

Abstract

Oxidative addition of aryl halides to Ni(0) is a ubiquitous elementary step in cross-coupling and related reactions, usually producing a square-planar Ni(II)–aryl intermediate. Here we show that a triphosphine ligand supports oxidative addition at a tris-ligated Ni(0) center to cleanly form stable five-coordinate Ni(II)–aryl compounds. Kinetic and computational studies support a concerted, two-electron mechanism rather than radical halogen abstraction. These results support the idea that oxidative addition to triphosphine Ni(0) species may be more generally involved in Ni/phosphine catalytic systems.
The oxidative addition of aryl (pseudo)halides to reduced group 10 metal centers is a common entry point to cross-coupling processes,1 which are ubiquitous synthetic tools in academic and industrial research. In this context, Pd catalysts bearing phosphine ligands are by far the most used.2,3 The oxidative addition of aryl halides on a Pd(0) center generally produces aryl Pd(II) intermediates through a well-documented two-electron mechanism.4−11 Nickel is receiving increasing attention as an alternative to palladium; not only is it less expensive but it also displays interesting reactivity patterns due to a lower electronegativity and the accessibility of odd-electron Ni(I) or Ni(III) species. This opens up radical pathways,12 facilitating the activation of challenging substrates such as alkyl halides and their efficient use in cross coupling.13,14 On the other hand, when aryl halides are used, Ni(I) species are often off-cycle catalyst deactivation products, as found for C–N coupling15 or trifluoromethylthiolation reactions,16 or even detrimental for catalysis, as observed for some Suzuki–Miyaura couplings.17
Active Ni-based precatalysts for cross-coupling reactions often bear monodentate or bidentate phosphine donor ligands.18 Hence, the mechanism of aryl–halide oxidative additions to phosphine-ligated Ni(0) has been placed under some scrutiny. trans-(PPh3)2Ni(Ar)(X) complexes can be generated by oxidative addition of the aryl halide to a Ni(PPh3)4 center,19,20 but these complexes are unstable in solution and readily decompose into Ni(I) species and biaryl byproducts, as recently reported by Baird and Budzelaar.21 The current mechanistic understanding of these reactions is largely based on seminal work by Kochi,22 who showed that the reaction of substituted aryl halides with Ni0(PEt3)4 leads to the oxidative addition products [NiII(PEt3)2(Ar)X] or paramagnetic [NiIX(PEt3)3] species. The solvent-caged radical pair [NiI(PEt3)3+ArX•–] was proposed as the common intermediate for both products (Figure 1a). This hypothesis was recently refined computationally by the Maseras group23 (Figure 1b). They propose that two distinct mechanisms generate the NiII and NiI products: NiII adducts are formed by oxidative addition via a SN2-type transition state, while NiI compounds are generated through an open-shell singlet halogen abstraction transition state.
Figure 1.

Mechanism of oxidative addition of aryl halides to Ni(0) phosphine complexes: (a) Kochi proposal; (b) Maseras and Nelson proposal; (c) our system bearing a pincer phosphine ligand.
Using bidentate phosphine ligands, both Ni(II)–aryl and Ni(I)–halide products can also be observed. Electron-rich trialkylphosphine ligands often yield well-defined Ni(II)–aryl compounds,24,25 but the wide-angle triarylphosphine XantPhos produces exclusively Ni(I)–halides together with biaryl.14 Recently, the Nelson group26 studied the oxidative addition of substituted aryl halides to [Ni0(COD)(dppf)]. The initial [(dppf)NiII(X)Ar] aryl products readily evolve to NiI species [(dppf)NiIX] with release of biaryl in the presence of the initial Ni0 complex. Due to the ortho effect,27 only the Ni(II) species bearing ortho-substituted σ-aryl ligands were transiently detectable in solution. Hazari also emphasized the importance of having at least one ortho substituent in the σ-aryl ligand for the synthesis of stable [NiII(Ar)X(dppf)] complexes used as precatalysts for Suzuki–Miyaura coupling reactions.28
Because of the electronic stability of the square-planar geometry for d8 centers, the observed Ni(II) products of oxidative addition generally bear two phosphine ligands.24,25 However, the proposals by Kochi22 and Maseras23 involves triphosphine Ni(0) complexes as the reactive species. In addition, kinetic studies suggest that the oxidative addition of aryl bromides to the tricoordinate (BINAP)Ni(η2-NCPh) proceeds without prior dissociation of PhCN.15 We reasoned that a tridentate phosphine ligand may stabilize tris-ligated intermediates and allow for the direct observation of the key addition step. Indeed, we show that a well-defined Ni0 complex supported by the bis(2-bis(p-tolyl)phosphinophenyl)phenylphosphine (PPPp-tol) ligand cleanly adds aryl halides to form stable pentacoordinate NiII σ-aryl complexes (Figure 1c). A concerted oxidative addition mechanism is proposed based on a kinetic study and structure–reactivity analysis supported by DFT studies.
Initially, the Ni(0) complex [(PPPp-tol)Ni(BPI)] (1, BPI = benzophenone imine, a labile coligand),29 was treated with the electron-deficient substrates 4-halotrifluorotoluenes in THF at room temperature to yield the oxidative addition products 2a–c in good yields (Scheme 1). Moreover, oxidative addition also proceeded in high yields with more challenging substrates such as 4-bromotoluene (Figures S11–S13) or 4-chlorotoluene (reaction at 80 °C, Figure S14).
Scheme 1. General Reaction of the Oxidative Addition of Para-Substituted Aryl Halides to [(PPPp-tolyl)Ni(BPI)].

The isolated iodo complex 2a displays two broad 31P NMR resonances at 82.57 (br, 1P) and 34.42 (br, 2P) ppm (Figure S4), while 2b,c display sharp triplets (2b, 86.3 ppm, 2JP,P = 32.5 Hz, 1P; 2c, 88.4 ppm, 2JP,P = 33.3 Hz, 1P) and doublets (2b, 37.1 2JP,P = 32.5 Hz, 2P; 2c, 36.9 ppm, 2JP,P = 33.3 Hz, 2P), indicating that all phosphorus atoms are bound to nickel (Figures S7 and S10). Singlet 19F resonances around −61.3 ppm confirm the presence of the aromatic moiety in 2a–c (Figures S6 and S9).
More structural insights are given by the X-ray crystal structure of 2a (Figure 2A), which reveals a pentacoordinate structure best described as a trigonal-bipyramidal (TBP) geometry (τ5 = 0.67),30 where the axial positions are occupied by the aryl ligand and the central P atom of the PPPp-tol ligand, the iodide ligand occupying an equatorial position. A previous study by Haupt31 described a pentacoordinate 2-hydroxyphenyl nickel complex bearing three monodentate trimethylphosphine ligands having a square-pyramidal (SP) geometry (τ5 = 0.29) with a greater Ni–C bond distance (2.004(8) Å) in comparison to that in 2a (1.944(9) Å). Additionally, crystallographically characterized square-planar aryl Ni(II) complexes,32−34 mostly synthesized by transmetalation, display slightly shorter Ni–C bond distances (1.896(1)–1.925(3) Å), except with the bulky Pad-DalPhos ligand (1.971(3) Å).35
Figure 2.

(A) Molecular structure of compound 2a in the crystal (50% probability level). Hydrogen atoms and the toluene solvent molecule are omitted for clarity. Selected bond distances (Å) and angles (deg): Ni1–I1 2.7144(12), Ni1–P1 2.185(3), Ni1–P2 2.148(2), Ni1–P3 2.232(2), Ni1–C47 1.944(9); P2–Ni1–P3 133.40(10), P1–Ni1–C47 173.5(3), P1–Ni1–I1 88.26(8). (B) Calculated TBP structure of compound 3. (C) Calculated SP structure of compound 3.
To assess whether the pentacoordinate structure is conserved in solution, the p-tolyl complex 3 was treated with the halide abstractor sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF). A new species was formed, presumably the cationic complex [(PPPp-tol)Ni(p-tolyl)]+, displaying sharp 31P NMR signals at 83.9 (t, 2JP,P = 22.0 Hz, 1P) and 48.7 (d, 2JP,P = 22.0 Hz, 2P) ppm (Figure S20). DFT calculations confirm a square-planar structure for [(PPPp-tol)Ni(p-tolyl)]+ (see the Supporting Information). The addition of tetrabutylammonium bromide to the complex [(PPPp-tol)Ni(p-tolyl)]+ regenerated complex 3 (Figure S21), confirming that the neutral, pentacoordinate structure is present in solution.
The structure of 3 has been further investigated with DFT (Figure 2B), yielding a distorted-trigonal-bipyramidal structure (τ5 = 0.6) with the aryl group in the axial position, similar to the experimental crystal structure of 2a. A square-pyramidal (τ5 = 0.12) structure with basal aryl and one of the two −P(p-tolyl)2 groups in the apical position lies 5.0 kcal mol–1 higher in energy (Figure 2C and Table S7). Geometry optimization of analogues of 3 in which the tridentate phosphine is replaced by three PH3, PMe3, or PPhMe2 ligands yields in each case a square-pyramidal structure very similar to that of the less stable isomer of the full system (Table S11), suggesting that the preference for the axial aryl position in 3 is due to the chelating structure of the triphosphine ligand.
Having established the nature of the oxidative addition products, we set out to investigate the mechanism by kinetic studies. 4-Bromotoluene (Scheme 1) was chosen as the model substrate. The kinetic profile of the reaction was followed by 31P NMR analysis using triphenylphosphine oxide as an internal standard (Figure 3). In the presence of a large excess of both the aryl bromide (21 equiv) and the BPI coligand (27 equiv), the consumption of complex 1 follows pseudo-first-order kinetics with an observed rate constant kOBS of 0.031 min–1.
Figure 3.
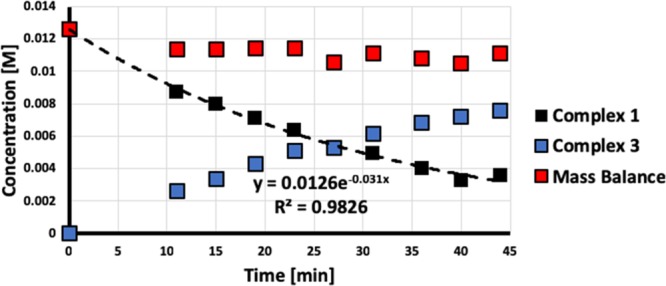
Kinetic profile for the reaction of 4-bromotoluene (21 equiv) with complex 1 in the presence of 27 equiv of BPI.
The partial orders with respect to 4-bromotoluene and BPI were determined from the dependence of kOBS on their concentrations. The reaction is first order in aryl bromide (Figure S24) and inverse first order in BPI (Figure S26), resulting in the rate law
| 1 |
This is consistent with a preequilibrium situation in which BPI and the aryl bromide compete for the vacant site prior to oxidative addition.15 Eyring analysis (T = 25–45 °C, Figure S28) yields the activation parameters ΔH⧧ = 16.5(0.7) kcal mol–1 and ΔS⧧ = −18(2) cal K–1 mol–1, resulting in ΔG⧧ = 21.9(1.3) kcal mol–1 at 298 K. The negative entropy of activation (ΔS⧧) and the global second order for the reaction are consistent with a bimolecular rate-limiting transition state.
The reaction is 4 times slower when toluene is used as solvent instead of THF (Figure S29), suggesting that the transition state is polarized, presumably with a partial negative charge on the aryl ligand. A Hammett analysis (Figure 4) shows good correlation of the values of log10(kX/kH) for the reaction of complex 1 with a series of para-substituted aryl bromides with σp– parameters; the use of standard σp values yields a lower correlation (Figure S34).36 This is consistent with a considerable increase in π-electron donation from the ring to the substituent at the TS; similar improved correlations with σp– have been noted for related oxidative addition processes.37 The positive ρ value (+2.6) is consistent with a polarized three-center (C–Ni–X) transition state.38
Figure 4.

Hammett plot for the reaction of para-substituted aryl bromides with complex 1.
The transformation of 1 to 3 appears clean except for the loss of a small amount at early times (Figure 3). An EPR analysis suggests that a small amount of NiI is present at the end of the reaction (Figure S36), but the constant mass balance argues against a concomitant comproportionation pathway. Instead, we tentatively attribute the observed formation of Ni(I) to the reaction of 1 with an oxidizing impurity at early stages. To assess the influence of NiI species on the kinetic profile, we synthesized the NiI complex 5 by comproportionation of Ni(cod)2 and NiBr2 in the presence of the ligand PPPp-tolyl. Its tetrahedral coordination geometry is apparent from an X-ray crystal structure (Figure S34). Virtually no change in the kinetic profile was observed in the presence of 10 mol % of NiI complex 5 (Figure S35), excluding a catalytic effect of the NiI species. Moreover, no reaction is observed between pure complex 5 and 4-bromotoluene under the same conditions (Figure S37). Additionally, the fact that virtually no NiI species is produced during the reaction of 1 with the aryl halide argues against the formation of a Kochi-type solvent-caged radical intermediate in this system, as it would likely lead to a mixture of NiII and NiI products.
DFT calculations show that the reaction of 1 with 4-(trifluoromethyl)phenyl bromide yielding 2b is highly exothermic (ΔG° = −22.8 kcal mol–1; Table S7, CF3 section), whereas bromine abstraction to yield PPPp-tolNiBr, BPI, and the CF3C6H4 radical is endothermic by 19.6 kcal mol–1 (Table S7). The exothermicity of the oxidative addition is in line with previous computational results with three monophosphine ligands, but bromide abstraction from PhBr starting from Ni(PMe3)4 was found to be much less unfavorable, at only +1.9 kcal mol–1.23 Regarding the reaction of 1 with p-tolyl bromide (Figure 5), DFT calculations suggest a mechanism involving initial substitution of BPI by 4-bromotoluene to form a metastable π complex lying 13.4 kcal mol–1 above the reactants, followed by oxidative addition. The mechanism of the substitution has not been studied, but a fully dissociative route is unlikely on the basis of the calculated high free energy of PPPp-tolNi (+22.9 kcal/mol; Table S7, Me section). Three TSs, with structures by and large similar to those reported by Maseras et al.,23 have been located, corresponding to oxidative addition leading to the observed product, its less stable isomer (this TS is not shown in Figure 5), and bromine atom abstraction. The TS for the observed oxidative addition has a calculated ΔG⧧ of 23.4 kcal mol–1, in good agreement with the experimental value of 21.9 kcal mol–1. The isomeric oxidative TS is 2.9 kcal mol–1 less stable (ΔG⧧ = 26.2 kcal mol–1), while the bromine abstraction TS is predicted to be even higher, at 30.6 kcal mol–1. A computed Hammett plot (Figure S39) for oxidative addition yields a ρ value of +3.8, somewhat higher than the experimental value of +2.6, while a similar plot for halide abstraction (Figure S40) shows low correlation and a very slightly positive slope (+0.9).
Figure 5.

Gibbs free energy profiles computed at the TPSSh-D3BJ/BBS (see the Supporting Information) level of theory for the oxidative addition and halide abstraction mechanisms. R = Tol.
In summary, a triphosphine pincer ligand promotes a clean oxidative addition of aryl halides to a Ni(0) center to form pentacoordinate arylnickel(II) species. These species are stable under the reaction conditions, even in the absence of ortho substituents. Kinetics and structure−activity relationships support a concerted process occurring through a polarized transition state. DFT calculations support this mechanistic description, accounting for both the free enthalpy of activation and substituent effects. In particular, the competing halogen atom abstraction is predicted to be significantly higher in energy and be much less sensitive to substituent effects. These observations support the idea that triphosphine nickel(0) species are competent at oxidative addition of aryl halides under mild conditions, contrasting with the often monoligated Pd(0) active species. Applications of this system to nickel-catalyzed cross coupling are currently being investigated in our laboratories.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 715060; P.M.P.-G., M.-E.M.), and from the NoNoMeCat Marie Skłodowska-Curie training networ funded by the European Union under the Horizon2020 Program (675020-MSCA-ITN-2015-ETN; A.D.). The X-ray diffractometer has been financed by The Netherlands Organization for Scientific Research (NWO). The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation Flanders (FWO) and the Flemish Government–department EWI.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.0c00060.
Accession Codes
CCDC 1977373–1977374 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
∥ P.M.P.-G. and A.D. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Labinger J. A. Tutorial on Oxidative Addition. Organometallics 2015, 34, 4784–4795. 10.1021/acs.organomet.5b00565. [DOI] [Google Scholar]
- Hartwig J. J.Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Behr A.; Neubert P.. Applied Homogenoeus Catalysis; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Fauvarque J.-F.; Pfluger F.; Troupel M. Kinetics of oxidative addition of zerovalent palladium to aromatic iodides. J. Organomet. Chem. 1981, 208, 419–427. 10.1016/S0022-328X(00)86726-1. [DOI] [Google Scholar]
- Amatore C.; Pfluger F. Mechanism of Oxidative Addition of Palladium(0) with Aromatic Iodides in Toluene, Monitored at Ultramicroelectrodes. Organometallics 1990, 9, 2276–2282. 10.1021/om00158a026. [DOI] [Google Scholar]
- Portnoy M.; Milstein D. Mechanism of Aryl Chloride Oxidative Addition to Chelated Palladium(0) Complexes. Organometallics 1993, 12, 1665–1673. 10.1021/om00029a026. [DOI] [Google Scholar]
- Hartwig J. F.; Paul F. Oxidative Addition of Aryl Bromide after Dissociation of Phosphine from a Two-Coordinate Palladium(0) Complex, Bis(tri-o-tolylphosphine)palladium(0). J. Am. Chem. Soc. 1995, 117, 5373–5374. 10.1021/ja00124a026. [DOI] [Google Scholar]
- Hartwig J. F.; Barrios-Landeros F. Distinct Mechanisms for the Oxidative Addition of Chloro-, Bromo- and Iodoarenes to a Bisphosphine Palladium(0) Complex with Hindered Ligands. J. Am. Chem. Soc. 2005, 127, 6944–6945. 10.1021/ja042959i. [DOI] [PubMed] [Google Scholar]
- Ariafard A.; Lin Z. Understanding the Relative Easiness of Oxidative Addition of Aryl and Alkyl Halides to Palladium(0). Organometallics 2006, 25, 4030–4033. 10.1021/om060236x. [DOI] [Google Scholar]
- McMullin C. L.; Jover J.; Harvey J. N.; Fey N. Accurate modelling of Pd(0) + PhX oxidative addition kinetics. Dalton Trans. 2010, 39, 10833–10836. 10.1039/c0dt00778a. [DOI] [PubMed] [Google Scholar]
- McMullin C. L.; Fey N.; Harvey J. N. Computed ligand effects on the oxidative addition of phenyl halides to phosphine supported palladium(0) catalyst. Dalton Trans. 2014, 43, 13545–13556. 10.1039/C4DT01758G. [DOI] [PubMed] [Google Scholar]
- Ananikov V.; Nickel P. The “Spirited Horse” of Transition Metal Catalysis. ACS Catal. 2015, 5, 1964–1971. 10.1021/acscatal.5b00072. [DOI] [Google Scholar]
- Hu X. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci. 2011, 2, 1867–1886. 10.1039/c1sc00368b. [DOI] [Google Scholar]
- Diccianni J. B.; Katigbak J.; Hu C.; Diao T. Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc. 2019, 141, 1788–1796. 10.1021/jacs.8b13499. [DOI] [PubMed] [Google Scholar]
- Ge S.; Green R. A.; Hartwig J. F. Controlling First-Row Catalysts: Amination of Aryl and Heteroaryl Chlorides and Bromides with Primary Aliphatic Amines Catalyzed by BINAP-Ligated Single-Component Ni(0) Complex. J. Am. Chem. Soc. 2014, 136, 1617–1627. 10.1021/ja411911s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G.; Kalvet I.; Englert U.; Schoenebeck F. Fundamental Studies and Development of Nickel-Catalyzed Trifluoromethylthiolation of Aryl Chlorides: Active Catalytic Species and Key Roles of Ligand and Traceless MeCN Additive Revealed. J. Am. Chem. Soc. 2015, 137, 4164–4172. 10.1021/jacs.5b00538. [DOI] [PubMed] [Google Scholar]
- Mohadjer Beromi M.; Nova A.; Balcells D.; Brasacchio A. M.; Brudvig G. W.; Guard L. M.; Hazari N.; Vinyard D. J. Mechanistic Study of an Improved Ni Precatalyst for Suzuki-Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc. 2017, 139, 922–936. 10.1021/jacs.6b11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazari N.; Melvin P. R.; Beromi M. M. Well-defined nickel and palladium precatalysts for cross-coupling. Nature Reviews Chemistry 2017, 1, 0025 10.1038/s41570-017-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein H. A.; Luscombe C. K. Externally Initiated Regioregular P3HT with Controlled Molecular Weight and Narrow Polydispersity. J. Am. Chem. Soc. 2009, 131, 12894–12895. 10.1021/ja9054977. [DOI] [PubMed] [Google Scholar]
- Reuther J. F.; Bhatt M. P.; Tian G.; Batchelor B. L.; Campos R.; Novak B. M. Controlled Living Polymerization of Carbodiimides Using Versatile, Air-Stable Nickel(II) Initiators: Facile Incorporation of Helical. Macromolecules 2014, 47, 4587–4595. 10.1021/ma5009429. [DOI] [Google Scholar]
- Manzoor A.; Wienefeld P.; Baird M. C.; Budzelaar P. H. M. Catalysis of Cross-Coupling and Homocoupling Reactions of Aryl Halides Utilizing Ni(0), Ni(I), and Ni(II) Precursors; Ni(0) Compounds as the Probable Catalytic Species but Ni(I) Compounds as Intermediates and Products. Organometallics 2017, 36, 3508–3519. 10.1021/acs.organomet.7b00446. [DOI] [Google Scholar]
- Tsou T. T.; Kochi J. K. Mechanism of oxidative addition. Reaction of nickel(0) complexes with aromatic halides. J. Am. Chem. Soc. 1979, 101 (21), 6319–6332. 10.1021/ja00515a028. [DOI] [Google Scholar]
- Funes-Ardoiz I.; Nelson J. D.; Maseras F. Halide Abstraction Competes with Oxidative Addition in the Reactions of Aryl Halides with [Ni(PMenPh(3-n))4]. Chem. - Eur. J. 2017, 23, 16728–16733. 10.1002/chem.201702331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinina G. G.; Brennessel W. W.; Miller J. L.; Vicic D. A. Exploring Trifluoromethylation Reactions at Nickel: A Structural and Reactivity Study. Organometallics 2008, 27, 3933–3938. 10.1021/om800300k. [DOI] [Google Scholar]
- Nicolas E.; Ohleier A.; D’Accriscio F.; Pécharman A.-E.; Demange M.; Ribagnac P.; Ballester J.; Gosmini C.; Mézailles N. (Diphosphine)Nickel”-Catalyzed Negishi Cross-Coupling: An Experimental and Theoretical Study. Chem. - Eur. J. 2015, 21, 7690–7694. 10.1002/chem.201500192. [DOI] [PubMed] [Google Scholar]
- Bajo S.; Laidlaw G.; Kennedy A. R.; Sproules S.; Nelson D. J. Oxidative Addition of Aryl Electrophiles to a Prototypical Nickel(0) Complex: Mechanism and Structure/Reactivity Relationships. Organometallics 2017, 36, 1662–1672. 10.1021/acs.organomet.7b00208. [DOI] [Google Scholar]
- Chatt J.; Shaw B. L. Alkyls and aryls of transition metals. Part III. Nickel (II) derivatives. J. Chem. Soc. 1960, 1718–1729. 10.1039/jr9600001718. [DOI] [Google Scholar]
- Mohadjer Beromi M.; Banerjee G.; Brudvig G. W.; Charboneau D. J.; Hazari N.; Lant H. M. C.; Mercado B. Q. Modifications to the Aryl Group of dppf-Ligated Ni σ-Aryl Precatalysts: Impact on Speciation and Catalytic Activity in Suzuki-Miyaura Coupling Reactions. Organometallics 2018, 37 (21), 3943–3955. 10.1021/acs.organomet.8b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsino A. F.; Gutiérrez del Campo M.; Lutz M.; Moret M.-E. Enhanced Catalytic Activity of Nickel Complexes of an Adaptive Diphosphine-Benzophenone Ligand in Alkyne Cyclotrimerization. ACS Catal. 2019, 9, 2458–2481. 10.1021/acscatal.8b05025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Veschoor G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2’-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc., Dalton Trans. 1984, 1349–1356. 10.1039/DT9840001349. [DOI] [Google Scholar]
- Klein H. F.; Dal A.; Jung T.; Flörke U.; Haupt H. J. Intermediates and Side-Reactions in the Synthesis of Molecular Diphenolatonickel Compounds Containing Trimethylphosphane Ligands. Eur. J. Inorg. Chem. 1998, 1998, 2027–2032. . [DOI] [Google Scholar]
- Park N. H.; Teverovskiy G.; Buchwald S. L. Development of an Air-Stable Nickel Precatalyst for the Amination of Aryl Chlorides, Sulfamates, Mesylates, and Triflates. Org. Lett. 2014, 16, 220–223. 10.1021/ol403209k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standley E. A.; Smith S. J.; Müller P.; Jamison T. F. A Broadly Applicable Strategy for Entry into Homogeneous Nickel(0) Catalysts from Air-Stable Nickel(II) Complexes. Organometallics 2014, 33, 2012–2018. 10.1021/om500156q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. S. K.; Lavoie C. M.; MacQueen P. M.; Ferguson M. J.; Stradiotto M. A Comparative Reactivity Survey of Some Prominent Biphosphine Nickel(II) Precatalyst in C-N Cross-Coupling. Organometallics 2016, 35, 3248–3254. 10.1021/acs.organomet.6b00650. [DOI] [Google Scholar]
- Lavoie C. M.; MacQueen P. M.; Rotta-Loria N. L.; Sawatzky R. S.; Borzenko A.; Chisholm A. J.; Hargreaves B. K. V.; McDonald R.; Ferguson M. J.; Stradiotto M. Challenging nickel-catalysed amine arylations enabled by tailored ancillary ligand design. Nat. Commun. 2016, 7, 11073. 10.1038/ncomms11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch H.; Leo A.; Taft R. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Konovalov A. I.; Lishchynsyi A.; Grushin V. V. Mechanism of Trifluoromethylation of Aryl Halides with CuCF3 and the Ortho Effect. J. Am. Chem. Soc. 2014, 136 (38), 13410–13425. 10.1021/ja507564p. [DOI] [PubMed] [Google Scholar]
- Foà M.; Cassar L. Oxidative addition of aryl halides to tris(triphenylphosphine)nickel(0). J. Chem. Soc., Dalton Trans. 1975, 2572–2576. 10.1039/dt9750002572. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.