

Abstract
Influenza A virus (IAV) infection-associated morbidity and mortality are a key global healthcare concern, necessitating the identification of novel therapies capable of reducing the severity of IAV infections. In this study, we show that the consumption of a low-carbohydrate, high-fat ketogenic diet (KD) protects mice from lethal IAV infection and disease. KD feeding resulted in an expansion of γδ T cells in the lung that improved barrier functions, thereby enhancing anti-viral resistance. Expansion of these protective γδ T cells required metabolic adaptation to a ketogenic diet, as neither feeding mice a high-fat high-carbohydrate diet nor providing chemical ketone body substrate that bypasses hepatic ketogenesis protected against infection. Therefore, KD mediated immune-metabolic integration represents a viable avenue towards preventing or alleviating influenza disease.
One Sentence Summary:
Ketogenic diet protects mice against influenza virus infection through γδ T cell expansion and metabolic adaptation.
Introduction
Respiratory influenza A virus (IAV) infections are a major source of human morbidity and mortality, causing over 20,000 deaths annually in the United States and incurring an economic burden in excess of $87 billion each year (1, 2). While an efficacious universal IAV vaccine is highly desirable and is under development (3, 4), in its absence, novel therapeutic approaches are vital for the treatment of influenza diseases. Such therapeutic strategies can entail either improvements in viral resistance or enhancement of disease tolerance that alleviates lethal consequences of the viral infections (5, 6). Disease tolerance strategies include providing energy substrates that aid in the metabolic adaptation required for host survival (7–9).
Inflammasome activation and neutrophil-mediated toxicity can promote tissue damage associated with IAV infections (10–12). In light of our recent findings that the ketone metabolite, β-hydroxybutyrate (BHB), inhibits NLRP3 inflammasome-dependent IL-1β secretion from neutrophils and macrophages (13, 14), we hypothesized that ketogenic diet (KD) might also reduce IAV disease severity. To probe the effects of KD-induced BHB production on IAV disease in a physiologically relevant setting, we conducted all experiments in myxovirus protein 1 (Mx1)-congenic C57BL/6 mice (hereafter referred to as Mx1 mice). Mx1 is a dynamin-like guanosine triphosphatase that is a critical interferon-inducible gene important for the control of primary IAV infections in mice (15, 16). Most inbred mouse strains, including C57BL/6 mice, however, lack a functional copy of the Mx1 gene (17). Whereas wild type C57BL/6 mice are extremely susceptible to IAV infection, succumbing to as few as 100 PFU of A/PR8 IAV, Mx1 mice are highly resistant to infection with doses over 106 PFU (10, 18, 19). Therefore, in this study, we use Mx1 mice to probe the impact of KD on influenza infection and disease in the setting of intact innate immunity.
Results
After intranasal challenge with IAV (108 pfu), Mx1 mice that had been fed KD for 7 days prior to infection were protected from body weight loss and had improved survival relative to mice on a normal chow diet (Figs. 1A, B). In addition, KD-fed mice were able to better maintain blood O2 saturation, suggesting improved gas exchange as compared to chow-fed mice (Fig. 1C). This observed protection was associated with improved antiviral resistance, as viral titers were significantly lower in the lungs of KD-fed mice (Fig. 1D). To gain insight into the mechanisms underlying KD-enhanced antiviral immunity in the lungs of these mice, we performed transcriptome analysis of infected whole lung tissue samples (fig. S1, Table S1). Notably, KD did not result in enhanced interferons (IFN) or IFN-stimulated genes (ISGs). Instead, Ingenuity Pathway Analysis (IPA) indicated enrichment in T cell activation pathways (fig. S1C). The results suggested that KD protects mice against IAV through non-conventional mechanisms with potential contribution from T cells early after IAV infection. Of the top 10 significantly regulated genes in KD-fed mice was a γδTCR gene segment (Tcrg-C1) and by comparing our dataset with ImmGen γδ T cell datasets (20) we identified four additional genes in this list that are highly associated with γδ T cells (Cxcr6, Blk, Cd163l1, Ccr4) (Fig. 1E). Indeed, by flow cytometry, we observed significant increases in the frequencies and absolute numbers of γδ T cells in the lungs of KD-fed mice (Fig. 1F–H), while no differences were found for other cell types tested (fig. S2A). Similar observations were also made in the bronchoalveolar lavage (BAL) (fig. S2B–D). Intracellular cytokine staining identified these γδ T cells to be an IL-17-, but not IFN-γ, competent subset (fig. S2E, F). Such IL-17-producing γδ T cells were recently shown to mediate neonatal influenza protection by inducing an ILC2/Treg tissue repair response (21) and are essential for control of pulmonary bacterial infections (22–24). We did not detect differences in BAL IL-17 (fig. S2G) and we observed lower BAL IFN-γ levels (fig. S2H) in KD-fed mice compared to chow-fed mice on day 3 post-infection. Although IPA indicated “Th2 Activation” as a significantly regulated pathway in our dataset (fig. S1C), our transcriptome analysis did not reveal KD-induced enhancement of type 2 immunity genes identified by Guo et al. (21) (fig. S2I).
Figure 1. γδ T cells are required for improved survival of KD-fed mice during IAV infection.
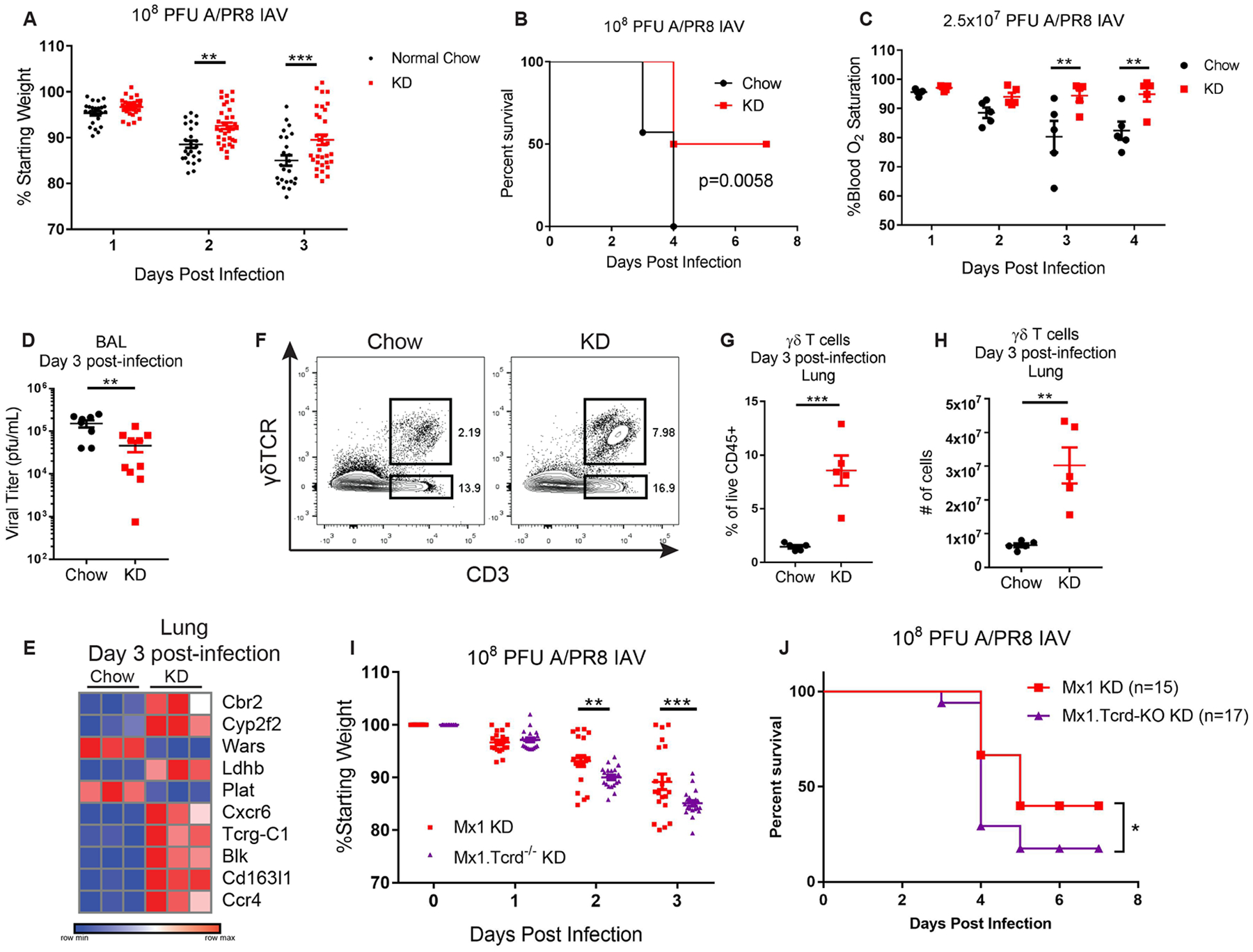
(A) Body weight change (pooled from 5 independent experiments for a total of n=25 chow and n=33 KD mice analyzed) and (B) survival after infection with 108 PFU A/PR8 IAV in chow (n=7) vs KD (n=10). Survival differences are representative of 3 independent experiments and were calculated by log-rank test. (C) Blood oxygen saturation kinetics after sub-lethal infection with 2.5×107 PFU A/PR8 IAV. N=5 mice/group. (D) BAL viral titers on day 3 post-infection with 108 PFU A/PR8 IAV. Pooled from 2 independent experiments (chow n=8, KD n=10). (E) Heat map of RNAseq top 10 differentially expressed genes (day 3 post-infection, based on adjusted p-value p<0.05) in IAV-infected whole lung tissue of KD- versus chow-fed mice. (F) Representative FACS plot of lung γδ T cells on day 3 post-infection. (G) Percentage and (H) absolute number of γδ T cells in lungs 3 days post-IAV infection. N=5 mice/group. (F-H) Data are representative of more than 5 independent experiments. (I) Body weight change after infection of Mx1 (n=21) versus Tcrd−/− Mx1 mice fed KD (n=20) with 108 pfu IAV. Pooled from 3 independent experiments. (J) Survival after infection of Mx1 (n=15) versus Tcrd−/− Mx1 mice fed KD (n=17) with 108 pfu IAV. Survival differences were calculated by Gehan-Breslow-Wilcoxon test. (I) For all graphs, each symbol represents an individual mouse and data are expressed as mean±SEM. Statistical differences were calculated by (A, C, I) paired 2-way ANOVA, and (D, G, H) unpaired t-test. *p<0.05, **p<0.01, ***p<0.001.
To assess the importance of γδ T cells in KD-mediated protection against influenza virus infection, we generated Tcrd−/− Mx1 mice and fed them either regular chow or KD prior to IAV infection. Notably, the protective effect of KD was diminished in Tcrd−/− Mx1 mice following lethal IAV infection (Fig. 1I, J). These data indicate that γδ T cells are required for KD-mediated protection against influenza disease. Of note, Tcrd−/− mice on KD did not exhibit complete lethality, suggesting multiple KD-induced physiological effects may synergize to improve IAV survival.
We considered the possibility that the enhanced body weight preservation in KD-fed mice might simply reflect the high caloric density of the diet (6.76kcal/g, 90% of calories from fat, <1% of calories from carbohydrate) compared to standard chow diet (3.1kcal/g, 18% of calories from fat, 58% of calories from carbohydrate). To test this, we compared the consequences of IAV infection in mice fed KD versus those fed standard high-fat diet (HFD; 5.21kcal/g, 60% of calories from fat, 20% of calories from carbohydrate) beginning one week prior to infection. Unlike KD-fed mice, HFD-fed mice lost body weight upon IAV infection at levels comparable to mice on regular chow (Fig. 2A). Surprisingly, HFD feeding also led to a significant increase in lung γδ T cells (Fig. 2B) that were also primed to produce IL-17 (Fig. 2C). Taken together these data show that high-fat high-carbohydrate western diet-induced expansion of γδ T cells is insufficient to confer protection, suggesting an important specificity for ketogenesis in protection against IAV infection.
Figure 2. High-fat content of KD is not sufficient to induce protective γδ T cells.
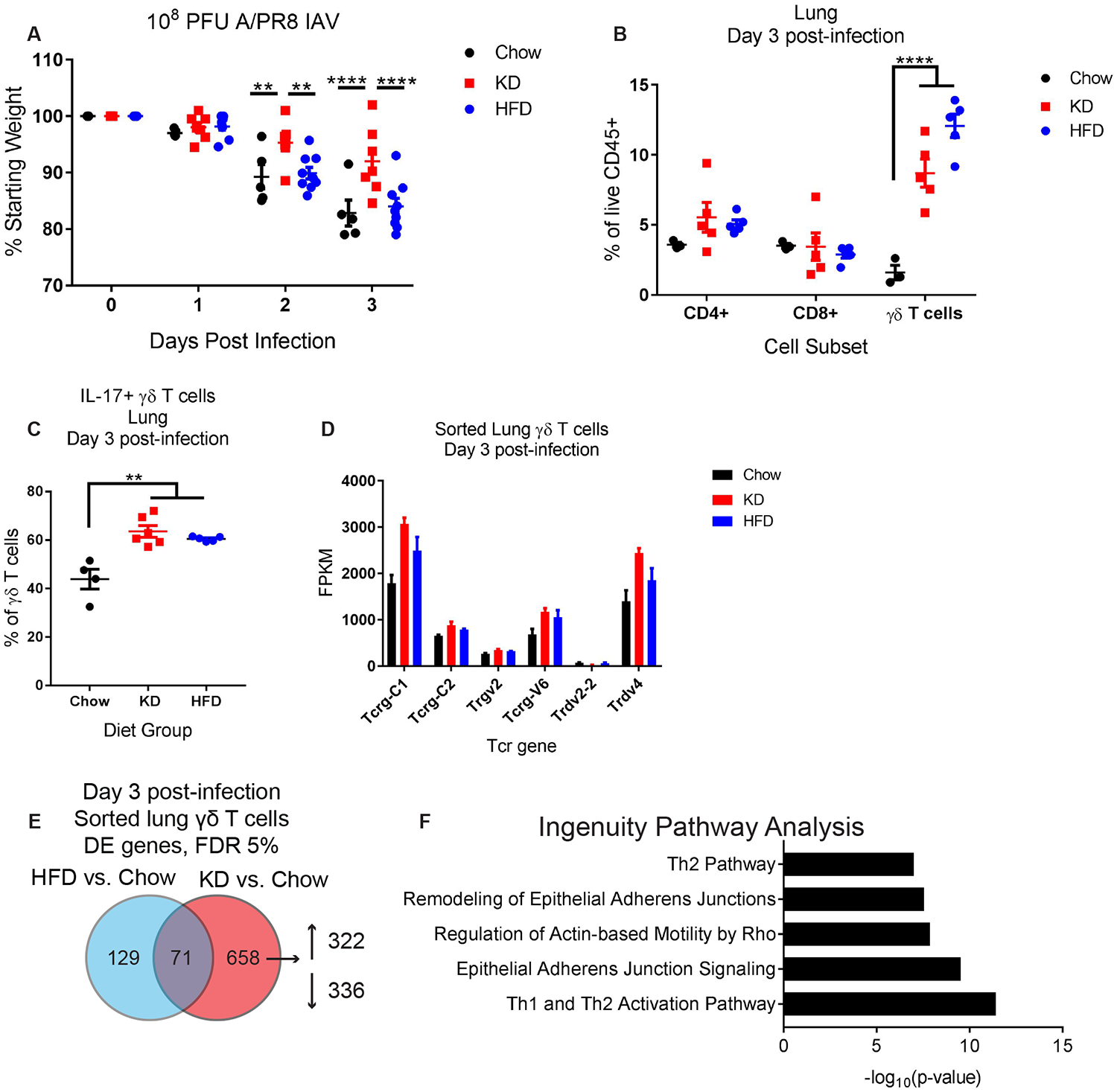
(A) Body weight change of chow (n=5), KD (n=7), and HFD-fed (n=9) mice after infection with 108 pfu IAV. (B) Lung γδ T cell abundance 3 days post-IAV infection in chow (n=3), KD (n=5), and HFD-fed (n=5) mice. (C) Frequency of γδ T cells from the lungs of chow (n=4), KD (n=6), and HFD-fed (n=5) mice that produce IL-17 after PMA+ionomycin stimulation ex vivo. (D) Bar graph of significantly regulated Tcrd and Tcrg gene segments. (E) Venn diagram of numbers of differentially expressed genes (FDR 5%) in Chow, KD, and HFD sorted lung γδ T cells on day 3 post-infection. (F) Ingenuity Pathway Analysis of genes selectively induced by KD compared to chow. (A-C) Data are representative of 3 independent experiments. For all graphs, each symbol represents an individual mouse. Data are expressed as mean±SEM. Statistical differences were calculated by (A) paired 2-way ANOVA, (B) 2-way ANOVA with Tukey’s correction for multiple comparisons, and (C) 1-way ANOVA with Tukey’s correction for multiple comparisons. **p<0.01, ****p<0.0001.
To identify the potential link between diet-induced γδ T cells and protection against IAV infection, we probed for unique features of KD-induced γδ T cell phenotypes by RNA sequencing analysis of sorted γδ T cells from the lungs of mice fed chow vs. HFD vs. KD beginning 1 week prior to IAV infection (fig. S3, Tables S2–S4). Certain γδ T cell receptor genes, including Tcrg-C1 and Trdv4, were highly expressed among the sorted lung γδ T cells and were increased by KD (Fig. 2C), which suggested rather than recruitment of a novel γδ T cell subset, protective functions of common γδ T cells were being enhanced. We discovered a relatively small list of genes (126 total) that were differentially regulated (FDR 10%) by KD as compared to both chow and HFD (fig. S4A). Notably, all these genes except for one (Tgtp1) were regulated by KD in the same direction compared to both chow and HFD (fig. S4B, C). We noted KD-specific enrichment for multiple differentially regulated cytokines, ISGs, and effector molecules in this list, including the up-regulation of Il-17a, Isg20, Ifit1bl1, Ifi204, Ctla4, Ccr1, Cxcr4 and the down-regulation of Il12rb2, P2rx7, and Ccr7. Ingenuity pathway analysis of the genes differentially regulated specifically by KD relative to normal chow (Fig. 2E) showed an enhancement of Th2-type responses and enrichment for several pathways involved in epithelial cell adhesion functions (Fig. 2F, Table S5). These data thus indicate that KD promotes the enrichment of a subset of lung γδ T cells with a unique genetic signature distinct from those induced by HFD.
We initially focused our analyses on day 3 post-infection because most lethality occurred 4 days post-infection (Fig. 1B). However, differences in body weight are apparent by day 2 post-infection. We therefore decided to assess early changes induced by KD. Notably, KD-mediated increases in lung γδ T cells were evident prior to infection (fig. S5A) and remained elevated early after infection (fig. S5B). Like day 3 post-infection, the expanded γδ T cells in the lungs 24hr post-infection were IL-17-competent and were mostly CD44+ CD27- (fig. S5C–E). Importantly, viral titers were similar in chow versus KD and HFD at this early time point following IAV infection (fig. S5F). Additionally, despite previous studies showing neutrophil recruitment by γδ T cells (25, 26), we found no differences in neutrophil or Ly6C+ monocyte recruitment in the BAL during KD or HFD feeding (fig. S6).
Next, we examined the mechanisms by which γδ T cells accumulate in the lungs in response to KD feeding. KD increased BrdU incorporation in γδ T cells (fig. S7A), suggesting increased proliferation of these cells. Blockade of Giα signaling via treatment with pertussis toxin (PTx) impeded γδ T cell expansion in KD-fed mice (fig. S7B). Together, these results suggest that both proliferation and chemokine-mediated recruitment drive the γδ T cell expansion in response to KD during IAV infection.
Given that BHB serves as the primary energy substrate during glucose deprivation and KD feeding (27), we next tested whether BHB itself was responsible for the γδ T cell increase in the lungs post IAV infection. To test this possibility, we fed mice 1,3-butanediol (BD), which increases circulating BHB levels without the requirement for mitochondrial fatty acid oxidation, a necessary metabolic switch induced by KD. While BD efficiently elevated blood BHB to levels comparable to KD-fed mice, surprisingly, it failed to induce γδ T cell expansion or to protect Mx1 mice from IAV-induced weight loss (Fig. 3A, B), indicating that BHB is not sufficient to mediate this phenotype. These data indicate that metabolic adaptation favoring enhanced fatty acid oxidation during KD feeding (28), rather than a simple increase in BHB levels, is required for the expansion of protective γδ T cells in the lung and the maintenance of body weight during IAV infection. In further support of this hypothesis, we examined proteins essential for oxidative mitochondrial metabolism in the lung and observed that KD-fed mice displayed higher expression of catalase, a key enzyme that protects against ROS damage, and 3-oxoacid CoA transferase 1 (Oxct1 or SCOT), a rate limiting enzyme in mitochondrial ketolysis. In addition, as compared to chow-fed mice, those fed KD also showed elevated expression of mitochondrial electron transport chain complexes in the lungs (Fig. 3C). Neither KD nor HFD altered ketone metabolism genes specifically in γδ T cells (Fig. 3D) and although KD induced gene signatures associated with increased oxidative phosphorylation metabolic programming, ketone metabolism pathways were not significantly altered by KD in sorted γδ T cells (fig. S3, Table S4). Together these data demonstrate that KD-dependent increased oxidative metabolism and improved redox balance in the lung is linked with γδ T cell expansion and enhanced survival in response to an otherwise lethal IAV infection.
Figure 3. Protective γδ T cell expansion requires metabolic adaptation to KD.
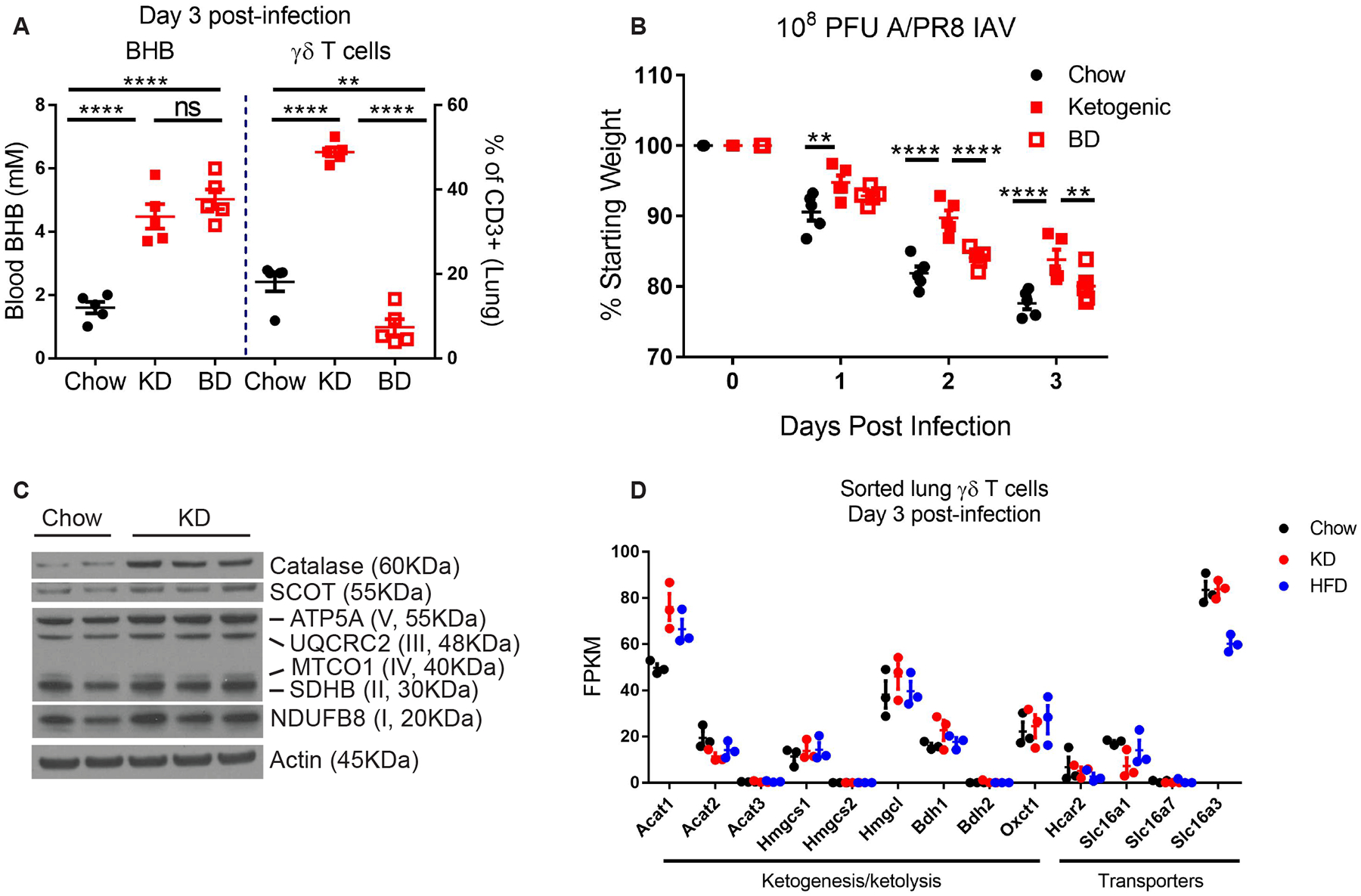
(A) Blood BHB and lung γδ T cells on day 3 post-IAV in mice fed chow (n=5) vs. KD (n=5) vs. 1,3-Butanediol (BD, n=5) beginning 1 week prior to infection. Statistical differences were calculated by 1-way ANOVA with Tukey’s correction for multiple comparisons (B) Body weights of IAV-infected mice fed chow (n=5), KD (n=5), or BD (n=5). Statistical differences were calculated by paired 2-way ANOVA with Tukey’s correction for multiple comparisons. (A-B) Data are representative of at least 2 independent repeats. (C) Western blot of mitochondrial oxidative metabolism proteins in whole lung tissue 3 days after IAV infection in chow and KD-fed mice. Each lane represents an individual mouse. (D) RNAseq expression data of ketone metabolism genes from sorted γδ T cells 3 days post-infection. For all graphs, each symbol represents an individual mouse. Data are expressed as mean±SEM. **p<0.01, ****p<0.0001.
To identify the molecular basis for γδ T cell-mediated protection against IAV lethality we performed RNAseq on IAV-infected lungs from KD-fed Tcrd−/− Mx1 mice. (fig. S8, Table S6). We identified a variety of genes significantly regulated by KD and manually clustered them based on γδ T cell-dependence (fig. S9). We analyzed the intersection of genes significantly regulated by KD that were also γδ T cell-dependent and identified 11 genes that met these criteria (Fig. 4A). This gene list indicated that KD, in a γδ T cell-dependent manner, increased protective airway secretory cells (Scgb1a1, Scgb3a2), scavenger receptors Cd163l1 (possibly due to its expression on γδ T cells) and Marco (Scgb3a2 binding partner). These genes also suggested that in a γδ T cell-dependent manner KD improved pulmonary metabolism of endogenous carbonyl compounds including ketones derived from lipid peroxidation (Cbr2), metabolism of toxic compounds (Cyp2f2, Cyp4f15), and increased tight junctions (Cldn10). This pattern of gene enrichment suggested enhanced secretory cell function in the lung in response to KD. Periodic acid–Schiff (PAS) staining to identify mucus-producing cells confirmed an increase in the numbers of mucus-producing cells in the airways of KD-fed mice that was dependent on γδ T cells (Fig 4B–H). Accordingly, expression of Scgb1a1, a specific marker of club cells and is a founding member of the secretoglobin family, correlated with decreasing viral titers in the lung (Fig. 4H).
Figure 4. KD-induced protective γδ T cells enhance lung barrier function.
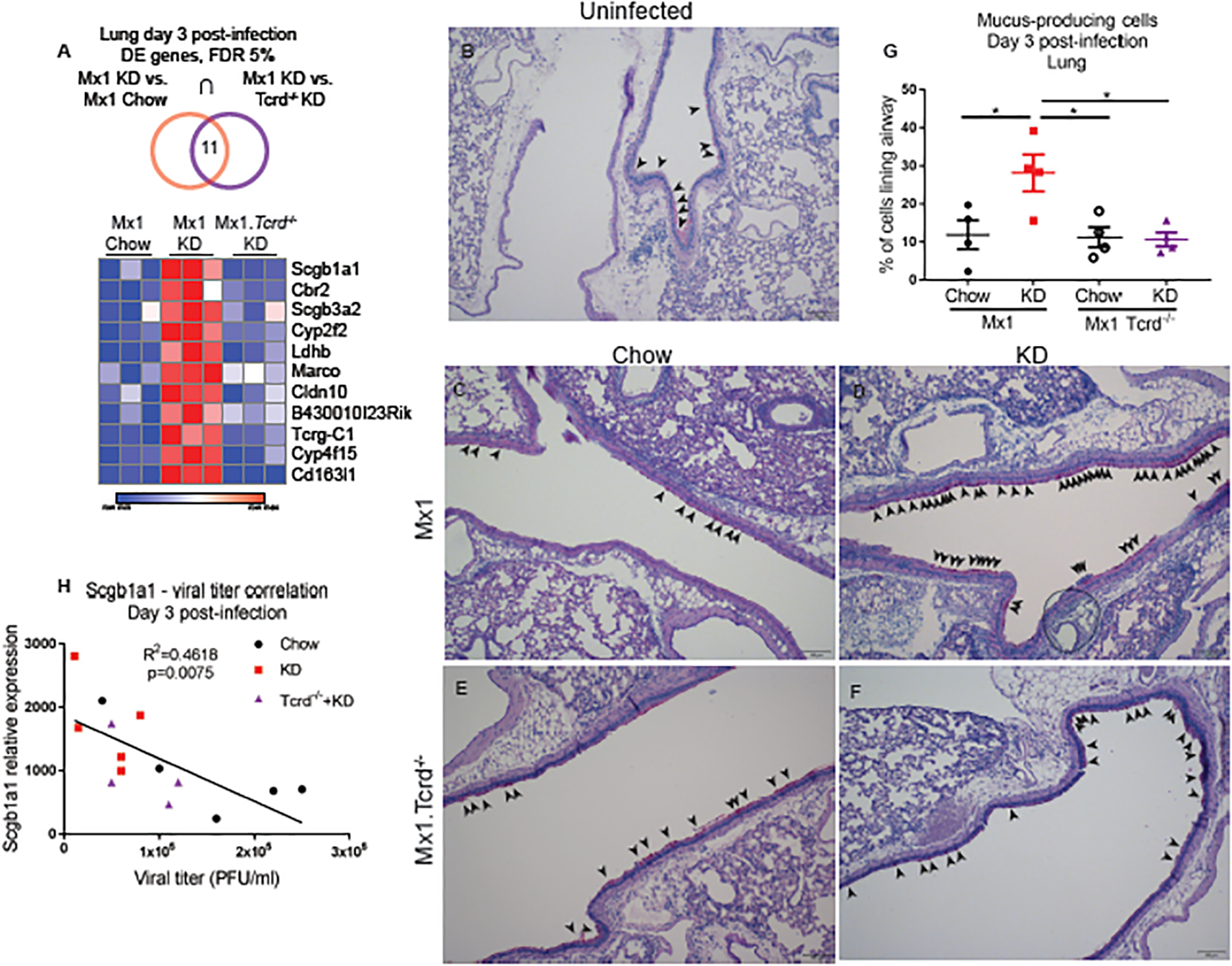
(A) Heat map of significantly regulated genes (based on adjusted p-value p<0.05) in both comparisons of Mx1 chow vs. Mx1 KD, and Mx1 KD vs. Mx1 Tcrd−/− KD. Genes are ranked from highest to lowest expression. (B-F) Representative images of lung airway PAS staining. Arrowheads indicate PAS-positive cells. Scale bars, 100um. (G) Quantification of PAS staining images. Statistical differences were calculated by 1-way ANOVA with Tukey’s correction for multiple comparisons. Data are represented as mean±SEM and n=4 mice/group. (H) Correlation analysis of lung Scgb1a1 expression with BAL viral titers collected on day 3 post-IAV infection. Statistics were calculated by linear regression pooled from chow (n=5), KD (n=5), and Mx1 Tcrd−/− KD (n=4). For all graphs, each symbol represents an individual mouse. *p<0.05.
Discussion
Our study found that KD feeding confers protection against influenza virus infection in Mx1 mice. KD increased the number of γδ T cells in the respiratory tract, and these γδ T cells were required to achieve the full protective effect of KD. The contributions of γδ T cells to lung homeostasis and IAV immunity remain incompletely explored. γδ T cells have previously been reported to increase late after IAV infection in mice (29). Human γδ T cells can expand in a TCR-independent manner in response to IAV, and the human Vγ9Vδ2 T cell subset has been shown to efficiently induce the cytolytic killing of IAV-infected A549 airway cells (30, 31). The precise mechanism by which KD-induced γδ T cells enhance lung barrier integrity will require further study.
How might γδ T cells protect the host against influenza virus challenge in response to KD? Our results may be explained by efficient killing of IAV-infected airway epithelial cells by the γδ T cells, which expand specifically in response to KD in mice, resulting in lower viral titers and overall better preservation of airway tissue integrity. However, the transcriptome data did not reveal any canonical cytotoxicity genes uniquely induced by KD (fig. S4). Alternatively, the γδ T cells induced by KD may enhance the barrier and innate defense functions of airway epithelial cells at baseline, thus allowing them to better respond upon IAV challenge, leading to observed lower viral titers. Consistent with this latter model, KD-fed mice increased mucus-producing cells in the airway in a γδ T cell-dependent manner. Additionally, RNAseq analysis of infected lung tissue revealed unique epithelial gene patterns that were associated with lower viral titer and improved survival that were lost in KD-fed Tcrd−/− mice.
Other studies have shown that epithelial γδ T cells improve lung integrity both in the context of bacterial infection and ozone exposure (32), while Tcrd−/− mice are more susceptible to mucosal injury in a DSS-colitis model (33). Collectively, these studies highlight a key role for γδ T cells in maintaining barrier integrity beyond their cytolytic potential. This possibility is further supported by a recent report that adipose tissue γδ T cells have critical protective functions (34). Our results showed the capacity of KD, but not HFD, in conferring protection against influenza virus infection. Further, KD-mediated protection relied on large part to the presence of γδ T cells. We propose that γδ T cells may serve as a modulator of epithelial functionality in response to diet and nutrient availability, by inducing changes in the epithelial cell differentiation and function for adaptation to the changing environment. Harnessing the beneficial effects of KD through γδ T cells may therefore offer a potential novel avenue for influenza disease prevention and treatment.
Materials and Methods
Study Design
The objective of this study was to determine how KD feeding affects host defense against lethal IAV infection. Mice were randomized to indicated diet groups for 1 week prior to infection and were subsequently monitored for clinical signs of disease and euthanized to assess the immune response against IAV. The different diets used are visibly different so investigator blinding during experiments was impossible, but genotypes and diet groups were deidentified during analysis to blind investigators during this phase of the experiments. Group sizes are indicated in respective figure legends. None of the data shown contained any statistical outliers. All experiments were repeated independently at least 3 times.
Mice
All mice were C57BL/6 carrying functional alleles of Mx1 as previously reported (35). Tcrd−/− mice (purchased from Jackson Labs) (36) were originally on the B6 background and then crossed with Mx1 mice in the Iwasaki lab to generate homozygous knockout mice for each genotype. Mice were housed in SPF conditions under normal 12hr light/dark cycles in the Yale animal facility. All procedures were approved by the Yale Institutional Animal Care and Use Committee.
Diets
All diets were provided ad libitum throughout experiments. Mice were maintained on standard vivarium chow (Envigo 2018S) until switched to respective experimental diets exactly 1 week prior to infection. Ketogenic diet (KD, D12369B) and high-fat diet (60%, HFD, D12492) were purchased from Research Diets. 1,3-butanediol diet (BD) was prepared by mixing standard chow (Purina 5002, 198g)+1,3-butanediol (Sigma, 80mL)+H2O (120mL)+Saccharine (Sigma, 2g) and replaced daily.
Infection and Treatments
Mice were anesthetized by i.p. injection of ketamine and xylazine and were then infected intranasally with 108 pfu A/PR/8/34 (H1N1) influenza virus in a total volume of 50ul. Mice were monitored daily for body weight changes. BHB was measured in whole blood by handheld Precision Xtra test strips. Mice were treated with 500ng of pertussis toxin (PTx) in 50ul PBS by daily i.p. injection immediately following infection. BrdU was provided in drinking water at 0.8mg/ml+5mM saccharin beginning 1 day prior to infection and changed every other day. Blood O2 saturation was measured by pulse oximetry (MouseOx Plus, Starr Life Sciences) in mice infected with a sublethal IAV dose (2.5×107 pfu) so that all mice would survive infection.
Measurement of Viral Titers
Bronchoalveolar lavage (BAL) was collected by washing the trachea and lungs 3 times with 1mL PBS+0.1% BSA. 10-fold serial dilutions of BAL were made in 100ul aliquots of PBS+0.1% BSA and used to inoculate Madin-Darby Canine kidney cells in 12-well plates. After 1 h of incubation, each well was overlaid with 1ml of agar medium for 48 h. Cell monolayers were then stained with 0.1 % crystal violet in 20 % ethanol and plaques were enumerated.
Western Blot
Whole lung tissue was snap frozen in liquid nitrogen and homogenized in RIPA buffer containing protease inhibitors. Protein concentration was measured by DC Protein Assay (Bio-rad) and equal amounts of protein were run on an SDS-PAGE gel and transferred to nitrocellulose membrane using standard techniques. Blots were probed with primary antibodies against Catalase (Sigma), SCOT (Proteintech), ETC complexes (Abcam, samples prepared according to manufacturer’s recommendation), and Actin (Cell Signaling). Incubation in secondary antibodies of the appropriate species (ThermoFisher) were followed by chemiluminescent visualization (Fisher, Bio-rad).
Flow Cytometry
Lungs were digested for 1 hour in RPMI+10% FCS+1mM CaCl2+1mM MgCl2+5mM HEPES+2.5% Collagenase D. Tissue was minced through a 70uM strainer to obtain a single cell suspension. Cells were stained with live/dead viability dye (Invitrogen) followed by surface staining for B220, CD11b, NK1.1, CD3, CD4, CD8, and γδTCR. BrdU incorporation was detected by BrdU staining using the BD BrdU flow kit staining protocol. For intracellular cytokine staining lung cells were stimulated at 37°C with Phorbol 12-myristate 13-acetate (PMA, 10ng/mL, Sigma) + ionomycin (1μM, Sigma) for 4 hours, and protein transport inhibitor (eBioscience) was added for the last 3 hours of stimulation. Intracellular IL-17 and IFNγ staining were detected using the BD fix/perm staining kit. Cell counts were obtained by normalizing to CountBright Absolute Counting Beads (ThermoFisher).
RNAseq
Whole transcriptome analysis was performed in whole lung tissue and also in FACS sorted γδ T cells from digested lung tissue. The quality of raw reads was assessed with FastQC (37). Raw reads were mapped to the GENCODE vM9 mouse reference genome (38) using STAR aligner (39) with the following options: --outFilterMultimapNmax 15 --outFilterMismatchNmax 6 --outSAMstrandField All --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM. The quality control of mapped reads was performed using in-house scripts that employ Picard tools (40). The list of rRNA genomic intervals that we used for this quality control was prepared on the basis of UCSC mm10 rRNA annotation file (41) and GENCODE primary assembly annotation for vM9 (38). rRNA intervals from these two annotations were combined and merged to obtain the final list of rRNA intervals. These intervals were used for the calculation of the percentage of reads mapped to rRNA genomic loci. Strand specificity of the RNA-Seq experiment was determined using an in-house script, on the basis of Picard (40)mapping statistics. Expression quantification was performed using RSEM (42). PCA was performed in R. On the basis of the PCA, we excluded 1 sample from the whole lung dataset, but none from the sorted γδ T cell dataset, from further analysis as an outlier. Gene differential expression was calculated using DESeq2 (43). Pathway analysis was done using Ingenuity Pathway Analysis (IPA, Qiagen) (44) and fgsea (fast GSEA) R-package (45) with the minimum of 15 and maximum of 500 genes in a pathway and with 1 million permutations. For the pathway analysis, we used the Canonical Pathways from the MSigDB C2 pathway set v6.1 (46, 47).
Quantitative PCR
Select genes of interest from RNAseq dataset in whole lung tissue were independently verified by quantitative RT-PCR. Lungs were snap frozen in liquid nitrogen and homogenized directly in RLT buffer for RNA extraction (Qiagen). RNA was quantified and cDNA was transcribed using iScript cDNA synthesis kit (Bio-Rad). Gene expression was measured by RT-PCR by ΔΔCt method and expressed relative to Hprt or Gapdh. Relative expression was used for correlation analyses.
Immunohistochemistry
Periodic acid-Schiff (PAS) staining was performed on formalin-fixed paraffin-embedded lung tissue sections. Sections were deparrafinized in xylenes and rehydrated in EtOH using standard procedures. PAS staining was performed exactly according to manufacturer’s protocol using the PAS staining kit (Sigma). Slides were then dehydrated in EtOH and cleared in xylenes for mounting. Images were acquired on a Keyence BZ-X700 microscope.
Statistical Analysis
All analyses, except for RNAseq, were performed using GraphPad Prism 7 software. To compare changes over time between different groups, paired 2-way ANOVA with Tukey’s correction for multiple comparisons was used to calculate statistical differences. Survival was based on 20% body weight loss and statistical significance was calculated by log-rank test. Statistical differences comparing 3 groups were calculated by 1-way ANOVA with Tukey’s correction for multiple comparisons. Unpaired two-tailed t-tests were used to calculate statistical differences between 2 groups. Correlation analyses were performed using linear regression. For all tests a p-value p≤0.05 was considered statistically significant. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Supplementary Material
Fig. S1. Bioinformatics workflow for whole lung RNAseq analysis.
Fig. S2. Quantification and phenotype of lung γδ T cells 3 days post-IAV infection.
Fig. S3. RNAseq bioinformatics of sorted lung γδ T cells 3 days post-IAV infection.
Fig. S4. Transcriptomic signature of KD-specific genes in lung γδ T cells 3 days post-IAV infection.
Fig. S5. γδ T cells expansion precedes IAV infection.
Fig. S6. Inflammatory myeloid cell infiltration is not affected by KD or HFD.
Fig. S7. Proliferation and recruitment drive KD-mediated γδ T cell expansion.
Fig. S8. Bioinformatics workflow for whole lung RNAseq analysis in Mx1 Tcrd−/− mice.
Fig. S9. γδ T cell-dependent regulation of KD-induced transcriptome changes.
Table S1. Significantly regulated pathways in whole lungs of chow vs KD mice 3 days post-IAV infection.
Table S2. Gene set enrichment analysis of sorted lung γδ T cells from HFD vs KD-fed mice.
Table S3. Gene set enrichment analysis of sorted lung γδ T cells from HFD vs chow-fed mice.
Table S4. Gene set enrichment analysis of sorted lung γδ T cells from KD vs chow-fed mice.
Table S5. KD-specific gene signature of lung γδ T cells.
Table S6. Significantly regulated pathways in whole lungs of Mx1 KD vs Mx1 Tcrd−/− KD mice 3 days post-IAV infection.
Acknowledgments:
We thank the members of Dixit and Iwasaki labs for critical discussion and feedback related to this project.
Funding: This work was in part supported by funding from the NIH (AI054359, R01EB000487, R01AI127429, R42AI120269 and R21AI131284; to AI). AI received support from the Claude D. Pepper Older Americans Independence Center at Yale University School of Medicine P30 AG021342 NIH/NIA. AI is an Investigator of the Howard Hughes Medical Institute. The Dixit lab is supported in part by NIH grants P01AG051459, AI105097, AG051459, AR070811 and the Glenn Foundation on Aging Research. ELG is supported by NIH grant K99AG058801.
Footnotes
Competing interests: Authors declare no competing interests.
Data and materials availability: RNA-seg data reported in this study have been deposited in the Gene Expression Omnibus database under the accession number GSE136536.
References and Notes:
- 1.Thompson WW et al. , Estimates of US influenza-associated deaths made using four different methods. Influenza Other Respir Viruses 3, 37–49 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molinari NA et al. , The annual impact of seasonal influenza in the US: measuring disease burden and costs. Vaccine 25, 5086–5096 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Ortiz JR et al. , Report on eighth WHO meeting on development of influenza vaccines that induce broadly protective and long-lasting immune responses: Chicago, USA, 23–24 August 2016. Vaccine 36, 932–938 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Paules CI, Marston HD, Eisinger RW, Baltimore D, Fauci AS, The Pathway to a Universal Influenza Vaccine. Immunity 47, 599–603 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Iwasaki A, Pillai PS, Innate immunity to influenza virus infection. Nat Rev Immunol 14, 315–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medzhitov R, Schneider DS, Soares MP, Disease tolerance as a defense strategy. Science 335, 936–941 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahill GF Jr., Veech RL, Ketoacids? Good medicine? Trans Am Clin Climatol Assoc 114, 149–161; discussion 162–143 (2003). [PMC free article] [PubMed] [Google Scholar]
- 8.Wang A et al. , Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 166, 1512–1525 e1512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman JC, Verdin E, Ketone bodies as signaling metabolites. Trends Endocrinol Metab 25, 42–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillai PS et al. , Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 352, 463–466 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brandes M, Klauschen F, Kuchen S, Germain RN, A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 154, 197–212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mauad T et al. , Lung pathology in fatal novel human influenza A (H1N1) infection. Am J Respir Crit Care Med 181, 72–79 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Goldberg EL et al. , beta-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep 18, 2077–2087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youm YH et al. , The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21, 263–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Verhelst J, Parthoens E, Schepens B, Fiers W, Saelens X, Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol 86, 13445–13455 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haller O, Staeheli P, Schwemmle M, Kochs G, Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol 23, 154–163 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O, Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8, 4518–4523 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P, Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J Gen Virol 93, 555–559 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Moritoh K et al. , Generation of congenic mouse strains by introducing the virus-resistant genes, Mx1 and Oas1b, of feral mouse-derived inbred strain MSM/Ms into the common strain C57BL/6J. Jpn J Vet Res 57, 89–99 (2009). [PubMed] [Google Scholar]
- 20.Heng TS, Painter MW, Immunological Genome Project C, The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091–1094 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Guo XJ et al. , Lung gammadelta T Cells Mediate Protective Responses during Neonatal Influenza Infection that Are Associated with Type 2 Immunity. Immunity 49, 531–544 e536 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirby AC, Newton DJ, Carding SR, Kaye PM, Evidence for the involvement of lung-specific gammadelta T cell subsets in local responses to Streptococcus pneumoniae infection. European journal of immunology 37, 3404–3413 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okamoto Yoshida Y et al. , Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. Journal of immunology 184, 4414–4422 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Simonian PL et al. , IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. Journal of immunology 182, 6540–6549 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan B, Chen F, Xu L, Xing J, Wang X, HMGB1-TLR4-IL23-IL17A axis promotes paraquat-induced acute lung injury by mediating neutrophil infiltration in mice. Sci Rep 7, 597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez-Valle F, Gray EE, Cyster JG, Inflammation induces dermal Vgamma4+ gammadeltaT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc Natl Acad Sci U S A 112, 8046–8051 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puchalska P, Crawford PA, Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab 25, 262–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy AR et al. , A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab 292, E1724–1739 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Carding SR et al. , Late dominance of the inflammatory process in murine influenza by gamma/delta + T cells. J Exp Med 172, 1225–1231 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y et al. , The interaction of influenza H5N1 viral hemagglutinin with sialic acid receptors leads to the activation of human gammadelta T cells. Cell Mol Immunol 10, 463–470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H et al. , Human Vgamma9Vdelta2-T cells efficiently kill influenza virus-infected lung alveolar epithelial cells. Cell Mol Immunol 10, 159–164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.King DP et al. , Cutting edge: protective response to pulmonary injury requires gamma delta T lymphocytes. J Immunol 162, 5033–5036 (1999). [PubMed] [Google Scholar]
- 33.Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R, Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A 99, 14338–14343 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kohlgruber AC et al. , gammadelta T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol 19, 464–474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horisberger MA, Staeheli P, Haller O, Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc Natl Acad Sci U S A 80, 1910–1914 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Itohara S et al. , T cell receptor delta gene mutant mice: independent generation of alpha beta T cells and programmed rearrangements of gamma delta TCR genes. Cell 72, 337–348 (1993). [DOI] [PubMed] [Google Scholar]
- 37.Andrews S, FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc, (2010).
- 38.Mudge JM, Harrow J, Creating reference gene annotation for the mouse C57BL6/J genome assembly. Mamm Genome 26, 366–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picard tools.
- 41.Karolchik D et al. , The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32, D493–496 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kramer A, Green J, Pollard J Jr., Tugendreich S, Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sergushichev A, An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, (2016). [Google Scholar]
- 46.Liberzon A et al. , Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Bioinformatics workflow for whole lung RNAseq analysis.
Fig. S2. Quantification and phenotype of lung γδ T cells 3 days post-IAV infection.
Fig. S3. RNAseq bioinformatics of sorted lung γδ T cells 3 days post-IAV infection.
Fig. S4. Transcriptomic signature of KD-specific genes in lung γδ T cells 3 days post-IAV infection.
Fig. S5. γδ T cells expansion precedes IAV infection.
Fig. S6. Inflammatory myeloid cell infiltration is not affected by KD or HFD.
Fig. S7. Proliferation and recruitment drive KD-mediated γδ T cell expansion.
Fig. S8. Bioinformatics workflow for whole lung RNAseq analysis in Mx1 Tcrd−/− mice.
Fig. S9. γδ T cell-dependent regulation of KD-induced transcriptome changes.
Table S1. Significantly regulated pathways in whole lungs of chow vs KD mice 3 days post-IAV infection.
Table S2. Gene set enrichment analysis of sorted lung γδ T cells from HFD vs KD-fed mice.
Table S3. Gene set enrichment analysis of sorted lung γδ T cells from HFD vs chow-fed mice.
Table S4. Gene set enrichment analysis of sorted lung γδ T cells from KD vs chow-fed mice.
Table S5. KD-specific gene signature of lung γδ T cells.
Table S6. Significantly regulated pathways in whole lungs of Mx1 KD vs Mx1 Tcrd−/− KD mice 3 days post-IAV infection.