

Abstract
Peroxiredoxins from the Prx1 subfamily (Prx) are moonlighting peroxidases that operate in peroxide signaling and are regulated by sulfinylation. Prxs offer a major model of protein–thiol oxidative modification. They react with H2O2 to form a sulfenic acid intermediate that either engages into a disulfide bond, committing the enzyme into its peroxidase cycle, or again reacts with peroxide to produce a sulfinic acid that inactivates the enzyme. Sensitivity to sulfinylation depends on the kinetics of these two competing reactions and is critically influenced by a structural transition from a fully folded (FF) to locally unfolded (LU) conformation. Analysis of the reaction of the Tsa1 Saccharomyces cerevisiae Prx with H2O2 by Trp fluorescence-based rapid kinetics revealed a process linked to the FF/LU transition that is kinetically distinct from disulfide formation and suggested that sulfenate formation facilitates local unfolding. Use of mutants of distinctive sensitivities and of different peroxide substrates showed that sulfinylation sensitivity is not coupled to the resolving step kinetics but depends only on the sulfenic acid oxidation and FF-to-LU transition rate constants. In addition, stabilization of the active site FF conformation, the determinant of sulfinylation kinetics, is only moderately influenced by the Prx C-terminal tail dynamics that determine the FF → LU kinetics. From these two parameters, the relative sensitivities of Prxs toward hyperoxidation with different substrates can be predicted, as confirmed by in vitro and in vivo patterns of sulfinylation.
Keywords: enzyme, thiol peroxidase, sulfenic acid, sulfinic acid, redox regulation, conformational transition, rapid kinetics
Introduction
Sulfinylation of cysteine (Cys) thiolate is increasingly recognized as important in cell regulation and signaling.1 Eukaryotic peroxiredoxins (Prxs) from the Prx1 subfamily are thiol peroxidases and the first enzyme family shown to be regulated by reversible Cys sulfinylation, an hitherto thought irreversible oxidative modification that coevolved with the sulfinic acid reductase sulfiredoxin (Srx).2−5 Prxs are major peroxide-reducing enzymes, reacting with substrates by attack of a peroxidatic Cys CP that becomes sulfenylated (CP–SOH). In the peroxidatic cycle, the CP–SOH condenses with the enzyme resolving Cys CR to form a disulfide bond, the reduction of which by thioredoxin (Trx) completes the cycle (Figure 1a). Alternatively, the CP–SOH can react with a second peroxide molecule to form a sulfinic acid (CP–SO2H), which inactivates the enzyme. Prxs inactivation by sulfinylation (also referred to as hyperoxidation) is thought as critical for allowing unimpeded H2O2 cell signaling, as if active, they would, by their high abundance and high substrate reactivity, quench the H2O2 signal.3,6 For instance, an Srx-dependent 24 h rhythmic sulfinylation of Prx3 occurs in mammalian mitochondria, which fosters H2O2 mitochondrial buildup and rhythmic cytosolic release, thereby contributing to establish the circadian nature of corticosteroid secretion.7 As a major target of Trx, Prx sulfinylation serves to free the function of Trx to other targets during severe oxidative stress.8 Prx sulfinylation also provides a gain of function by stabilizing its oligomeric structure, thereby switching it into a molecular chaperone9 or fostering protein interactions.10
Figure 1.

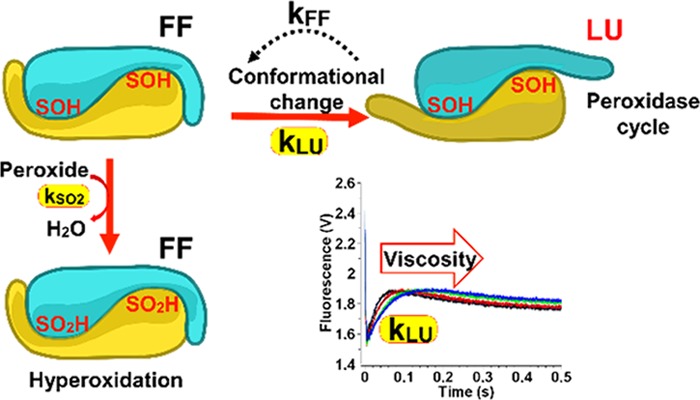
(a) Prx1 catalytic cycle. The Prx1 peroxidase cycle and hyperoxidation mechanism are shown at the level of the Prx1 dimeric unit to highlight the conformational state [fully folded (FF) and locally unfolded (LU)] of the active site identified by the peroxidatic Cys CP and C-terminal tail identified by resolving Cys CR. The rate constants of peroxide reduction kSOH, sulfinylation kSO2, resolution kSS, and FF–LU transition kFF and kLU are indicated. (b) Zoom into the catalytic site of Tsa1. The subunit containing the CP is shown in blue, and the subunit containing the CR is shown in yellow (PDB code 3SBC). Residues CP and CR are shown in yellow, W83 and W173 in orange, and W161 in red. Within the C-terminal tail, residues A177 and A178 are highlighted in green and the Y190-F191 motif in purple.
Prokaryotic Prxs are resistant to hyperoxidation, whereas eukaryotic ones, especially the Prx1 subfamily, are sensitive.3 Further, within sensitive Prxs, a large range of sulfinylation sensitivity levels exists, as for instance the higher sensitivity of human Prx2 relative to Prx1 and Prx3.11,12 These differences may indicate differential enzyme regulation depending on the concentration of H2O2.13 Karplus and collaborators identified structural signatures that distinguish hyperoxidation-sensitive Prxs from the resistant ones, one of which is a Tyr-Phe motif located in the C-terminal helix, and proposed a mechanistic model of how these features generate sensitivity3 (Figure 1). Prxs are obligate symmetrical homodimers in which both subunits contribute to the assembly of each of the two active sites by providing either the CP or the C-terminal tail-CR to one site (Figure 1b). In the Karplus model, reduced Prx exists in a fully folded (FF) conformation that stabilizes an active site, endowing CP with extraordinary H2O2 reactivity. A conformational transition from the FF to a locally unfolded (LU) state, in which the helix carrying CP and the enzyme C-terminal tail partially unfold, enables CP–SOH and CR condensation into a disulfide bond. When the enzyme is in the FF state, however, the CP–SOH can further react with H2O2 to generate sulfinic acid. Enzyme sulfinylation and the conformational FF–LU transition equilibrium appear positioned as two reciprocally exclusive competing events. In this model, the Tyr-Phe C-terminal helix motif, by stabilizing to the FF conformation, generates sulfinylation sensitivity3,14−16 (Figure 1b).
Kinetic and mass spectrometry-based evaluation of Prx intrinsic hyperoxidation sensitivity has so far relied on the hypothesis that the FF–LU transition is in rapid equilibrium, that is, that the FF → LU and LU → FF processes occur much faster than the resolving and hyperoxidation steps.11,12 A steady-state sensitivity index Chyp1%, defined as the concentration of H2O2 required to oxidize 1% of the sites in one enzyme catalytic cycle, has been derived from the above mechanistic model as a function of the kinetic rate constants of the disulfide formation step, kSS; the hyperoxidation step, kSO2; and the FF–LU equilibrium constant KLU(17) (Figure 1a). These methods enabled the identification of other structural determinants of hyperoxidation sensitivity in the C-terminal tail, in the CP environment and in the dimer–dimer interface.18−20 In addition, pre-steady-state analyses using rapid kinetic approaches allowed resolution of the Prx catalytic steps for sulfenic acid and disulfide formation13,21,22 and, recently, for hyperoxidation.12 However, assessing nonchemical steps within a catalytic cycle is challenging, and thus few studies have provided direct information on FF → LU and LU → FF transition kinetics,23 which is critical to understand the origin of Prx sulfinylation sensitivity.
We address here the hyperoxidation mechanism of the major Saccharomyces cerevisiae Prx1-type enzyme Tsa1 by pre-steady-state and steady-state kinetics using Trp fluorescence, circular dichroism (CD), and in vivo analysis. The reaction of Tsa1 with peroxides monitored in single turnover using a stopped-flow apparatus showed multiphasic kinetics, of which one is kinetically distinct from the resolving step and can be assigned to a conformation change associated with the FF–LU transition, thus suggesting that the FF–LU transition does not fulfill the proposed rapid equilibrium hypothesis. Using this approach, mutants of distinctive sensitivities, and the comparison of H2O2 and organic peroxide substrates, we established that hyperoxidation sensitivity is independent of the resolving step kinetics, allowing us to refine the hyperoxidation index Chyp1%, which in fact appears to only depend on the sulfinylation and FF-to-LU transition rate constants. Our results also suggest that the molecular determinants that control each of these two parameters are distinct. We now can calculate Chyp1%, a critical parameter for in vivo modeling of redox regulation. Understanding the sulfinylation mechanism is also relevant in view of the recently discovered new sulfiredoxin sulfinylated substrates.1
Results
Disulfide Bond Formation Is Kinetically Resolved from a Conformational Process in Tsa1 Peroxidase Cycle
To kinetically resolve the catalytic steps, the reaction of Tsa1 with H2O2 was explored by monitoring the change of enzyme Trp fluorescence over time under single turnover conditions in the absence of Trx. Tsa1 contains three Trp residues, one located close to the active site (W83), another in helix α5 preceding the C-terminal tail (W161), and the last in the C-terminal tail close to the resolving Cys (W173) (Figure 1b). The intrinsic sensitivity of Trp fluorescence to alterations of the redox state of Tsa1, with 62 and 124% emission change for the CP–CR disulfide (Tsa1SS) and hyperoxidized (Tsa1SO2) forms, respectively, relative to the reduced enzyme (Tsa1red), provided a powerful reaction-monitoring probe (Figure 2a). The reaction of Tsa1 (5 μM) with H2O2 generated three-exponential kinetics, fast (phase 1) and slow (phase 3) ones, both displaying a decrease in fluorescence, which flanked the remaining one (phase 2) characterized by a fluorescence increase (Figure 2b). At lower amounts of Tsa1 and H2O2, the phase 1 rate constant appeared linearly dependent on the H2O2 concentration (Figure 2d,e), indicating a second-order, bimolecular process, which could thus correspond either to H2O2 binding, a process expected to be reversible, or to the irreversible Cys CP attack on H2O2.22 However, the low y-intercept of the k1obs/H2O2 plot (Figure 2e), which implied an essentially irreversible process, and the absence of signal with the CP to Ser mutant Tsa1C48S, which indicated a requirement of Cp (Figure S1), suggest that the kinetic phase 1 reflects the CP attack on H2O2 and concomitant CP–SOH intermediate formation. The phase 1 second-order rate constant k1 of 9.7 × 107 M–1 s–1 (Table 1) fits previously reported values.24 In contrast, phases 2 and 3 were characterized by H2O2-independent observed rate constants k2 and k3, respectively (Figure 2c), and therefore reflect first-order, monomolecular events that do not directly involve H2O2 as a reactant. As a first approach to assign the second and third phases to mechanistic steps, we analyzed by mass spectrometry the nature of redox products obtained after completion of the reaction of reduced Tsa1 with 10 μM H2O2 (Figure 2f). Dimeric Tsa1SS was by far the major species, thus indicating that no hyperoxidation occurred in these conditions. We thus hypothesized that phases 2 and 3, which are characterized by rate constants k2 of 64 s–1 and k3 of 4.6 s–1 (Table 1), reflect either a conformational change associated with the FF-to-LU transition or CP–CR disulfide formation.
Figure 2.

Wild-type Tsa1 oxidizes H2O2 by three-phase kinetics. (a) Trp fluorescence emission spectra of reduced (black), disulfide (red), and hyperoxidized (blue) Tsa1 (2 μM) after excitation at 295 nm. (b) Pre-steady-state kinetics for the reaction of Tsa1 (5 μM) with H2O2 (10 μM, light gray; 25 μM, gray; and 50 μM, black) monitored by Trp fluorescence. The excitation wavelength is set at 295 nm, and the signal is collected above 320 nm. The time courses are shown in log time scale to highlight the three phases and fitted against a three-exponential equation (red lines). In the inset, time courses are shown in linear X-scale. Each curve is the average of six runs. (c) Second-order plots and linear fits of the observed rate constants k1obs (circles, black line), k2 (squares, red line), and k3 (diamond, green line) vs H2O2 concentration. Very similar results were obtained using the untagged native Tsa1, showing that the N-terminal His tag has no impact on Tsa1 catalysis. (d) Pre-steady-state kinetics for the reaction of Tsa1 (0.5 μM) with low H2O2 concentrations (from 0.5 to 5 μM) monitored by Trp fluorescence. Only phases 1 and 3 are observable in these conditions. Each trace is the average of six runs and is fitted according to a biphasic equation (red line). (e) Precise determination of k1 by a second-order plot and linear fit of the fast phase rate constants k1obs measured in (d). (f) Superimposed deconvoluted mass spectra of the Tsa1 redox species before and after 5 s reaction with 10 μM H2O2 followed by acid quenching.
Table 1. Steady-State, Pre-Steady-State, and Hyperoxidation Kinetics Parameters of Wild-Type and Mutant Tsa1 with H2O2.
| H2O2 |
||||||
|---|---|---|---|---|---|---|
| Tsa1 | Tsa1Y190G F191G | Tsa1A177S A178D | wild-type | Tsa1W161F | ||
| ksteadystateapparent (s–1) | 2.4 ± 0.1 | 2.6 ± 0.1 | 2.4 ± 0.2 | global fit | 2.8 ± 0.5 | |
| Chyp1% (μM) | X | 2620 ± 220 | 730 ± 100 | 440 ± 90 | ||
| k1 (M–1 s–1) | 1.7 × 107a | (4 ± 1.4) × 107 | (9.7 ± 0.4) × 107 | kSOH | (1.0 ± 0.01) × 108 | (6.7 ± 0.4) × 107 |
| k2 (s–1) | 216 ± 41 | 64 ± 5 | kLU | 65 ± 1 | 37 ± 1 | |
| k3 (s–1) | 3 ± 0.1 | 5.3 ± 0.6 | 4.6 ± 0.3 | kSS | 5.8 ± 0.1 | 7.0 ± 0.6 |
| k4 (M–1 s–1)b | 0 | (1.0 ± 0.4) × 103 | (2.1 ± 0.2) × 103 | kSO2 | (2.9 ± 0.1) × 103 | (2.9 ± 0.6) × 103 |
Up to 50 μM.
Measured on C171A mutants. Data are reported as the mean value obtained from two independent experiments performed on distinct protein productions ± standard deviation (s.d.).
To test whether phases 2 or 3 could be attributed to a conformational transition, we repeated the stopped-flow experiment in the presence of increasing concentration of saccharose, a viscogen expected to slow down protein motions without interfering with chemical processes.25,26 Conditions were chosen to ensure kinetic resolution of the three phases, that is, 5 μM Tsa1 and 10 μM H2O2. The efficiency of the stopped-flow mixer in viscous solutions was verified by mixing Trp and up to 30% saccharose (Figure S2). As shown in Figure 3a, increasing saccharose from 0 to 30% significantly reduced k2 by 75%, without altering k1obs and k3 or only very slightly. Similar results were obtained with fructose or sorbitol as viscogen (Figure S3). To establish whether phase 2 corresponded to a conformation change linked to the FF-to-LU transition, we analyzed the behavior of the Y190G-F191G Tsa1 mutant (Tsa1Y190G F191G), which is expected to exist primarily in the LU conformation due to C-terminal α helix destabilization.3 The conformational state of the protein was first assessed by circular dichroism (Figure 3b). The spectrum of reduced Tsa1, which exists in the FF conformation,24 exhibited three positive maxima at 262, 287, and 296 nm. The spectrum of Tsa1SS, which adopts the LU conformation, exhibited a minimum at 277 nm and a positive maximum at 295 nm, thus showing much larger difference upon oxidation compared to human Prx1 (hPrx1) and human Prx2 (hPrx2).12,16 Further, the reduced Tsa1Y190G F191G spectrum was similar to the Tsa1SS one and only slightly altered upon H2O2 oxidation, which is consistent with the LU conformation, as previously observed for hPrx2.16 Similar results were obtained in the far-UV CD Tsa1 spectra (Figure S4). Kinetics of Tsa1Y190G F191G oxidation by H2O2 revealed a biphasic profile, the first phase of which has a H2O2-dependent rate constant k1 of 1.7 × 107 M–1 s–1 up to 50 μM, and then, it reaches a plateau at ∼120 s–1, likely corresponding to CP–SOH formation (Figure 3c). The second slower process was characterized by a fixed rate constant of 3.0 ± 0.1 s–1 close to the k3 value measured for the wild type (Figure 3c and Table 1). However, the viscogen-sensitive phase 2 was absent here, strongly suggesting that in the wild-type enzyme this phase reflects a protein structural motion associated with the FF–LU transition.
Figure 3.

Attribution of phase 2 to a conformational transition. (a) Effect of saccharose (0% black, 10% red, 20% green, and 30% blue) on the reaction of Tsa1 (5 μM) with H2O2 (10 μM) monitored as in Figure 2, fitted against a three-exponential equation (red lines). Inset: effect of saccharose concentration on rate constants k1obs, k2, and k3 normalized to 0% saccharose. The stopped-flow mixer efficiency in viscous solutions was established by mixing Trp and up to 30% saccharose, which showed no artifactual effects on the dilution kinetics (Figure S2). (b) Near-UV circular dichroism spectra of 50 μM wild-type Tsa1 (plain) and Tsa1Y190G F191G (dashed line) under the reduced (black) and disulfide (red) forms. Measurements were performed in a 1 cm cuvette in a phosphate 10 mM and NaF 100 mM buffer (pH 7) and are the average of three records. (c) Pre-steady-state kinetics for the reaction of Tsa1 (5 μM) with H2O2 (5, 10, 25, 50, 100, 200, 400, and 800 μM, light gray to black) monitored as in Figure 2, fitted against a biexponential equation (red lines). Inset: second-order plots and linear fits of the observed rate constants for the fast phase k1obs (circles, black line) and slow phase k3 (diamond, green line). (d) Pre-steady-state kinetics for the reaction of Tsa1 (50 μM) with H2O2 (100 μM) monitored by a near-UV CD signal at 270 nm. The trace is the average of 50 runs, and the first-order fit is shown in red. (e) Effect of saccharose (0% black, 15% red, and 30% blue) on the reaction of human Prx1 (5 μM) with H2O2 (10 μM) monitored as in Figure 2 and fitted against a biexponential equation (red or black lines). Inset: effect of saccharose concentration on the rate constant of the increasing phase normalized to 0% saccharose.
To further support this conclusion, we independently monitored the FF–LU transition using the large near-UV CD signal change observed between Tsa1 and Tsa1SS at 270 nm. The low sensitivity of the CD signal in stopped-flow mode required use of 50 μM wild type reduced Tsa1 and 100 μM H2O2, which prevented visualizing the first phase of predicted very fast observed rate constant of 104 s–1. Under these conditions, the near-UV CD-monitored reaction obeyed monophasic kinetics characterized by a rate constant of 54 ± 2 s–1 close to the k2 value of 64 s–1 measured using Trp fluorescence, thus suggesting that the same process is monitored (Figure 3d). Overall, these data support that phase 2 reflects a conformation change linked to the FF–LU transition. Since dimeric Tsa1SS is the final product of the reaction in these conditions (Figure 2f), phase 3 must then correspond to the formation of the CP–CR disulfide bond.
To extend these results, we compared the kinetics of H2O2 reduction of human Prx1 (hPrx1) and Tsa1 by Trp fluorescence. The hPrx1 reaction with 10 μM H2O2 obeyed biphasic kinetics (Figure 3e) as reported,12 which consists in a fast decreasing phase equivalent to Tsa1 phase 1 (sulfenic acid formation) and a second increasing phase of rate constant 10.8 s–1 previously attributed to CP–CR disulfide formation.12 The latter rate constant value was strongly dependent on saccharose concentration, indicating that, as for Tsa1, the kinetics of this phase is at least in part controlled by a process associated with a conformation change (Figure 3e, inset). Thus, in contrast to Tsa1, the hPrx1 FF–LU associated process and disulfide bond formation are not resolved and hPrx1 phase 2 thus kinetically reflects the conformational event.
Tsa1 Hyperoxidation Sensitivity and Conformation Change Kinetics Are Correlated
To correlate the kinetics of the above individual events with the sensitivity to hyperoxidation, we used the method developed by Nelson,17 which provides a measure of the hyperoxidation sensitivity index Chyp1% from the fraction of hyperoxidized enzyme per catalytic cycle in multiple turnover conditions, that is, in the steady state in the presence of the Trx system. Deviation from linear kinetics resulting from enzyme oxidative inactivation is monitored here by the consumption of reduced nicotinamide adenine dinucleotide phosphate (NADPH) at 340 nm in a NADPH/Tsa1/Trx/Trx reductase coupled assay (Figure 4a). As shown previously,17 the deduced rate constant of inactivation normalized to the peroxidase turnover rate constant (finact) was linearly dependent on H2O2 concentration, yielding a slope value that reflected the sensitivity to hyperoxidation (Figure 4b) and provided the Chyp1% value, that is, the concentration of H2O2 required to hyperoxidize 1% of sites in one cycle. For hPrx1, we measured a Chyp1% of 80 μM, in good correlation with the reported value of 62 μM.17 Surprisingly, Tsa1 appeared 9-fold less sensitive than hPrx1 (Figure 4b), with a Chyp1% of 730 μM (Table 1).
Figure 4.
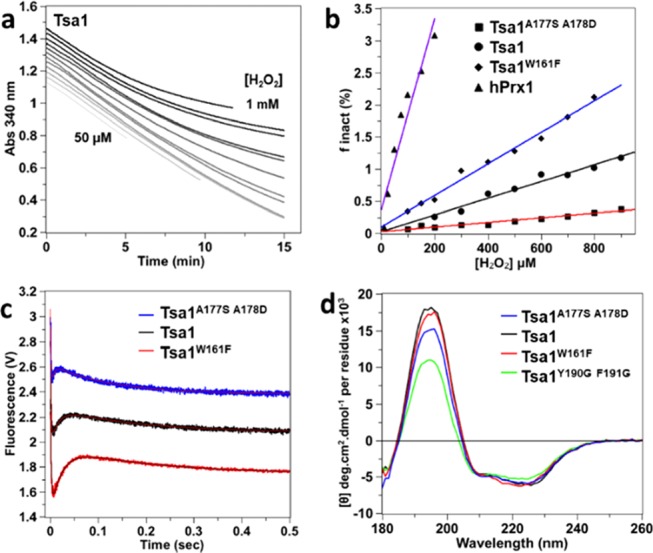
Steady-state hyperoxidation sensitivity of wild-type and mutant Tsa1 with H2O2. (a) Steady-state kinetics for the determination of hyperoxidation sensitivity of Tsa1 monitored by consumption of NADPH (200 μM) at 340 nm in the presence of thioredoxin reductase (0.25 μM), Trx (5 μM), Tsa1 (1 μM), and variable amounts of H2O2 (from 50, 100, 150, 200, 300, etc. to 1 mM) in TK buffer. The time courses have been shifted on the y axis for clarity. (b) Secondary plot of the inactivated fraction finact per turnover deduced from (a) vs H2O2 concentration. The hyperoxidation index Chyp1% is deduced from the slope of the linear fit for wild type (black circles, black line fit), mutants Tsa1W161F (black diamonds, blue line fit) and Tsa1A177S A178D (black squares, red line fit), and hPrx1 (black triangles, purple line fit). Data are the mean of two independent experiments. (c) Pre-steady-state kinetics for the reaction of Tsa1A177S A178D, Tsa1, or Tsa1W161F (5 μM, top to bottom) with H2O2 (10 μM) monitored by Trp fluorescence as in Figure 2b, fitted against a three-exponential equation (red or black line). Time courses have been shifted on the y axis for clarity. (d) Far-UV CD spectra of 5 μM Tsa1 (black), Tsa1W161F (red), Tsa1A177S A178D (blue), and Tsa1Y190G F191G (green) under the reduced state. Measurements were performed in a 0.01 cm flat cell in phosphate (10 mM) NaF (100 mM) buffer (pH 7) and are the average of three records.
To identify mechanistic determinants of hyperoxidation sensitivity, we designed Tsa1 mutations located close to the C-terminal region that are expected to alter Chyp1%. The W161F mutant (Tsa1W161F) modifies the C-terminus of helix α5. The A177S-A178D mutant (Tsa1A177S A178D) replaces some known determinants of Chyp1% by the residues present at the same location in hPrx1,19 and the above-mentioned Tsa1Y190G F191G adopts mostly the LU conformation (Figures 1b and 3b). All mutants had wild-type peroxidase activity, reducing H2O2 with a steady-state rate constant of ca. 2.4–2.8 s–1 (Table 1). Tsa1Y190G F191G was not sensitive to hyperoxidation (Figure S5), as expected, whereas Tsa1W161F and Tsa1A177S A178D were more and less sensitive than the wild type, with Chyp1% of 440 and 2620 μM, respectively (Figure 4b and Table 1). Under single turnover conditions, these two mutants obeyed triphasic kinetics (Figure 4c), with rate constants k1 of 6.7 × 107 and 4 × 107 M–1 s–1 and k3 of 7.0 and 5.3 s–1, respectively, close to the wild type (Table 1, Figures S6, and S7). In contrast, their k2 were inversely correlated with hyperoxidation sensitivity, with values of 37 and 216 s–1 for Tsa1W161F and Tsa1A177S A178D, respectively, relative to the wild type (64 s–1). In keeping with the Chyp1% and k2 correlation, as the most hyperoxidation-sensitive enzyme, hPrx1 yielded a k2 of 10.2 s–1. Further, the presence of saccharose caused changes of Tsa1 mutant kinetics similar to those seen with wild type, supporting that phase 2 indeed identifies a conformational event also in both mutants (Figures S8 and S9). In addition, the far-UV CD spectra of reduced Tsa1W161F had a wild-type profile, whereas Tsa1A177S A178D displayed an intermediate signature between the FF and LU conformations, which suggested displacement of the equilibrium toward the LU conformation in the reduced state (Figure 4d). The inverse correlation seen between the sensitivity to hyperoxidation and the rate constant k2 thus suggests a direct competition between hyperoxidation and the FF–LU conformational transition, also excluding an influence of the kinetics of CP–CR disulfide formation on the sensitivity to hyperoxidation.
Direct Assessment of the Kinetics of Tsa1 Hyperoxidation
To complete dissection of the hyperoxidation mechanism, we sought to assess the sulfinylation rate constant (Figure 1a). To directly measure this parameter, we used the Tsa1C171A mutant, which, by lacking CR, does not form the catalytic disulfide bond and thus could provide a means of isolating the reaction of H2O2 with the CP–SOH intermediate. Upon reacting Tsa1C171A with H2O2 (>25 μM) under single turnover conditions, Trp fluorescence increased during 5 s with a biphasic kinetics. Here, the reaction of H2O2 with CP (phase 1) was too fast to be observed, since Tsa1C171A has a wild-type reactivity toward H2O2 (k1 = 7.1 × 107 M–1 s–1) (Figure S10). The first phase of small amplitude had an H2O2-independent rate constant of 2–3 s–1 and corresponds to a process that remains to be attributed. The second phase (phase 4) was slower, of larger but fixed amplitude, and had an H2O2-dependent rate constant k4obs, which indicated a bimolecular reaction involving H2O2, characterized by a second-order value of 2.1 × 103 M–1 s–1 (Table 1 and Figure 5a,b). Phase 4 suggested the occurrence of sulfinylation, based on its increasing fluorescence signal and on the increased fluorescence seen for sulfinylated Tsa1SO2 (Figure 2a). Furthermore, mass spectrometry identified Tsa1C171ASO2 as the major redox species upon completion of a reaction with 200 μM H2O2 (Figure 5c). These data thus indicate that phase 4 reflects the Tsa1C171A–SOH reaction with H2O2 that leads to enzyme hyperoxidation. The deduced rate constant of Tsa1C171A hyperoxidation k4 of 2.1 × 103 M–1 s–1 was in agreement with those obtained for human Prx1 and Prx2,12 suggesting similar kinetics of hyperoxidation of Prx1-type Prxs from diverse origins. When combined to C171A, the W161F (Tsa1W161F C171A) and A177S-A178D (Tsa1C171A A177S A178D) Tsa1 mutants had a similar hyperoxidation behavior to Tsa1C171A, characterized by rate constants k4 of 2.9 × 103 and 1.0 × 103 M–1 s–1, respectively (Figure 5b and Table 1).
Figure 5.

Direct observation of hyperoxidation kinetics with C171A mutants. (a) Pre-steady-state kinetics for the reaction of Tsa1C171A (5 μM) with H2O2 (25, 50, 200, 400, and 800 μM as indicated) monitored by Trp fluorescence, fitted against a biexponential equation (red lines). Phase 4 is labeled. The * indicates a small, faster phase that is not attributed. (b) Second-order plot and linear fit of the slow phase observed rate constant k4obs (deduced from panel (a) fits) against H2O2 concentration, for Tsa1C171A (black circles, black fit), Tsa1W161F C171A (black diamonds, black fit), and Tsa1C171A A177S A178D (black squares, red fit). (c) Deconvoluted mass spectra of the Tsa1C171A redox species before and after 5 s reaction with 200 μM H2O2, followed by acid quenching.
High Peroxide Level or Reactivity Recapitulates the Full Hyperoxidation Mechanism
To integrate all kinetic steps within the full mechanism, we then used conditions that allow hyperoxidation in single turnover for the wild-type enzyme to be observed. According to the hypothesis of a direct competition between hyperoxidation and the FF-to-LU transition, higher H2O2 concentrations should overcome the kinetic barrier of the conformation step, with phase 2 becoming a composite of the fluorescence signals of the two competing reactions. Tsa1 stopped-flow kinetic series were performed with increasing concentrations of H2O2, up to 10 mM. We observed that the phase 2 amplitude increased with H2O2, suggesting that this phase must at least in part incorporate an event generating a more fluorescent species, presumably the Tsa1SO2 (Figure 6a). These data were globally fitted against a model (Figure 6a,b) based on the Prx catalytic cycle (Figure 1), which returned intrinsic kLU, kSS, and kSO2 rate constants close to the k2, k3, and k4 observed values, respectively, obtained by direct fitting (Table 1). This confirmed that the hyperoxidation rate constant k4 obtained using the C171A mutants can be used as an estimate for kSO2. Furthermore, adding an additional step in which the Tsa1–SOH in the LU form reacts with H2O2 did not improve the fit, as expected. In addition, making the Tsa1–SOH LU-to-FF reaction reversible returned a very low rate constant value, kFF, and did not improve the fit either. These data suggest that the rapid equilibrium hypothesis does not apply as they imply that, although intrinsically reversible, the Tsa1–SOH FF-to-LU transition nevertheless behaves as a quasi-irreversible process in the catalytic cycle.
Figure 6.

Kinetic integration of all mechanistic steps. (a) Pre-steady-state kinetics for the reaction of Tsa1 (5 μM) with increasing H2O2 as indicated, monitored by Trp fluorescence as in Figure 2b. The data are globally fitted (red lines) using Kintek Explorer software and the model and equation shown in (b). It must be noted that kLU and kSS behaved as dependent parameters in the fit, although the concordance between kLU and the value deduced from CD monitoring (65 vs 54 s–1) validated the global fit results. (b) Kinetic model and equation used for Tsa1 reaction global fitting. Fluorescence intensity factors for Tsa1SS and Tsa1SO2 were fixed at 62 and 124% of the Tsa1–SH value (p) (based on Figure 2a) and intensity factors for Tsa1FF–SOH (q) and Tsa1LU–SOH (r) were fit to 63 and 66% of the Tsa1–SH (p) value, respectively. The kSOH was fixed at the k1 value (9.7 × 104 M–1 s–1) measured at low Tsa1 concentration in Figure 2d,e. The parameter C was adjusted to account for the background fluorescence. The fit was not improved by fitting individual background values for each H2O2 concentration. (c) Secondary plot and linear fit of the Tsa1 inactivated fraction per turnover finact with tBOOH (squares, deduced from Figure S12a, black fit) and CuOOH (triangle, deduced from Figure S12b, black fit), compared with H2O2 (black circles, red fit from Figure 4b). The hyperoxidation index Chyp1% is deduced from the slope of the linear fits. Data are the mean of two independent experiments. (d) Second-order plots and linear fits of the observed rate constant k2 vs peroxide concentration, deduced by fit of pre-steady-state kinetics for the reaction of Tsa1 (5 μM) with tBOOH (squares, blue line) and CuOOH (triangles, red line) from Figure S13 against a three-exponential equation. (e) Determination of the rate constant k4 for the Tsa1C171A reaction with tBOOH and CuOOH. Second-order plots and linear fits of the rate constant k4obs against peroxide concentration, obtained from pre-steady-state kinetics of the reaction with tBOOH (squares, black fit) and CuOOH (triangle, black fit), compared to H2O2 (circles, red fit, from Figure 5b).
To further assess this interpretation, simulations of the kinetics of Tsa1SO2 formation were performed based on the model from Figure 6b and compared to the observed values (Figure S11). Combined with the results from CD and fluorescence-based kinetics, these simulations support that for the Tsa1FF–SOH ⇄ Tsa1LU–SOH equilibrium, the forward and reverse rate constants are consistent with kLU of 65 s–1 and an upper kFF value of 0.6 s–1, respectively. Since the kLU values are of comparable order of magnitude to those of the disulfide formation and hyperoxidation rate constants, these results support the notion that the rapid equilibrium hypothesis likely does not apply in this case.
Using alternate organic peroxide substrates with high reactivity should similarly impact hyperoxidation sensitivity by increasing the competition against the conformation step. We found that Tsa1 was much more sensitive to hyperoxidation by organic peroxides, with Chyp1% of 63 μM for tert-butyl hydroperoxide (tBOOH) and 11 μM for cumene hydroperoxide (CuOOH), that is, 12- and 66-fold lower, respectively, relative to H2O2 (Figures 6c, S12a,b, and Table 2). As observed with H2O2, Trp fluorescence monitoring of the reduction of tBOOH by Tsa1 displayed a three-phase kinetics (Figure S13a), characterized by k1 of 1.7 × 107 M–1 s–1 that remained much higher than the subsequent steps (Figure S13a, inset). Further, the saccharose-sensitive phase 2 could be assigned to the FF–LU-linked step (Figure S14a,b). Phase 2 amplitude increased with the tBOOH concentration, likely due to the combination of the FF/LU event and the formation of Tsa1SO2, as observed for the H2O2 data set. If the two reactions were in competition, as in the proposed model, the observed rate constant for this phase should increase linearly with the peroxide concentration, with a slope corresponding to kSO2. The observed k2 indeed increased linearly with the concentration of both tBOOH and CuOOH, further supporting the direct competition mechanism (Figure 6d). Linear fit yielded y-intercept values of 59 and 57 s–1, respectively, corresponding to kLU, unchanged relative to H2O2, and slopes of 8.8 × 103 and 7.4 × 104 M–1 s–1, respectively, corresponding to kSO2. In support of this interpretation, tBOOH and CuOOH hyperoxidized Tsa1C171A with rate constants k4 of 9.5 × 103 and 3.9 × 104 M–1 s–1 in the same range, respectively (Figure 6e and Table 2). Therefore, in addition to the enzyme intrinsic FF–LU transition kinetics, the hyperoxidation sensitivity is also a function of CP–SOH reactivity, that is, the hyperoxidation rate constant kSO2.
Table 2. Calculated and Experimental Hyperoxidation Sensitivity of Tsa1, Human Prxs, and AhpC.
| Prx | Tsa1A177S A178D | Tsa1 | Tsa1W161F | Tsa1 | Tsa1 |
|---|---|---|---|---|---|
| peroxide | H2O2 | H2O2 | H2O2 | tBOOH | CuOOH |
| kLU (s–1) | 216 ± 42 | 64 ± 5 | 37 ± 1 | 59 ± 6 | 57 ± 5 |
| k4a (M–1 s–1) | 103 ± 400 | (2.1 × 103) ± 200 | (2.9 × 103) ± 70 | (9.5 × 103) ± 200 | (39 × 103) ± 500 |
| calculated Chyp1% (μM) | 2160 ± 1280 | 305 ± 53 | 128 ± 7 | 62 ± 8 | 16 ± 1 |
| experimental Chyp1% (μM) | 2620 ± 220 | 730 ± 100 | 440 ± 93 | 63 ± 15 | 11 ± 2 |
| Prx | hPrx1 | hPrx1 | hPrx2 | hPrx3 | AhpC |
|---|---|---|---|---|---|
| peroxide | H2O2 | ONOOH | H2O2 | H2O2 | H2O2 |
| kLU (s–1) | 10.8 ± 2 | 10.8 ± 2 | 0.2–0.612,13,16 | 2011 | >205 calculated |
| k4a (M–1 s–1) | 1.77 × 103 12 | 2.8 × 105 12 | (1.73–1.97) × 103 12,16 | 2 × 103 estimated | 41012 |
| calculated Chyp1% (μM) | 61 ± 13 | 0.4 | 1–3 | 100 | |
| experimental Chyp1% (μM) | 80 ± 5/5018 | 518 | 12727 | >500018 |
Measured on C171A mutants. Data are reported as the mean value obtained from two independent experiments performed on distinct protein productions ± s.d.
Integration of Kinetic Mechanism and Steady-State Hyperoxidation Sensitivity
Can we predict Prx hyperoxidation sensitivity based on kinetics rate constants? The method of Nelson17 measures Chyp1% based on the assumption that the FF–LU transition behaves as a rapid equilibrium in the catalytic cycle, but our results suggest that the Tsa1–SOH FF–LU transition does not fit the rapid equilibrium hypothesis. If this is true, the fraction of hyperoxidized enzyme per catalytic cycle finact can be defined as follows
with ROOH standing for the peroxide substrate, PrxFF–SOH for the sulfenic acid intermediate in the FF conformation, and PrxLU–SOH for the sulfenic acid intermediate in the LU conformation. Furthermore, considering the FF-to-LU transition as practically irreversible nullifies the kFF[PrxLU–SOH] term, simplifying the equation to
and
| 1 |
if inactivation occurs in less than 5% of the enzyme molecules, which comply to the conditions used in this study.3 Thus, as with the Nelson method, finact is expected to be linearly dependent on the peroxide concentration but with a slope equal to kSO2/kLU (eq 1). A summary of the kLU and k4 (used as an estimate of kSO2 for enzymes possessing the CR) rate constants measured for Tsa1 and a comparison of the Chyp1% value calculated from these values with the experimental ones (Figure 4b) showed a good agreement for wild-type Tsa1, Tsa1 mutants, H2O2, and organic peroxides (Table 2 and Figure S15). In the case of human Prxs, as the disulfide formation step was not kinetically resolved from the rate-limiting FF-to-LU transition (Figure 3e), we used the reported rate constants of the resolving step11,12 as proxy of kLU. Based on published human Prxs kSO2 experimental or estimated values,11,12 the hyperoxidation sensitivity index Chyp1% is again predicted with a good agreement compared to the experimental values published with H2O2 (Table 2 and Figure S15). This interpretation also explains the results obtained by Randall et al.,16 using the equation “direct Chyp1%” = 0.01 × kres/kSO2, with kres corresponding to the observed resolving phase rate constant. These comparisons further support a mechanistic model in which the degree of hyperoxidation sensitivity is controlled by the FF-to-LU transition kinetics of the sulfenic intermediate and not by the disulfide formation event. In the case of AhpC, the reverse calculation using a Chyp1% > 5 mM18 and a kSO2 of 410 M–1 s–1 16 gave an estimation of the FF-to-LU rate constant >205 s–1, similar to the Tsa1A177S A178D mutant.
Relative Hyperoxidation Sensitivity of Prxs in Vivo
To assay Prx sulfinylation in vivo, we monitored the differential migration of reduced vs sulfinylated Prx seen upon alkylation with methyl-PEG (24)-maleimide (mPEG). mPEG adds 1239.44 Da per modified residue and can only alkylate reduced, but not sulfinylated, Cys residues, thereby differentiating the reduced from the sulfinylated enzyme. We first compared the reactivity of Myc-tagged and untagged Tsa1, using a Myc-specific and a human-Prx antibody, the latter also reacting with Tsa1, and although it produces multiple nonspecific signals, it allows quantification (Figure S16a). Upon mPEG alkylation of the reduced enzyme, migration of either enzyme form was up-shifted to a molecular size corresponding to the addition of two mPEG moieties (2-mPEG), which lines up with the two Tsa1 Cys residues CP and CR. When cells were exposed to H2O2, the 2-mPEG-modified band disappeared upon increasing H2O2 concentration, with appearance of a new lower-migrating band, which corresponds to Tsa1 modified by one mPEG, presumably at CR, as carrying CP in the sulfinate form, as confirmed by immunoblotting with an anti-SO2/3 antibody (Figure S16b). The Tsa1 sulfinylation level can thus be determined by the relative intensity ratio of the 1-mPEG and 2-mPEG Tsa1 bands (SO2/SO2 + SH, Figure S16c), irrespective of absolute levels of the enzyme. The compared sulfinylation profiles of Myc-Tsa1 and Tsa1 showed that although they displayed similar sulfinylation sensitivity, both reaching 50% saturation at 200 μM H2O2, Myc-Tsa1 sulfinylation was delayed at H2O2 <100 μM, indicating that the N-terminal Myc tag alters enzyme reactivity (Figure S16b).
We thus used untagged Tsa1 to next compare the enzyme sulfinylation profile in response to a 5 min cell exposure to H2O2 and organic peroxides (Figure 7a). Sulfinylation reached 50% at the doses of 75, 125, and 200 μM, for CuOOH, tBOOH, and H2O2, respectively, consistent with in vitro data that indicated a much higher hyperoxidation sensitivity toward organic peroxides, relative to H2O2. We next compared the in vivo sulfinylation sensitivity of Tsa1A177S A178D and hPrx1 to that of Tsa1. As expected from the in vitro kinetics, Tsa1A177S A178D required much higher doses of H2O2 to reach 50% sulfinylation (500 μM), relative to Tsa1 (200 μM) (Figure 7d,e). As expected too, hPrx1 was far more reactive, reaching 50% sulfinylation at 100 μM H2O2, also displaying a non-negligible basal sulfinylation, ca. 20%, presumably caused by endogenous H2O2. Western blot of the same lysates with the anti-PrxSO2/3, although less quantitative, provided results similar to those of the mPEG procedure (Figure 7b,e). It is worth noting that full 100% enzyme sulfinylation was never reached, also plateauing at a higher value with tBOOH and CuOOH, relative to H2O2, and at a much lower value in the case of the Tsa1A177S A178D mutant, which presumably result from the assay integrating both intrinsic enzyme reactivity rates and the cellular peroxide degradation, as best exemplified with the Tsa1A177S A178D mutant. In summary, the sulfinylation parameters measured in vitro are valid in the cellular context.
Figure 7.

Hyperoxidation of wild-type and mutants Tsa1 with H2O2 and tBOOH in S. cerevisiae. (a–c) Tsa1 is more reactive toward organic peroxides than with H2O2 in vivo. Tsa1 hyperoxidation in cells exposed to H2O2, tBOOH, and CuOOH, during 5 min at the indicated concentrations. Thiols were derivatized by N-ethylmaleimide (NEM) or mPEG, as indicated, after reduction with dithiothreitol (DTT), as described in methods. (d–f) Comparison of the H2O2 reactivity of Prx1, Tsa1, and Tsa1A177S A178D in vivo. Thiols were derivatized by NEM or mPEG, as indicated, after reduction with DTT, as described in methods, using cell lysates of Δtsa1 expressing human Prx1, Tsa1, or Tsa1A177S A178D and exposed to H2O2 at the indicated concentration. (a, d) Western blot of reduced (−SH) (2 × mPEG) and hyperoxidized (−SO2H) (1 × mPEG) forms of Tsa1 (indicated by black arrows), revealed with an anti-Prx1 antibody. (b, e) Western blot of the Prx SO2H form using a Prx anti-SO2/3 antibody and the cell lysates used in (a) and (c), respectively. (c, f) Quantification of the degree of oxidation (SO2H/SH + SO2H) vs peroxide concentration.
Discussion
The mechanistic foundations of Prx1-type sulfinylation were established by the seminal work of Wood, Poole, and Karplus in a model that integrated three parameters as determinants of the sensitivity to hyperoxidation.3 By Trp fluorescence-based rapid kinetics of the S. cerevisiae Prx1-type Tsa1, we have identified a conformational event linked to the FF-to-LU transition that is kinetically distinct from the recycling step. This finding establishes that hyperoxidation sensitivity is dictated by only two parameters, the sulfinylation step per se and the FF → LU rate constants (Figure 1a). Accordingly, formation of Tsa1SO2 and the conformational FF-to-LU transition appear as two reciprocally exclusive competing paths, with the “kinetic pause” that enables Tsa1SO2 formation occurring prior to the conformational transition. A slow FF-to-LU transition, as it happens in hPrx1, however becomes rate-limiting for the subsequent resolving step (Figure 3e). In addition, our data suggest that the stability of the active site FF conformation, which sets the sulfinylation kinetics, is only moderately influenced by the C-terminal tail conformation, which sets FF → LU kinetics.
This model is supported by several lines of experimental evidence. First, using a wide range of H2O2 concentrations relevant to hyperoxidation, Tsa1 reaction kinetics could be globally fitted according to the mechanism corresponding to Figure 1a. Importantly, global fitting and simulations suggest that once the CP–SOH intermediate is formed, the FF-to-LU transition is practically not reversible, as initially thought. Indeed, based on the model from Figure 6b, simulations suggest an upper kFF value of 0.6 s–1 for the reverse Tsa1FF–SOH ⇄ Tsa1LU–SOH reaction, giving an equilibrium constant KLUSOH = kLU/kFF > 108 (Figure S11). Since in the reduced state Tsa1–SH the FF conformation is favored, this therefore suggests that sulfenate formation has a profound effect on the FF/LU equilibrium in triggering local unfolding. This unexpected finding in fact fits ultrahigh-resolution structure analysis of reaction snapshots obtained with the robust bacterial PrxQ. These data suggested that the rate constant kLU is higher than kFF, based on the fact that the nascent sulfenate in the FF state forms in a high-energy structure, which promotes local unfolding by destabilization of the active site.28 As already suggested in this study,28 we propose that a slower FF-to-LU local unfolding process, as the one measured here for the sensitive Tsa1 and hPrx1, is due to an active site adjustment that accommodates sulfenate formation and movement. Second, use of our refined model to derive the steady-state hyperoxidation sensitivity, Chyp1%, predicts kLU and kSO2 as the only determinant kinetic steps, in good agreement with the Chyp1% experimentally measured values (Figure S15). Third, a global fit returned a rate constant of H2O2-dependent hyperoxidation kSO2 similar to the value measured directly using the Tsa1C171A mutant, in the 103 M–1 s–1 range.
The much higher hyperoxidation rate constants of Prx, relative to other redox-sensitive proteins,29 suggest that the active site in the Prx–SOH FF conformation favors hyperoxidation by activating H2O2, as it does for activating the initial reaction of CP with H2O2.30 The similarity of the kSO2 for H2O2 measured for Tsa1 and human Prxs12 is consistent with the high conservation of Prxs active sites. The hyperoxidation rate constants measured with tBOOH and CuOOH were surprisingly much higher than those of H2O2. These organic peroxides may establish interactions in the vicinity of the FF active site by virtue of their aliphatic or aromatic moieties, respectively, which could favor the positioning of the peroxide function relative to the sulfenate. Conversely, the tBOOH kSOH value was slightly lower than the very high, close to the diffusion limit, kSOH value measured with H2O2, which could be a consequence of the organic peroxide bulkier structure relative to H2O2, reducing active site accessibility.
We characterized the Tsa1 A177S-A178D mutation, which substitutes these two residues in Tsa1 by those present at the same location in hPrx1 initially identified as C-terminal tail-determinants influencing Chyp1%.19 We found that this mutant had wild-type values for kSOH and for the amplitude of phase 1 but paradoxically a CD profile indicative of a FF–LU equilibrium shifted toward the LU conformation. Such a shifted FF–LU equilibrium would have been expected to decrease kSOH, if indeed the C-terminal helix contributes to active site stability and hence to CP H2O2 reactivity.30 This result can be explained by two nonmutually exclusive hypotheses: (i) The equilibrium shifted in favor of the LU conformation is a rapid equilibrium, ensuring nonlimiting LU → FF displacement upon H2O2 reaction with Tsa1FF–SH. This is consistent with the high conformational exchange rate measured for PrxQ.23 (ii) The Tsa1A177S A178D mutant exists in a mixed conformation, maintaining the active site in an FF conformation competent for a highly efficient CP attack on H2O230 and destabilizing to some extent the C-terminal tail, as observed in some structures of AhpC.31 In the Tsa1A177S A178D mutant, as in wild-type Tsa1, sulfenic acid formation would be the trigger of the active site transition to the LU state, then “pushing” the C-terminal tail toward a full LU form and shifting the FF/LU equilibrium in favor of LU, which is consistent with the conclusions obtained by Perkins et al. on AhpC.31 Similar interpretation may apply for the Tsa1Y190G F191G mutant CD spectrum that displayed the full LU signature, but its kinetics paradoxically showed only a 6-fold decrease of k1 (1.7 × 107 M–1 s–1, second-order kinetics up to 50 μM H2O2) relative to the wild type. In this mutant, the absence of the buttressing effect of the C-terminal helix favors faster local active site unfolding by the formed CP–SOH, thereby disfavoring hyperoxidation, while the C-terminal tail remaining in the LU conformation prevents phase 2 occurrence. These two examples support the notion that the unfolding of the C-terminal tail is not sufficient to induce the FF-to-LU transition at the active site, in agreement with previous studies.31 We thus propose that molecular determinants of hyperoxidation sensitivity fall into two classes: those stabilizing the FF active site conformation and thus favoring high peroxide CP reactivity (kSO2 and necessarily kSOH) and those influencing the C-terminal tail flexibility that determine kLU (Figure 8).
Figure 8.
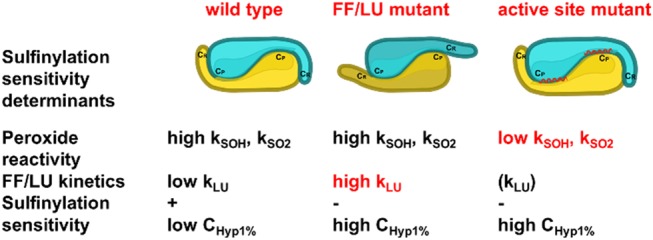
Scheme illustrating the two proposed classes of Prx sulfinylation determinants. Mutants FF/LU affecting the C-terminal tail flexibility have high FF-to-LU kinetics, retain high peroxide reactivity, and are not sensitive to sulfinylation; mutants affecting the active site structure have poor peroxide reactivity and are predicted to be poorly sensitive to sulfinylation, irrespective of the FF–LU kinetics.
How do the in vitro Prx hypersensitivity parameters described here translate into biological contexts? To answer this question, we monitored Prx sulfinylation levels after a short, 5 min peroxide exposure. We observed a H2O2-dose response effect, consistent with the intracellular H2O2 level dependence of the kinetics of sulfinylation. Under these conditions, the hyperoxidation parameters established in vitro were predictive of in vivo relative sensitivities of the enzyme sets toward hyperoxidation, when comparing both H2O2 and organic peroxides, which have distinct kSO2 and Prxs with distinct kLU. The much higher sensitivity of hPrx1 relative to Tsa1, which displayed a basal level of sulfinylation when expressed in S. cerevisiae, is consistent with the measured lower Chyp1% of hPrx1 and might reflect the much lower intracellular peroxide levels of mammalian cells relative to yeast.32,33
In conclusion, our work provides a quantitative basis to predict in vivo relative hyperoxidation sensitivities for different Prx types, based on enzymatic parameters determined in vitro. It sets the bases for new Prxs structure–function studies and to approach the mechanism of the regulation of other proteins by reversible Cys sulfinylation, which is more common than initially thought.1
Materials and Methods
Chemicals
All chemicals were of reagent grade and were used without additional purification. Tris was from VWR (West Chester, PA). Tris, (2-carboxyethyl)phosphine hydrochloride (TCEP), tert-butyl hydroperoxide (tBOOH), cumene hydroperoxide (CuOOH), saccharose, Trp, NaF, KCl, and MgCl2 were from Merck (Darmstadt, Germany). NADPH was obtained from Roche (Basel, Switzerland), and dithiothreitol (DTT) and ammonium sulfate were from Euromedex (Souffelweyersheim, France). Hydrogen peroxide (H2O2) was from Acros Organics (Geel, Belgium). Peroxide stock concentrations were measured accurately by the peroxidase enzymatic coupled assay using Tsa1/Trx/Trx reductase/NADPH, following the total NADPH consumption at 340 nm (ε340 = 6200 M–1 cm–1).
Recombinant Protein Preparation
Recombinant thioredoxin1 (Trx), Trx reductase from Escherichia coli, and wild-type and mutant His-tagged Tsa1 (Tsa1) from S. cerevisiae were produced and purified following the experimental procedures previously described.34−36 Tsa1 mutants were generated by standard polymerase chain reaction (PCR) site-directed mutagenesis and sequenced to confirm that no mutations had been introduced in the amplification reactions.
The pET28bHT-hPrx1 plasmid encoding the N-terminal His tag fusion protein of human Prx1 was obtained by cloning the prdx1 open reading frame amplified by PCR (GC-rich system, Roche Applied Science, Basel, Switzerland) using a complementary DNA (cDNA) clone from Homo sapiens (RZPD, German science center for genome research, clone IRAUp969E034) as template into the pET28b(+) plasmid between the NdeI and SacI sites. The forward primer contained a NdeI restriction site, and the reverse primer contained a SacI restriction site (sequences of oligonucleotides not shown). E. coli C41(DE3) [F–ompT hsdSB (rB–mB–) gal dcm (DE3)] transformants containing the pET28bHT-hPrx1 plasmid were grown by overnight culture at 37 °C for 24 h in the autoinducible ZYM-5052 medium37 supplemented with kanamycin (50 mg L–1). Cells were harvested by centrifugation, resuspended in a minimal volume of buffer B (20 mM sodium phosphate, 500 mM NaCl, pH 7.5), and disrupted by sonication. hPrx1 contained in the soluble fraction was purified on a Ni-Sepharose column equilibrated with buffer B plus 50 mM imidazole, connected to an ÄKTA Avant system (GE Healthcare, France) and eluted by a 0.5 M imidazole step. At this stage, wild-type and mutant proteins were pure as checked by electrophoresis on 12.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel followed by Coomassie Brilliant Blue R-250 staining and by electrospray mass spectrometry analyses. After overnight dialysis in a 20 mM sodium phosphate and 100 mM NaCl, pH 7.5, buffer, purified hPrx1 was stored at −80 °C in the presence of 10 mM DTT and was stable for several weeks under these conditions.
The plasmid pET20bTsa1 encoding the S. cerevisiae 2-Cys-Prx Tsa1 (referred to as native Tsa1) was obtained by cloning the Tsa1 open reading frame amplified by PCR using S. cerevisiae W303 genomic DNA as template into the pET20b plasmid between the NdeI and SacI sites, as described for hPrx1. The recombinant protein was produced as described for hPrx1. For native Tsa1 purification, the protein contained in the soluble fraction of the cellular extract was precipitated by ammonium sulfate at 55% saturation, followed by hydrophobic chromatography on a phenyl-Sepharose column (Amersham Biosciences) equilibrated with a 20 mM sodium phosphate and 100 mM NaCl, pH 7, buffer plus 1 M ammonium sulfate, eluted with a linear 1–0 M ammonium sulfate gradient. Final purification was achieved by anion exchange chromatography on a Q-Sepharose column equilibrated with buffer A, by an elution by a 0–1 M NaCl linear gradient. The protein was characterized and stored as described for hPrx1.
Immediately before use, the proteins were incubated with 20 mM TCEP for 20 min on ice, followed by desalting in TK buffer (50 mM Tris, 100 mM KCl, pH 7) on a PD-10 column. Protein monomer concentration was determined spectrophotometrically using a molar extinction coefficient of 29 500 M–1 cm–1 for Tsa134 and 18 450 M–1 cm–1 for hPrx1. Preparation of Tsa1SO2 was performed as previously described34 and of Tsa1SS by addition of 1.2 equiv of H2O2 to reduced Tsa1 in solution. The intrinsic fluorescence emission spectra of Tsa1 (2 μM) were recorded on a SAFAS Xenius fluorimeter at an excitation wavelength of 295 nm using a photomultiplier voltage of 450 V.
Stopped-Flow Rapid Kinetics
The reaction of wild-type and mutant Tsa1s with peroxide substrates was followed in single turnover conditions by monitoring intrinsic fluorescence intensity at 25 °C in buffer TK on an SX19MV-R stopped-flow apparatus (Applied Photophysics) equipped with a 5 μL cell, fitted for fluorescence measurements, with excitation wavelength set at 295 nm and emitted light collected above 320 nm using a cutoff filter. One syringe contained Tsa1 (5 μM, final concentration after mixing), and the other syringe contained the peroxide substrate. Equal volumes of each syringe were rapidly mixed to start the reaction. An average of at least six runs was recorded for each concentration of peroxide. The data set obtained at variable peroxide concentrations was fitted against multiexponential equation using Pro-Data viewer (Applied Photophysics) or SciDavis 1.2 software. For experiments in the presence of viscogen, the desired concentration (10–20 or 30%) was added to buffer TK.
Steady-State Tsa1 Peroxidase Activity
Tsa1 peroxidase activity was measured in TK buffer using the Trx/Trx reductase/NADPH coupled assay (1 μM Trx reductase, 200 μM NADPH, 150 μM Trx) with 100 μM H2O2, started by addition of 0.5 or 1 μM Tsa1 at 25 °C. Initial rate measurements were carried out on a UVmc2 spectrophotometer (Safas, Monaco) by following the decrease of absorbance at 340 nm due to the consumption of NADPH. A blank measurement recorded in the absence of Tsa1 was systematically deduced from the assay to account for nonspecific oxidation of Trx or Trx reductase.
Steady-State Hyperoxidation Sensitivity
The hyperoxidation sensitivity index Chyp1% was measured using a method adapted from Nelson et al.17 Tsa1 (1 μM) peroxidase activity was measured as for the steady-state assay, in the presence of 5 μM Trx, 0.25 μM Trx reductase, 200 μM NADPH, and variable peroxide substrate in buffer TK by monitoring absorbance at 340 nm over 15 min on a UVmc2 spectrophotometer (Safas, Monaco). As Tsa1 becomes hyperoxidized over the course of the reaction (with a fraction inactivated at each turnover), the absorbance trace deviates from linear kinetics. The rate constant of inactivation kinact was deduced from the slope of the kinetic trace first-order derivative and divided by the initial rate constant measured at time 0 to give the fraction of inactivation per turnover finact. Because the conditions were chosen to limit the hyperoxidized fraction to less than 5% (Figures 4b, 5b, and 6c), it increases linearly with the peroxide concentration.17 The reciprocal of the slope of finact vs peroxide gives the peroxide concentration virtually required to hyperoxidize 100% of Tsa1 in one turnover. It is divided by 100 to give Chyp1%, corresponding to the peroxide concentration at which 1% of the enzyme molecules will be oxidatively inactivated per turnover.
Circular Dichroism
CD spectra of wild-type and mutant Tsa1s were recorded at 25 °C on a Chirascan Plus spectrometer (Applied Photophysics, U.K.). Far-UV measurements were carried out in a 0.01 cm pathlength flat quartz cell at 50 μM Tsa1 in a 10 mM sodium phosphate and 100 mM sodium fluoride, pH 7, buffer. For near-UV measurements, the same conditions were used, using a 1 cm pathlength cuvette. Scans were recorded with 1 nm steps from 260 to 180 nm (far-UV) or 320 to 250 nm (near-UV), and each experiment was averaged over three scans. The kinetics of the reaction of Tsa1 (50 μM) with H2O2 (100 μM) was monitored using the CD signal at 270 nm on the same apparatus coupled with a stopped-flow module. The bandwidth was opened to 4 nm to increase the detected light intensity. An average of 50 measurements was acquired to minimize the signal/noise ratio.
Yeast Strains, Plasmids, and Growth Media
The S. cerevisiae strains used in this study are derivatives of BY4741 (MATa his3Δ1leu2Δ0met15Δ0ura3Δ0)38 and RDKY3615 (MATa, ura3-52, leu2Δ1, trp1Δ63, his3Δ200) and listed in Table 3. Cells were grown at 30 °C in synthetic minimal media (SD) (0.67% yeast nitrogen base w/o amino acids, 2% glucose) supplemented with the appropriate amino acid or YPD (1% yeast extract, 2% peptone, and 2% glucose). The plasmids used in this study are pRS316-Myc-Tsa14 and pRS316-Tsa1 A177S-A178D that was generated by subcloning of the ORF of pET28b-Tsa1 A177S-A178D between XbaI and SacI into pRS316.
Table 3. S. cerevisiae Strains.
mPEG Differential Cysteine Derivatization Procedure To Monitor Peroxiredoxin Sulfenylation in Vivo
Yeast cells (10 mL) grown to an OD600 nm of 0.5 were exposed to H2O2, tBOOH, and CuOOH at the indicated concentration for 5 min. Trichloroacetic acid (TCA) (100%) was added to the cell culture to a final concentration of 20%. The cell culture was centrifuged at 6000g for 5 min at 4 °C. Pellets were washed with 20% TCA, and cells were lysed with glass beads in 0.2 mL of TCA (20%). Lysates were pelleted down by centrifugation at 14 000g for 15 min at 4 °C. Pellets were washed twice with acetone; dried; solubilized in 0.2 mL of a buffer containing 10 mM DTT, cOmplete mini ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Roche) and 25 μg mL–1 phenylmethylsulfonylfluoride, 2% SDS, and 100 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (Hepes) (pH 7.4); and incubated at 25 °C for 30 min. Samples were precipitated by 20% TCA and centrifuged at 14 000g for 15 min at 4 °C. After two washs with acetone, the dried pellets were solubilized in 0.1 mL of a buffer containing 10 mM methyl-PEG (24)-maleimide (Thermo Fisher) or 50 mM NEM, cOmplete of mini EDTA-free protease inhibitor cocktail (Roche) and 25 μg mL–1 phenylmethylsulfonylfluoride, 2% SDS, and 100 mM Hepes (pH 7.4) and incubated at 25 °C for 60 min. Protein samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE) as described,42 and proteins were immune-detected with an anti-Prx1 (Santa Cruz Biotechnology, ref sc-137222), anti-Myc (9E10), or Prx anti-SO2/3 antibody kindly provided by S.G. Rhee.
Acknowledgments
The authors gratefully acknowledge Dr. H. Mazon for help with mass spectrometry analyses, Dr. F. Talfournier for insightful discussions, and J. Charbonnel for efficient technical help. This work was supported by ANR grant ANR-17-CE11-0034-02 to M.B.T. and S.R.-C., Ligue contre le Cancer to S.R.-C., the University de Lorraine and Région Grand Est. The authors acknowledge support of the group by the “Impact Biomolecules” project of the “Lorraine Université d’Excellence” (Investissements d’avenir-ANR). This work was granted access to the Biophysics and Structural Biology Core Facility of UMS2008 IBSLor CNRS-UL-INSERM and mass spectrometry platform SCMS of Université de Lorraine.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.9b04471.
Complementary kinetic experiments (Figures S1–S3, S5–S10, and S12–S14); far-UV CD spectra of wild-type Tsa1 and Tsa1Y190G F191G under the reduced and disulfide forms (Figure S4); simulation of the kinetics of formation of Tsa1SO2 (Figure S11); comparison of the calculated and experimental Chyp1% for wild-type and mutant Tsa1s (Figure S15); validation of the in vivo procedure to monitor Prx hyperoxidation (Figure S16) (PDF)
Author Present Address
∥ GlaxoSmithKline Vaccines, 20 Avenue Fleming, 1300 Wavre, Belgium (S.B.).
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Akter S.; Fu L.; Jung Y.; Conte M. L.; Lawson J. R.; Lowther W. T.; Sun R.; Liu K.; Yang J.; Carroll K. S. Chemical Proteomics Reveals New Targets of Cysteine Sulfinic Acid Reductase. Nat. Chem. Biol. 2018, 14, 995–1004. 10.1038/s41589-018-0116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevallet M.; Wagner E.; Luche S.; van Dorsselaer A.; Leize-Wagner E.; Rabilloud T. Regeneration of Peroxiredoxins during Recovery after Oxidative Stress. Only Some Overoxidized Peroxiredoxins Can Be Reduced during Recovery After Oxidative Stress. J. Biol. Chem. 2003, 278, 37146–37153. 10.1074/jbc.M305161200. [DOI] [PubMed] [Google Scholar]
- Wood Z. A.; Poole L. B.; Karplus P. A. Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science 2003, 300, 650–653. 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Biteau B.; Labarre J.; Toledano M. B. ATP-Dependent Reduction of Cysteine–Sulphinic Acid by S. cerevisiae Sulphiredoxin. Nature 2003, 425, 980–984. 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- Nelson K. J.; Knutson S. T.; Soito L.; Klomsiri C.; Poole L. B.; Fetrow J. S. Analysis of the Peroxiredoxin Family: Using Active-Site Structure and Sequence Information for Global Classification and Residue Analysis. Proteins: Struct., Funct., Bioinf. 2011, 79, 947–964. 10.1002/prot.22936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourquet S.; Huang M. E.; D’Autreaux B.; Toledano M. B. The Dual Functions of Thiol-Based Peroxidases in H2O2 Scavenging and Signaling. Antioxid. Redox Signaling 2008, 10, 1565–1576. 10.1089/ars.2008.2049. [DOI] [PubMed] [Google Scholar]
- Kil I. S.; Lee S. K.; Ryu K. W.; Woo H. A.; Hu M.-C.; Bae S. H.; Rhee S. G. Feedback Control of Adrenal Steroidogenesis via H2O2-Dependent, Reversible Inactivation of Peroxiredoxin III in Mitochondria. Mol. Cell 2012, 46, 584–594. 10.1016/j.molcel.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Day A. M.; Brown J. D.; Taylor S. R.; Rand J. D.; Morgan B. A.; Veal E. A. Inactivation of a Peroxiredoxin by Hydrogen Peroxide Is Critical for Thioredoxin-Mediated Repair of Oxidized Proteins and Cell Survival. Mol. Cell 2012, 45, 398–408. 10.1016/j.molcel.2011.11.027. [DOI] [PubMed] [Google Scholar]
- Jang H. H.; Lee K. O.; Chi Y. H.; Jung B. G.; Park S. K.; Park J. H.; Lee J. R.; Lee S. S.; Moon J. C.; Yun J. W.; et al. Two Enzymes in One: Two Yeast Peroxiredoxins Display Oxidative Stress-Dependent Switching from a Peroxidase to a Molecular Chaperone Function. Cell 2004, 117, 625–635. 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Turner-Ivey B.; Manevich Y.; Schulte J.; Kistner-Griffin E.; Jezierska-Drutel A.; Liu Y.; Neumann C. A. Role for Prdx1 as a Specific Sensor in Redox-Regulated Senescence in Breast Cancer. Oncogene 2013, 32, 5302–5314. 10.1038/onc.2012.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peskin A. V.; Dickerhof N.; Poynton R. A.; Paton L. N.; Pace P. E.; Hampton M. B.; Winterbourn C. C. Hyperoxidation of Peroxiredoxins 2 and 3. Rate Constants for the Reactions of the Sulfenic Acid of the Peroxidatic Cysteine. J. Biol. Chem. 2013, 288, 14170–14177. 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla Rizza J.; Randall L. M.; Santos J.; Ferrer-Sueta G.; Denicola A. Differential Parameters between Cytosolic 2-Cys Peroxiredoxins, PRDX1 and PRDX2: PRDX1, PRDX2 Differential Parameters. Protein Sci. 2019, 28, 191–201. 10.1002/pro.3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portillo-Ledesma S.; Randall L. M.; Parsonage D.; Dalla Rizza J.; Karplus P. A.; Poole L. B.; Denicola A.; Ferrer-Sueta G. Differential Kinetics of Two-Cysteine Peroxiredoxin Disulfide Formation Reveal a Novel Model for Peroxide Sensing. Biochemistry 2018, 57, 3416–3424. 10.1021/acs.biochem.8b00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo K. H.; Lee S.; Jeong S. Y.; Kim E. T.; Kim H. J.; Kim K.; Song K.; Chae H. Z. Regulation of Thioredoxin Peroxidase Activity by C-Terminal Truncation. Arch. Biochem. Biophys. 2002, 397, 312–318. 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- Jara M.; Vivancos A. P.; Hidalgo E. C-Terminal Truncation of the Peroxiredoxin Tpx1 Decreases Its Sensitivity for Hydrogen Peroxide without Compromising Its Role in Signal Transduction. Genes Cells 2008, 13, 171–179. 10.1111/j.1365-2443.2007.01160.x. [DOI] [PubMed] [Google Scholar]
- Randall L. M.; Dalla Rizza J.; Parsonage D.; Santos J.; Mehl R. A.; Lowther W. T.; Poole L. B.; Denicola A. Unraveling the Effects of Peroxiredoxin 2 Nitration; Role of C-Terminal Tyrosine 193. Free Radical Biol. Med. 2019, 141, 492–501. 10.1016/j.freeradbiomed.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. J.; Parsonage D.; Karplus P. A.; Poole L. B.. Evaluating Peroxiredoxin Sensitivity Toward Inactivation by Peroxide Substrates. In Methods Enzymology; Cadenas E.; Packer L., Eds.; Hydrogen Peroxide and cell signaling, Part B; Academic Press: Amsterdam, The Netherlands, 2013; Vol. 527, pp 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc J. A.; Nelson K. J.; Haynes A. C.; Lee J.; Reisz J. A.; Graff A. H.; Clodfelter J. E.; Parsonage D.; Poole L. B.; Furdui C. M.; Lowther W. T. Novel Hyperoxidation Resistance Motifs in 2-Cys Peroxiredoxins. J. Biol. Chem. 2018, 293, 11901–11912. 10.1074/jbc.RA117.001690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poynton R. A.; Peskin A. V.; Haynes A. C.; Lowther W. T.; Hampton M. B.; Winterbourn C. C. Kinetic Analysis of Structural Influences on the Susceptibility of Peroxiredoxins 2 and 3 to Hyperoxidation. Biochem. J. 2016, 473, 411–421. 10.1042/BJ20150572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamariah N.; Sek M. F.; Eisenhaber B.; Eisenhaber F.; Grüber G. Transition Steps in Peroxide Reduction and a Molecular Switch for Peroxide Robustness of Prokaryotic Peroxiredoxins. Sci. Rep. 2016, 6, 37610 10.1038/srep37610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. J.; Perkins A.; Van Swearingen A. E. D.; Hartman S.; Brereton A. E.; Parsonage D.; Salsbury Jr.; Freddie R.; Karplus P. A.; Poole L. B. Experimentally Dissecting the Origins of Peroxiredoxin Catalysis. Antioxid. Redox Signaling 2018, 521–536. 10.1089/ars.2016.6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsonage D.; Nelson K. J.; Ferrer-Sueta G.; Alley S.; Karplus P. A.; Furdui C. M.; Poole L. B. Dissecting Peroxiredoxin Catalysis: Separating Binding, Peroxidation, and Resolution for a Bacterial AhpC. Biochemistry 2015, 54, 1567–1575. 10.1021/bi501515w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ådén J.; Wallgren M.; Storm P.; Weise C. F.; Christiansen A.; Schröder W. P.; Funk C.; Wolf-Watz M. Extraordinary Ms–Ms Backbone Dynamics in Arabidopsis Thaliana Peroxiredoxin Q. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 1880–1890. 10.1016/j.bbapap.2011.07.011. [DOI] [PubMed] [Google Scholar]
- Tairum C. A.; Santos M. C.; Breyer C. A.; Geyer R. R.; Nieves C. J.; Portillo-Ledesma S.; Ferrer-Sueta G.; Toledo J. C.; Toyama M. H.; Augusto O.; Netto L. E. S.; de Oliveira M. A. Catalytic Thr or Ser Residue Modulates Structural Switches in 2-Cys Peroxiredoxin by Distinct Mechanisms. Sci. Rep. 2016, 6, 33133 10.1038/srep33133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhtina M.; Lee S.; Wang Y.; Dunlap C.; Lamarche B.; Tsai M.-D. Use of Viscogens, DNTPαS, and Rhodium(III) as Probes in Stopped-Flow Experiments To Obtain New Evidence for the Mechanism of Catalysis by DNA Polymerase β. Biochemistry 2005, 44, 5177–5187. 10.1021/bi047664w. [DOI] [PubMed] [Google Scholar]
- Rahuel-Clermont S.; Bchini R.; Barbe S.; Boutserin S.; André I.; Talfournier F. Enzyme Active Site Loop Revealed as a Gatekeeper for Cofactor Flip by Targeted Molecular Dynamics Simulations and FRET-Based Kinetics. ACS Catal. 2019, 1337–1346. 10.1021/acscatal.8b03951. [DOI] [Google Scholar]
- Haynes A. C.; Qian J.; Reisz J. A.; Furdui C. M.; Lowther W. T. Molecular Basis for the Resistance of Human Mitochondrial 2-Cys Peroxiredoxin 3 to Hyperoxidation. J. Biol. Chem. 2013, 288, 29714–29723. 10.1074/jbc.M113.473470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A.; Parsonage D.; Nelson K. J.; Ogba O. M.; Cheong P. H.-Y.; Poole L. B.; Karplus P. A. Peroxiredoxin Catalysis at Atomic Resolution. Structure 2016, 24, 1668–1678. 10.1016/j.str.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner J. J.; Parsons Z. D.; Cummings A. H.; Zhou H.; Gates K. S. Redox Regulation of Protein Tyrosine Phosphatases: Structural and Chemical Aspects. Antioxid. Redox Signaling 2011, 15, 77–97. 10.1089/ars.2010.3611. [DOI] [PubMed] [Google Scholar]
- Hall A.; Nelson K.; Poole L. B.; Karplus P. A. Structure-Based Insights into the Catalytic Power and Conformational Dexterity of Peroxiredoxins. Antioxid. Redox Signaling 2011, 15, 795–815. 10.1089/ars.2010.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins A.; Nelson K. J.; Williams J. R.; Parsonage D.; Poole L. B.; Karplus P. A. The Sensitive Balance between the Fully Folded and Locally Unfolded Conformations of a Model Peroxiredoxin. Biochemistry 2013, 52, 8708–8721. 10.1021/bi4011573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B. K.; Sikes H. D. Quantifying Intracellular Hydrogen Peroxide Perturbations in Terms of Concentration. Redox Biol. 2014, 2, 955–962. 10.1016/j.redox.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domènech A.; Ayté J.; Antunes F.; Hidalgo E. Using in Vivo Oxidation Status of One- and Two-Component Redox Relays to Determine H2O2 Levels Linked to Signaling and Toxicity. BMC Biol. 2018, 16, 61. 10.1186/s12915-018-0523-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel X.; Béchade G.; Kriznik A.; Van Dorsselaer A.; Sanglier-Cianferani S.; Branlant G.; Rahuel-Clermont S. Evidence for the Formation of a Covalent Thiosulfinate Intermediate with Peroxiredoxin in the Catalytic Mechanism of Sulfiredoxin. J. Biol. Chem. 2008, 283, 22371–22382. 10.1074/jbc.M800493200. [DOI] [PubMed] [Google Scholar]
- Mössner E.; Huber-Wunderlich M.; Glockshuber R. Characterization of Escherichia coli Thioredoxin Variants Mimicking the Active-Sites of Other Thiol/Disulfide Oxidoreductases. Protein Sci. 1998, 7, 1233–1244. 10.1002/pro.5560070519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulrooney S. B. Application of a Single-Plasmid Vector for Mutagenesis and High-Level Expression of Thioredoxin Reductase and Its Use to Examine Flavin Cofactor Incorporation. Protein Expression Purif. 1997, 9, 372–378. 10.1006/prep.1996.0698. [DOI] [PubMed] [Google Scholar]
- Studier F. W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expression Purif. 2005, 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Brachmann C. B.; Davies A.; Cost G. J.; Caputo E.; Li J.; Hieter P.; Boeke J. D. Designer Deletion Strains Derived from Saccharomyces Cerevisiae S288C: A Useful Set of Strains and Plasmids for PCR-Mediated Gene Disruption and Other Applications. Yeast 1998, 14, 115–132. 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Schmidt K. H.; Pennaneach V.; Putnam C. D.; Kolodner R. D.. Analysis of Gross-Chromosomal Rearrangements in Saccharomyces cerevisiae. Methods in Enzymology; DNA Repair, Part B; Academic Press, 2006; Vol. 409, pp 462–476. [DOI] [PubMed] [Google Scholar]
- Noichri Y.; Palais G.; Ruby V.; D’Autreaux B.; Delaunay-Moisan A.; Nyström T.; Molin M.; Toledano M. B. In Vivo Parameters Influencing 2-Cys Prx Oligomerization: The Role of Enzyme Sulfinylation. Redox Biol. 2015, 6, 326–333. 10.1016/j.redox.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraqui I.; Faye G.; Ragu S.; Masurel-Heneman A.; Kolodner R. D.; Huang M.-E. Human Peroxiredoxin PrxI Is an Orthologue of Yeast Tsa1, Capable of Suppressing Genome Instability in Saccharomyces Cerevisiae. Cancer Res. 2008, 68, 1055–1063. 10.1158/0008-5472.CAN-07-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaunay A.; Isnard A.-D.; Toledano M. B. H2O2 Sensing through Oxidation of the Yap1 Transcription Factor. EMBO J. 2000, 19, 5157–5166. 10.1093/emboj/19.19.5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.