

Abstract
The recent pandemic associated with SARS-CoV-2, a virus of the Coronaviridae family, has resulted in an unprecedented number of infected people. The highly contagious nature of this virus makes it imperative for us to identify promising inhibitors from pre-existing antiviral drugs. Two druggable targets, namely 3C-like proteinase (3CLpro) and 2′-O-ribose methyltransferase (2′-O-MTase) were selected in this study due to their indispensable nature in the viral life cycle. 3CLpro is a cysteine protease responsible for the proteolysis of replicase polyproteins resulting in the formation of various functional proteins, whereas 2′-O-MTase methylates the ribose 2′-O position of the first and second nucleotide of viral mRNA, which sequesters it from the host immune system. The selected drug target proteins were screened against an in-house library of 123 antiviral drugs. Two promising drug molecules were identified for each protein based on their estimated free energy of binding (ΔG), the orientation of drug molecules in the active site and the interacting residues. The selected protein-drug complexes were then subjected to MD simulation, which consists of various structural parameters to equivalently reflect their physiological state. From the virtual screening results, two drug molecules were selected for each drug target protein [Paritaprevir (ΔG = −9.8 kcal/mol) & Raltegravir (ΔG = −7.8 kcal/mol) for 3CLpro and Dolutegravir (ΔG = −9.4 kcal/mol) and Bictegravir (ΔG = −8.4 kcal/mol) for 2′-OMTase]. After the extensive computational analysis, we proposed that Raltegravir, Paritaprevir, Bictegravir and Dolutegravir are excellent lead candidates for these crucial proteins and they could become potential therapeutic drugs against SARS-CoV-2.
Communicated by Ramaswamy H. Sarma
Keywords: Drug repurposing, SARS-CoV-2, 3C-like proteinase, 2′-O-ribose methyltransferase, docking, MD simulation
1. Introduction
Members of the family Coronaviridae are enveloped and maintain a single-strand, positive-sense RNA genome ranging from 26 to 32 kb in length (Su et al., 2016). They can be classified into four genera: alpha, beta, delta and gamma, out of which alpha and beta coronaviruses (CoVs) are known to infect humans (de Wilde et al., 2018). They are circulated among humans, other mammals, and birds and can cause respiratory, enteric, hepatic and neurologic diseases (Weiss & Leibowitz, 2011; Zhu et al., 2020). Even though the majority of human coronavirus infections are mild, the epidemics of two betacoronaviruses (βCoV), namely Severe Acute Respiratory Syndrome coronavirus (SARS-CoV) and Middle East Respiratory Syndrome coronavirus (MERS-CoV), has caused more than 10,000 cumulative cases in the past two decades, with mortality rates of 10% for SARS-CoV and 37% for MERS-CoV (World Health Organization, 2019). In late December 2019, several local health facilities reported groups of patients with pneumonia of unknown cause, and they were found to be epidemiologically connected with the Huanan wholesale seafood market in Wuhan, Hubei province, China (Huang et al., 2020). Next-generation sequencing analysis from bronchoalveolar lavage fluid samples and cultured isolates from the patients indicated a novel human-infecting coronavirus (Huang et al., 2020), officially named Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (World Health Organization, 2020a). Also, the World Health Organisation (WHO) has officially named the disease caused by SARS-CoV-2 as coronavirus disease 2019 (COVID-19) (World Health Organization, 2020a).
As of 29 March 2020, there have been more than 652,000 confirmed cases of COVID-19 across 177 countries/regions, with 30,300 fatalities. China, Italy and Spain have been the most affected countries with combined fatalities of more than 19,100 patients (Johns Hopkins CSSE, 2020). On 11 March, the WHO has officially declared COVID-19 as a pandemic (World Health Organization, 2020c). Earlier, on 30 January 2020, the World Health Organisation (WHO) declared SARS-CoV-2 pandemic as a Public Health Emergency of International Concern (World Health Organization, 2020e). According to the Centers for Disease Control and Prevention (CDC) and the WHO, the global death rate has been 3.4% (World Health Organization, 2020b), but it varies significantly with the age of the patients (Centers for Disease Control and Prevention, 2020). In order to control the menace caused by SARS-CoV-2, we suggest two of its indispensable druggable targets, namely 3C-like proteinase and 2′-O-ribose methyltransferase (Benkert et al., 2011; Yang et al., 2003). 3C-like proteinase (3CLpro) is a cysteine protease present in the Coronavirus replicase polyprotein. This protease plays a central role in viral replication and transcription functions through extensive proteolysis of two replicase polyproteins, pp1a and pp1ab (Yang et al., 2003). It cleaves at least 11 inter-domain sites on the pp1a and pp1ab polyproteins to generate individual functional proteins, including an RNA-directed RNA polymerase, a helicase, an exoribonuclease, an endoribonuclease and a 2′-O-ribose methyltransferase. Also, it is excised from polyproteins by its proteolytic activity and forms a homodimer with one active site per subunit (Yang et al., 2003). All of these features of 3CLpro makes it a principal drug target for SARS-CoV-2.
Another crucial protein responsible for viral replication and expression in host cells is non-structural protein 16 (nsp16) or 2′-OMTase. Most viral mRNAs possess a 5′-terminal cap structure (m7GpppN) which is essential for efficient splicing, nuclear export, translation and stability (Chen et al., 2011). This structure undergoes methylation catalysed by 2′-O-ribose methyltransferase at the ribose 2′-O position of the first and second nucleotide of the mRNA(Chen et al., 2011; Lugari et al., 2010). Nsp 16 encodes the 2′-OMTase which provides the viral mRNA with the ability to camouflage and obscure itself from the host cell, thus preventing recognition and activation of the host immune response which is essential for successful viral infection (Menachery et al., 2014). This protein can, therefore, act as another potential drug target for the SARS-CoV-2.
The present study utilises the systematic drug repurposing approach to identify antiviral drugs which can act as promising inhibitors against 3CLpro and 2′-OMTase and also understand their inhibitory mechanism through extensive in silico approaches. We have employed molecular docking and molecular dynamics (MD) simulation studies to calculate various structural parameters including the estimated binding free energy (ΔG) of the drugs, their estimated inhibition constant (Ki), Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), Principal Component Analysis (PCA) and the intermolecular hydrogen bonds (H-bonds) for free and drug bounded protein molecules.
2. Material and methods
2.1. Homology modelling and model evaluation
The amino acid sequence of SARS-CoV-2 orf1ab polyprotein was retrieved from National Centre for Biotechnological Information (NCBI) (Tax id: 2697049, Accession No: QHD43415). It was then subjected to pairwise sequence alignment against 3CLpro and 2′-OMTase from its close relative SARS-CoV (tax id: 694009) to figure out their amino acid sequence in SARS-CoV-2. Homology modelling of these proteins was performed using SWISS-MODEL (Waterhouse et al., 2018) with the user template option. Template structures were selected from Protein BLAST (Camacho et al., 2009) against Protein Data Bank (PDB) (Berman et al., 2000) based on query coverage, percentage of sequence identity and crystal structure resolution. Energy minimisation of modelled proteins was achieved using YASARA (Krieger et al., 2009) and GalaxyWEB (Ko et al., 2012) web servers. Model evaluation was performed before and after minimisation using Structure Analysis and Verification Server (SAVES) v5.0 meta server (UCLA MBI), which includes ERRAT (Colovos & Yeates, 1993), Verify-3D (Luthy et al., 1992) and PROCHECK (Laskowski et al., 1993) programmes.
2.2. Screening and selection of antiviral drugs against 3CLpro and 2′-OMTase
PyRx virtual screening software v0.8 (http://pyrx.sourceforge.net/downloads) was used for the screening of antiviral drugs against 3CLpro and 2′-OMTase proteins. One hundred twenty-three entries belonging to the category of antiviral drugs were downloaded from DrugBank database (Wishart et al., 2018) and were subsequently converted into a single structure data file (SDF) library (see supplementary data, Table 3). AutoDockVina (Trott & Olson, 2009) docking wizard inbuilt in PyRx was utilised for all of the docking calculations. In PyRx, blind docking with the exhaustiveness 32 option was used to carry out the virtual screening and docking of antiviral drugs against 3CLpro (Grid Box: center_x = 75.98, center_y = −15.53, center_z = 18.01, size_x = 70.46, size_y = 90.53, size_z = 61.34) and 2′-OMTase (Grid Box: center_x = 64.12, center_y = 70.35, center_z = 71.46, size_x = 67.11, size_y = 66.13, size_z = 68.36). Once we got the results obtained from virtual screening, we selected two drug molecules for each protein based on the estimated binding free energy (ΔG), estimated inhibition constant (Ki), their orientation at the catalytic site and the interacting residues. The selected protein-drug complexes were further subjected to MD simulations for understanding the structural stability of protein-drug complexes at the long-interval.
Table 3.
Interaction details of Paritaprevir and Raltegravir with 3CLpro.
| S/No | DrugBank ID | HB | D (Å) | Pi-SR | D (Å) | vdWISR |
|---|---|---|---|---|---|---|
| 1. | Paritaprevir (DB09297) | Ser46 | 2.33 | Met49 Leu50 Cys145 Pro168 | 5.27 3.85, 4.61 4.60 3.64 | Thr24, Thr25, Leu27, His41, Cys44, Thr45, Glu47, Phe140, Gly143, Glu166, Gln189 |
| 2. | Raltegravir (DB06817) | Thr24 Ser46 Asn142 Gly143 Glu166 | 2.53 2.47 2.39 2.11 2.59 | Thr25 Met49 Met165 | 3.93 4.87 5.14 | Thr26, Leu27, His41, Cys44, Thr45, Phe140, Ser144, Cys145 |
Hydrogen Bond residues (HB), Distance (D), Pi-Interaction Sharing Residues (Pi-SR), and Van der Waals Interaction Sharing Residues (vdWISR).
2.3. Molecular dynamics (MD) simulations
To make the docking results mimic the physiological state of protein molecules, the free proteins and the best protein-drug complexes were subjected to MD simulation in an explicit solvent model. MD simulation was performed using GROMACS 2019 (Abraham et al.) with GROMOS96 43a1 force field parameters (Chiu et al., 2009). The topology of the drug molecules was created using PRODRG webserver (Schuttelkopf & van Aalten, 2004). The simulation of all of the proteins and protein-drug complexes were run for a period of 100 ns. To make the system electrostatically neutral, counter ions were added to the protein-drug complexes. The complexes were solvated within 10 Å SPC/E water cube (Berendsen et al., 1987). The protein-drug complexes were minimised in multiple steps using steepest descent method, where minimisation of the whole system, water cube and non-heavy atoms of the complexes were accomplished. The entire systems were then progressively heated up to 300 K on a time scale of 100 ps. The equilibration steps were performed in two different phases, one with constant pressure and temperature (NPT) and the other with steady volume and temperature (NVT). Various structural parameters, like Root Mean Square Deviation (RMSD), Root Mean Square Fluctuations (RMSF), Radius of Gyration (Rg), Principal Component Analysis (PCA) based on essential dynamics (ED) approach, Inter-molecular Hydrogen Bonding (H-bonding) and Solvent-Accessible Surface Area (SASA) were calcualted as a function of time to explore the structural behaviour of the proteins and protein-drug complexes.
3. Results and discussion
3.1. Homology modeling and model evaluation
Homology modelling is one of the standard in silico procedures, which allows predicting the three-dimensional structure of a protein molecule based on experimentally solved homologous structures known as structural templates (Bordoli et al., 2009). Sequence identity and Query coverage are two crucial parameters to identify the suitable structural templates for homology modelling technique. Moreover, the 3D structure of the target protein is essential to explore its function (Micheletti, 2013). The 3D models of 3CLpro and 2′-OMTase were predicted using SWISS-MODEL (Waterhouse et al., 2018). For the best results, we used the user-defined template option present in SWISS-MODEL. Protein BLAST (Camacho et al., 2009) results were taken into account for the selection of suitable template structures. The X-ray crystal structure of 3CLpro from Human SARS coronavirus (SARS-CoV) solved at 1.9 Å (PDB ID: 1UJ1) resolution was selected as the template for SARS-CoV-2 3CLpro. For 2′-OMTase, we used a 2 Å resolution X-ray crystal structure of nsp10/nsp16 complex (PDB ID: 3R24) of SARS-CoV. The template (PDB ID: 1UJ1) was selected because it has 96.08% sequence identity and 100% query coverage towards SARS-CoV-2 3CLpro. Also, the template (PDB ID: 3R24) has a sequence identity of 93.29% and query coverage of 100% against SARS-CoV-2. In SWISS-MODEl, Global Model Quality Estimation (GMQE) (Waterhouse et al., 2018) score of 0.99 out of 1 and QMEAN (Benkert et al., 2011) score of 0.09 was achieved for 3CLpro, whereas for 2′-OMTase, GMQE score was 0.97 out of 1, and QMEAN score was −2.57. These quality estimations depict the reliability of the modelled protein structure, target–template alignment and the template search method. It is also worth noting that 3CLpro makes a homodimeric structure, but since only one protomer is active in each dimer, we only modelled the single protomer, and further used it for in silico interaction or molecular docking and MD simulation studies (Chen et al., 2006).
Once the models were obtained, they were evaluated using SAVES v5.0 metaserver based on ERRAT, Verify 3D and PROCHECK. The models were further refined and minimised using YASARA Energy Minimization Server and GalaxyWEB servers. The final model of 3CLpro had a Verify 3D score of 100% and ERRAT quality factor of 98.19. In the case of PROCHECK, 93.9% residues were present in the most favoured regions of Ramachandran Plot (Ramachandran et al., 1963), and more importantly, only one residue (Tyr154) was present in the disallowed regions. In the case of 2′-OMTase, the final model had a Verify 3D score of 87.33%, and ERRAT quality factor of 94.50. According to PROCHECK, 90% of the residues were in the most favoured regions of the Ramachandran plot, and notably, no residues were present in the disallowed regions. We used PDBsum (Laskowski et al., 2018) to calculate the overall average G-Factor score, which is 0.12 for both 3CLpro and 2′-OMTase, which also reflected the accuracy of the refined modelled structure (see supplementary data, Tables 1 and 2).
Table 1.
Selected drug molecules obtained from virtual screening of 3CLpro against the in-house library of anti-viral drugs.
| S/No | Drug Bank ID | EBE (kcal/mol) | EIC (M) | Molecular formula | Chemical scheme |
|---|---|---|---|---|---|
| 1 | Paritaprevir (DB09297) | −9.8 | 65.52 nM | C40H43N7O7S | 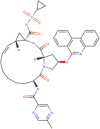 |
| 2 | Raltegravir (DB06817) | −7.8 | 1.916 µM | C20H21FN6O5 | 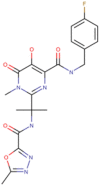 |
EBE: Estimated Binding Free Energy; EIC: Estimated Inhibition Constant.
Table 2.
Selected drug molecules obtained from virtual screening of 2′-OMTase against the in-house library of anti-viral drugs.
| S/No | Drug Bank ID | EBE (kcal/mol) | EIC(M) | Molecular formula | Chemical scheme |
|---|---|---|---|---|---|
| 1 | Dolutegravir (DB08930) | −9.4 | 128.7 nM | C20H19F2N3O5 | 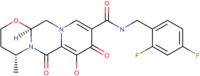 |
| 2 | Bictegravir (DB11799) | −8.4 | 0.696 µM | C21H18F3N3O5 | 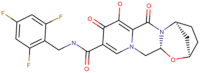 |
EBE: Estimated Binding Free Energy; EIC: Estimated Inhibition Constant.
3.2. Screening of anti-viral drugs against 3CLpro and 2′-OMTase
Virtual screening is one of the essential tools in drug designing and discovery process as it allows the prediction of the conformation and orientation of a ligand or drug molecule within the binding site of a target molecule (Kitchen et al., 2004). Also, the virtual screening tools employ scoring functions to predict the strength of the protein-ligand interactions, generating a score for each docked pose. Subsequently, the docked poses are ranked based on the score provided by the scoring function to identify the pose that most closely resembles the actual binding mode of the ligand (Gimeno et al., 2019). PyRx was used for the virtual screening of antiviral drugs against 3CLpro and 2′-OMTase proteins. One hundred twenty-three compounds belonging to the category of antiviral drugs were downloaded from DrugBank (Wishart et al., 2018) and were subsequently converted into a single SDF library. This in-house SDF library was used to screen both the proteins. Two drugs were selected against each protein based on their estimated free energy of binding (ΔG), binding site orientation and the protein residues with which they were found to be interacting with drug molecules. In the case of 3CLpro (see Table 1), we chose two molecules, namely Paritaprevir (DB09297) and Raltegravir (DB06817). Similarly, for 2′-OMTase (see Table 2), the best two drugs were found to be Dolutegravir (DB08930) and Bictegravir (DB11799).
3.3. Binding pattern of drug molecules against 3CLpro and 2′-OMTase
3.3.1. 3CLpro of SARS-CoV-2
As SARS-CoV 3CLpro (PDB ID: 1UJ1), the predicted model also forms a homo-dimer with both protomers (signified as ‘A’ and ‘B’) oriented nearly at right angles to each other (see supplementary data, Figure 1). Each monomer of the modelled structure is composed of three distinct globular domains (see Figure 1). Domains I comprises residues 8–101, domain II includes residues 102–184 and domain III consists of residues 201–303 and is attached with domain II through an extended loop region (residues 185–200). Domain I and Domain II form a chymotrypsin fold, whereas domain III is required for the homodimer assembly. The substrate-binding site is positioned in the cleft of domain I and domain II and is composed of a Cys145-His41 catalytic dyad. The substrate-binding pocket consists of His41, Phe140, Asn142, Gly143, Ser144, Cys145, Tyr161, His163, Glu166 and His172 residues (Yang et al., 2003).
Figure 1.

Interaction of drugs with 3CLpro (Domain I-Red, Domain II-Purple, Domain III-Cyan, Extended loop-Yellow). (a) Three dimensional representation of 3CLpro active site residues interacting with Paritaprevir (DB09297) (Green). (b) Two dimensional representation of 3CLpro active site residues interacting with Paritaprevir (Green) via Van der Waals interactions (slightly green colour), hydrogen bonds (dark green colour), and pi-interactions (light pink colour). (c) Three dimensional representation of 3CLpro active site residues interacting with Raltegravir (DB06817) (Orange). (d) Two dimensional representation of 3CLpro active site residues interacting with Raltegravir(Orange).
The estimated binding free energy (ΔG) of Paritaprevir against 3CLpro was observed to be −9.8 kcal/mol, and the estimated inhibitory constant was 65.52 nM. On the other hand, the binding affinity (ΔG) of Raltegravir was measured to be −7.8 kcal/mol, and the inhibitory constant was 1.916 µM. Both Paritaprevir and Raltegravir were found to be interacting with the active site residues (including the Cys145-His41 catalytic dyad) (see Table 3) and interacting with the substrate-binding pocket (see Figure 1). The binding free energy of 3CLpro with other drug molecules has been given in supplementary information.
3.3.2. 2′-OMTase of SARS-CoV-2
The predicted model of 2′-O methyltransferase (2′-OMTase) (see Figure 2) comprises of a core MTase domain, S-Adenosyl methionine (SAM) binding region and a cap-binding groove. The core MTase domain is found to comprise residues 30–209 of nsp16 as found in SARS-CoV (Chen et al., 2011). 2′-OMTase catalyses the transfer of SAM methyl group by relying on a conserved K-D-K-E tetrad within the substrate-binding pocket for activity (Menachery et al., 2014). The surface provided by K-D-K-E (Lys-46, Asp-130, Lys-170 and Glu-203) motif present at the bottom of the central groove might bind the first adenine nucleotide conserved as the acceptor of methyl group during methylation (Chen et al., 2011).
Figure 2.

Interaction of drugs with 2′-OMTase (MTase Domain-Purple). (a) Three dimensional representation of 2′-OMTase active site residues interacting with Dolutegravir (DB08930) (Sand colour). (b) Two dimensional representation of 2′-OMTase active site residues interacting with Dolutegravir (Sand colour) via Van der Waals interactions (slightly green colour), hydrogen bonds (dark green colour), and pi-interactions (light pink colour, orange colour). (c) Three dimensional representation of 2′-OMTase active site residues interacting with Bictegravir (DB11799) (Yellow). (d) Two dimensional representation of 2′-OMTase active site residues interacting withBictegravir (Yellow).
In the case of 2′-OMTase, Dolutegravir showed an estimated binding free energy (ΔG) of −9.4 kcal/mol, and the inhibition constant was 128.7 nM. Besides, Bictegravir conferred an estimated binding free energy (ΔG) of −8.4 kcal/mol against 2′-OMTase, and the estimated inhibition constant was 0.696 µM. Moreover, both Dolutegravir and Bictegravir were found to be actively interacting with the catalytic site residues (see Table 4) and interacting with the substrate-binding pocket (see Figure 2). The binding free energy of 2′-OMTase with other drug molecules has been given in supplementary information.
Table 4.
Interaction details of Dolutegravir and Raltegravir with 2′-OMTase.
| S/No | DrugBank ID | HB | D (Å) | Pi-SR | D (Å) | vdWISR |
|---|---|---|---|---|---|---|
| 1 | Dolutegravir (DB08930) | Asp130 Tyr132 Lys170 | 2.45 2.94 1.95, 2.55 | Met42 Leu100 Met131 Pro134 | 5.45 3.49 5.83 4.73 | Asn43, Lys46, Gly71, Gly73, Ser74, Asp75, Asp99, Asn101, Glu203 |
| 2 | Bictegravir (DB11799) | Asp130 Lys170 Asn198 | 2.43 2.28 2.15 | Met42 Leu100 Met131 Pro134 | 4.34 4.46, 5.02 4.73 5.12, 5.17 | Asn43, Lys46, Gly71, Asp99, Asn101, Tyr132, Asp133 |
Hydrogen Bond residues (HB), Distance (D), Pi-Interaction Sharing Residues (Pi-SR), and Van der Waals Interaction Sharing Residues (vdWISR).
According to the results obtained from electrostatic surface potential, all of the drug molecules were found interacting mostly with the negatively charged residues of both 3CLpro (see Figure 3) and 2′-OMTase (see Figure 4), which revealed that these molecules were oriented in the active site of both the proteins.
Figure 3.

The electrostatic surface potential interaction of Paritaprevir (DB09297) (Green) and Raltegravir (DB06817) (Orange) bound to 3CLpro. The zoomed view is representing the active site cleft.
Figure 4.

The electrostatic surface potential interaction of Dolutegravir (DB08930) (Sand) and Bictegravir (DB11799) (Yellow) bound to 2′-OMTase. The zoomed view is representing the active site cleft.
3.4. Molecular dynamics (MD) simulation
Molecular dynamics simulation is used to study the physical transitions of atoms as well as entire macromolecules and are effectively adopted to comprehend the structure-to-function relevance of a given macromolecule (Ambure et al., 2019; Amera et al., 2019; Hospital et al., 2015). Moreover, it can also be employed to illustrate the strength, pattern, and properties of protein-drug interactions and the dynamic conformational changes of macromolecule experiences under various physiological conditions (Amera et al., 2019). Different structural parameters, including RMSD, RMSF, Rg, PCA (based on ED approach), Intermolecular H-bondsand SASA, were evaluated as a function of time. To elucidate the conformational stability, dynamics, structural stability, folding properties and compactness of protein-drug complexes, MD simulations were performed for a period of 100 ns.
3.4.1. Root Mean Square Deviation analysis
RMSD is essential to quantify the structural stability of a protein or protein-drug complexes within a regular time frame. RMSD analysis depicted that unbound 3CLpro started stabilising after 15 ns, and it maintained stability until 90 ns. The RMSD pattern varied slightly during the final 10 ns. Besides, the 3CLpro-Raltegravir complex also attained stability at around 10 ns, and it maintained it until 90 ns. The RMSD again decreased during the final 10 ns (90–100 ns). In contrast, 3CLpro-Paritaprevir complex gained stability at around 20 ns, retained that stability until 70 ns except for one minor dip at about 50 ns. After 70 ns, the average RMSD declined until the end of the simulation cycle (see Figure 5a). An average RMSD value of 0.295 nm, 0.361 nm and 0.401 nm was obtained for unbound 3CLpro, 3CLpro-Raltegravir and 3CLpro-Paritaprevir complexes, respectively. Overall results explained that these two drug molecules did not significantly influence the structural stability of 3CLpro, although they nearly maintained the structural integrity, particularly Raltegravir. Based on the average RMSD values obtained from this analysis, the 3CLpro-Raltegravir complex was found to be closer to the drug-free form of 3CLpro.
Figure 5.

Analysis of Molecular Dynamics Simulation results of free 3CLpro (Black), 3CLpro-Raltegravir complex (Red) and 3CLpro-Paritaprevir complex (turquoise). (a) Root Mean Square Deviation (RMSD), (b) Root Mean Square Fluctuation (RMSF), (c) Radius of Gyration (Rg), (d) Solvent Accessible Surface Area (SASA).
RMSD analysis of the free form of 2′-OMTase depicted that the protein molecule achieved stability at around 40 ns, and it almost maintained its stability until 100 ns. The maximum RMSD fluctuations were recorded in the first 20 ns only. There were minor variations at around 60 ns, but apart from that, the protein was quite stable until the end of the simulation cycle. The 2′-OMTase-Dolutegravir complex attained stability at around 20 ns and maintained that stability until 90 ns, except a minor drop at approximately 70 ns. The average fluctuations spiked a little bit in the final 10 ns (90–100 ns) of this simulation. The 2′-OMTase-Bictegravir complex also took 40 ns to attain stability, but it also maintained it until the end of the simulation cycle (40–100 ns). It showed a minor dip at 60 ns, but it was able to gain its stability back at around 65 ns. As the free protein, the maximum fluctuations of protein-drug complexes were recorded in the first 20 ns only, after that, all of them took a steady path (see Figure 6a) and maintained convergence state until the end of the simulation period. However, in the case 2′-OMTase-Dolutegravir complex, slight changes were observed at 90–100 ns. An average RMSD value of 0.299 nm, 0.393 nm and 0.410 nm was obtained for unbound 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex, respectively. As evident from the average RMSD values, both of the drug complexes were very comparable to each other, but the 2′-OMTase-Dolutegravir complex was found closer to the free protein molecule.
Figure 6.

Analysis of Molecular Dynamics Simulation results of free 2′-OMTase (Blue), 2′-OMTase-Bictegravir complex (Orange) and 2′-OMTase-Dolutegravir complex (Dark Green). (a) Root Mean Square Deviation (RMSD), (b) Root Mean Square Fluctuation (RMSF), (c) Radius of Gyration (Rg), (d) Solvent Accessible Surface Area (SASA).
3.4.2. Root Mean Square Fluctuation analysis
RMSF is an essential structural parameter to identify the flexible and rigid regions of the protein structure. It is a measure of deviations of a particle from its original position. Moreover, it can also be used to identify the flexible residues in the protein, and thus it enables us to explore the conformational flexibility of the protein structure. The RMSF analysis was estimated for C-alpha atom of each residue representing the average displacement of each atom.
In the case of 3CLpro, we observed the highest fluctuations in loop heavy domain II and domain III. The loop regions are very flexible elements of protein molecules, and their flexibility is essential to accommodate the drug at the binding site appropriately. Besides, the 3CLpro-Raltegravir complex was found to be showing more significant fluctuations in comparison to 3CLpro-Paritaprevir complex (see Figure 5b). The average RMSF values of 3CLpro, 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex were found to be 0.170 nm, 0.182 nm and 0.184 nm, respectively. Additionally, the majority of the protein residues were stable with RMSF values smaller than 0.3 nm. Also, the catalytic dyad (His41 and Cys145) participating in interactions with the drug molecules remained highly stable throughout the MD simulation.
In the case of 2′-OMTase, the fluctuations were observed throughout the entire protein. 2′-OMTase also has an extensively looped structure, and as previously explained, we did expect to see significant fluctuations across the molecule. Moreover, both protein-drug complexes were also highly flexible (see Figure 6b). The average RMSF values of 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex were found to be 0.155 nm, 0.161 nm and 0.165 nm, respectively. Even though the average fluctuations of both protein-drug complexes were very similar, the 2′-OMTase-Dolutegravir complex was consistently found to be showing the most significant fluctuations. Besides, the conserved K-D-K-E (Lys-46, Asp-130, Lys-170 and Glu-203) remained remarkably stable throughout the MD simulation.
3.4.3. Radius of gyration (Rg) analysis
The radius of gyration is a valuable tool to understand the folding properties and compactness of protein and protein-drug complexes. It can also be used to elucidate the influence a drug molecule exerts leading to conformational changes in protein structure. A relatively high Rg value indicates a protein molecule with loose packing, whereas a smaller Rg value indicates a protein structure with tight packing. This analysis is also used to understand whether the drug molecules maintained the folding behaviour of protein or not.
In the case of 3CLpro, both of the protein-drug complexes were found to be more compact than the unbound protein molecule (see Figure 5c). The average Rg value of 3CLpro, 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex was found to be 2.165 nm, 2.116 nm and 2.144 nm, respectively. We also observed a few minor variations in Rg, which could be attributed to the conformational shifts that resulted from the changes in the secondary structures of protein during the MD simulation.
In the case of 2′-OMTase, both the protein-drug complexes were slightly less compact in comparison to the free 2′-OMTase. The free protein, as well as the protein-drug complexes, attained stability at 30 ns and remained stable until the end (see Figure 6c). The average Rg values of 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex were found to be 1.796 nm, 1.799 nm and 1.805 nm, respectively. Most of the fluctuations in the Rg values were recorded before 20 ns which again supported that these drug molecules stayed strongly bound to the active site and they helped to maintain the stability and compactness of the protein structure.
3.4.4. Intermolecular H-bonding
Hydrogen bonding is among the most crucial parameters to understand the binding affinity of drug molecules towards a protein molecule. Formation or deformation of H-bonds is a necessary consideration during MD simulations. A large number of H-bonds present in between protein and drug molecule signifies a strong binding affinity.
In the case of 3CLpro, we observed the maximum number of hydrogen bonds in the 3CLpro-Paritaprevir complex (six intermolecular H-bonds) followed by 3CLpro-Raltegravir complex (five intermolecular H-bonds). The average value of intermolecular H-bonds were 2 for both 3CLpro-Paritaprevir complex and 3CLpro-Raltegravir complex, respectively (see Figure 7).This results explained that both the drug molecules interacted effectively towards the active site of 3CLpro with a significant number of hydrogen bonds.
Figure 7.

Intermolecular hydrogen bonds between the drugs and 3CLpro; 3CLpro-Paritaprevir complex (turquoise), 3CLpro-Raltegravir complex (Red).
In the case of 2′-OMTase, we observed a maximum number of hydrogen bonds in the 2′-OMTase-Dolutegravir complex (six intermolecular H-bonds) followed by 2′-OMTase-Bictegravir complex (four intermolecular H-bonds). The average value of intermolecular H-bonds were 2 and 1 for 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex, respectively (see Figure 8). From the results of intermolecular hydrogen bonding, Dolutegravir was found to be interacting with a higher number of hydrogen bonds towards 2′-OMTase than the other drug molecule.
Figure 8.

Intermolecular hydrogen bonds between the drugs and 2′-OMTase; 2′-OMTase-Bictegravir complex (Orange), 2′-OMTase-Dolutegravir complex (Dark Green).
3.4.5. Solvent accessible surface area (SASA)
Additionally, we also performed a SASA analysis of all of the proteins and protein-drug complexes. This analysis is useful in understanding the solvent behaviour (Hydrophilic or Hydrophobic) of a protein molecule as well as protein-drug complexes.
No major differences were observed in the SASA profiles of 3CLpro and its protein-drug complexes (see Figure 5d). The average values of SASA for the free 3CLpro, 3CLpro-Paritaprevir complex and 3CLpro-Raltegravir complex were 133.89 nm2, 131.79 nm2 and 131.26 nm2, respectively.
The average SASA of 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex were found to be 126.77 nm2, 130.116 nm2 and 131.03 nm2, respectively. The results suggested that both of the proteins-drug complexes were impressively stable after the binding of drug molecules to their active sites (see Figure 6d).
3.4.6. Principal component analysis (PCA)
PCA of MD simulations is a standard technique widely used to identify the essential motion of protein molecules. The PCA was employed to reveal the biologically significant and relevant motions from the global trajectories of unbound proteins as well as the protein-drug complexes using Essential Dynamics (ED) approach. ED analyses the combined fluctuations of the most unsteady regions of protein molecules into two variables, namely Principal Component 1 (PC1) and Principal Component 2 (PC2), which represents the majority of fluctuations observed in MD simulation.
PCA graph of unbound 3CLpro, 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex revealed that the protein molecule was slightly more flexible in the presence of both drug molecules (see Figure 9). The trace of covariance matrix for 3CLpro, 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex was found to be 115.482 nm2, 136.366 nm2 and 132.059 nm2, respectively. The 3CLpro-Paritaprevir complex was found to be occupying lesser conformational space than the 3CLpro-Raltegravir complex. This result, along with previous analyses such as RMSD, RMSF, Rg and SASA additionally supported that Raltegravir and also Paritaprevir could be the most promising candidates which might be inhibiting the function of 3CLpro.
Figure 9.

Principal Component Analysis (PCA) of free 3CLpro (Black), 3CLpro-Paritaprevir complex (turquoise) and 3CLpro-Raltegravir complex (Red).
PCA of free 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex depicted that the protein, as well as the protein-drug complexes, were occupying almost equal conformational space (see Figure 10). Trace of the covariance matrix for 2′-OMTase, 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex was found to be 106.463 nm2, 123.878 nm2 and 117.187 nm2, respectively. Together with all of the MD simulation analysis, PCA supported that Dolutegravir and Bictegravir could be the most promising drugs capable of inhibiting the function of 2′-OMTase.
Figure 10.

Principal Component Analysis (PCA) of free 2′-OMTase (Blue), 2′-OMTase-Dolutegravir complex (Dark Green) and 2′-OMTase-Bictegravir complex (Orange).
The average values of the structural parameters including RMSD, RMSF, Rg, SASA, PCA and Intermolecular H-bonding has been described in Table 5 (3CLpro) and Table 6 (2′-OMTase).
Table 5.
Time averaged structural properties obtained from MD simulation of 3CLpro.
| Analysis | 3CLpro | 3CLpro-Raltegrevir | 3CLpro-Paritaprevir |
|---|---|---|---|
| Average RMSD (nm) | 0.295 | 0.361 | 0.401 |
| Average RMSF (nm) | 0.170 | 0.182 | 0.184 |
| Average Rg (nm) | 2.165 | 2.116 | 2.144 |
| PCA (Trace of Covariance Matrix nm2) | 115.482 | 136.366 | 132.059 |
| Average Intermolecular Hydrogen Bonds (Protein-Ligand) | – | 2 | 2 |
| Average SASA (nm2) | 133.89 | 131.26 | 131.79 |
Table 6.
Time averaged structural properties obtained from MD simulation of 2′-OMTase.
| Analysis | 2′-OMTase | 2′-OMTase-Dolutegravir | 2′-OMTase-Bictegravir |
|---|---|---|---|
| Average RMSD (nm) | 0.299 | 0.393 | 0.410 |
| Average RMSF (nm) | 0.155 | 0.161 | 0.165 |
| Average Rg (nm) | 1.796 | 1.799 | 1.805 |
| PCA (Trace of Covariance Matrix nm2) | 106.463 | 123.878 | 117.187 |
| Average Intermolecular Hydrogen Bonds (Protein-Ligand) | – | 2 | 1 |
| Average SASA (nm2) | 126.77 | 130.116 | 131.03 |
3.4.7. Comparative analysis of pre and post MD simulation
Structural superposition before and after the MD simulation of drug-free 3CLpro structures showed an RMSD of 2.41 Å. It indicates that the MD simulated 3CLpro structure was consistent and geometrically well optimised, which did not deviate, in comparison to the initial protein structure. Moreover, we also performed the structural superimposition analysis of drug bounded 3CLpro structures which explained that the RMSD before and after MD simulation of 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex was reliable and stable with the values of 2.15 Å and 2.47 Å, respectively (see Figure 11).
Figure 11.

Structural superimposition of the initial and MD simulated free 3CLpro, 3CLpro-Raltegravir complex and 3CLpro-Paritaprevir complex.
Structural superposition of pre and post MD simulation of drug-free 2′-OMTase structures showed an RMSD of 2.115 Å. Besides, we also carried out the same analysis for both drug bounded structures of 2′-OMTase. Also, we performed the same structural superimposition analysis of drug bounded structures 2′-OMTase, which explained that the RMSD before and after MD simulation of 2′-OMTase-Dolutegravir complex and 2′-OMTase-Bictegravir complex was reliable and stable with the values of 1.767 Å and 2.568 Å, respectively (see Figure 12).
Figure 12.

Structural superimposition of the initial and MD simulated free 2′-OMTase, 2′-OMTase-Bictegravir complex and 2′-OMTase-Dolutegravir complex.
This is additionally supported that these two drugs were oriented in the active site of both proteins throughout the 100 ns MD simulations.
4. Conclusion
The current study aimed to screen pre-existing antiviral drugs and come up with potential antidotes against SARS-CoV-2. The advantages of drug repurposing (also known as drug repositioning, drug reprofiling and drug retasking) include: the risk of the drug failing the safety and toxicity tests is much reduced, reduced costs during clinical trials and finally, significantly reduced time scale required for drug development (Pushpakom et al., 2019). Developing novel drugs or vaccines against SARS-CoV-2 will demand a lot of time, which we cannot afford at present. SARS-CoV-2 is spreading at a rapid rate; therefore, it is rational to look for pre-existing drugs and formulations, which can control this highly contagious virus. The precautionary steps taken by governments all over the world include social distancing, quarantines and lockdowns, which themselves have adverse psychological and economic repercussions (Madhav et al., 2017). The WHO also recognises the vast potential of drug repurposing approach, and on 18 March 2020, they announced ‘SOLIDARITY’ (World Health Organization, 2020d) global mega trial of the four most promising COVID-19 treatments. In the present study, we have identified two drugs against 3CLpro (Raltegravir and Paritaprevir) and two drugs against 2′-OMTase (Bictegravir and Dolutegravir), which are supposed to act as potential inhibitors against COVID-19. The proposed drug candidates obtained from this computational study could be efficacious in developing a much-needed threauptics against SARS-CoV-2, and it will further strengthen the concept of drug repurposing. However, being not able to replicate the physiological conditions entirely is one of the main drawbacks of computational biology (Gimeno et al., 2019; Marklund & Benesch, 2019) and therefore, further in vivo/in vitro validations are required to confirm the findings of this study. Raltegravir (Markowitz et al., 2007), Bictegravir (Sax et al., 2017) and Dolutegravir (Cahn et al., 2013; Walmsley et al., 2013) belong to the family of integrase strand transfer inhibitors (INSTI). They are already being used in antiretroviral therapy (ART) against the Human Immuno-deficiency Virus (HIV) infections. The final drug, Paritaprevir(Feld et al., 2016; Sulkowski et al., 2015), is being used to treat chronic Hepatitis C Virus (HCV) infections. It suppresses viral replication by inhibiting the non-structural 3/4 A protease of HCV (Carrion et al., 2014). Finally, the findings of this study might help develop antiviral therapeutics against SARS-CoV-2, expediting medical countermeasure advancement and should serve to persuade researchers and physicians to strengthen their efforts on discovering novel uses for existing medications.
Supplementary Material
Glossary
Abbreviations
- 3CLpro
3C-like proteinase
- 2′-O-MTase
2′-O-ribose methyltransferase
- SARS-CoV-2
Acknowledgements
Dr. Amit Kumar Singh thanks the Department of Science and Technology (DST) and Indian National Science Academy (INSA), New Delhi, India. Gizachew Muluneh Amera thanks the College of Natural Science, Wollo University, Dessie, Ethiopia for the sponsorship. The authors also thank to Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethical standards
Ethical standards are compulsory for studies relating to human and animal subjects.
References
- Abraham M. J., Lindahl D., van der Spoel E., Hess B., & the GROMACS development team (2014). GROMACS User Manual version, 5(2), 1–298.
- Ambure P., Bhat J., Puzyn T., & Roy K. (2019). Identifying natural compounds as multi-target-directed ligands against Alzheimer’s disease: An in silico approach. Journal of Biomolecular Structure and Dynamics, 37(5), 1282–1306. doi: 10.1080/07391102.2018.1456975 [DOI] [PubMed] [Google Scholar]
- Amera G. M., Khan R. J., Pathak A., Jha R. K., Muthukumaran J., & Singh A. K. (2019). Screening of promising molecules against MurG as drug target in multi-drug-resistant-Acinetobacter baumannii – Insights from comparative protein modeling, molecular docking and molecular dynamics simulation. Journal of Biomolecular Structure and Dynamics, 1–23. doi: 10.1080/07391102.2019.1700167 [DOI] [PubMed] [Google Scholar]
- Amera G. M., Khan R. J., Pathak A., Kumar A., & Singh A. K. (2019). Structure based in-silico study on UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase (MurE) from Acinetobacter baumannii as a drug target against nosocomial infections. Informatics in Medicine Unlocked, 16, 100216. doi: 10.1016/j.imu.2019.100216 [DOI] [Google Scholar]
- Benkert P., Biasini M., & Schwede T. (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics, 27(3), 343–350. doi: 10.1093/bioinformatics/btq662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C., Grigera J. R., & Straatsma T. P. (1987). The missing term in effective pair potentials. The Journal of Physical Chemistry, 91(24), 6269–6271. doi: 10.1021/j100308a038 [DOI] [Google Scholar]
- Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., & Bourne P. E. (2000). The Protein Data Bank. Nucleic Acids Research, 28(1), 235–242. doi: 10.1093/nar/28.1.235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L., Kiefer F., Arnold K., Benkert P., Battey J., & Schwede T. (2009). Protein structure homology modeling using SWISS-MODEL workspace. Nature Protocols, 4(1), 1–13. doi: 10.1038/nprot.2008.197 [DOI] [PubMed] [Google Scholar]
- Cahn P., Pozniak A. L., Mingrone H., Shuldyakov A., Brites C., Andrade-Villanueva J. F., Richmond G., Buendia C. B., Fourie J., Ramgopal M., Hagins D., Felizarta F., Madruga J., Reuter T., Newman T., Small C. B., Lombaard J., Grinsztejn B., Dorey D., … Min S. (2013). Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: Week 48 results from the randomised, double-blind, non-inferiority SAILING study. The Lancet, 382(9893), 700–708. doi: 10.1016/S0140-6736(13)61221-0 [DOI] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., & Madden T. L. (2009). BLAST+: Architecture and applications. BMC Bioinformatics, 10(1), 421. doi: 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrion A. F., Gutierrez J., & Martin P. (2014). New antiviral agents for the treatment of hepatitis C: ABT-450. Expert Opinion on Pharmacotherapy, 15(5), 711–716. doi: 10.1517/14656566.2014.889116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. (2020). Severe outcomes among patients with coronavirus disease 2019 (COVID-19) — United States, February 12–March 16, 2020. [DOI] [PMC free article] [PubMed]
- Chen H., Wei P., Huang C., Tan L., Liu Y., & Lai L. (2006). Only one protomer is active in the dimer of SARS 3C-like proteinase. Journal of Biological Chemistry, 281(20), 13894–13898. doi: 10.1074/jbc.M510745200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Su C., Ke M., Jin X., Xu L., Zhang Z., Wu A., Sun Y., Yang Z., Tien P., Ahola T., Liang Y., Liu X., & Guo D. (2011). Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2’-O-methylation by nsp16/nsp10 protein complex. PLoS Pathogens, 7(10), e1002294. doi: 10.1371/journal.ppat.1002294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S.-W., Pandit S. A., Scott H. L., & Jakobsson E. (2009). An improved united atom force field for simulation of mixed lipid bilayers. The Journal of Physical Chemistry B, 113(9), 2748–2763. doi: 10.1021/jp807056c [DOI] [PubMed] [Google Scholar]
- Colovos C., & Yeates T. O. (1993). Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Science, 2(9), 1511–1519. doi: 10.1002/pro.5560020916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wilde A. H., Snijder E. J., Kikkert M., & van Hemert M. J. (2018). Host factors in coronavirus replication. Current Topics in Microbiology and Immunology, 419, 1–42. doi: 10.1007/82_2017_25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld J. J., Moreno C., Trinh R., Tam E., Bourgeois S., Horsmans Y., Elkhashab M., Bernstein D. E., Younes Z., Reindollar R. W., Larsen L., Fu B., Howieson K., Polepally A. R., Pangerl A., Shulman N. S., & Poordad F. (2016). Sustained virologic response of 100% in HCV genotype 1b patients with cirrhosis receiving ombitasvir/paritaprevir/r and dasabuvir for 12weeks. Journal of Hepatology, 64(2), 301–307. doi: 10.1016/j.jhep.2015.10.005 [DOI] [PubMed] [Google Scholar]
- Gimeno A., Ojeda-Montes M., Tomás-Hernández S., Cereto-Massagué A., Beltrán-Debón R., Mulero M., Pujadas G., & Garcia-Vallvé S. (2019). The light and dark sides of virtual screening: What is there to know? International Journal of Molecular Sciences, 20(6), 1375. doi: 10.3390/ijms20061375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A., Goñi J. R., Orozco M., & Gelpí J. L. (2015). Molecular dynamics simulations: Advances and applications. Advances and Applications in Bioinformatics and Chemistry, 8, 37–47. doi: 10.2147/AABC.S70333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., … Cao B. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. The Lancet, 395(10223), 497–506. doi: 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns Hopkins CSSE. (2020). Coronavirus COVID-19 global cases by Johns Hopkins CSSE Retrieved March 28, 2020 from https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
- Kitchen D. B., Decornez H., Furr J. R., & Bajorath J. (2004). Docking and scoring in virtual screening for drug discovery: Methods and applications. Nature Reviews Drug Discovery, 3(11), 935–949. doi: 10.1038/nrd1549 [DOI] [PubMed] [Google Scholar]
- Ko J., Park H., Heo L., & Seok C. (2012). GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Research, 40(W1), W294–W297. doi: 10.1093/nar/gks493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., & Karplus K. (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins: Structure, Function, and Bioinformatics, 77(S9), 114–122. doi: 10.1002/prot.22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A., Jabłońska J., Pravda L., Vařeková R. S., & Thornton J. M. (2018). PDBsum: Structural summaries of PDB entries. Protein Science., 27(1), 129–134. doi: 10.1002/pro.3289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski R. A., MacArthur M. W., Moss D. S., & Thornton J. M. (1993). PROCHECK: A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography, 26(2), 283–291. doi: 10.1107/S0021889892009944 [DOI] [Google Scholar]
- Lugari A., Betzi S., Decroly E., Bonnaud E., Hermant A., Guillemot J.-C., Debarnot C., Borg J.-P., Bouvet M., Canard B., Morelli X., & Lécine P. (2010). Molecular mapping of the RNA cap 2’-O-methyltransferase activation interface between severe acute respiratory syndrome coronavirus nsp10 and nsp16. Journal of Biological Chemistry, 285(43), 33230–33241. doi: 10.1074/jbc.M110.120014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthy R., Bowie J. U., & Eisenberg D. (1992). Assessment of protein models with three-dimensional profiles. Nature, 356(6364), 83–85. doi: 10.1038/356083a0 [DOI] [PubMed] [Google Scholar]
- Madhav N., Oppenheim B., & Gallivan M. (2017). Disease control priorities: Improving health and reducing poverty (3rd ed). The International Bank for Reconstruction and Development. [PubMed] [Google Scholar]
- Marklund E. G., & Benesch J. L. P. (2019). Weighing-up protein dynamics: The combination of native mass spectrometry and molecular dynamics simulations. Current Opinion in Structural Biology, 54, 50–58. doi: 10.1016/j.sbi.2018.12.011 [DOI] [PubMed] [Google Scholar]
- Markowitz M., Nguyen B.-Y., Gotuzzo E., Mendo F., Ratanasuwan W., Kovacs C., Prada G., Morales-Ramirez J. O., Crumpacker C. S., Isaacs R. D., Gilde L. R., Wan H., Miller M. D., Wenning L. A., & Teppler H. (2007). Rapid and durable antiretroviral effect of the HIV-1 integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: Results of a 48-week controlled study. JAIDS Journal of Acquired Immune Deficiency Syndromes, 46(2), 125–133. doi: 10.1097/QAI.0b013e318157131c [DOI] [PubMed] [Google Scholar]
- Menachery V. D., Debbink K., & Baric R. S. (2014). Coronavirus non-structural protein 16: Evasion, attenuation, and possible treatments. Virus Research, 194, 191–199. doi: 10.1016/j.virusres.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D., Yount B. L., Josset L., Gralinski L. E., Scobey T., Agnihothram S., Katze M. G., & Baric R. S. (2014). Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2’-o-methyltransferase activity. Journal of Virology, 88(8), 4251–4264. doi: 10.1128/JVI.03571-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheletti C. (2013). Comparing proteins by their internal dynamics: Exploring structure-function relationships beyond static structural alignments. Physics of Life Reviews, 10(1), 1–26. doi: 10.1016/j.plrev.2012.10.009 [DOI] [PubMed] [Google Scholar]
- Pushpakom S., Iorio F., Eyers P. A., Escott K. J., Hopper S., Wells A., Doig A., Guilliams T., Latimer J., McNamee C., Norris A., Sanseau P., Cavalla D., & Pirmohamed M. (2019). Drug repurposing: Progress, challenges and recommendations. Nature Reviews Drug Discovery, 18(1), 41–58. doi: 10.1038/nrd.2018.168 [DOI] [PubMed] [Google Scholar]
- Ramachandran G. N., Ramakrishnan C., & Sasisekharan V. (1963). Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology, 7(1), 95–99. doi: 10.1016/S0022-2836(63)80023-6 [DOI] [PubMed] [Google Scholar]
- Sax P. E., Pozniak A., Montes M. L., Koenig E., DeJesus E., Stellbrink H.-J., Antinori A., Workowski K., Slim J., Reynes J., Garner W., Custodio J., White K., SenGupta D., Cheng A., & Quirk E. (2017). Coformulated bictegravir, emtricitabine, and tenofovir alafenamide versus dolutegravir with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection (GS-US-380–1490): A randomised, double-blind, multicentre, phase 3, non-inferiority trial. The Lancet, 390(10107), 2073–2082. doi: 10.1016/S0140-6736(17)32340-1 [DOI] [PubMed] [Google Scholar]
- Schuttelkopf A. W., & van Aalten D. M. (2004). PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D Biological Crystallography, 60(8), 1355–1363. doi: 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
- Su S., Wong G., Shi W., Liu J., Lai A. C. K., Zhou J., Liu W., Bi Y., & Gao G. F. (2016). Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends in Microbiology., 24(6), 490–502. doi: 10.1016/j.tim.2016.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulkowski M. S., Eron J. J., Wyles D., Trinh R., Lalezari J., Wang C., Slim J., Bhatti L., Gathe J., Ruane P. J., Elion R., Bredeek F., Brennan R., Blick G., Khatri A., Gibbons K., Hu Y. B., Fredrick L., Schnell G., … Podsadecki T. (2015). Ombitasvir, paritaprevir co-dosed with ritonavir, dasabuvir, and ribavirin for hepatitis C in patients co-infected with HIV-1: A randomized trial. JAMA, 313(12), 1223–1231. doi: 10.1001/jama.2015.1328 [DOI] [PubMed] [Google Scholar]
- Trott O., & Olson A. J. (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. doi: 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UCLA MBI The structure analysis and verification server Retrieved from https://servicesn.mbi.ucla.edu/SAVES/
- Walmsley S. L., Antela A., Clumeck N., Duiculescu D., Eberhard A., Gutiérrez F., Hocqueloux L., Maggiolo F., Sandkovsky U., Granier C., Pappa K., Wynne B., Min S., & Nichols G. (2013). Dolutegravir plus abacavir–lamivudine for the treatment of HIV-1 infection. New England Journal of Medicine, 369(19), 1807–1818. doi: 10.1056/NEJMoa1215541 [DOI] [PubMed] [Google Scholar]
- Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., & Schwede T. (2018). SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Research, 46(W1), W296–W303. doi: 10.1093/nar/gky427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S. R., & Leibowitz J. L. (2011). Coronavirus pathogenesis. Advances in Virus Research, 81, 85–164. doi: 10.1016/b978-0-12-385885-6.00009-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S., Feunang Y. D., Guo A. C., Lo E. J., Marcu A., Grant J. R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., … Wilson M. (2018). DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Research, 46(D1), D1074–D1082. doi: 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2003, December). Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003 Retrieved March 28, 2020 from http://www.who.int/csr/sars/country/table2004_04_21/en/index.html
- World Health Organization. ( 2019, November). Middle East respiratory syndrome coronavirus (MERS-CoV) Retrieved March 28, 2020 from https://www.who.int/emergencies/mers-cov/en/
- World Health Organization (2020a). Naming the coronavirus disease (COVID-19) and the virus that causes it Retrieved March 28, 2020 from https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it
- World Health Organization (2020b). WHO Director-General’s opening remarks at the media briefing on COVID-19 – 3 March 2020 Retrieved March 28, 2020 from https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19—3-march-2020
- World Health Organization. (2020c). WHO Director-General’s opening remarks at the media briefing on COVID-19 – 11 March 2020. Retrieved March 28, 2020 from https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19—11-march-2020
- World Health Organization. (2020. d). WHO Director-General’s opening remarks at the media briefing on COVID-19 – 18 March 2020. Retrieved March 28, 2020.
- World Health Organization (2020. e). Statement on the second meeting of the International Health Regulations (2005) Emergency Committee regarding the outbreak of novel coronavirus (2019-nCoV) Retrieved March 28, 2020 from https://www.who.int/news-room/detail/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov)
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., & Rao Z. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proceedings of the National Academy of Sciences of Sciences, 100(23), 13190–13195. doi: 10.1073/pnas.1835675100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., & Tan W. (2020). A novel coronavirus from patients with pneumonia in China, 2019. New England Journal of Medicine, 382, 727–733. doi: 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.