

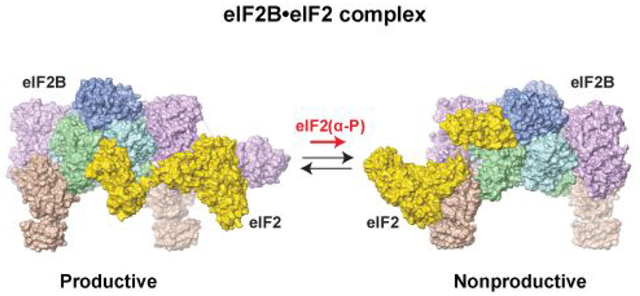
Abstract
The eukaryotic translation initiation factor eIF2 is a GTPase, which brings the initiator Met-tRNAi to the ribosome as the eIF2-GTP•Met-tRNAi ternary complex (TC). TC regeneration is catalyzed by the guanine nucleotide exchange factor (GEF) eIF2B. eIF2 phosphorylation by several stress-induced kinases converts it into a competitive inhibitor of eIF2B. Inhibition of eIF2B activity lowers cellular TC concentrations, which in turn triggers the Integrated Stress Response (ISR). Depending on its degree of activation and duration, ISR protects the cell from the stress or can itself induce apoptosis. ISR dysregulation is a causative factor in the pathology of multiple neurodegenerative disorders, while ISR inhibitors are neuroprotective. The realization that eIF2B is a promising therapeutic target has triggered significant interest in its structure and the mechanisms of its action and regulation. Recently, four groups published the Cryo-EM structures of eIF2B with its substrate eIF2 and/or its inhibitor, phosphorylated eIF2 (eIF2(α-P)). While all three structures of the nonproductive eIF2B•eIF2(α-P) complex are similar to each other, there is a sharp disagreement between the published structures of the productive eIF2B•eIF2 complex: one group reports a structure similar to that of the nonproductive complex, whereas two others observe a vastly different eIF2B•eIF2 complex. Here, we discuss the recent reports on the structure, function, and regulation of eIF2B; the pre-clinical data on the use of ISR inhibitors for treatment of neurodegenerative disorders; and how the new structural and biochemical information can inform and influence the use of eIF2B as a therapeutic target.
Keywords: translation initiation, neurodegenerative disorders, eIF2B, eIF2, GEF, ISR
Graphical Abstract
Protein expression is a central process in the cell, responsible for a significant portion of its energy needs. Eukaryotic translation initiation factor 2B (eIF2B) is a focal point of regulation of protein synthesis. It is the guanine nucleotide exchange factor (GEF) specific for eIF2, the GTPase that brings Met-tRNAi to the 43S translation pre-initiation complex (PIC) as part of the eIF2-GTP•Met-tRNAi ternary complex (TC). Upon start codon selection, eIF2 hydrolyzes GTP and the resulting eIF2-GDP dissociates from the PIC in complex with its GTPase-activating protein (GAP), eIF5.3–5 eIF2B converts eIF2-GDP back to eIF2-GTP, which binds Met-tRNAi, forming a new TC (Figure 1).3, 6, 7 eIF2 consists of α, β, and γ subunits, with eIF2γ being the actual GTPase. eIF2B has five subunits, α-ε. eIF2Bγ and ε form the catalytic subcomplex (eIF2Bcat), and eIF2Bα, β, and δ form the regulatory subcomplex (eIF2Breg).8
Figure 1. Regulation of eIF2B and the Integrated Stress Response (ISR).
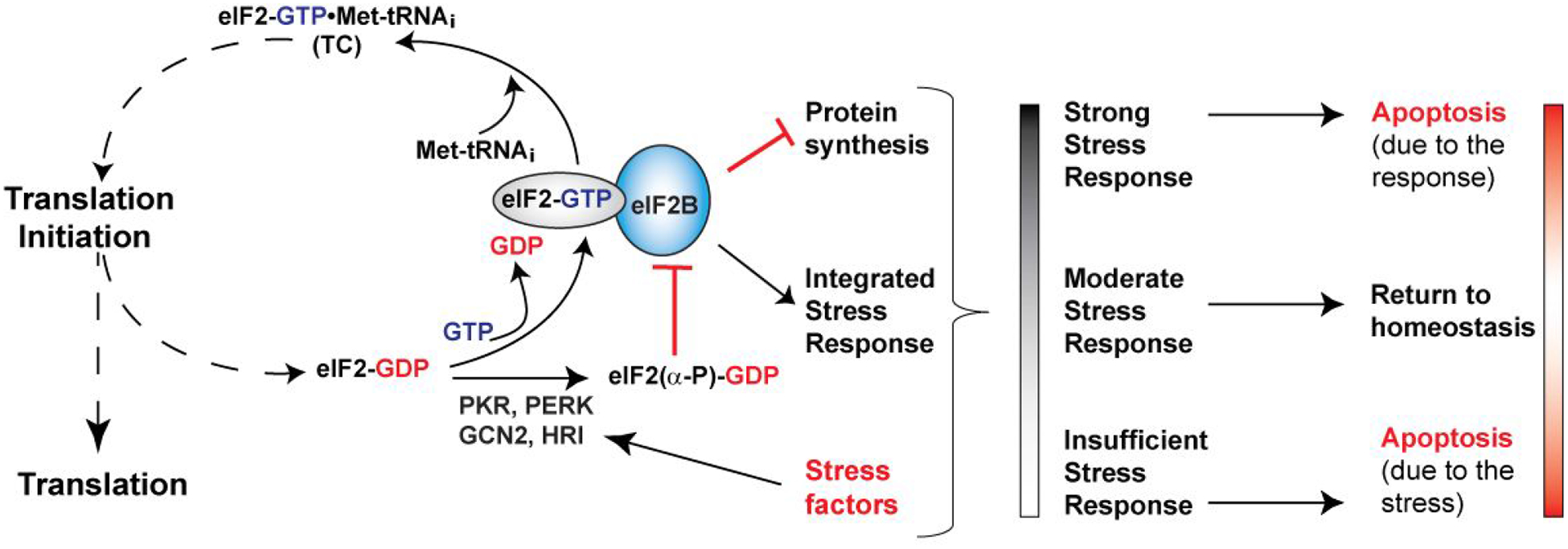
eIF2 brings the Met-tRNAi to the ribosomal translation initiation complex, in the form of the eIF2-GTP•Met-tRNAi ternary complex (TC). Upon start codon recognition, eIF2 hydrolyzes GTP, and eIF2-GDP is released. eIF2B catalyzes nucleotide exchange and Met-tRNAi binding to form a new TC. Phosphorylation of the α-subunit of eIF2 by several stress-activated kinases turns eIF2-GDP from substrate into an inhibitor of eIF2B. Inhibition of eIF2B activity triggers the ISR, which involves both pro-apoptotic and pro-survival pathways. The ultimate fate of the cell depends on the interplay between the stress, the pro-survival and pro-apoptotic branches of the ISR, and other stress responses in the cell. The stress response is usually proportional to the stress and self-contained through negative feedback. In the absence of adequate stress response, the stress factors can cause cell damage and death. At the opposite end of the spectrum, stress response that is too strong and/or prolonged can itself cause apoptosis.
Historically, eIF2B had been considered a “simple” GEF that converts the substrate eIF2-GDP into the product eIF2-GTP. However, this notion has been challenged in recent years. eIF2-GDP is released from the ribosome in complex with eIF5, and eIF2B has to displace eIF5 before it can promote GDP dissociation from eIF2. Therefore, eIF5 acts as GDP dissociation inhibitor (GDI), and eIF2B acts as GDI dissociating factor (GDF).9–11 Thus, at present, it is established that eIF2 is channeled from the PIC, through eIF5, to eIF2B. Work in the 1980s and 1990s indicated that eIF2-GTP is a tightly bound reaction intermediate, whereas the real product is the TC. It was also suggested that the TC itself may be transferred from eIF2B to the PIC.14, 15 Together with a recent report that eIF5 and eIF2B compete for the TC,17 there is now a significant body of evidence that eIF2 is channeled in a full cycle from the PIC to eIF5, to eIF2B, and then back to eIF5 and the PIC (for a detailed discussion of the thermodynamics of eIF2B catalysis and channeling see ref.18).
The eIF2B catalytic activity is regulated by phosphorylation of its substrate eIF2; phosphorylation of eIF2B itself, as well as by small molecules, such as nucleotides and cofactors, binding to eIF2B. Phosphorylation of eIF2α at S51 by any one of four kinases, PKR, PERK, GCN2, and HRI, in response to various stress factors, converts eIF2 into eIF2(α-P), which functions as a competitive inhibitor of eIF2B (Figure 1). Inhibition of eIF2B activity not only slows down protein synthesis, but also turns on translation of a set of mRNAs encoding transcription factors and other proteins, which in turn triggers the Integrated Stress Response (ISR). ISR involves both pro-survival pathways, aimed at restoring homeostasis (e.g. PERK activation in response to ER stress) and pro-apoptotic pathways, leading to apoptosis (e.g. PKR activation by viral infection).19–23 The fate of the cell depends on the magnitude and duration of the stress response, as well as on the interplay between the ISR and other factors (Figure 1).
Yeast have only one of the four eIF2α kinases, GCN2. Genetic studies in Saccharomyces cerevisiae (S. cerevisiae) have identified mutations in a number of proteins, including eIF2B and eIF2, that disrupt the cell response to amino acid starvation (reviewed in refs.20, 21). Mutations that inhibit the induction of ISR in response to amino acid depletion are called General amino acid control nonderepressible (Gcn−). Gcn− mutations have been found in all three eIF2Breg subunits (eIF2Bα, β, and δ), as well as eIF2α, and more recently eIF2β.24 Mutations that cause ISR induction in the absence of amino acid starvation are called General amino acid control derepressed (Gcd−). Gcd− mutations have been found in all five eIF2B subunits and all three eIF2 subunits (reviewed in refs.20, 21).
Dysregulation of ISR plays a causative role in a number of neurodegenerative disorders.19–23 PERK activation mediates neuronal cell death in prion disease, Alzheimer’s disease, and other disorders. Accordingly, PERK inhibitors are neuroprotective in animal models of these diseases, but cause pancreatic and liver toxicity. The small molecule ISR inhibitor (ISRIB) promotes memory formation in mice and is neuroprotective, without serious toxicity.25–30 Mutations in eIF2B subunits cause the neurodegenerative disorder leukoencephalopathy with vanishing white matter (VWM), also known as Childhood Ataxia with CNS hypomyelination (CACH),22, 31, 32 whose mechanisms are not well understood, but appear to affect eIF2B stability and/or activity.1, 33–36 The realization that eIF2B is a promising therapeutic target has triggered significant interest in its structure and mechanisms of action and regulation, and it is the subject to active research by a number of groups.
eIF2B structure and function
Stoichiometry and architecture of eIF2B
Since it was first characterized, eIF2B had been considered to be an α/β/γ/δ/ε heteropentamer of approximately 300 kDa, which appeared to be supported by Size Exclusion Chromatography and Analytical Ultracentrifugation,14, 37–39 although some authors observed a much larger complex.40 The first structures of eIF2B subunits were the crystal structures of human eIF2Bα41 and of the catalytic C-terminal domain (CTD) of yeast42 and human43 eIF2Bε. The folds of all individual subunits were known, because the eIF2B α, β, and δ subunits are homologous to each other and to the ribose-1,5-bisphosphate isomerase (RBPI) and related enzymes, while the γ and ε subunits are homologous to each other and to sugar-nucleotidyl transferases (reviewed in3). However, the overall eIF2B architecture remained unknown, and all available data was being viewed through the prism of a pentameric eIF2B.
In 2014, multiple groups reported evidence that eIF2B is in fact a 600 kDa heterodecamer, composed of two copies each of the α, β, γ, δ, and ε subunits.33, 36, 44 However, the structure of the eIF2B decamer remained elusive. A number of eIF2Bγε homologs are tetramers (reviewed in ref.3) and it was thus logical to suggest that if eF2B is a decamer, then eIF2Bγ and ε are likely to form a tetramer. This idea appeared to be supported by MS and MS/MS data, in which the subcomplexes observed upon partial disruption of the eIF2B decamer indicated the existence of an eIF2Bγ2ε2 core.44 However, in retrospect, these results turned out to be an artefact of the MS experiments. The Proud group was the first to report that in mammalian eIF2B, eIF2Bα promotes decamer formation by bringing two eIF2Bβγδε tetramers together.36 Our work indicated that the eIF2B regulatory subcomplex is a hexamer composed of an α2 homodimer and two βδ heterodimers;33 however, the proposed overall architecture of the eIF2Bα2(βδ)2 hexamer turned out to be incorrect (see below). In 2015, Kuhle and co-authors reported the crystal structure of the eIF2Bβ/δ subcomplex from a thermophilic yeast, which forms an eIF2B(βδ)2 tetramer, and provided experimental evidence confirming that the eIF2B regulatory α/β/δ subcomplex (eIF2Breg) forms an eIF2Bα2(βδ)2 hexamer, as had been proposed previously. Using the structure of the hexameric Thermococcus kodakarensis RBPI (tkRBPI) as a template, the authors proposed the first correct atomic model for the structure of eIF2Breg.34
The structure of eIF2B
The first crystal structure of the complete eIF2B decamer, from Schizosaccharomyces pombe (S. pombe), was published in 20161 (Figure 2). The only portion of eIF2B missing from the structure was the catalytic eIF2Bε-CTD, which is connected to the rest of the protein through a flexible linker. eIF2Breg forms an eIF2Bα2(βδ)2 hexamer, with one eIF2Bγε catalytic subcomplex bound to each eIF2Bβδ dimer. Directed crosslinking showed that eIF2α binds in the pocket formed by the NTDs of eIF2Bα, β, and δ, whereas eIF2γ binds to eIF2Bγ and ε. Some of the eIF2Bε residues crosslinked to eIF2γ were in the vicinity of the functionally important NF motif.45 Phosphorylation of eIF2α caused reduction in crosslinking of eIF2γ to eIF2Bε, but not to eIF2Bγ. There was also complete loss of two of the cross-links between eIF2α and eIF2Bβ, while crosslinking of eIF2α to eIF2Bα and δ was not affected.1
Figure 2. Crystal structure of S. pombe eIF2B.1.
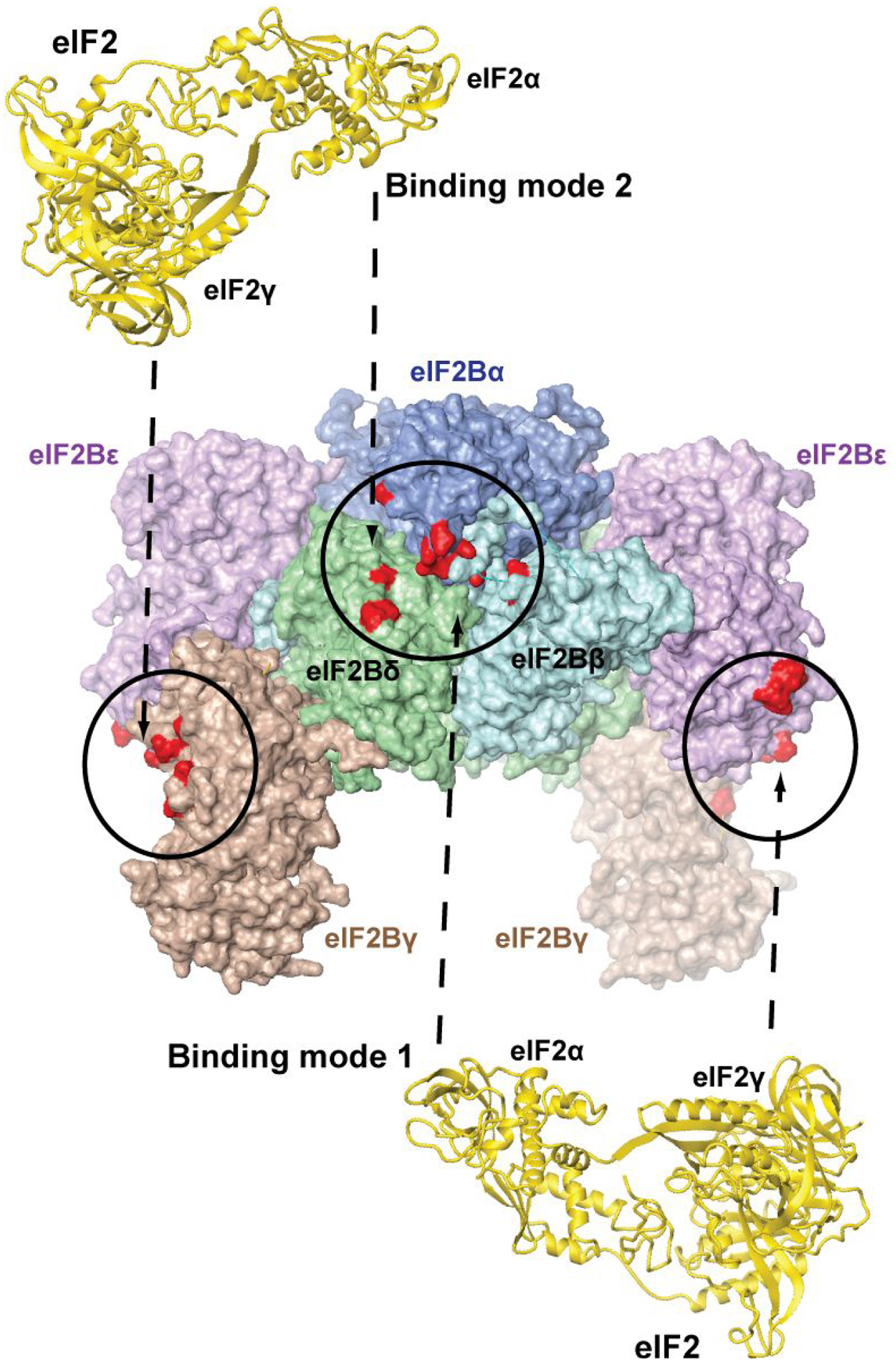
The structure of eIF2B is shown in surface representation. The individual eIF2B subunits are labeled. Sites of cross-linking to eIF2γ (left and right) and eIF2α (center) and are colored red and circled. The second eIF2α-binding pocket (not visible) is on the opposite face of the complex. The two alternative binding modes of eIF2 (shown in gold ribbon), involving the visible eIF2α-binding pocket on the front, are illustrated with dashed arrows above and below the eIF2B structure, and numbered as in the text. Note that the same two alternative eIF2 binding modes are possible on the opposite face of eIF2B, but not shown for clarity.
The eIF2B structure and the crosslinking results were consistent with, and helped explain the phenotypes of a number of Gcd− and Gcn− mutations in yeast. However, they also posed new important questions about the mode(s) of eIF2 binding to eIF2B.
- First, there are two eIF2α-binding pockets on opposite sides of eIF2Breg, and two eIF2γ-binding surfaces, one on each of the two eIF2Bγε dimers. Due to the symmetry in eIF2B, this creates two alternative possibilities: (1) eIF2α could approach the eIF2Bα/β/δ pocket between eIF2Bβ and δ, with eIF2γ oriented toward one of the two eIF2Bγε dimers (Binding mode 1, Figure 2, bottom); or (2) eIF2α could approach the eIF2Bα/β/δ pocket between eIF2Bα and δ, with eIF2γ oriented toward the other eIF2Bγε dimer, on the opposite side of eIF2B (Binding mode 2, Figure 2, top).
- Second, the overall size of eIF2 does not allow it to reach simultaneously all eIF2B surfaces it cross-links to.1 If eIF2α is bound to eIF2Bβ and δ (Binding mode 1, Figure 2, bottom), eIF2γ can contact eIF2Bε, including the NF motif, but not the eIF2Bγ surface, where crosslinks were also observed. Conversely, if eIF2α is bound to eIF2Bα and δ (Binding mode 2, Figure 2, top), eIF2γ can contact eIF2Bγ, but not eIF2Bε. Again, two possible scenarios could be envisioned. eIF2α and eIF2γ binding to eIF2B could be mutually exclusive;1 however, that would be at odds with some of the available genetic data. Alternatively, eIF2γ would need to adopt an extended conformation;46 however, such conformation has not been observed in any available eIF2 structure.
Furthermore, the changes in cross-linking patterns indicated that phosphorylation affects the binding mode and/or the overall conformation of eIF2. The loss of two crosslinks between eIF2α and eIF2Bβ could be due to local changes around P-S51; whereas the lower crosslinking efficiency between eIF2γ and eIF2Bε could be explained by changes in the conformation of eIF2 and/or its mode of binding to the eIF2Bγε platform.1, 46 As we describe below, the reality turned out to be far more complex and unexpected. The first structures of human eIF2B were solved by Cryo-EM, in complex with ISRIB.47, 48 The structures were very similar to the crystal structure of S. pombe eIF2B, with ISRIB stabilizing the decameric complex by “stapling” the two eIF2Bβδ dimers together.47, 48
Structures of the eIF2B•eIF2 complexes and the mechanism of eIF2B action
The Cryo-EM structures of the eIF2B complexes with the substrate, eIF2, and the inhibitor, eIF2(α-P)-GDP, were recently published almost simultaneously by several groups.2, 12, 13, 16 The structure of the enzyme - inhibitor complex, eIF2B•eIF2(α-P)2, 13, 16 (Figure 3B) resembles the eIF2 Binding mode 2, shown in Figure 2, top. An important feature of the complex is the remodeling of the protein surface around S51 in eIF2α upon phosphorylation. While phospho-S51 (P-S51) is buried, it forms a network of electrostatic intramolecular interactions with R63, K86, and D68, which likely promotes the deep insertion of the P-S51-adjacent Arginine-rich loop into the eIF2Bα-δ cavity. This finding was unexpected because it had been assumed that P-S51 directly contacts eIF2B. Previous NMR Chemical Shift Perturbation (CSP) analysis of the S51D mutation data at high salt concentration showed modest chemical shift changes that did not indicate major conformational rearrangement.49 A more recent CSP analysis at physiological salt showed large chemical shift changes as a result of the S51D mutation, and even larger changes as a result of S51 phosphorylation, indicative of significant conformational changes;46 however, the nature of these changes had remained unknown until now because the CSP assay does not provide structural information.
Figure 3. Structure of the eIF2B complexes with eIF2 and eIF2(α-P).
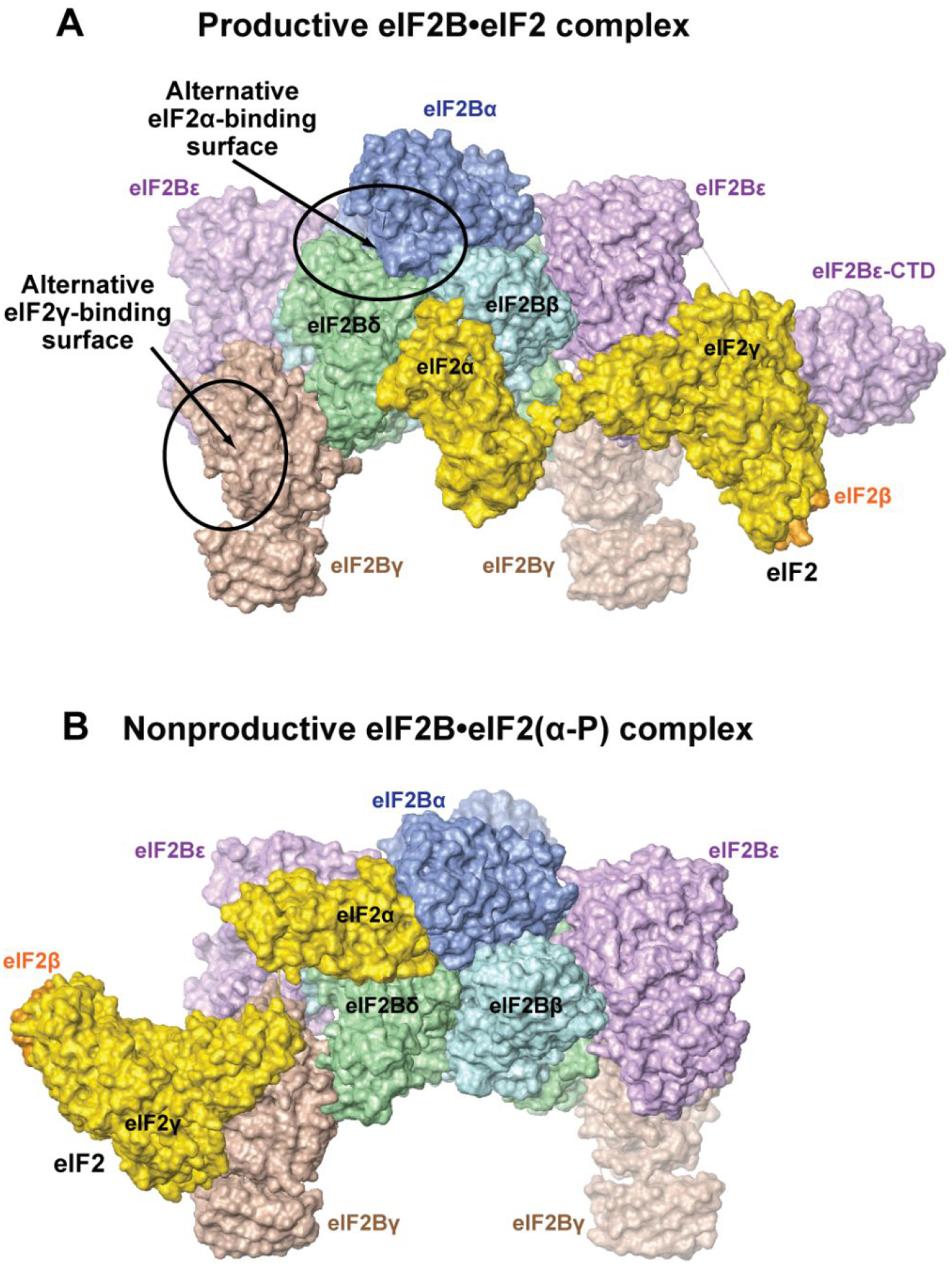
A. Structure of the productive eIF2B•eIF2 complex.2 eIF2B is in approximately the same orientation as in Figure 2. eIF2α and γ are colored gold; the visible portion of eIF2β is orange. The structure is similar to the structure of the eIF2B•eIF2 complex in reference12.The alternative eIF2α- and eIF2γ-binding surfaces observed in the structure of the eIF2B•eIF2 complex from reference13 are circled and labeled. Note that the eIF2B•eIF2 structure in reference13 is similar to the structure of the nonproductive eIF2B•eIF2(α-P) complex2, 13, 16 (panel B).
B. Structure of the nonproductive eIF2B•eIF2(α-P) complex.2 The orientation and coloring are the same as in panel A. The structure is similar to the structures of the eIF2B•eIF2(α-P) complex in references13, 16, and to the structure of the eIF2B•eIF2 complex in reference13.
Surprisingly, the structures of the enzyme - substrate complex, eIF2B•eIF2, differ vastly.2, 12, 13 In two of the eIF2B•eIF2 structures, eIF2γ binds primarily to eIF2Bε, including the surface surrounding the NF motif, and eIF2α contacts primarily eIF2Bβ and δ in the pocket of the regulatory eIF2Bα2(βδ)2 subcomplex (eIF2Breg)2, 12 (Figure 3A), resembling the eIF2 Binding mode 1, shown in Figure 2, bottom. The G domain and Domain III of eIF2γ are sandwiched between eIF2Bε-NTD and -CTD. The catalytic domain, eIF2Bε-CTD, binds to eIF2γ via its functionally important N-terminal surface45 and is well resolved in the structure (Figure 3A). The Switch 1 region in eIF2γ adopts an open conformation, which is likely to induce GDP release.2, 12 In contrast, the eIF2B•eIF2 complex structure reported by the Pavitt group13 is similar to that of the nonproductive eIF2B•eIF2(α-P) complex2, 13, 16 (Figure 3B), where eIF2 is rotated ~180°, with eIF2γ contacting primarily to eIF2Bγ, and eIF2α binding primarily to eIF2Bα and δ in the pocket on the opposite side of eIF2Breg. eIF2Bε-CTD is not resolved in this complex.13 In the crystal structure of a complex of S. cerevisiae eIF2α and S. pombe eIF2B, the eIF2α binding is also similar to that in the nonproductive complex.2 Thus, while phosphorylated eIF2α has only been observed in Binding mode 2 (Figure 2, top), unphosphorylated eIF2α appears able to bind to the eIF2Bα/β/δ pocket in either of the two alternative binding modes shown in Figure 2.
It is highly unlikely for the discrepancy between the reported eIF2B•eIF2 structures to be due to the source of the proteins (S. cerevisiae for the Pavitt group and human for the Ito and Walter/Frost groups), or one of the two alternative structures to be an experimental artefact. Comparing the eIF2B•eIF2 structures (Figure 3) to the cross-links between S. pombe eIF2 and eIF2B1 (Figure 2) shows that while the contact interfaces in both structures are consistent with a subset of the cross-links between eIF2B and eIF2, neither structure alone can explain the cross-linking data in full (note that the two alternative binding modes shown with dashed arrows in Figure 2 correspond to the two structures in Figure 3). Instead, only a combination of the two alternative complexes can account for all the cross-links. Therefore, we favor the explanation that the eIF2B•eIF2 complex exists in two drastically different alternative states, one productive (Figure 3A) and the other nonproductive (Figure 3B), with eIF2α phosphorylation shifting the equilibrium toward the nonproductive state.2 It is thus possible that differences in the methods used for sample preparation and experimental conditions have favored either one of the two alternative conformations of the eIF2B•eIF2 complex, or the other.
The productive complex structure shown in Figure 3A is supported by the observation that mutations in the eIF2α-contacting surfaces of eIF2Bβ and δ affected the catalytic activity of eIF2B.12 It also helps explain the phenotypes of Gcd− and Gcn− mutations in S. cerevisiae.50–55 E143 and L144 in eIF2Bβ (corresponding to the site of the E164A/I165A Gcd− mutation in S. cerevisiae eIF2Bβ) are at the interface with eIF2α in the productive complex (Figure 4, right). The E164A/I165A Gcd− mutation in S. cerevisiae eIF2Bβ is synthetic lethal with the Y81S and R88T Gcn− mutations in eIF2α. The corresponding residues in human eIF2α contact eIF2Bα in the nonproductive complex (Figure 4, left), but also contact eIF2Bβ and δ in the productive complex (Figure 4, right), which helps explain their synthetic lethality with the Gcd− mutation in eIF2Bβ. Furthermore, Q130 in human eIF2Bβ (corresponding to the site of the lethal R151A mutation in S. cerevisiae eIF2Bβ) is also at the interface with eIF2α interface in the productive complex, which explains how the R151A mutation destabilizes eIF2 binding to eIF2B.55 As expected, surface-exposed Gcn− mutations in eIF2Bα and δ map to the interface with eIF2α in the nonproductive complex (Figure 4, left). The only Gcd− mutation in S. cerevisiae eIF2Bα at the interface with eIF2α in the nonproductive complex is D71N, corresponding to E70 in human eIF2Bα (Figure 4, left), which suggests that D71N could act by increasing the eIF2B affinity for eIF2α in the nonproductive complex.
Figure 4. eIF2α binding in the productive and nonproductive eIF2B•eIF2 complexes.
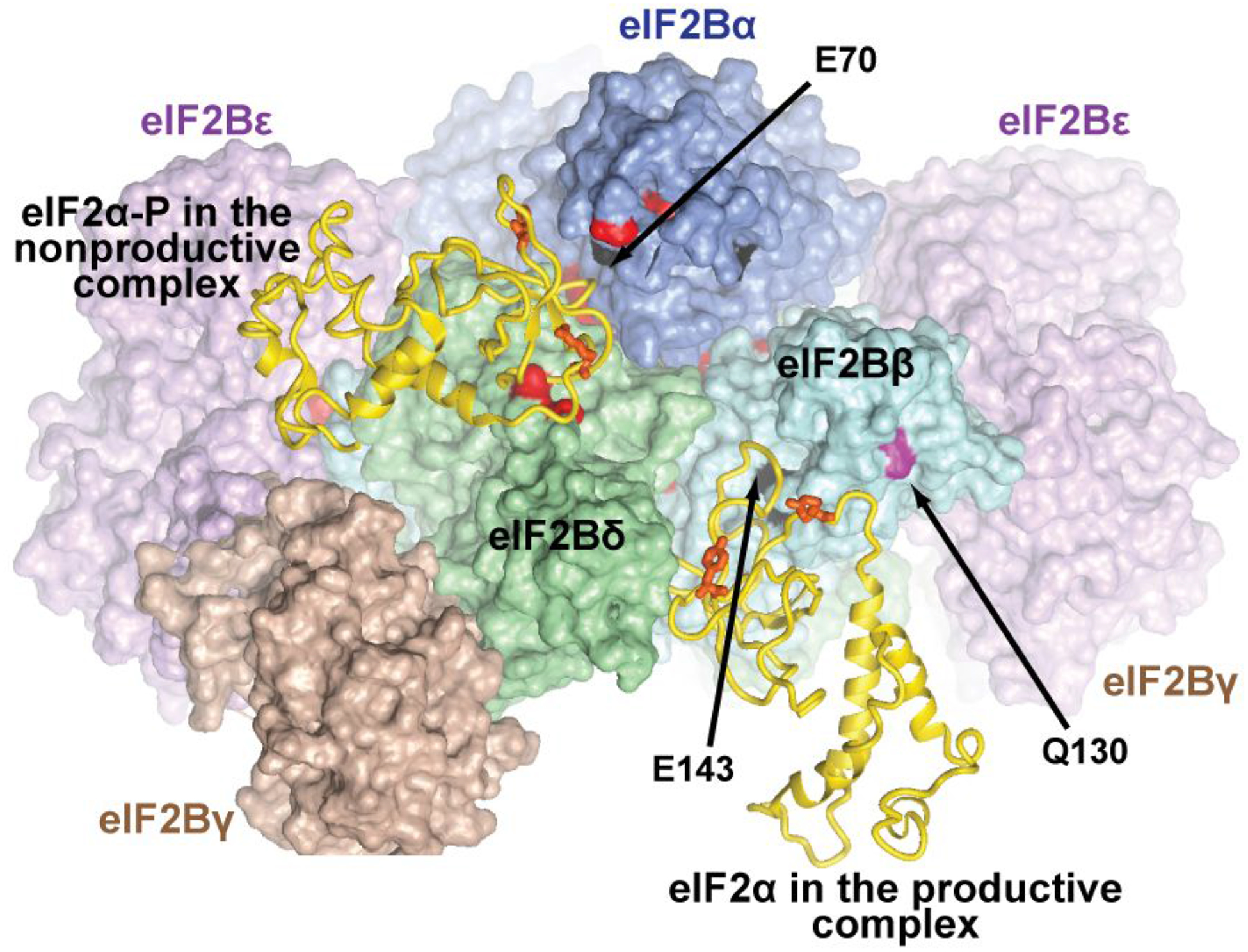
eIF2B is in a similar orientation to that in Figure 2. The coloring is as in Figure 2, but with more aggressive shading, to zoom in on the eIF2α interface. More distant portions of the complex are invisible due to shading or are cut out. eIF2α-NTD from the nonproductive eIF2B•eIF2(α-P) complex (left) and from the productive eIF2B•eIF2 complex (right)2 are shown as gold ribbon. The rest of eIF2 is not shown. Residues in eIF2B subunits corresponding to sites of Gcd− mutations in S. cerevisiae are colored black; residues corresponding to sites of Gcn− mutations in S. cerevisiae are colored red; Q130 in human eIF2Bβ, corresponding to the site of a lethal mutation in S. cerevisiae is colored magenta. Residues discussed in the text are labeled. The residues in human eIF2α, corresponding to the sites of the Y81S and R88T Gcn− mutations in S. cerevisiae eIF2α, are shown as orange sticks (not labeled).
It should also be noted that a large number of both Gcn− and Gcd− mutations in eIF2Bα, β, and δ are buried at the interfaces between eIF2B subunits. The phenotypes of such Gcd− mutations could be explained with affecting eIF2B stability and/or activity. The phenotypes of Gcn− mutations at the interface between eIF2Bα and the rest of eIF2B could be explained by loss of eIF2Bα, because α-less eIF2B is resistant to inhibition by eIF2(α-P).56, 57 However, the presence of Gcn− mutations at other intersubunit interfaces indicates that subtle changes in intersubunit orientations could affect the equilibrium between the productive and nonproductive states. The questions surrounding the structure(s) of the eIF2B•eIF2 complex can be resolved, e.g. by introducing mutations at interfaces between eIF2B and eIF2 that are unique to one of the structures (Figure 3) and testing their effects on eIF2B binding to eIF2 and eIF2(α-P).
Regulation of eIF2B activity
New structural insights
The new insights obtained from the structures of the eIF2B•eIF2 complexes point to new, previously unanticipated regulatory mechanisms. In principle, the eIF2B activity can be regulated in two non-mutually exclusive ways: i) by modulating the catalytic activity of eIF2B in the productive complex; and/or ii) by shifting the equilibrium between the productive and nonproductive states, changing the fraction of eIF2B available to perform catalysis. It is not possible for two eIF2 molecules to bind simultaneously to the same eIF2Bγε platform in the two different orientations, shown in Figure 3, because there is steric overlap between eIF2γ in the two alternative binding modes.2 The equilibrium between the productive and nonproductive eIF2B•eIF2 complexes would be a function of the relative affinities of eIF2 in the two binding modes. The notion that there is equilibrium between two alternative eIF2B•eIF2 complexes, one productive and one nonproductive, opens up the very intriguing possibility that phosphorylation of eIF2α may not be the only regulatory mechanism that acts by changing this equilibrium (discussed in more detail below). Any ligand or modification that differentially affects the affinity of eIF2B for eIF2 in the two alternative complexes would change the equilibrium between them. The interaction between eIF2B and eIF2 involves several distinct interfaces, most of which are drastically different between the two alternative states, and likely have different affinities:
eIF2α binding in the pocket on eIF2Bα/β/δ: contacting either eIF2Bβ and δ (Figure 3A), or eIF2Bα and δ (Figure 3B). When part of eIF2, unphosphorylated eIF2α can bind to eIF2Bα/β/δ in both orientations (Figure 3A and B); however, free eIF2α, even when not phosphorylated, seems to favor binding to the pocket on eIF2Bα/β/δ in the orientation seen in Figure 3B.2 Therefore, it appears that the eIF2α binding affinity is stronger in the nonproductive complex, even when not phosphorylated.
eIF2γ binding to the eIF2Bγε platform: contacting either eIF2Bε (Figure 3A), or eIF2Bγ (Figure 3B). It is presently not known whether eIF2γ has higher affinity for eIF2Bγ or for eIF2Bε, but it is likely that only the interaction with eIF2Bε allows productive binding to the catalytic eIF2Bε-CTD (see 3, below).
eIF2γ binding to the catalytic eIF2Bε-CTD. Since this interaction is only observed in the productive complex (Figure 3A), and since it is required for catalysis,58 it is likely stronger in the productive complex, and could even be absent in the nonproductive complex.
Binding of the eIF2β N-terminal tail (NTT) to eIF2Bε-CTD.59 eIF2β-NTT and eIF2Bε-CTD are likely to interact with each other in both the productive and the nonproductive complexes because both eIF2β-NTT and the linker between eIF2Bε-CTD and the rest of eIF2Bε are long and flexible (neither of them is visible in either the productive or the nonproductive complex) and place little restrictions on the ability of eIF2β-NTT and eIF2Bε-CTD to reach each other. While it is difficult to know whether the eIF2β-NTT - eIF2Bε-CTD interaction is stronger in the productive or in the nonproductive complex, the position of eIF2Bε-CTD in the productive complex, right next to the site where eIF2β attaches to eIF2γ (Figure 3A), indicates likely synergy between the interactions of eIF2Bε-CTD with eIF2β-NTT and with eIF2γ.
How does eIF2 phosphorylation change the equilibrium between the productive and nonproductive states?
The new structures of the eIF2B•eIF2 complexes show that phosphorylated eIF2α appears incompatible with the orientation shown in Figure 3A.2, 12 Phosphorylation destabilizes the intramolecular contacts between eIF2α-NTD and -CTD,46, 54 which would also promote eIF2α binding to eIF2Bα/β/δ in the preferred orientation, where eIF2α contacts eIF2Bα and δ (Figure 3B), directing eIF2γ toward eIF2Bγ, and thus also favor the nonproductive state. Therefore, it appears that eIF2α phosphorylation shifts the equilibrium toward the nonproductive complex by both stabilizing the nonproductive complex and destabilizing the productive complex. Steric clashes would prevent simultaneous binding to the same eIF2Bγε platform of eIF2(α-P) in the nonproductive orientation (Figure 3B), and eIF2 in the productive orientation (Figure 3A).2 As can be seen in Figure 4, there is no obvious steric clash between eIF2α and eIF2α-P bound in the eIF2Bα/β/δ pocket. If binding of eIF2α and eIF2α-P to the pocket is strongly anti-cooperative, then one eIF2(α-P) molecule could be sufficient to inhibit an entire eIF2B decamer. Therefore, it is of great interest whether unphosphorylated eIF2α can bind to the same pocket where phosphorylated eIF2 binds to.
Do other regulators of eIF2B act by modulating the equilibrium between the productive and nonproductive states?
In addition to eIF2α phosphorylation, eIF2B activity is regulated by phosphorylation of eIF2B itself and by a number of small molecules in the cell, such as ATP, AMP, and NADPH (reviewed in ref. 3). The mechanism of action of eIF2Bε phosphorylation by GSK-3β, or of the other regulators is not known. As described above, the eIF2B•eIF2 complex appears to be in equilibrium between a productive and nonproductive states, with eIF2(α-P) shifting the equilibrium toward the nonproductive state. It is, therefore, likely that other regulators of eIF2B activity also act by influencing this equilibrium. If this is indeed the case, such regulators must also affect the degree of inhibition by eIF2(α-P). If a regulator has lower affinity for eIF2B•eIF2(α-P)-GDP than for eIF2B•eIF2, then based on thermodynamic coupling, the regulator•eIF2B complex has lower affinity for eIF2(α-P)-GDP, which would reduce inhibition by eIF2(α-P)-GDP (by raising its KI). Likewise, if a regulator shifts the equilibrium of the eIF2B•eIF2 complex toward the nonproductive state, it would also increase the eIF2B affinity for eIF2(α-P)-GDP and thus increase inhibition by eIF2(α-P)-GDP (by lowering its KI). There are at present no reports for such an interplay between a regulator and eIF2(α-P)-GDP. However, a recent study offers some indirect evidence that eIF2B phosphorylation by GSK-3β could increase the inhibitory effect of eIF2(α-P)-GDP, because inactivation of GSK-3β and eIF2Bε dephosphorylation correlated with increased eIF2B activity, even in the presence of high levels of eIF2(α-P).60
ISRIB mechanisms of action
ISRIB is one of the best-known regulators of eIF2B activity.29, 61 It binds directly to eIF2B and was found to stimulate its activity at least in part by promoting formation of the eIF2B holoenzyme, the eIF2B(αβγδε)2 decamer.47, 48 Whether this is indeed the main ISRIB mechanism of action in vivo depends on whether eIF2B decamerization is rate-limiting (whether there are enough eIF2B subcomplexes in the cell that ISRIB can act on). In mammals, eIF2B cellular concentrations vary depending on cell type but most estimates are > 100 nM (reviewed in ref.18). At such concentrations the equilibrium should be shifted heavily toward formation of eIF2B decamers.47, 62 The decamer is less stable at 5–20 nM concentrations, typically used in vitro; which could result in lower eIF2B enzymatic activity, and exaggerate the observed defects in decamerization, as well as the stimulating effect of ISRIB. However, such conditions could also be more representative of eIF2B concentration and activity in terminally differentiated cells with lower translation rates, or in cells, where a large fraction of eIF2B is bound to the inhibitor eIF2(α-P)-GDP, and the concentration of free active eIF2B is low.
ISRIB also promoted formation of eIF2B(βγδε)2 octamers in the absence of eIF2Bα.47, 48 α-less eIF2B has been reported to be enzymatically active,56, 57 although the mammalian eIF2B(βγδε)2 octamer appears to be less active than the eIF2B holoenzyme.63 In contrast, no such drop in activity was observed in yeast,56 possibly because the yeast α-less eIF2B seems to form a stable octamer.44 More importantly, α-less eIF2B is not inhibited by eIF2α phosphorylation.56, 57 The new structures of the eIF2B complexes with eIF2 (substrate) and eIF2(α-P) (inhibitor) offer the structural basis for these findings. In the inhibited eIF2B•eIF2(α-P) complex, eIF2Bα mediates important contacts with eIF2α. In contrast, in the productive eIF2B•eIF2 complex, eIF2α only contacts eIF2Bβ and δ from two different eIF2Bβδ dimers, but not eIF2Bα. Therefore, an eIF2B(βγδε)2 octamer is sufficient for the interaction with eIF2, and eIF2Bα is not required for eIF2B activity. However, the mammalian eIF2B octamer is less stable than the decamer, and eIF2Bα should stimulate eIF2B activity indirectly by stabilizing the complex between the two eIF2Bβγδε tetramers.
The idea that ISRIB stimulates the formation of eIF2B(βγδε)2 octamers that are both active and resistant to eIF2(α-P) inhibition is very intriguing, if eIF2B sub-complexes are indeed present in vivo. Support for the existence of such sub-complexes in human cells comes from a recent study of eIF2B bodies (large cytoplasmic multiprotein assemblies observed by fluorescence), which reported evidence that such eIF2B sub-complexes may indeed exist in vivo, and that ISRIB appears to promote their formation.64 The authors observed larger eIF2B bodies with high degree of co-localization of all subunits, as well as small bodies, most of which had only detectable eIF2Bγ and ε (co-localization of all subunits did not reach 100% even for the large eIF2B bodies, which could be due to epitope accessibility, sensitivity, or other issues). They also found that ISRIB caused increased eIF2Bδ localization to the small eIF2B bodies, which coincided with increased cycling of eIF2 through these bodies that was not inhibited by eIF2(α-P). The ISRIB effect on eIF2Bδ localization was abrogated by a mutation in eIF2Bδ that prevents ISRIB binding. eIF2Bβ was not observed in the small bodies, which is surprising for several reasons: eIF2Bβ and δ form a stable heterodimer, required for eIF2Bδ stability;55, 65 ISRIB “staples” two eIF2Bβδ dimers together through contacts with both eIF2Bβ and δ;47, 48 and last but not least, eIF2Bβ and δ together form the eIF2α-binding site in the productive eFI2B:eIF2 complex.2, 12 It is not clear at this time whether the inability to observe eIF2Bβ in small bodies was due to its actual absence, or to technical difficulties, such as the relevant epitopes being sterically blocked.64 This uncertainty notwithstanding, the report indicates that eIF2B sub-complexes lacking eIF2Bα do exist in vivo and that ISRIB is able to promote their formation and activity. The same is likely to be true also for the majority of eIF2B molecules, which are free and not part of these bodies.
It is currently not known with certainty whether ISRIB affects the equilibrium between the productive and nonproductive eIF2B•eIF2 complexes, or the eIF2B catalytic activity in the productive complex. However, there is some experimental evidence suggesting that ISRIB could act by destabilizing eIF2(α-P)-GDP binding,28, 62 in addition to its effect on decamerization.
eIF2B as a therapeutic target
eIF2B and the Integrated Stress Response
As described above, regulation of eIF2B activity is at the center of the Integrated Stress Response (ISR) (Figure 1). The degree of ISR activation determines its effects on translation of individual mRNAs. For example, induction of full-blown ISR lowers the eIF2-GTP•Met-tRNAi ternary complex (TC) concentration to levels that cause reduced overall protein synthesis, while turning on translation of a set of mRNAs through a process called translation reinitiation. Individual mRNAs vary vastly in their sensitivity to reduced TC concentrations and a number of mRNAs coding for house-keeping proteins or proteins required to mediate the stress response continue being translated during ISR, some of them possibly also via TC-independent translation (reviewed in refs.7, 20, 66, 67). A more modest ISR induction may have little effect on bulk translation, while still lowering TC concentration enough to turn on translation of a subset of all the mRNAs whose translation is stimulated by full-blown ISR. An interesting example can be found in ref.60, where ISR is part of a chain of events involved in axon directional migration during neural wiring. Localized translation in the axon tip was reported to induce ISR, which in turn promotes expression of a specific set of proteins. In this system, the ISR was found to be transient, thanks at least in part to activation of eIF2B by inhibition of the GSK-3β kinase and eIF2Bε dephosphorylation. Despite significant eIF2α phosphorylation, there was no inhibition of global protein synthesis or increase in translation of other ISR-inducible proteins, such as the activating transcription factor 4 (ATF4).60
While ISR is triggered by a wide range of stress factors and signals that converge on eIF2B, it is hardly ever the only response to a given stress factor. Instead, ISR operates in conjunction with other cell responses specific to the particular stress. To fully understand each process, one needs to look at the interplay between the stressor itself, the ISR, and the other cellular responses to the stress. For instance, accumulation of unfolded proteins in the ER (ER stress), in the absence of adequate response, causes cell dysfunction and ultimately leads to apoptosis. The ER stress triggers the unfolded protein response (UPR), which has three branches, mediated by the inositol-requiring protein-1 (IRE1), the activating transcription factor 6 (ATF6), and PERK. Of these, PERK activates the ISR, while IRE-1 and ATF6 activate other signaling pathways in the cell. The cumulative effect of all three branches of the UPR, together with other processes in the cell, is reducing the load of unfolded proteins in the ER and restoring proper cell function. While the stress responses include activation of certain pro-apoptotic pathways, the interplay among pro-survival and pro-apoptosis signals and the negative feedback built into the system ensure cell survival and return to homeostasis in the majority of the cases (reviewed in ref.64). However, uncontrolled stress response that is too strong and/or too prolonged can itself lead to cell death. This is the case in a number of neurodegenerative disorders, where PERK-mediated ISR causes apoptosis and accordingly, PERK inhibitors are neuroprotective.26, 27 Thus, in the case of ER stress, the stress response needs to be strong enough to protect the cell from the stress, but not too strong, which would itself cause apoptosis (see Figure 1, above). The opposite is true in the case of viral infection, where the desired outcome is apoptosis, mediated at least in part by PKR activation. For successful infection, the virus must avoid eliciting a full-blown stress response and apoptosis, while moderate ISR can even be beneficial for the virus, because it helps the cell coping with the stress caused by the mass production of viral components. The same scenarios are observed in cancer. ISR is often needed for cancer cell survival and growth, and its inhibition has been reported to cause cancer cell death.68 At the same time, the cytotoxic effects of some anticancer drugs are mediated by induction of a strong ISR, leading to apoptosis, and ISR suppression in this case confers resistance to the anticancer treatment.69
Use of ISR inhibitors in neurodegenerative disorders
The reports that ISR is chronically activated in prion disease, and that PERK inhibitors are neuroprotective, identified ISR as a promising therapeutic target in this and other neurodegenerative disorders. However, PERK inhibitors are associated with pancreatic toxicity. Then came the breakthrough discovery of ISRIB,29 which inhibits ISR to a lesser degree than PERK inhibitors, but is nevertheless neuroprotective, without serious side effects.25 As discussed above, ISRIB does not inhibit eIF2α phosphorylation. Instead, it stimulates eIF2B activity and reduces the inhibitory effect of eIF2α-P at intermediate levels of eIF2α phosphorylation, but is ineffective when a large fraction of eIF2α is phosphorylated. The list of animal models of neurodegenerative disorders, for which ISRIB and other ISR inhibitors have been found to be neuroprotective, has been continuously growing and, besides Prion Disease, now includes Alzheimer’s Disease,70 Frontotemporal Dementia,71 Parkinson’s Disease,72 Traumatic Brain Injury,73 VWM,62, 74 and Down Syndrome.75 Recently, the Mental deficiency, Epilepsy, Hypogenitalism, Microcephaly, and Obesity (MEHMO) syndrome, a rare X-linked intellectual disability, caused by mutations in eIF2γ, was added to the list of diseases for which ISR inhibitors appear promising. It was reported that ISR is constitutively activated in MEHMO patient-derived induced pluripotent stem cells (iPSCs), and that ISRIB rescued the cell growth, translational, and neuronal differentiation defects associated with a MEHMO mutation in eIF2γ.76
Due to its low solubility, ISRIB is not being considered as a potential drug in humans, but rather as a proof of principle in animal models. Most of the activity currently is focused on developing suitable ISRIB derivatives and on finding new drugs that inhibit ISR. An ISRIB analog with better solubility was recently shown to be neuroprotective in an animal model of VWM; however, unlike ISRIB, it had serious side effects.77 It is not clear whether the toxicity is a unique feature of this particular ISRIB analog, or is more generally due to its higher solubility, which could increase its bioavailability in certain organs to undesired levels. A screen of a library of FDA-approved drugs for ISR inhibitors yielded two promising candidates: trazodone, an antidepressant, and dibenzoylmethane, an anti-cancer drug candidate. Both of these drugs were neuroprotective in mouse models of Prion Disease and Frontotemporal Dementia. Like ISRIB, these drugs inhibited ISR downstream from eIF2α phosphorylation, although their targets remain unknown.78 While these results are promising, other authors have cautioned that animal studies may not translate into success in humans. In particular, clinical studies with trazodone have not shown clear indication of improvement of cognitive function in Alzheimer’s disease patients,79 although this could have been due to the dosage and duration used.80
In conclusion, the new structures of the eIF2B•eIF2 complexes now allow the rational design of drugs that specifically inhibit or stimulate ISR. Depending on the disease being targeted, the goal of therapeutic ISR manipulation would be either stimulation or suppression. ISR is vital for every cell in the body; therefore, powerful ISR inhibition is likely to have undesired side effects. For example, PERK inhibitors are neuroprotective in animal models of a number of neurodegenerative disorders, but cause pancreatic and liver toxicity, whereas ISRIB, which only moderately suppresses ISR, is neuroprotective without serious side effects.25–30 Another potential issue is that, even if an ISR inhibitor can be neuroprotective at doses that do not cause serious side effects, in the long term the stress itself could cause cell dysfunction or death. Therefore, it appears that the most promising approach, at least with respect to neurodegenerative disorders, is to explore combination therapies with drugs that target the cause of the stress and ISR inhibitors that reduce the excessive stress response.
Acknowledgments
We thank Boriana Marintcheva for helpful discussions.
Funding
This work was supported by the National Institutes of Health [GM095720 to A.M.], Japan Society for the Promotion of Science (Grant-in-Aid for Scientific Research [B], [JP19H03172 to T.I.]), Japan Agency for Medical Research and Development (Platform Project for Supporting Drug Discovery and Life Science Research [Basis for Supporting Innovative Drug Discovery and Life Science Research] [JP19am0101082]), and RIKEN (the Integrated life science research to challenge super aging society and the Pioneering Project “Dynamic Structural Biology”).
References
- [1].Kashiwagi K, Takahashi M, Nishimoto M, Hiyama TB, Higo T, Umehara T, Sakamoto K, Ito T, and Yokoyama S (2016) Crystal structure of eukaryotic translation initiation factor 2B, Nature 531, 122–125. [DOI] [PubMed] [Google Scholar]
- [2].Kashiwagi K, Yokoyama T, Nishimoto M, Takahashi M, Sakamoto A, Yonemochi M, Shirouzu M, and Ito T (2019) Structural basis for eIF2B inhibition in integrated stress response, Science 364, 495–499. [DOI] [PubMed] [Google Scholar]
- [3].Marintchev A, and Wagner G (2004) Translation initiation: structures, mechanisms and evolution, Q Rev Biophys 37, 197–284. [DOI] [PubMed] [Google Scholar]
- [4].Pestova TV, Lorsch JR, and Hellen CUT (2007) The Mechanism of Translation Initiation in Eukaryotes, In Translational Control in Biology and Medicine (Mathews MB, Sonenberg N, Hershey JWB, Ed.), pp 87–128, Cold Spring Harbor Laboratory Press, Cold Spring harbor, NY. [Google Scholar]
- [5].Sonenberg N, and Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets, Cell 136, 731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hinnebusch AG (2014) The scanning mechanism of eukaryotic translation initiation, Annu Rev Biochem 83, 779–812. [DOI] [PubMed] [Google Scholar]
- [7].Jackson RJ, Hellen CU, and Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation, Nat Rev Mol Cell Biol 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pavitt GD, Ramaiah KV, Kimball SR, and Hinnebusch AG (1998) eIF2 independently binds two distinct eIF2B subcomplexes that catalyze and regulate guanine-nucleotide exchange, Genes & development 12, 514–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jennings MD, and Pavitt GD (2010) eIF5 has GDI activity necessary for translational control by eIF2 phosphorylation, Nature 465, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jennings MD, Zhou Y, Mohammad-Qureshi SS, Bennett D, and Pavitt GD (2013) eIF2B promotes eIF5 dissociation from eIF2*GDP to facilitate guanine nucleotide exchange for translation initiation, Genes & development 27, 2696–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Singh CR, Lee B, Udagawa T, Mohammad-Qureshi SS, Yamamoto Y, Pavitt GD, and Asano K (2006) An eIF5/eIF2 complex antagonizes guanine nucleotide exchange by eIF2B during translation initiation, The EMBO journal 25, 4537–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kenner LR, Anand AA, Nguyen HC, Myasnikov AG, Klose CJ, McGeever LA, Tsai JC, Miller-Vedam LE, Walter P, and Frost A (2019) eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response, Science 364, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Adomavicius T, Guaita M, Zhou Y, Jennings MD, Latif Z, Roseman AM, and Pavitt GD (2019) The structural basis of translational control by eIF2 phosphorylation, Nat Commun 10, 2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Salimans M, Goumans H, Amesz H, Benne R, and Voorma HO (1984) Regulation of protein synthesis in eukaryotes. Mode of action of eRF, an eIF-2-recycling factor from rabbit reticulocytes involved in GDP/GTP exchange, Eur J Biochem 145, 91–98. [DOI] [PubMed] [Google Scholar]
- [15].Gross M, Rubino MS, and Hessefort SM (1991) The conversion of eIF-2.GDP to eIF-2.GTP by eIF-2B requires Met-tRNA(fMet), Biochem Biophys Res Commun 181, 1500–1507. [DOI] [PubMed] [Google Scholar]
- [16].Gordiyenko Y, Llacer JL, and Ramakrishnan V (2019) Structural basis for the inhibition of translation through eIF2alpha phosphorylation, Nat Commun 10, 2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jennings MD, Kershaw CJ, Adomavicius T, and Pavitt GD (2017) Fail-safe control of translation initiation by dissociation of eIF2alpha phosphorylated ternary complexes, Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bogorad AM, Lin KY, and Marintchev A (2018) eIF2B Mechanisms of Action and Regulation: A Thermodynamic View, Biochemistry 57, 1426–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dever TE, Dar AC, and Sicheri F (2007) The eIF2α kinases, In Translational Control in Biology and Medicine (Mathews MB, Sonenberg N, Hershey JWB, Ed.), pp 319–344, Cold Spring Harbor Laboratory Press, Cold Spring harbor, NY. [Google Scholar]
- [20].Hinnebusch AG (2005) Translational regulation of GCN4 and the general amino acid control of yeast, Annu Rev Microbiol 59, 407–450. [DOI] [PubMed] [Google Scholar]
- [21].Hinnebusch AG, Dever TE, and Asano K (2007) Mechanism of translation initiation in the yeast Saccharomyces cerevisiae, In Translational Control in Biology and Medicine (Mathews MB, Sonenberg N, Hershey JWB, Ed.), pp 225–268, Cold Spring Harbor Laboratory Press, Cold Spring harbor, NY. [Google Scholar]
- [22].Ron D, and Harding HP (2007) eIF2α phosphorylation in cellular stress responses and disease, In Translational Control in Biology and Medicine (Mathews MB, Sonenberg N, Hershey JWB, Ed.), pp 345–368, Cold Spring Harbor Laboratory Press, Cold Spring harbor, NY. [Google Scholar]
- [23].Wek RC, Jiang HY, and Anthony TG (2006) Coping with stress: eIF2 kinases and translational control, Biochem Soc Trans 34, 7–11. [DOI] [PubMed] [Google Scholar]
- [24].Jennings MD, Kershaw CJ, White C, Hoyle D, Richardson JP, Costello JL, Donaldson IJ, Zhou Y, and Pavitt GD (2016) eIF2beta is critical for eIF5-mediated GDP-dissociation inhibitor activity and translational control, Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, Ortori CA, Barrett DA, Fromont C, Fischer PM, Harding HP, Ron D, and Mallucci GR (2015) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity, Cell Death Dis 6, e1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, and Mallucci GR (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice, Sci Transl Med 5, 206ra138. [DOI] [PubMed] [Google Scholar]
- [27].Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, and Mallucci GR (2012) Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration, Nature 485, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sekine Y, Zyryanova A, Crespillo-Casado A, Fischer PM, Harding HP, and Ron D (2015) Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound, Science 348, 1027–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, Okreglak V, Ashkenazi A, Hann B, Nader K, Arkin MR, Renslo AR, Sonenberg N, and Walter P (2013) Pharmacological brake-release of mRNA translation enhances cognitive memory, Elife 2, e00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sidrauski C, Tsai JC, Kampmann M, Hearn BR, Vedantham P, Jaishankar P, Sokabe M, Mendez AS, Newton BW, Tang EL, Verschueren E, Johnson JR, Krogan NJ, Fraser CS, Weissman JS, Renslo AR, and Walter P (2015) Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response, Elife 4, e07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bugiani M, Boor I, Powers JM, Scheper GC, and van der Knaap MS (2010) Leukoencephalopathy with vanishing white matter: a review, J Neuropathol Exp Neurol 69, 987–996. [DOI] [PubMed] [Google Scholar]
- [32].Fogli A, and Boespflug-Tanguy O (2006) The large spectrum of eIF2B-related diseases, Biochem Soc Trans 34, 22–29. [DOI] [PubMed] [Google Scholar]
- [33].Bogorad AM, Xia B, Sandor DG, Mamonov AB, Cafarella TR, Jehle S, Vajda S, Kozakov D, and Marintchev A (2014) Insights into the architecture of the eIF2Balpha/beta/delta regulatory subcomplex, Biochemistry 53, 3432–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuhle B, Eulig NK, and Ficner R (2015) Architecture of the eIF2B regulatory subcomplex and its implications for the regulation of guanine nucleotide exchange on eIF2, Nucleic acids research 43, 9994–10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Richardson JP, Mohammad SS, and Pavitt GD (2004) Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity, Molecular and cellular biology 24, 2352–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wortham NC, Martinez M, Gordiyenko Y, Robinson CV, and Proud CG (2014) Analysis of the subunit organization of the eIF2B complex reveals new insights into its structure and regulation, FASEB J 28, 2225–2237. [DOI] [PubMed] [Google Scholar]
- [37].Konieczny A, and Safer B (1983) Purification of the eukaryotic initiation factor 2-eukaryotic initiation factor 2B complex and characterization of its guanine nucleotide exchange activity during protein synthesis initiation, The Journal of biological chemistry 258, 3402–3408. [PubMed] [Google Scholar]
- [38].Panniers R, and Henshaw EC (1983) A GDP/GTP exchange factor essential for eukaryotic initiation factor 2 cycling in Ehrlich ascites tumor cells and its regulation by eukaryotic initiation factor 2 phosphorylation, The Journal of biological chemistry 258, 7928–7934. [PubMed] [Google Scholar]
- [39].Goss DJ, Parkhurst LJ, Mehta HB, Woodley CL, and Wahba AJ (1984) Studies on the role of eukaryotic nucleotide exchange factor in polypeptide chain initiation, The Journal of biological chemistry 259, 7374–7377. [PubMed] [Google Scholar]
- [40].Kimball SR, Everson WV, Myers LM, and Jefferson LS (1987) Purification and characterization of eukaryotic initiation factor 2 and a guanine nucleotide exchange factor from rat liver, The Journal of biological chemistry 262, 2220–2227. [PubMed] [Google Scholar]
- [41].Hiyama TB, Ito T, Imataka H, and Yokoyama S (2009) Crystal structure of the alpha subunit of human translation initiation factor 2B, J Mol Biol 392, 937–951. [DOI] [PubMed] [Google Scholar]
- [42].Boesen T, Mohammad SS, Pavitt GD, and Andersen GR (2004) Structure of the catalytic fragment of translation initiation factor 2B and identification of a critically important catalytic residue, The Journal of biological chemistry 279, 10584–10592. [DOI] [PubMed] [Google Scholar]
- [43].Wei J, Jia M, Zhang C, Wang M, Gao F, Xu H, and Gong W (2010) Crystal structure of the C-terminal domain of the varepsilon subunit of human translation initiation factor eIF2B, Protein Cell 1, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gordiyenko Y, Schmidt C, Jennings MD, Matak-Vinkovic D, Pavitt GD, and Robinson CV (2014) eIF2B is a decameric guanine nucleotide exchange factor with a gamma2epsilon2 tetrameric core, Nat Commun 5, 3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gomez E, and Pavitt GD (2000) Identification of domains and residues within the epsilon subunit of eukaryotic translation initiation factor 2B (eIF2Bepsilon) required for guanine nucleotide exchange reveals a novel activation function promoted by eIF2B complex formation, Molecular and cellular biology 20, 3965–3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bogorad AM, Lin KY, and Marintchev A (2017) Novel mechanisms of eIF2B action and regulation by eIF2alpha phosphorylation, Nucleic acids research 45, 11962–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, Frost A, and Walter P (2018) Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule, Science 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, Harding HP, Allen F, Parts L, Fromont C, Fischer PM, Warren AJ, and Ron D (2018) Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B, Science 359, 1533–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ito T, Marintchev A, and Wagner G (2004) Solution structure of human initiation factor eIF2alpha reveals homology to the elongation factor eEF1B, Structure 12, 1693–1704. [DOI] [PubMed] [Google Scholar]
- [50].Hannig EM, Williams NP, Wek RC, and Hinnebusch AG (1990) The translational activator GCN3 functions downstream from GCN1 and GCN2 in the regulatory pathway that couples GCN4 expression to amino acid availability in Saccharomyces cerevisiae, Genetics 126, 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Vazquez de Aldana CR, Dever TE, and Hinnebusch AG (1993) Mutations in the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2 alpha) that overcome the inhibitory effect of IF-2 alpha phosphorylation on translation initiation, Proceedings of the National Academy of Sciences of the United States of America 90, 7215–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Vazquez de Aldana CR, and Hinnebusch AG (1994) Mutations in the GCD7 subunit of yeast guanine nucleotide exchange factor eIF-2B overcome the inhibitory effects of phosphorylated eIF-2 on translation initiation, Molecular and cellular biology 14, 3208–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pavitt GD, Yang W, and Hinnebusch AG (1997) Homologous segments in three subunits of the guanine nucleotide exchange factor eIF2B mediate translational regulation by phosphorylation of eIF2, Molecular and cellular biology 17, 1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, and Hinnebusch AG (2001) Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation, Molecular and cellular biology 21, 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dev K, Qiu H, Dong J, Zhang F, Barthlme D, and Hinnebusch AG (2010) The beta/Gcd7 subunit of eukaryotic translation initiation factor 2B (eIF2B), a guanine nucleotide exchange factor, is crucial for binding eIF2 in vivo, Molecular and cellular biology 30, 5218–5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang W, and Hinnebusch AG (1996) Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2, Molecular and cellular biology 16, 6603–6616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kimball SR, Fabian JR, Pavitt GD, Hinnebusch AG, and Jefferson LS (1998) Regulation of guanine nucleotide exchange through phosphorylation of eukaryotic initiation factor eIF2alpha. Role of the alpha- and delta-subunits of eiF2b, The Journal of biological chemistry 273, 12841–12845. [DOI] [PubMed] [Google Scholar]
- [58].Alone PV, and Dever TE (2006) Direct binding of translation initiation factor eIF2gamma-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2B epsilon, The Journal of biological chemistry 281, 12636–12644. [DOI] [PubMed] [Google Scholar]
- [59].Asano K, Krishnamoorthy T, Phan L, Pavitt GD, and Hinnebusch AG (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bepsilon, GTPase-activating and GDP-GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2, The EMBO journal 18, 1673–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Cagnetta R, Wong HH, Frese CK, Mallucci GR, Krijgsveld J, and Holt CE (2019) Noncanonical Modulation of the eIF2 Pathway Controls an Increase in Local Translation during Neural Wiring, Molecular cell 73, 474–489 e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Anand AA, and Walter P (2020) Structural insights into ISRIB, a memory-enhancing inhibitor of the integrated stress response, FEBS J 287, 239–245. [DOI] [PubMed] [Google Scholar]
- [62].Wong YL, LeBon L, Edalji R, Lim HB, Sun C, and Sidrauski C (2018) The small molecule ISRIB rescues the stability and activity of Vanishing White Matter Disease eIF2B mutant complexes, Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Williams DD, Price NT, Loughlin AJ, and Proud CG (2001) Characterization of the mammalian initiation factor eIF2B complex as a GDP dissociation stimulator protein, The Journal of biological chemistry 276, 24697–24703. [DOI] [PubMed] [Google Scholar]
- [64].Hodgson RE, Varanda BA, Ashe MP, Allen KE, and Campbell SG (2019) Cellular eIF2B subunit localization: implications for the integrated stress response and its control by small molecule drugs, Mol Biol Cell 30, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wortham NC, Stewart JD, Harris S, Coldwell MJ, and Proud CG (2016) Stoichiometry of the eIF2B complex is maintained by mutual stabilization of subunits, Biochem J 473, 571–580. [DOI] [PubMed] [Google Scholar]
- [66].Jackson RJ, Hellen CU, and Pestova TV (2012) Termination and post-termination events in eukaryotic translation, Adv Protein Chem Struct Biol 86, 45–93. [DOI] [PubMed] [Google Scholar]
- [67].Gunisova S, Hronova V, Mohammad MP, Hinnebusch AG, and Valasek LS (2018) Please do not recycle! Translation reinitiation in microbes and higher eukaryotes, FEMS Microbiol Rev 42, 165–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, Hsieh AC, Cunningham JT, Truillet C, Tameire F, Evans MJ, Evans CP, Yang JC, Hann B, Koumenis C, Walter P, Carroll PR, and Ruggero D (2018) Development of a stress response therapy targeting aggressive prostate cancer, Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, Rubinstein JL, Schimmer AD, Ketela T, and Chan SM (2019) Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response, Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- [70].Hosoi T, Kakimoto M, Tanaka K, Nomura J, and Ozawa K (2016) Unique pharmacological property of ISRIB in inhibition of Abeta-induced neuronal cell death, J Pharmacol Sci 131, 292–295. [DOI] [PubMed] [Google Scholar]
- [71].Radford H, Moreno JA, Verity N, Halliday M, and Mallucci GR (2015) PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia, Acta Neuropathol 130, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Celardo I, Costa AC, Lehmann S, Jones C, Wood N, Mencacci NE, Mallucci GR, Loh SH, and Martins LM (2016) Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease, Cell Death Dis 7, e2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chou A, Krukowski K, Jopson T, Zhu PJ, Costa-Mattioli M, Walter P, and Rosi S (2017) Inhibition of the integrated stress response reverses cognitive deficits after traumatic brain injury, Proceedings of the National Academy of Sciences of the United States of America 114, E6420–E6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Abbink TEM, Wisse LE, Jaku E, Thiecke MJ, Voltolini-Gonzalez D, Fritsen H, Bobeldijk S, Ter Braak TJ, Polder E, Postma NL, Bugiani M, Struijs EA, Verheijen M, Straat N, van der Sluis S, Thomas AAM, Molenaar D, and van der Knaap MS (2019) Vanishing white matter: deregulated integrated stress response as therapy target, Ann Clin Transl Neurol 6, 1407–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhu PJ, Khatiwada S, Cui Y, Reineke LC, Dooling SW, Kim JJ, Li W, Walter P, and Costa-Mattioli M (2019) Activation of the ISR mediates the behavioral and neurophysiological abnormalities in Down syndrome, Science 366, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Young-Baird SK, Lourenco MB, Elder MK, Klann E, Liebau S, and Dever TE (2020) Suppression of MEHMO Syndrome Mutation in eIF2 by Small Molecule ISRIB, Molecular cell 77, 875–886 e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Wong YL, LeBon L, Basso AM, Kohlhaas KL, Nikkel AL, Robb HM, Donnelly-Roberts DL, Prakash J, Swensen AM, Rubinstein ND, Krishnan S, McAllister FE, Haste NV, O’Brien JJ, Roy M, Ireland A, Frost JM, Shi L, Riedmaier S, Martin K, Dart MJ, and Sidrauski C (2019) eIF2B activator prevents neurological defects caused by a chronic integrated stress response, Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.[] Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, Smith E, Ortori CA, Barrett DA, Bushell M, and Mallucci GR (2017) Repurposed drugs targeting eIF2α-P-mediated translational repression prevent neurodegeneration in mice, Brain 140, 1768–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Camargos EF, and Nobrega OT (2017) Trazodone to change the risk of neurodegeneration: bedside to bench, Brain 140, e47. [DOI] [PubMed] [Google Scholar]
- [80].Halliday M, and Mallucci GR (2017) Reply: Trazodone to change the risk of neurodegeneration: bedside to bench, Brain 140, e48. [DOI] [PubMed] [Google Scholar]