the desolvation penalty calculated for the respective complexes shows a strong inverse correlation with vacuum stacking interactions of optimized heteroaromatic cores. This can be of great interest in the initial phase of a drug design process where vacuum stacking interactions are used to optimize the scaffold of a lead compound.
In a protein environment, the experimental geometries are not always optimized, and further substitutions on the heteroaromatics and the interactions in the binding pocket make it harder to use vacuum stacking interactions alone as a predicative tool in drug design. We clearly see in the studied FBLD campaign that stacking alone does not tell the whole story, because hydration properties influence complex binding. Vacuum stacking interactions mainly align the dipoles, resulting in geometries that are not entirely favorable, as shown by the optimization of L19 with and without implicit solvents. Additionally, calculation of the desolvation penalty allows a comparison of substituted heteroaromatics rather than just the core. Calculations of complexed geometries revealed that the GIST calculations of stacked fragments do not only correlate well with the experimental KI values but also show the preferred environment of a substituted heteroaromatic compound within the binding pocket.
Acknowledgments
This work was supported by the Austrian Science Fund (FWF) P30565 and P30737 and an Erwin Schrödinger fellowship J-4150 (granted to M.S.). Computational results have been achieved using the high-performance computing infrastructure LEO of the University of Innsbruck.
Glossary
Abbreviations
- FBLD
fragment-based lead design
- GAFF
General AMBER Force Field
- GIST
Grid Inhomogeneous Solvation Theory
- MD
Molecular Dynamics
- SMILES
Simplified Molecular Input Line Entry System
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.9b01165.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Bissantz C.; Kuhn B.; Stahl M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53 (14), 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn B.; Gilberg E.; Taylor R.; Cole J.; Korb O. How Significant Are Unusual Protein-Ligand Interactions? Insights from Database Mining. J. Med. Chem. 2019, 62 (22), 10441–10455. 10.1021/acs.jmedchem.9b01545. [DOI] [PubMed] [Google Scholar]
- Paulini R.; Müller K.; Diederich F. Orthogonal Multipolar Interactions in Structural Chemistry and Biology. Angew. Chem., Int. Ed. 2005, 44 (12), 1788–1805. 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]
- Kenny P. W.; Montanari C. A. Inflation of Correlation in the Pursuit of Drug-Likeness. J. Comput.-Aided Mol. Des. 2013, 27 (1), 1–13. 10.1007/s10822-012-9631-5. [DOI] [PubMed] [Google Scholar]
- Kenny P. W. The Nature of Ligand Efficiency. J. Cheminf. 2019, 11 (1), 8. 10.1186/s13321-019-0330-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings1PII of Original Article: S0169–409X(96)00423–1. The Article Was Originally Published in Advanced Drug Delivery Reviews 23 (1997) 3–25.1. Adv. Drug Delivery Rev. 2001, 46 (1), 3–26. 10.1016/S0169-409X(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Meyer E. A.; Castellano R. K.; Diederich F. Interactions with Aromatic Rings in Chemical and Biological Recognition. Angew. Chem., Int. Ed. 2003, 42 (11), 1210–1250. 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- Feher M.; Schmidt J. M. Property Distributions: Differences between Drugs, Natural Products, and Molecules from Combinatorial Chemistry. J. Chem. Inf. Comput. Sci. 2003, 43 (1), 218–227. 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- Taylor R. D.; MacCoss M.; Lawson A. D. G. Rings in Drugs. J. Med. Chem. 2014, 57 (14), 5845–5859. 10.1021/jm4017625. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A.A Synopsis of the Properties and Applications of Heteroaromatic Rings in Medicinal Chemistry. Advances in Heterocyclic Chemistry; Elsevier, 2017. [Google Scholar]
- Hunter C. A.; Sanders J. K. The Nature of. Pi.-. Pi. Interactions. J. Am. Chem. Soc. 1990, 112 (14), 5525–5534. 10.1021/ja00170a016. [DOI] [Google Scholar]
- Dougherty D. A. The Cation-π Interaction. Acc. Chem. Res. 2013, 46 (4), 885–893. 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallivan J. P.; Dougherty D. A. Cation-π Interactions in Structural Biology. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (17), 9459–9464. 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder M.; Kuhn B.; Diederich F. Efficient Stacking on Protein Amide Fragments. ChemMedChem 2013, 8 (3), 397–404. 10.1002/cmdc.201200512. [DOI] [PubMed] [Google Scholar]
- Wallnoefer H. G.; Fox T.; Liedl K. R.; Tautermann C. S. Dispersion Dominated Halogen-π Interactions: Energies and Locations of Minima. Phys. Chem. Chem. Phys. 2010, 12 (45), 14941–14949. 10.1039/c0cp00607f. [DOI] [PubMed] [Google Scholar]
- Graton J.; Le Questel J.-Y.; Maxwell P.; Popelier P. Hydrogen-Bond Accepting Properties of New Heteroaromatic Ring Chemical Motifs: A Theoretical Study. J. Chem. Inf. Model. 2016, 56 (2), 322–334. 10.1021/acs.jcim.5b00574. [DOI] [PubMed] [Google Scholar]
- Bootsma A. N.; Doney A. C.; Wheeler S. E. Predicting the Strength of Stacking Interactions between Heterocycles and Aromatic Amino Acid Side Chains. J. Am. Chem. Soc. 2019, 141 (28), 11027–11035. 10.1021/jacs.9b00936. [DOI] [PubMed] [Google Scholar]
- Bootsma A. N.; Wheeler S. E. Converting SMILES to Stacking Interaction Energies. J. Chem. Inf. Model. 2019, 59 (8), 3413–3421. 10.1021/acs.jcim.9b00379. [DOI] [PubMed] [Google Scholar]
- Loeffler J. R.; Schauperl M.; Liedl K. R. Hydration of Aromatic Heterocycles as Adversary of π-Stacking. J. Chem. Inf. Model. 2019, 59, 4209. 10.1021/acs.jcim.9b00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A.; Mahato J.; Dixit M.; Patwari G. N. Progressive Hydrophobicity of Fluorobenzenes. J. Phys. Chem. B 2019, 123 (47), 10083–10088. 10.1021/acs.jpcb.9b08057. [DOI] [PubMed] [Google Scholar]
- Sledz P.; Caflisch A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. 10.1016/j.sbi.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Nguyen C. N.; Kurtzman Young T.; Gilson M. K. Grid Inhomogeneous Solvation Theory: Hydration Structure and Thermodynamics of the Miniature Receptor Cucurbit[7]Uril. J. Chem. Phys. 2012, 137, 044101. 10.1063/1.4733951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen C. N.; Cruz A.; Gilson M. K.; Kurtzman T. Thermodynamics of Water in an Enzyme Active Site: Grid-Based Hydration Analysis of Coagulation Factor Xa. J. Chem. Theory Comput. 2014, 10 (7), 2769–2780. 10.1021/ct401110x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZMAP 1.2.1.4; OpenEye Scientific Software: Santa Fe, NM, 2015.
- Cappel D.; Sherman W.; Beuming T.. Calculating Water Thermodynamics in the Binding Site of Proteins - Applications of WaterMap to Drug Discovery https://www.ingentaconnect.com/contentone/ben/ctmc/2017/00000017/00000023/art00004 (accessed Jul 10, 2019). 10.2174/1568026617666170414141452. [DOI] [PubMed]
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for Molecular Recognition Exploiting Hydrophobic Enclosure in Protein-Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer M.; Oostenbrink C.. Water in Protein Hydration and Ligand Recognition. J. Mol. Recognit. 2019, 32 ( (12), ), 10.1002/jmr.2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balius T. E.; Fischer M.; Stein R. M.; Adler T. B.; Nguyen C. N.; Cruz A.; Gilson M. K.; Kurtzman T.; Shoichet B. K. Testing Inhomogeneous Solvation Theory in Structure-Based Ligand Discovery. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6839–E6846. 10.1073/pnas.1703287114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler D. Interfaces and the Driving Force of Hydrophobic Assembly. Nature 2005, 437 (7059), 640–647. 10.1038/nature04162. [DOI] [PubMed] [Google Scholar]
- Krimmer S. G.; Cramer J.; Schiebel J.; Heine A.; Klebe G. How Nothing Boosts Affinity: Hydrophobic Ligand Binding to the Virtually Vacated S1′ Pocket of Thermolysin. J. Am. Chem. Soc. 2017, 139 (30), 10419–10431. 10.1021/jacs.7b05028. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang R.; Yang C.-Y.; Wang S. Analysis of Ligand-Bound Water Molecules in High-Resolution Crystal Structures of Protein-Ligand Complexes. J. Chem. Inf. Model. 2007, 47 (2), 668–675. 10.1021/ci6003527. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.; Shoichet B. K. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J. Chem. Inf. Model. 2010, 50 (9), 1561–1573. 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- Browning C.; Martin E.; Loch C.; Wurtz J.-M.; Moras D.; Stote R. H.; Dejaegere A. P.; Billas I. M. L. Critical Role of Desolvation in the Binding of 20-Hydroxyecdysone to the Ecdysone Receptor. J. Biol. Chem. 2007, 282 (45), 32924–32934. 10.1074/jbc.M705559200. [DOI] [PubMed] [Google Scholar]
- Schuetz D. A.; Richter L.; Amaral M.; Grandits M.; Grädler U.; Musil D.; Buchstaller H.-P.; Eggenweiler H.-M.; Frech M.; Ecker G. F. Ligand Desolvation Steers On-Rate and Impacts Drug Residence Time of Heat Shock Protein 90 (Hsp90) Inhibitors. J. Med. Chem. 2018, 61 (10), 4397–4411. 10.1021/acs.jmedchem.8b00080. [DOI] [PubMed] [Google Scholar]
- Shimokhina N.; Bronowska A.; Homans S. W. Contribution of Ligand Desolvation to Binding Thermodynamics in a Ligand-Protein Interaction. Angew. Chem., Int. Ed. 2006, 45 (38), 6374–6376. 10.1002/anie.200602227. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Pan A. C.; Arlow D. H.; Borhani D. W.; Maragakis P.; Shan Y.; Xu H.; Shaw D. E. Pathway and Mechanism of Drug Binding to G-Protein-Coupled Receptors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (32), 13118–13123. 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong C. W. The Effect of Desolvation on the Binding of Inhibitors to HIV-1 Protease and Cyclin-Dependent Kinases: Causes of Resistance. Bioorg. Med. Chem. Lett. 2016, 26 (15), 3705–3713. 10.1016/j.bmcl.2016.05.080. [DOI] [PubMed] [Google Scholar]
- Misra R. N.; Rawlins D. B.; Xiao H.; Shan W.; Bursuker I.; Kellar K. A.; Mulheron J. G.; Sack J. S.; Tokarski J. S.; Kimball S. D.; et al. 1H-Pyrazolo[3,4-b]Pyridine Inhibitors of Cyclin-Dependent Kinases. Bioorg. Med. Chem. Lett. 2003, 13 (6), 1133–1136. 10.1016/S0960-894X(03)00034-9. [DOI] [PubMed] [Google Scholar]
- Biela A.; Khayat M.; Tan H.; Kong J.; Heine A.; Hangauer D.; Klebe G. Impact of Ligand and Protein Desolvation on Ligand Binding to the S1 Pocket of Thrombin. J. Mol. Biol. 2012, 418 (5), 350–366. 10.1016/j.jmb.2012.01.054. [DOI] [PubMed] [Google Scholar]
- Verteramo M. L.; Stenström O.; Ignjatović M. M.; Caldararu O.; Olsson M. A.; Manzoni F.; Leffler H.; Oksanen E.; Logan D. T.; Nilsson U. J.; et al. Interplay between Conformational Entropy and Solvation Entropy in Protein-Ligand Binding. J. Am. Chem. Soc. 2019, 141, 2012–2026. 10.1021/jacs.8b11099. [DOI] [PubMed] [Google Scholar]
- Kolář M.; Fanfrlík J.; Hobza P. Ligand Conformational and Solvation/Desolvation Free Energy in Protein-Ligand Complex Formation. J. Phys. Chem. B 2011, 115 (16), 4718–4724. 10.1021/jp2010265. [DOI] [PubMed] [Google Scholar]
- Ladbury J. E. Just Add Water! The Effect of Water on the Specificity of Protein-Ligand Binding Sites and Its Potential Application to Drug Design. Chem. Biol. 1996, 3 (12), 973–980. 10.1016/S1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- de Beer S.; Vermeulen N.; Oostenbrink C. The Role of Water Molecules in Computational Drug Design. Curr. Top. Med. Chem. 2010, 10 (1), 55–66. 10.2174/156802610790232288. [DOI] [PubMed] [Google Scholar]
- Raman E. P.; Yu W.; Lakkaraju S. K.; MacKerell A. D. Inclusion of Multiple Fragment Types in the Site Identification by Ligand Competitive Saturation (SILCS) Approach. J. Chem. Inf. Model. 2013, 53 (12), 3384–3398. 10.1021/ci4005628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller C. E.; Raman E. P.; MacKerell A. D.; Guvench O. Site Identification by Ligand Competitive Saturation (SILCS) Simulations for Fragment-Based Drug Design. Methods Mol. Biol. 2015, 1289, 75–87. 10.1007/978-1-4939-2486-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seco J.; Luque F. J.; Barril X. Binding Site Detection and Druggability Index from First Principles. J. Med. Chem. 2009, 52 (8), 2363–2371. 10.1021/jm801385d. [DOI] [PubMed] [Google Scholar]
- Alvarez-Garcia D.; Barril X. Molecular Simulations with Solvent Competition Quantify Water Displaceability and Provide Accurate Interaction Maps of Protein Binding Sites. J. Med. Chem. 2014, 57 (20), 8530–8539. 10.1021/jm5010418. [DOI] [PubMed] [Google Scholar]
- Alvarez-Garcia D.; Barril X. Relationship between Protein Flexibility and Binding: Lessons for Structure-Based Drug Design. J. Chem. Theory Comput. 2014, 10 (6), 2608–2614. 10.1021/ct500182z. [DOI] [PubMed] [Google Scholar]
- Majeux N.; Scarsi M.; Apostolakis J.; Ehrhardt C.; Caflisch A. Exhaustive docking of molecular fragments with electrostatic solvation. Proteins: Struct., Funct., Genet. 1999, 37 (1), 88–105. . [DOI] [PubMed] [Google Scholar]
- Jeon J.; Kim H. J. A Continuum Theory of Solvation in Quadrupolar Solvents. II. Solvation Free Energetics, Dynamics, and Solvatochromism. J. Chem. Phys. 2003, 119 (16), 8626–8635. 10.1063/1.1605377. [DOI] [Google Scholar]
- Krammer A.; Kirchhoff P. D.; Jiang X.; Venkatachalam C. M.; Waldman M. LigScore: A Novel Scoring Function for Predicting Binding Affinities. J. Mol. Graphics Modell. 2005, 23 (5), 395–407. 10.1016/j.jmgm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Rees D. C. The Rise of Fragment-Based Drug Discovery. Nat. Chem. 2009, 1 (3), 187–192. 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- Recht M. I.; Sridhar V.; Badger J.; Bounaud P.-Y.; Logan C.; Chie-Leon B.; Nienaber V.; Torres F. E. Identification and Optimization of PDE10A Inhibitors Using Fragment-Based Screening by Nanocalorimetry and X-Ray Crystallography. J. Biomol. Screening 2014, 19, 497. 10.1177/1087057113516493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Xu M.; Liu G.; Christoffersen C. T.; Wang M. Discovery of Novel PDE10 Inhibitors by a Robust Homogeneous Screening Assay. Acta Pharmacol. Sin. 2013, 34 (8), 1116–1120. 10.1038/aps.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuffenhauer A.; Ertl P.; Roggo S.; Wetzel S.; Koch M. A.; Waldmann H. The Scaffold Tree - Visualization of the Scaffold Universe by Hierarchical Scaffold Classification. J. Chem. Inf. Model. 2007, 47 (1), 47–58. 10.1021/ci600338x. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90 (2), 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Huber R. G.; Margreiter M. A.; Fuchs J. E.; von Grafenstein S.; Tautermann C. S.; Liedl K. R.; Fox T. Heteroaromatic π-Stacking Energy Landscapes. J. Chem. Inf. Model. 2014, 54 (5), 1371–1379. 10.1021/ci500183u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2017. [Google Scholar]
- Beljonne D.; Cornil J.; Silbey R.; Millié P.; Brédas J. L. Interchain Interactions in Conjugated Materials: The Exciton Model versus the Supermolecular Approach. J. Chem. Phys. 2000, 112 (10), 4749–4758. 10.1063/1.481031. [DOI] [Google Scholar]
- Liedl K. R. Dangers of Counterpoise Corrected Hypersurfaces. Advantages of Basis Set Superposition Improvement. J. Chem. Phys. 1998, 108 (8), 3199–3204. 10.1063/1.475715. [DOI] [Google Scholar]
- Weininger D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28 (1), 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graphics Modell. 2006, 25 (2), 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25 (9), 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Cerutti D. S.; Cheatham T. E.; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Greene D.; Homeyer N.; et al. AMBER 2017; University of California: San Francisco, 2017. [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79 (2), 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Wallnoefer H. G.; Liedl K. R.; Fox T. A Challenging System: Free Energy Prediction for Factor Xa. J. Comput. Chem. 2011, 32 (8), 1743–1752. 10.1002/jcc.21758. [DOI] [PubMed] [Google Scholar]
- Wienen-Schmidt B.; Jonker H. R. A.; Wulsdorf T.; Gerber H.-D.; Saxena K.; Kudlinzki D.; Sreeramulu S.; Parigi G.; Luchinat C.; Heine A.; et al. Paradoxically, Most Flexible Ligand Binds Most Entropy-Favored: Intriguing Impact of Ligand Flexibility and Solvation on Drug-Kinase Binding. J. Med. Chem. 2018, 61, 5922–5933. 10.1021/acs.jmedchem.8b00105. [DOI] [PubMed] [Google Scholar]
- Schauperl M.; Podewitz M.; Waldner B. J.; Liedl K. R. Enthalpic and Entropic Contributions to Hydrophobicity. J. Chem. Theory Comput. 2016, 12, 4600–4610. 10.1021/acs.jctc.6b00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehl A.; Hu H.; Maeda S.; Zhang Y.; Qu Q.; Paggi J. M.; Latorraca N. R.; Hilger D.; Dawson R.; Matile H.; et al. Structure of the M-Opioid Receptor-G i Protein Complex. Nature 2018, 558 (7711), 547. 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105 (8), 2999–3094. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.