

Abstract
A cobalt-catalyzed intramolecular silylperoxidation reaction was developed that allows for the conversion of unsaturated diisopropylsilyl ethers to 3-sila-1,2,4-trioxepanes. Reduction of the peroxide unit of the 3-sila-1,2,4-trioxepane yields six-membered ring diisopropylsilylene acetals.
Graphical Abstract
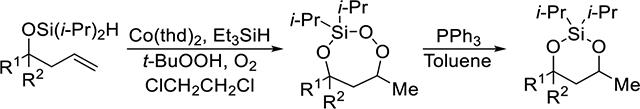
Selective for intramolecular silylperoxidation reaction
Flexible method for synthesis of 3-sila-1,2,4-trioxepanes or silylene acetals
INTRODUCTION
The cobalt-catalyzed oxygenation of alkenes is an effective reaction for the synthesis of silyl peroxides,1 but can also be applied to the synthesis of a variety of endoperoxides,2 including 1,2-dioxolanes,3,4,5 1,2-dioxanes,4,6 1,2-dioxepanes,7 and 1,2,4-trioxolanes.8,9 The reaction, which converts alkenes to silyl peroxides through a peroxyl radical intermediate,1,10,11 uses a cobalt(II) catalyst, molecular oxygen, and a silane reducing agent. Depending on the choice of substrate, the peroxyl radical intermediate can react intramolecularly, forming endoperoxides instead. Treatment of either a 1,4- or 1,5-diene4,12 with the cobalt-catalyzed oxygenation conditions resulted in the formation of 1,2-dioxolanes and 1,2-dioxanes, respectively (Scheme 1, eq 1). Similar reactivity was observed with vinylcyclopropanes, which formed 1,2-dioxolanes (Scheme 1, eq 2).3,12 In this Article, we report that unsaturated diisopropylsilyl ethers can undergo an intramolecular silylperoxidation reaction to form 3-sila-1,2,4-trioxepanes (Scheme 1, eq 3).13 Upon reduction of the peroxide unit using triphenylphosphine, the corresponding silylene-protected 1,3-diol was formed.
Scheme 1. Co-Catalyzed Endoperoxide Formation.
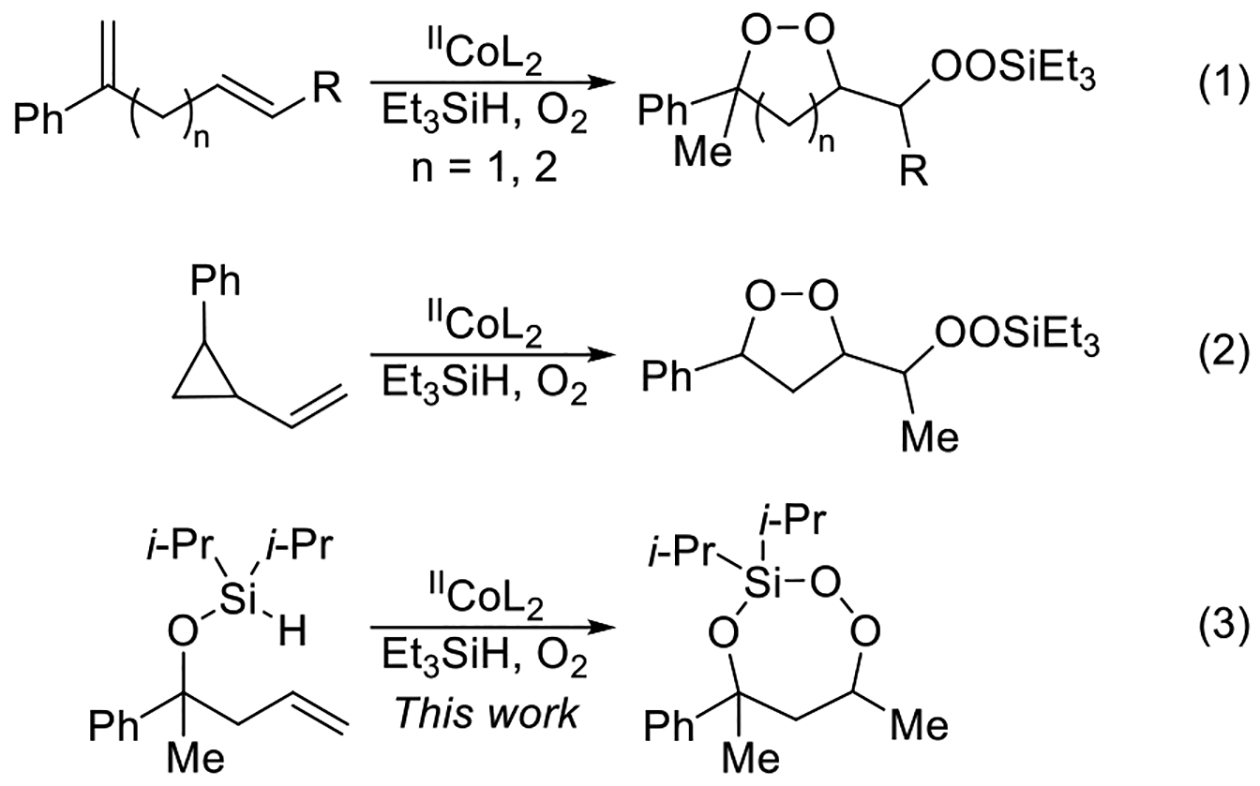
RESULTS AND DISCUSSION
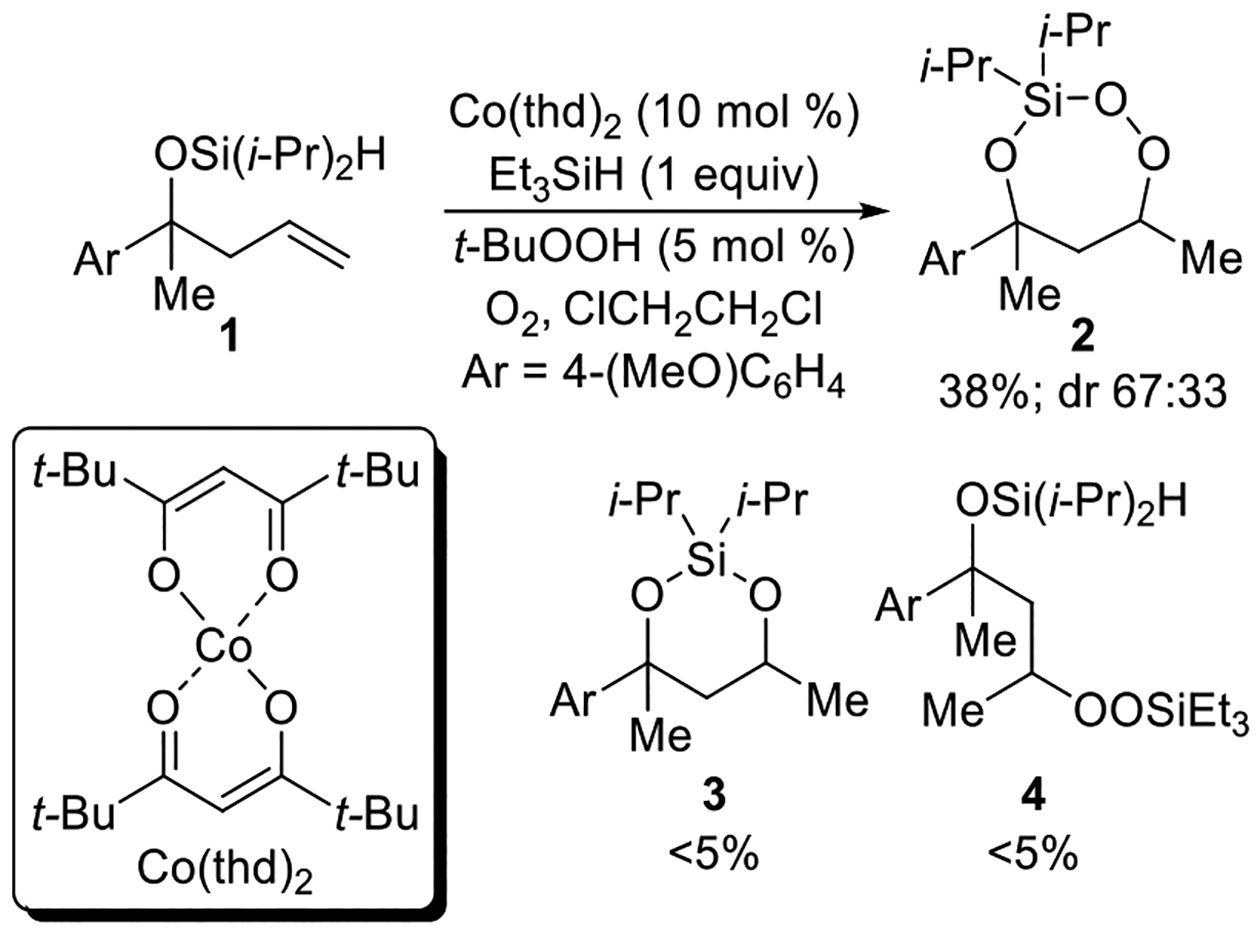
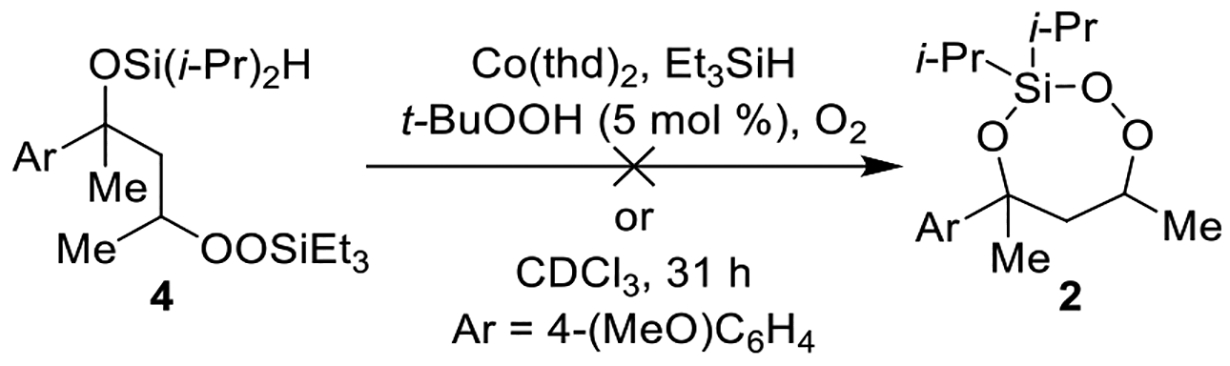
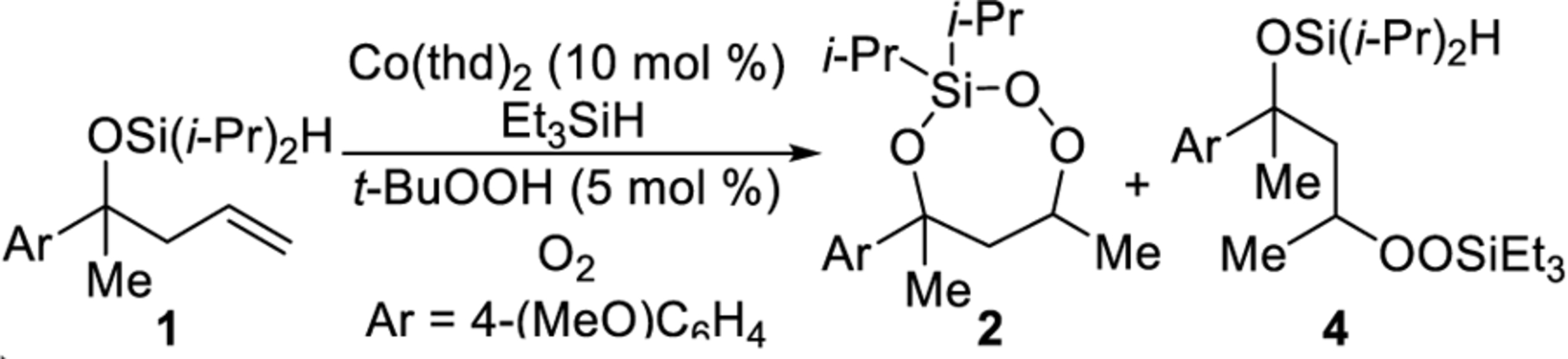
During our investigation into new applications of the cobalt-catalyzed oxygenation of alkenes, we found that intramolecular silylperoxidation can be achieved. When unsaturated diisopropylsilyl ether 1 was subjected to cobalt-catalyzed oxygenation (Scheme 2), after less than an hour, 3-sila-1,2,4-trioxepane 2 was formed as the major product. In addition, trace amounts of diisopropylsilylene acetal 3 and linear silyl peroxide 4 were detected by 1H NMR spectroscopy. Formation of 3-sila-1,2,4-trioxepane 2 as the major product was not anticipated because the silylation step of the cobalt-catalyzed oxygenation of alkenes is generally sensitive to both the steric and electronic environment of the silane.14,15 In this case, however, the diisopropylsilyl group was preferentially incorporated into the peroxide. The above observation was significant because, for comparison, the synthesis of a triisopropylsilyl peroxide using the cobalt-catalyzed oxygenation of alkenes is very difficult considering the poor reactivity of triisopropylsilane.14,16
Scheme 2. Products of Co-Catalyzed Intramolecular Silylperoxidation of Unsaturated Diisopropylsilyl Ethers.
The yield of the 3-sila-1,2,4-trioxepane was found to be sensitive to the choice of the added silane and cobalt catalyst. When unsaturated diisopropylsilyl ether 5 was subjected to cobalt-catalyzed oxygenation using Co(acac)2, triethylsilane, and catalytic tert-butyl hydroperoxide (Table 1, entry 1), 3-sila-1,2,4-trioxepane 6 was isolated in 21% yield. Triethylsilane and tert-butyl hydroperoxide were required to initiate the reaction rapidly.3 The use of a more sterically bulky silane (Table 1, entries 2 and 3) or one with electron withdrawing groups (Table 1, entry 4) gave only trace product formation after 20 h. The poor reactivity of these silanes can be explained by considering that formation of the reactive cobalt-hydride species is sensitive to the choice of silane.3,14 When Co(thd)2 was used instead of Co(acac)2, decreased reaction times and increased yields were observed (Table 1, entries 6–8). The combination of Co(thd)2 with catalytic phenylsilane (Table 1, entry 7) or stoichiometric triethylsilane (Table 1, entry 8) resulted in the greatest yields of the 3-sila-1,2,4-trioxepane. Optimized reaction conditions were achieved using 10 mol % Co(thd)2 and one equivalent of triethylsilane,17 which yielded the 3-sila-1,2,4-trioxepane 2 in 74% yield, as determined by 1H NMR spectroscopy (Table 1, entry 8). The isolated yield of 2, however, was only 38% (Table 1, entry 8), suggesting that the 3-sila-1,2,4-trioxepane was not stable to column chromatography.18
Table 1.
Optimization of Co-Catalyzed Intramolecular Silylperoxidation of Unsaturated Diisopropylsilyl Ethers
 | |||||
|---|---|---|---|---|---|
| Entry | R | IICoL2 (mol %) | Silane (equiv) | time (h) | yielda (NMR) |
| 1 | Ph | acac (20) | Et3SiH (2) | 1.5 | 21 %b |
| 2 | Ph | acac (20) | (i-Pr)3SiH (2.5) | 20 | trace |
| 3 | Ph | acac(20) | Ph3SiH (2.5) | 20 | trace |
| 4 | Ph | acac (20) | (EtO)3SiH (2.5) | 20 | trace |
| 5 | Ph | acac (20) | Ph2MeSiH (2) | 3 | 34% |
| 6 | Ph | thd (12)c | Ph2MeSiH (1) | 2 | 67% |
| 7 | 4-(MeO)C6H4 | thd (16) | PhSiH3(0.16) | 0.75 | 74% |
| 8 | 4-(MeO)C6H4 | thd (10) | Et3SiH(1) | 0.75 | 74% (38%)b |
Yield determined by 1H NMR spectroscopy with mesitylene as the internal standard.
Isolated yield.
thd = 2,2,6,6-tetramethyl-3,5-heptanedionato.
1,2, R = 4-(MeO)C6H4; 5,6, R = Ph.
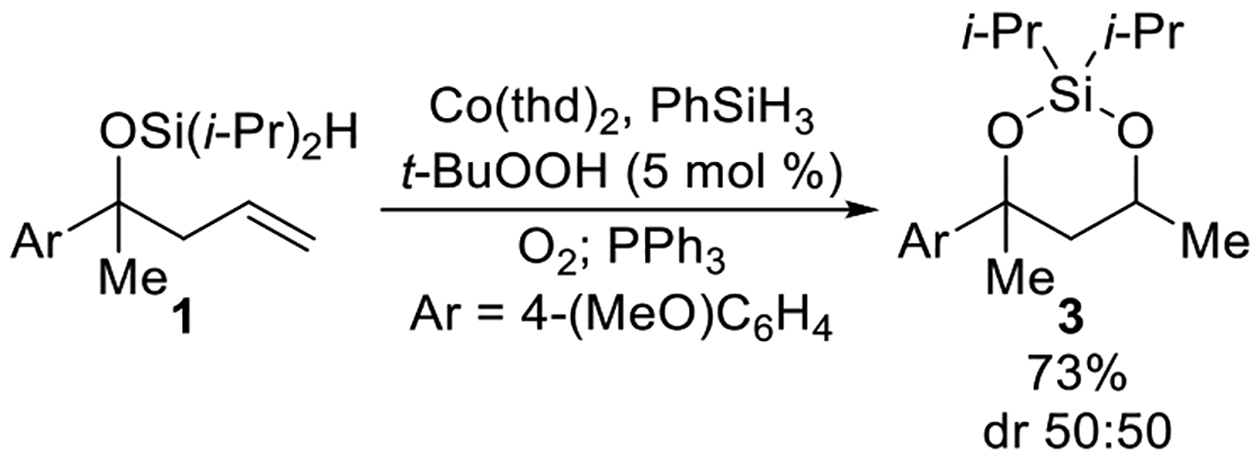
Reducing the 3-sila-1,2,4-trioxepane to the corresponding diisopropylsilylene acetal provided a product that was more stable to purification.19,20 After the cobalt-catalyzed oxygenation was completed, the peroxide unit of 3-sila-1,2,4-trioxepane 2 was reduced using triphenylphosphine.21,22 The above modification to the procedure resulted in a 73% isolated yield of diisopropylsilylene acetal 3 over two steps (Scheme 3). When the cobalt-catalyzed oxygenation was performed using triethylsilane instead of phenylsilane, the isolated yield of diisopropylsilylene acetal 3 decreased slightly to 60% due to the formation of trace amounts of linear silyl peroxide 4 (Scheme 2).14,23 Linear silyl peroxide 4 can be difficult to separate from diisopropylsilylene acetal 3.
Scheme 3. Formation of Diisopropylsilylene Acetal 3 from 3-Sila-1,2,4-Trioxepane 2.
Several control experiments yielded insight into the reaction mechanism. The first experiment was aimed at determining whether the 3-sila-1,2,4-trioxepane could form through a mechanism involving migration of the peroxide between the silicon atoms (Scheme 4). When linear silyl peroxide 4 was resubjected to the reaction conditions, none of the 3-sila-1,2,4-trioxepane 2 was observed by 1H NMR spectroscopy after 19 h. Even when a pure sample of linear silyl peroxide 4 was allowed to stand in deuterated chloroform, formation of 3-sila-1,2,4-trioxepane 2 was not observed.
Scheme 4. Exchange of Silanes Not Observed.
Other control experiments were designed to determine the role of triethylsilane. One hypothesis was that the triethylsilane catalyzed the initial formation of the cobalt-hydride species. When unsaturated diisopropylsilyl ether 1 was subjected to the reaction conditions without the addition of triethylsilane, formation of 3-sila-1,2,4-trioxepane 2 occurred slowly over the course of 19 h, with a final yield of 37% by 1H NMR spectroscopy (Table 2, entry 1). With the addition of one equivalent of triethylsilane, the reaction was completed in 0.5 h with a 74% yield of 3-sila-1,2,4-trioxepane 2 by 1H NMR spectroscopy (Table 2, entry 2). These data suggested that triethylsilane was necessary to catalyze the formation of the cobalt-hydride species and that the diisopropylsilyl group alone was not kinetically competent enough to do so. Even with a large excess of triethylsilane (Table 2, entries 3 and 4), formation of 3-sila-1,2,4-trioxepane 2 was competitive with the formation of linear silyl peroxide 4.
Table 2.
Equivalents of Triethylsilane and Effect on Product Distribution
 | |||||
|---|---|---|---|---|---|
| Entry | Et3SiH (equiv) | time (h) | conversion of 1 | yield 2a (NMR) | yield 4a (NMR) |
| 1 | - | 19 | 68% | 37% | - |
| 2 | 1 | 0.5 | 100% | 74% | 5% |
| 3 | 10 | 0.5 | 100% | 43% | 39% |
| 4 | 20 | 0.5 | 100% | 37% | 50% |
Yield determined by 1H NMR spectroscopy with mesitylene as the internal standard.
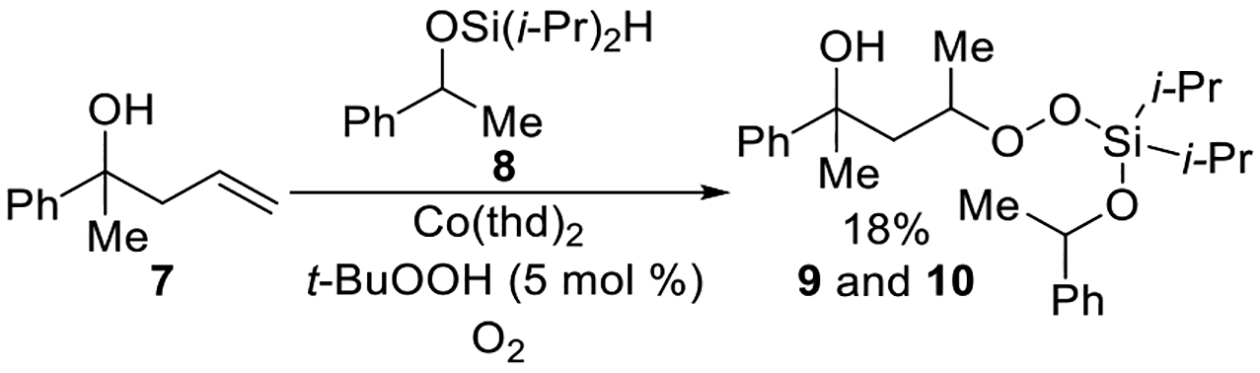
Further evidence of triethylsilane’s role was observed when alcohol 7 and diisopropylsilane 8 were subjected to the cobalt-catalyzed oxygenation (Scheme 5). The purpose of this experiment was to confirm that the diisopropylsilyl group alone cannot sufficiently initiate the reaction. With two equivalents of diisopropylsilane 8, even after 22 h, the conversion of 7 was low and the yield of diastereomers 9 and 10 was 18% (Scheme 5). This observation, combined with the data in Table 2, entry 1, indicates that the addition of triethylsilane is required to initiate the reaction.
Scheme 5. Co-Catalyzed Silylperoxidation Using Diisopropylsilane 8.
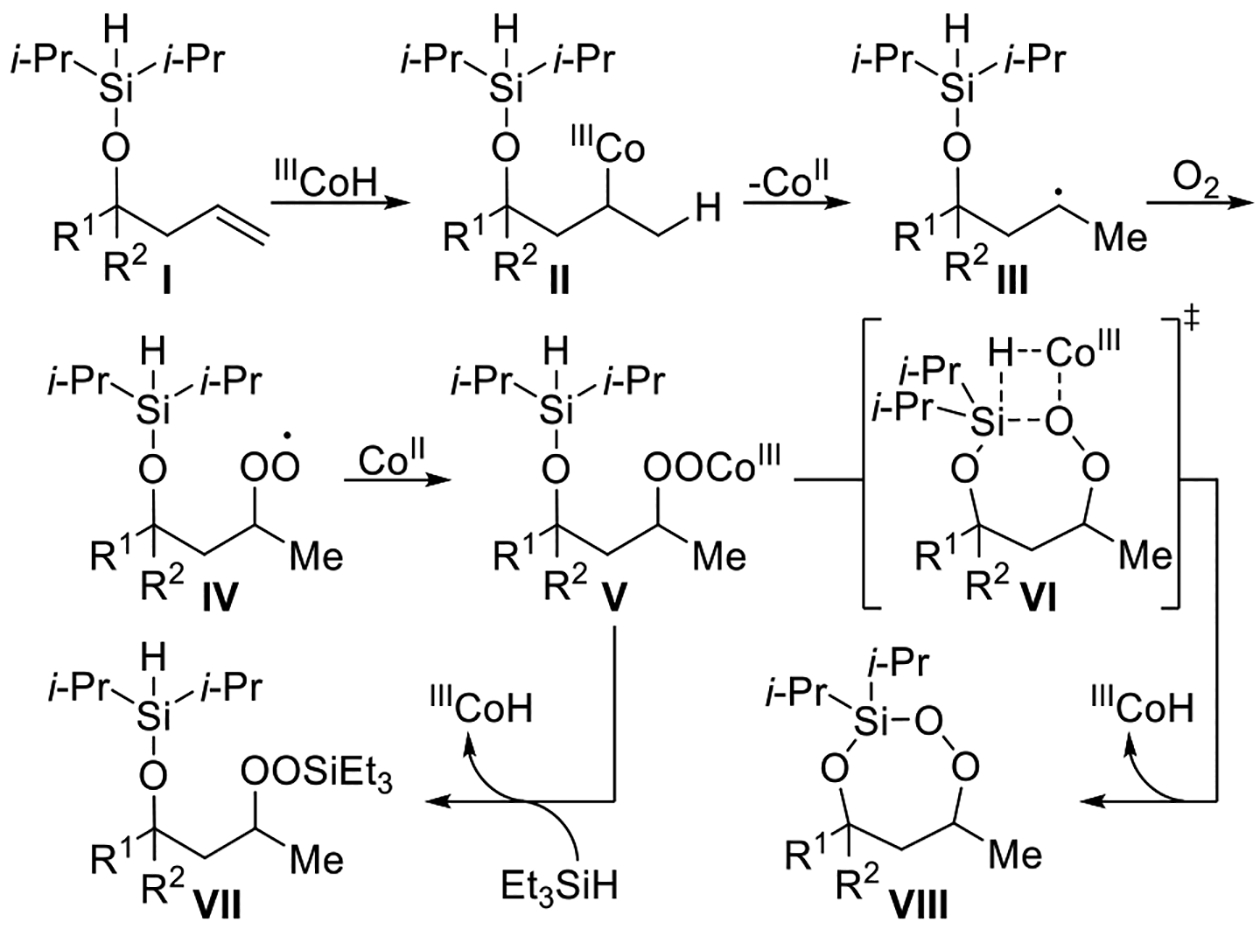
A mechanism can be proposed that is consistent with the above observations (Scheme 6). The first step in this mechanism is the formation of a cobalt-hydride species, which is catalyzed by tert-butyl hydroperoxide24 and triethylsilane.3,25 Addition of the cobalt-hydride species across the double bond of a molecule of unsaturated diisopropylsilyl ether I, followed by dissociation of the cobalt catalyst from II, would form alkyl radical III. The addition of cobalt-hydride occurs in such a way as to generate the more substituted radical intermediate.3,14 Alkyl radical III can react with molecular oxygen, forming peroxyl radical IV, which, upon coordination to cobalt, yields Co(III)–peroxyl intermediate V.4 Co(III)– peroxyl intermediate V can react by one of two ways. The first pathway involves an intramolecular transmetalation reaction (VI) between Co(III)–peroxyl intermediate V and the diisopropylsilyl group,3 resulting in formation of 3-sila-1,2,4-trioxepane VIII. The formation of other seven-membered ring products involving a transition state analogous to VI have been reported.26,27 The alternative pathway is also a transmetalation reaction, but instead occurs between Co(III)–peroxyl intermediate V and triethylsilane, yielding linear silyl peroxide VII. The mechanism of formation of 3-sila-1,2,4-trioxepane VIII is different from that of other intramolecular endoperoxide forming reactions (Scheme 1, eqs 1 and 2) in that product formation occurs from Co(III)–peroxyl intermediate V instead of peroxyl radical IV. It was suspected that because triethylsilane was only incorporated into less than 5% of the product (Scheme 2), any remaining triethylsilane either remained unreacted28 or was oxidized.3
Scheme 6. Proposed Mechanism for the Co-Catalyzed Intramolecular Silylperoxidation of Unsaturated Diisopropylsilyl Ethers.
The cobalt-catalyzed intramolecular formation of 3-sila-1,2,4-trioxepanes was general for several unsaturated diisopropylsilyl ethers (Table 3).29 Unsaturated silyl ethers 1 and 12–15 (Table 3)30 were synthesized by addition of a Grignard reagent to a carbonyl compound followed by silylation of the resulting alcohol, as outlined for diisopropylsilyl ether 15 in Scheme 7. There was a correlation between the degree of substitution of the alkene substrate and its reactivity in the intramolecular silylperoxidation.4 When mono-substituted alkenes 1, 12, and 13 were subjected to the cobalt-catalyzed oxygenation, the reaction was completed in two hours or less (Table 3, entries 1–3). Di-substituted alkene 14 required heating and a longer reaction time, ultimately yielding diisopropylsilylene acetal 18 in 39% (Table 3, entry 4). The added substitution at the allylic position of unsaturated silyl ether 15 also resulted in the need for heating and a longer reaction time (Table 3, entry 5). Unsaturated silyl ether 15 (Table 3, entry 5) gave the highest diastereomeric ratio out of all of the unsaturated silyl ethers tested. Generally, when a more sterically hindered alkene undergoes cobalt-catalyzed oxygenation, diastereoselectivity increases.31,32,33
Table 3.
Unsaturated Silyl Ether Screen
 | |||||
|---|---|---|---|---|---|
| Entry | unsaturated silyl ethera | IICoL2 (mol %) | time (h) | silylene acetal | yield |
| 1 | 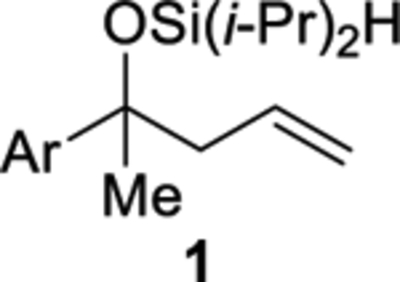 |
Co(thd)2 (10) | 0.5 | 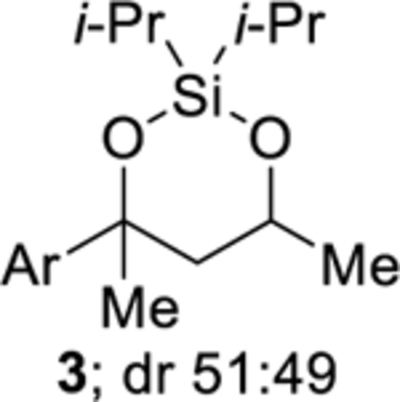 |
60% |
| 2 | 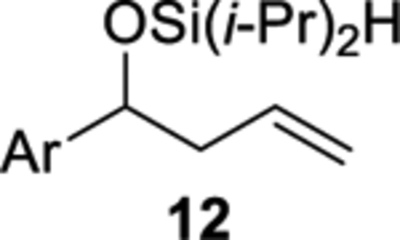 |
Co(thd)2 (10) | 1 | 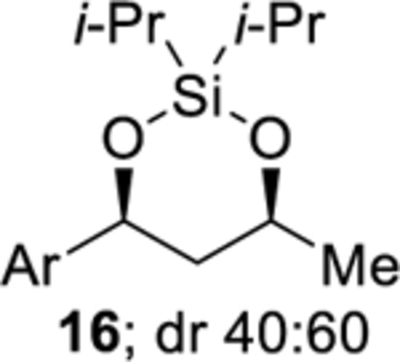 |
15%c |
| 3 |  |
Co(acac)2 (40) | 2 | 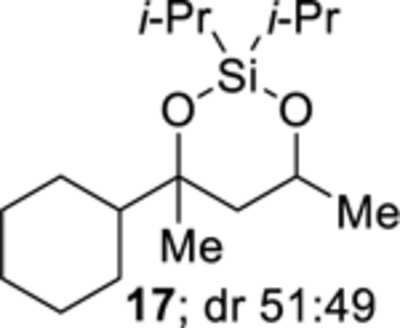 |
62% |
| 4 | 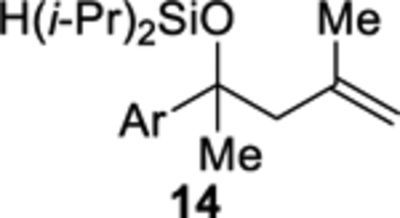 |
Co(acac)2 (40) | 3b | 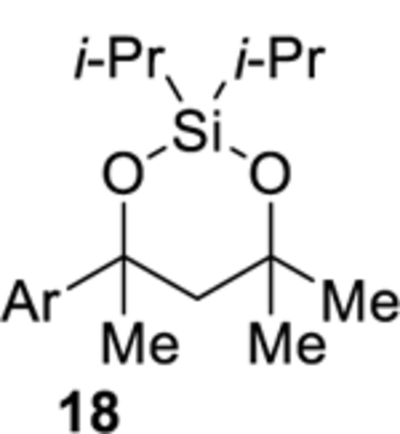 |
39% |
| 5 | 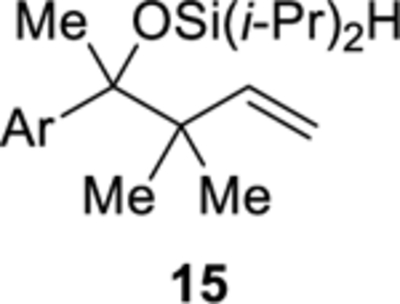 |
Co(acac)2 (40) | 5b | 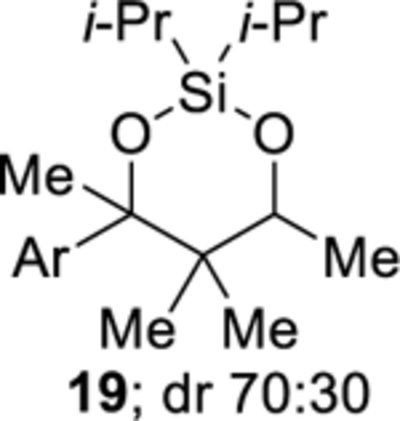 |
32% |
Ar = 4-(MeO)C6H4.
Reaction heated to 40 °C for this time.
1,3-Diol 20 was formed in 21% yield as a 67:33 mixture of diastereomers.
Scheme 7. Synthesis of Unsaturated Diisopropylsilyl Ethers.
In some examples (Table 3, entries 3–5), Co(acac)2 was used instead of Co(thd)2 to facilitate the purification process. During the course of the cobalt-catalyzed oxygenation, Co(thd)2 was converted to Co(thd)3,34,35 which can be difficult to separate from the diisopropylsilylene acetal product during column chromatography. Difficulties with the purification process using similar cobalt catalysts has been reported.36 Even though Co(acac)2 improves the purification process, Co(thd)2 was the better catalyst for these cobalt-catalyzed intramolecular silylperoxidation reactions. Compared to Co(acac)2, Co(thd)2 generally resulted in better yields of the 3-sila-1,2,4-trioxepane, shorter reaction times, and better conversion of the starting material.
The degree of substitution of the diisopropylsilyl ether affected the stability of the products from the cobalt-catalyzed intramolecular silylperoxidation. For example, secondary benzylic silyl ether 12 yielded the least amount of diisopropylsilylene acetal 16 (Table 3, entry 2).37 Diisopropylsilylene acetal 16 was also found to be sensitive to purification. Prior to chromatography, analysis of the crude reaction mixture of diisopropylsilylene acetal 16 showed a diastereomeric ratio of 40:60, however, after purification, only the minor diastereomer of 16 was isolated. By comparing the 1H and 13C NMR chemical shifts and coupling constants of diisopropylsilylene acetal 16 to structurally similar diisopropylsilylene acetals,38 it was determined that the syn diastereomer of 16 was isolated. Decomposition of diisopropylsilylene 16 had occurred during chromatography as evident by the formation of 1,3-diol 20 as a mixture of diastereomers (Table 3, entry 2). Decreased stability of benzylic diisopropylsilylene acetals has been previously observed.39
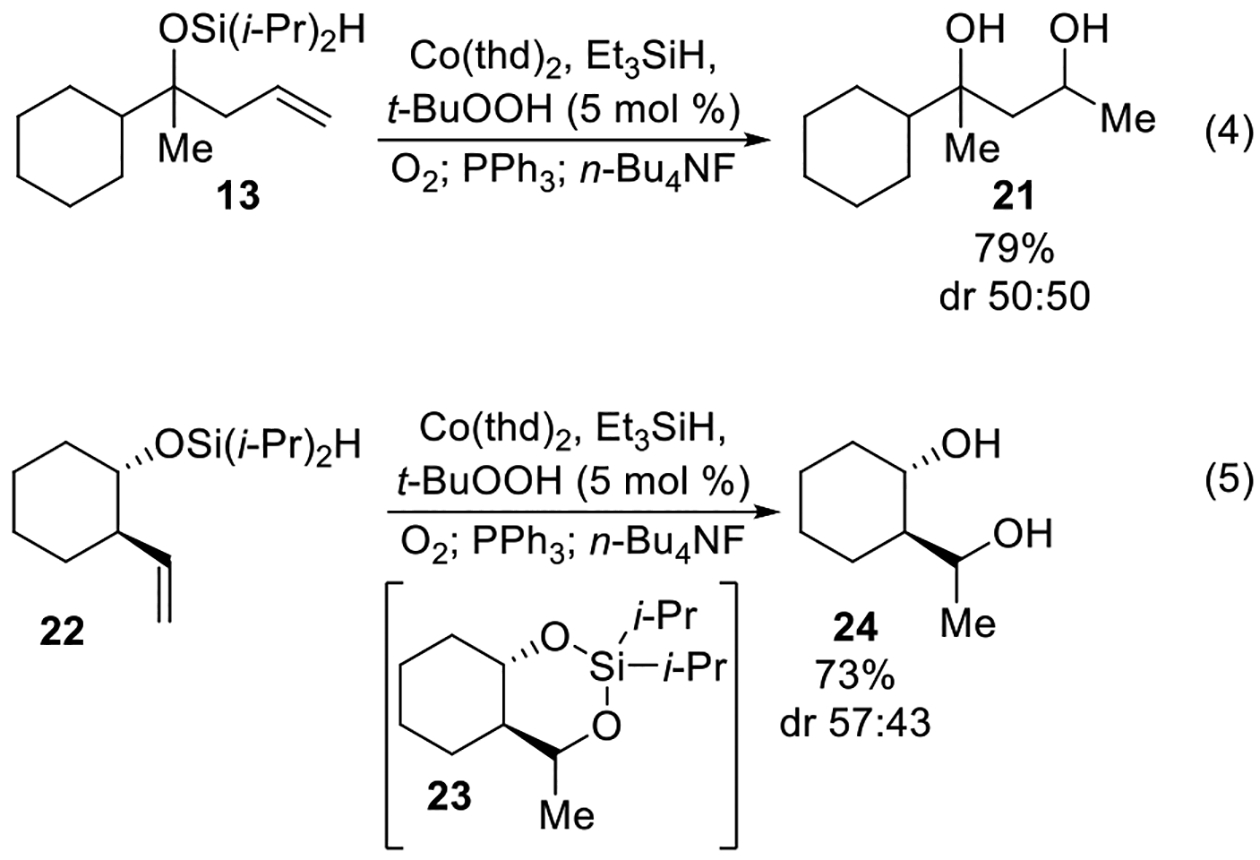
Removal of the diisopropylsilylene acetal protecting group using tetrabutylammonium fluoride yielded the corresponding diol, which was generally easier to purify. For example, attempts to purify diisopropylsilylene acetal 17 by chromatography (Table 3, entry 3), when the cobalt-catalyzed oxygenation was performed using Co(thd)2, were unsuccessful because of trace amounts of Co(thd)3 co-eluting with diisopropylsilylene acetal 17. By contrast, when the crude diisopropylsilylene acetal was treated with tetrabutylammonium fluoride (Scheme 8, eq 4), 1,3-diol 21 was isolated in 79% yield over three steps (Scheme 8, eq 4). Similarly, diisopropylsilylene acetal 23 was unstable to column chromatography and could not be purified (Scheme 8, eq 5). Treatment of a crude diisopropylsilylene acetal 23 with tetrabutylammonium fluoride yielded 1,3-diol 24 in 73% over three steps.
Scheme 8. Deprotection of Diisopropylsilylene Acetals.
The cobalt-catalyzed oxygenation of unsaturated diisopropylsilyl ethers could also be applied to the synthesis of five-membered ring diisopropylsilylene acetals (Scheme 9, eq 6). Unsaturated diisopropylsilyl ether 25 formed five-membered diisopropylsilylene acetal 26 (Scheme 9, eq 6) upon treatment with the cobalt-catalyzed oxygenation and subsequent reduction of the peroxide. This product, however, was not stable to purification by column chromatography.20 Its formation was suggested by 29Si NMR spectroscopy, which showed that not only had the chemical shift of diisopropylsilylene acetal 26 changed compared to that of the starting material (25), but also that this new chemical shift was within the range of other five-membered ring silylene acetals (Scheme 9, eq 6).40,41 These observations were confirmed by demonstrating that treatment of diisopropylsilylene acetal 26 with tetrabutylammonium fluoride yielded the corresponding 1,2-diol (27) in 19% over three steps (Scheme 9, eq 6).
Scheme 9. Ring Size Screen for the Co-Catalyzed Intramolecular Silylperoxidation of Unsaturated Diisopropylsilyl Ethers.
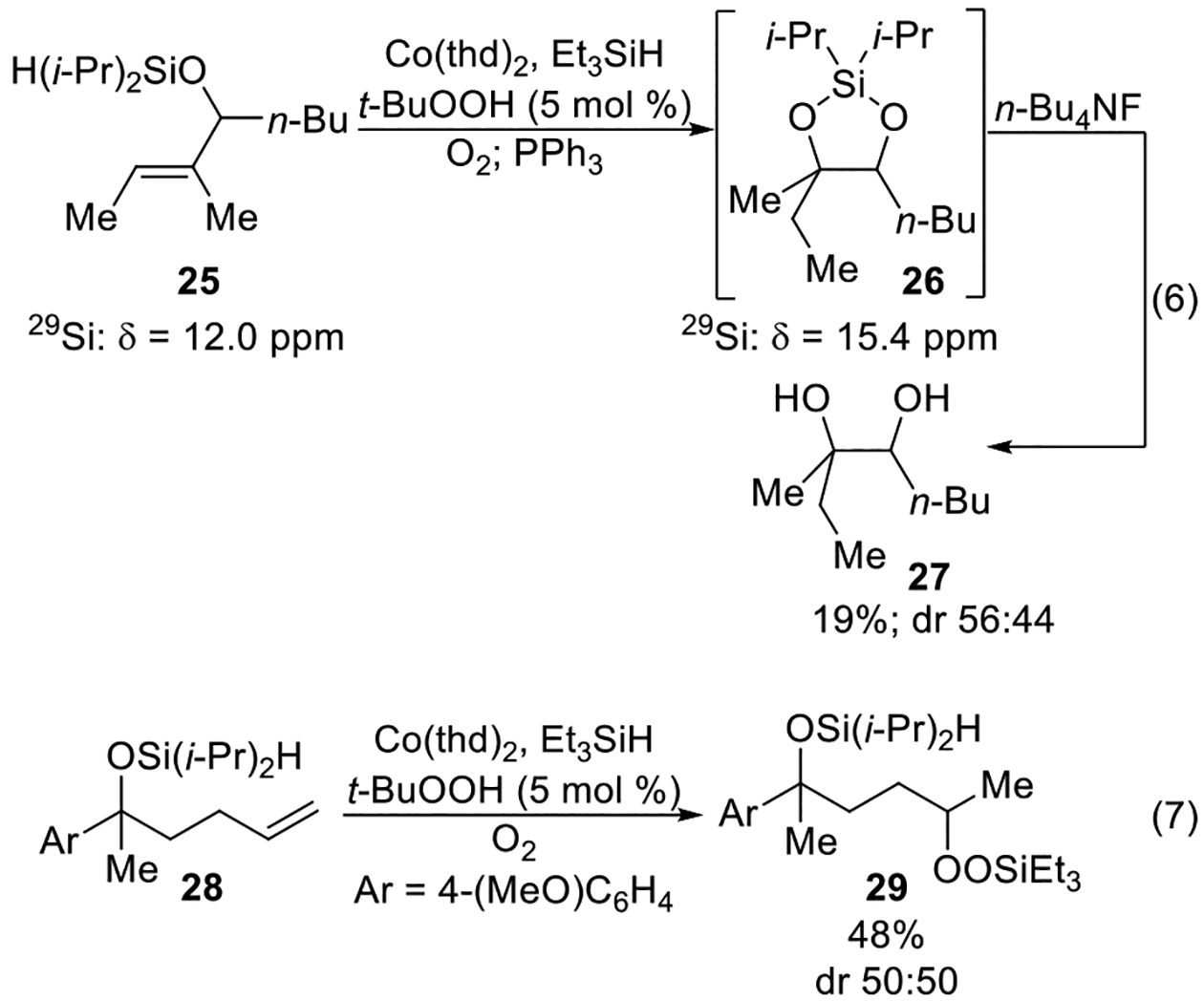
Synthesis of a seven-membered ring diisopropylsilylene acetal was not successful (Scheme 9, eq 7). When unsaturated diisopropylsilyl ether 28 was subjected to the cobalt-catalyzed oxygenation, no cyclization product was observed. Instead, only linear silyl peroxide 29 (Scheme 9, eq 7) was formed. As the distance between the alkene and diisopropylsilyl group increases, formation of the linear silyl peroxide becomes the favored reaction pathway instead of intramolecular cyclization (Scheme 6).
CONCLUSION
In summary, we have demonstrated that an intramolecular silylperoxidation reaction can be applied to the synthesis of 3-sila-1,2,4-trioxepanes from unsaturated diisopropylsilyl ethers. With the use of triphenylphosphine, the peroxide unit of the 3-sila-1,2,4-trioxepane can be reduced to yield diisopropylsilylene acetals. A mechanism has been suggested in which the addition of triethylsilane was necessary in order to initiate the reaction. The key intermediate of this reaction is a Co(III)–peroxyl intermediate, which can either react intra- or intermolecularly. Intramolecular formation of the 3-sila-1,2,4-trioxepane is favored over the intermolecular reaction, which forms the triethylsilyl peroxide instead. Formation of the 3-sila-1,2,4-trioxepane does not involve a silane-exchange pathway, but, an intramolecular transmetalation reaction cannot be ruled out. More sterically hindered alkenes yielded less of the diisopropylsilylene acetal. Removal of the diisopropylsilylene acetal group was achieved using tetrabutylammonium fluoride, which yields the corresponding diol.
EXPERIMENTAL SECTION
General Information.
All 1H and 13C NMR spectra were recorded at ambient temperature at 400, 500, and 600 MHz, or 100, 125, and 150 MHz, respectively. The data were reported as follows: chemical shifts reported in parts per million from residual solvent peaks (1H NMR CDCl3 δ 7.26, C6D6 δ 7.16; 13C NMR CDCl3 δ 77.23, C6D6 δ 128.4) on the δ scale, multiplicity (s, singlet; br, broad; d, doublet; t, triplet; q, quartet; quint, quintet; dd, doublet of doublets; m, multiplet), coupling constants (hertz), and integration. The multiplicity of carbon peaks was determined using HSQC or DEPT experiments. Infrared (IR) spectra were recorded using attenuated total reflectance (ATR). High-resolution mass spectra (HRMS) were recorded using a time-of-flight spectrometer with atmospheric-pressure chemical ionization (APCI) or electrospray ionization (ESI) ionization sources. Liquid chromatography was performed using forced flow (flash chromatography) of the indicated solvent system on silica gel (SiO2) 60 Å (230–400 mesh) unless noted, otherwise, aluminium oxide (Al2O3), neutral, Brockmann I, particle size 50–200 μm, 60 Å was used. THF and Et2O were dried and degassed using a solvent purification system before being used. All reactions were performed under a nitrogen atmosphere in glassware that had been flame-dried under vacuum unless otherwise stated. All reactions that were performed under an oxygen atmosphere used glassware that was not flame-dried. Unless otherwise noted, all reagents were commercially available. Prenylmagnesium chloride was prepared by known methods.42 Co(acac)2 was prepared by known methods.43 The following alcohols were prepared by known methods: 2-phenylpent-4-en-2-ol (7),44 1-(4-methoxyphenyl)but-3-en-1-ol,45 2-(4-methoxyphenyl)hex-5-en-2-ol,46 trans-2-vinylcyclohexan-1-ol,47 and (E)-3-methyloct-2-en-4-ol.48 Diisopropylsilyl ether 8 was prepared by known methods.49
Synthesis of Bis(2,2,6,6-tetramethyl-3,5-heptanedionato)cobalt(II) (Co(thd)2).
The synthesis of Co(thd)2 was performed using a modified reported procedure.34 Ammonium hydroxide (50% v/v aqueous solution, 0.63 mL, 8.4 mmol) was added dropwise to a solution of cobalt(II) chloride hexahydrate (1.00 g, 4.20 mmol) and 2,2,6,6-tetramethyl-3,5-heptanedione (1.74 mL, 8.41 mmol) in 50% aqueous ethanol (12 mL). The resulting reaction mixture was stirred for 1 h. Distilled H2O was added (23 mL) and, after 1 h, the resulting reaction mixture was poured through Büchner funnel and the filter cake washed with distilled H2O (3 × 40 mL). The resulting solid was spread onto a watch glass and allowed to dry at ambient temperature for 18 h. Co(thd)2 was isolated as a pink solid (1.18 g, 66%) and used without further purification.
Synthesis of Alcohols.
2-(4-Methoxyphenyl)pent-4-en-2-ol.
To a solution of ketone 11 (0.500 g, 3.33 mmol) in THF (5.0 mL) at 0 °C was added a solution of allylmagnesium chloride in THF (2.0 M, 1.8 mL, 3.7 mmol) dropwise. The resulting reaction mixture was allowed to warm to room temperature over 30 min. Saturated aqueous NH4Cl (5.0 mL) was added and the mixture was extracted with diethyl ether (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. 2-(4-Methoxyphenyl)pent-4-en-2-ol was isolated as a colorless oil (0.607 g, 95%) and used without further purification. The spectroscopic data are consistent with the data reported:50 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.8, 2H), 6.88 (d, J = 8.7, 2H), 5.64 (m, 1H), 5.13 (m, 1H), 5.10 (s, 1H), 3.80 (s, 3H), 2.66 (dd, J = 13.5, 6.7, 1H), 2.49 (dd, J = 13.7, 8.3, 1H), 2.04 (s, 1H), 1.53 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.5, 140.1, 134.1, 126.2, 119.5, 113.6, 73.5, 55.4, 48.7, 30.1.
2-Cyclohexylpent-4-en-2-ol.
To a solution of acetylcyclohexane (0.436 mL, 3.17 mmol) in THF (4.7 mL) at 0 °C was added a solution of allylmagnesium chloride in THF (2.0 M, 1.7 mL, 3.5 mmol) dropwise. The resulting reaction mixture was allowed to warm to room temperature over 30 min. Saturated aqueous NH4Cl (5.0 mL) was added and the mixture was extracted with diethyl ether (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes:EtOAc = 90:10) afforded 2-cyclohexylpent-4-en-2-ol (0.389 g, 73%) as a yellow oil. The spectroscopic data are consistent with the data reported:50 1H NMR (400 MHz, CDCl3) δ 5.89 (m, 1H), 5.18–5.07 (m, 2H), 2.23 (m, 2H), 1.87–1.64 (m, 5H), 1.39 (s, 1H), 1.36–1.13 (m, 4H), 1.09 (s, 3H), 1.06–0.93 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ 134.4, 118.8, 74.1, 47.7, 44.5, 27.8, 27.1, 26.97, 26.93, 26.8, 23.9.
2-(4-Methoxyphenyl)-3,3-dimethylpent-4-en-2-ol.
To a solution of ketone 11 (0.300 g, 2.00 mmol) in THF (6.1 mL) at −78 °C was added a solution of prenylmagnesium chloride in THF (0.4 M, 5.5 mL, 2.2 mmol) dropwise. The resulting reaction mixture was allowed to warm to room temperature over 40 min. Saturated aqueous NH4Cl (6.0 mL) was added and the mixture was extracted with diethyl ether (3 × 6 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes:EtOAc = 90:10) afforded 2-(4-methoxyphenyl)-3,3-dimethylpent-4-en-2-ol (0.324 g, 74%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.8, 2H), 6.84 (d, J = 8.8, 2H), 5.96 (dd, J = 17.6, 10.9, 1H), 5.13–5.00 (m, 2H), 3.81 (s, 3H), 1.88 (s, 1H), 1.56 (s, 3H), 1.00 (s, 3H), 0.98 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.3 (C), 145.5 (CH), 137.8 (C), 128.4 (CH), 113.9 (CH2), 112.6 (CH), 77.5 (C), 55.4 (CH3), 44.7 (C), 25.7 (CH3), 22.9 (CH3), 22.6 (CH3); IR (ATR) 3505, 2977, 1509, 1246, 1175, 1033, 830 cm−1; HRMS (ESI) m/z calcd for C14H20NaO2 (M + Na)+ 243.1356, found 243.1351.
2-(4-Methoxyphenyl)-4-methylpent-4-en-2-ol.
To a solution of ketone 11 (0.300 g, 2.00 mmol) in THF (6.0 mL) at 0 °C was added a solution of 2-methylallylmagnesium chloride in THF (0.5 M, 4.4 mL, 2.2 mmol) dropwise. The resulting reaction mixture was allowed to warm to room temperature over 1 h. Saturated aqueous NH4Cl (6.0 mL) was added and the mixture was extracted with diethyl ether (3 × 6 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes:EtOAc = 80:20) afforded 2-(4-methoxyphenyl)-4-methylpent-4-en-2-ol (0.270 g, 66%) as a colorless oil. The spectroscopic data are consistent with the data reported:51 1H NMR (400 MHz, CDCl3) δ 7.36 (d, J = 8.8, 2H), 6.86 (d, J = 8.8, 2H), 4.89 (m, 1H), 4.74 (m 1H), 3.80 (s, 3H), 2.61 (d, J = 13.3, 1H), 2.49 (d, J = 13.4, 1H), 2.29 (s, 1H), 1.54 (s, 3H), 1.41 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.4, 142.9, 140.6, 126.1, 115.7, 113.5, 73.1, 55.4, 52.3, 30.9, 24.5.
Representative Procedure for the Silylation of Alcohols.
Diisopropyl((2-(4-methoxyphenyl)pent-4-en-2-yl)oxy)silane (1).
To solution of 2-(4-methoxyphenyl)pent-4-en-2-ol (0.500 g, 2.60 mmol) and 1H-imidazole (0.354 g, 5.20 mmol) in THF (7.9 mL) was added diisopropylchlorosilane (0.488 mL, 2.86 mmol) dropwise. After 20 min, the reaction mixture was poured through a glass frit and the filter cake was washed with hexanes (3 × 8 mL). The filtrate was concentrated in vacuo and purification by flash chromatography (hexanes:EtOAc = 95:5) afforded diisopropylsilyl ether 1 (0.633 g, 79%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.34 (d, J = 8.9, 2H), 6.85 (d, J = 8.9, 2H), 5.62 (m, 1H), 4.97 (s, 1H), 4.94 (m, 1H), 4.34 (s, 1H), 3.81 (s, 3H), 2.56 (m, 2H), 1.62 (s, 3H), 1.08–0.88 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.3 (C), 140.1 (C), 134.9 (CH), 126.8 (CH), 117.5 (CH2), 113.2 (CH), 76.5 (C), 55.4 (CH3), 50.4 (CH2), 28.2 (CH3), 18.1 (CH3), 17.9 (CH3), 17.83 (CH3), 17.77 (CH3), 13.6 (CH), 13.3 (CH); IR (ATR) 2941, 2864, 2097, 1512, 1248, 1001, 829, 805 cm−1; Anal. Calcd. for C18H30O2Si: C, 70.53; H, 9.87. Found: C, 70.83; H, 10.08.
Diisopropyl((2-phenylpent-4-en-2-yl)oxy)silane (5).
Diisopropylsilyl ether 5 was prepared using the representative procedure for the silylation of alcohols using alcohol 7 (0.300 g, 1.85 mmol), 1H-imidazole (0.252 g, 3.70 mmol), and diisopropylchlorosilane (0.379 mL, 2.22 mmol) in THF (5.6 mL). The reaction time was 45 min. Purification by flash chromatography (hexanes:EtOAc = 95:5) afforded diisopropylsilyl ether 5 (0.273 g, 53%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 7.3, 2H), 7.22 (t, J = 7.3, 2H), 7.12 (t, J = 7.3, 1H), 5.53 (m, 1H), 4.89–4.82 (m, 2H), 4.29 (s, 1H), 2.49 (m, 2H), 1.56 (s, 3H), 1.02–0.83 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 147.9 (C), 134.7 (CH), 127.9 (CH), 126.7 (CH), 125.6 (CH), 117.6 (CH2), 76.9 (C), 50.3 (CH2), 28.3 (CH3), 18.2 (CH3), 17.9 (CH3), 17.84 (CH3), 17.76 (CH3), 13.6 (CH), 13.3 (CH); IR (ATR) 2942, 2864, 2098, 1462, 1071, 999, 698 cm−1; HRMS (ESI) m/z calcd for C17H26NaSi (M + Na – H2O)+ 281.1696, found 281.1707.
Diisopropyl((1-(4-methoxyphenyl)but-3-en-1-yl)oxy)silane (12).
Diisopropylsilyl ether 12 was prepared using the representative procedure for the silylation of alcohols using alcohol 1-(4-methoxyphenyl)but-3-en-1-ol (0.500 g, 2.81 mmol), 1H-imidazole (0.382 g, 5.61 mmol), and diisopropylchlorosilane (0.526 mL, 3.09 mmol) in THF (8.5 mL). The reaction time was 35 min. Purification by flash chromatography (hexanes:EtOAc = 95:5) afforded diisopropylsilyl ether 12 (0.525 g, 64%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.22 (d, J = 8.8, 2H), 6.85 (d, J = 8.8, 2H), 5.74 (m, 1H), 5.04–4.97 (m, 2H), 4.67 (t, J = 6.3, 1H), 4.14 (s, 1H), 3.80 (s, 3H), 2.53 (m, 1H), 2.42 (m, 1H), 1.07–0.87 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.9 (C), 136.6 (C), 135.2 (CH), 127.5 (CH), 117.2 (CH2), 113.6 (CH), 76.9 (CH), 55.4 (CH3), 45.1 (CH2), 17.8 (CH3), 17.7 (CH3), 17.6 (CH3), 17.4 (CH3), 12.8 (CH), 12.7 (CH); IR (ATR) 2941, 2864, 2091, 1512, 1246, 1000, 830, 798 cm−1; HRMS (ESI) m/z calcd for C17H27O2Si (M + H − H2O)+ 291.1775, found 291.1771. Anal. Calcd. for C17H28O2Si: C, 69.81; H, 9.65. Found: C, 69.83; H, 9.71.
((2-Cyclohexylpent-4-en-2-yl)oxy)diisopropylsilane (13).
Diisopropylsilyl ether 13 was prepared using the representative procedure for the silylation of alcohols using 2-cyclohexylpent-4-en-2-ol (0.389 g, 2.31 mmol), 1H-imidazole (0.314 g, 4.62 mmol), and diisopropylchlorosilane (0.434 mL, 2.54 mmol) in THF (7.0 mL). The reaction time was 1 h. Purification by flash chromatography (hexanes) afforded diisopropylsilyl ether 13 (0.505 g, 77%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.84 (m, 1H), 5.04 (m, 2H), 4.35 (s, 1H), 2.33 (dd, J = 13.9, 7.3, 1H), 2.25 (dd, J = 13.9, 7.4, 1H), 1.83–1.63 (m, 5H), 1.32 (m, 1H), 1.17 (s, 3H), 1.17–0.85 (m, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ 135.3 (CH), 117.1 (CH2), 77.4 (C), 47.2 (CH2), 44.9 (CH), 27.3 (CH3), 27.2 (CH3), 27.03 (CH3), 27.02 (CH3), 26.9 (CH3), 24.8 (CH2), 18.2 (CH2), 18.1 (CH2), 17.84 (CH2), 17.82 (CH2), 13.72 (CH), 13.67 (CH); IR (ATR) 2926, 2863, 2091, 1462, 1152, 1000, 814 cm−1; HRMS (ESI) m/z calcd for C17H34KOSi (M + K)+ 321.2011, found 321.2020. Anal. Calcd. for C17H34OSi: C, 72.27; H, 12.13. Found: C, 72.55; H, 12.33.
Diisopropyl((2-(4-methoxyphenyl)-4-methylpent-4-en-2-yl)oxy)silane (14).
Diisopropylsilyl ether 14 was prepared using the representative procedure for the silylation of alcohols using 2-(4-methoxyphenyl)-4-methylpent-4-en-2-ol (0.268 g, 1.30 mmol), 1H-imidazole (0.177 g, 2.60 mmol), and diisopropylchlorosilane (0.244 mL, 1.43 mmol) in THF (3.9 mL). The reaction time was 35 min. Purification by flash chromatography (hexanes:EtOAc = 97:3) afforded diisopropylsilyl ether 14 (0.334 g, 80%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.35 (d, J = 8.9, 2H), 6.83 (d, J = 8.9, 2H), 4.74 (m, 1H), 4.56 (m, 1H), 4.27 (s, 1H), 3.80 (s, 3H), 2.56 (d, J = 13.4, 1H), 2.46 (d, J = 13.4, 1H), 1.67 (s, 3H), 1.45 (s, 3H), 1.09–0.86 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.4 (C), 142.8 (C), 140.3 (C), 127.0 (CH), 114.9 (CH2), 113.1 (CH), 76.9 (C), 55.4 (CH3), 54.1 (CH2), 28.0 (CH3), 24.5 (CH3), 18.1 (CH3), 17.93 (CH3), 17.87 (CH3), 17.8 (CH3), 13.5 (CH), 13.4 (CH); IR (ATR) 2941, 2864, 2099, 1611, 1511, 1248, 1097, 829 cm−1. Anal. Calcd. for C19H32O2Si: C, 71.19; H, 10.06. Found: C, 71.48; H, 10.32.
Diisopropyl((2-(4-methoxyphenyl)-3,3-dimethylpent-4-en-2-yl)oxy)silane (15).
Diisopropylsilyl ether 15 was prepared using the representative procedure for the silylation of alcohols using 2-(4-methoxyphenyl)-3,3-dimethylpent-4-en-2-ol (0.320 g, 1.45 mmol), 1H-imidazole (0.396 g, 5.81 mmol), and diisopropylchlorosilane (0.546 mL, 3.20 mmol) in THF (4.4 mL). The reaction time was 18 h. Purification by flash chromatography (hexanes:EtOAc = 98:2) afforded diisopropylsilyl ether 15 (0.345 g, 71%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.8, 2H), 6.79 (d, J = 8.8, 2H), 5.95 (dd, J = 17.8, 10.8, 1H), 4.92 (m, 2H), 4.26 (s, 1H), 3.81 (s, 3H), 1.63 (s, 3H), 1.15 (m, 6H), 1.01–0.84 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.2 (C), 146.0 (CH), 138.0 (C), 129.0 (CH), 112.2 (CH2), 112.1 (CH), 80.8 (C), 55.3 (CH3), 45.9 (C), 24.1 (CH3), 22.9 (CH3), 22.6 (CH3), 18.4 (CH3), 18.1 (CH3), 18.0 (CH3), 17.9 (CH3), 13.8 (CH), 13.6 (CH); IR (ATR) 2942, 2864, 2098, 1510, 1248, 1097, 831, 807 cm−1. Anal. Calcd. for C20H34O2Si: C, 71.80; H, 10.24. Found: C, 71.54; H, 10.09.
Diisopropyl(((1R*,2S*)-2-vinylcyclohexyl)oxy)silane (22).
Diisopropylsilyl ether 22 was prepared using the representative procedure for the silylation of alcohols using trans-2-vinylcyclohexan-1-ol (0.134 g, 1.06 mmol), 1H-imidazole (0.145 g, 2.12 mmol), and diisopropylchlorosilane (0.199 mL, 1.17 mmol) in THF (3.2 mL). The reaction time was 20 min. Purification by flash chromatography (hexanes:EtOAc = 99:1 → hexanes:EtOAc = 97:3) afforded diisopropylsilyl ether 22 (0.115 g, 45%) as a colorless oil: 1H NMR (500 MHz, CDCl3) δ 5.87 (m, 1H), 5.00 (m, 2H), 4.18 (s, 1H), 3.34 (m, 1H), 1.97 (m, 2H), 1.74 (m, 2H), 1.63 (m, 1H), 1.33–1.84 (m, 4H), 1.03–0.94 (m, 14H); 13C{1H} NMR (125 MHz, CDCl3) δ 142.0 (CH), 114.4 (CH2), 77.2 (CH), 49.9 (CH), 35.3 (CH2), 30.9 (CH2), 25.2 (CH2), 24.9 (CH2), 17.81 (CH3), 17.76 (CH3), 17.73 (CH3), 17.66 (CH3), 12.9 (CH); IR (ATR) 2932, 2863, 2090, 1462, 1085, 1001, 907, 733 cm−1; HRMS (ESI) m/z calcd for C14H27Si (M + H − H2O)+ 223.1877, found 223.1880. Anal. Calcd. for C14H28OSi: C, 69.93; H, 11.74. Found: C, 69.67; H, 11.92.
(E)-Diisopropyl((3-methyloct-2-en-4-yl)oxy)silane (25).
Diisopropylsilyl ether 25 was prepared using the representative procedure for the silylation of alcohols using (E)-3-methyloct-2-en-4-ol (0.394 g, 2.77 mmol), 1H-imidazole (0.378 g, 5.55 mmol), and diisopropylchlorosilane (0.521 mL, 3.05 mmol) in THF (8.4 mL). The reaction time was 50 min. Purification by flash chromatography (pentanes) afforded diisopropylsilyl ether 25 (0.487 g, 68%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.37 (q, J = 6.8, 1H), 4.09 (s, 1H), 3.95 (t, J = 6.8, 1H), 1.59 (d, J = 6.8, 3H), 1.57–1.43 (m, 5H), 1.34–1.12 (m, 4H), 1.05–0.93 (m, 14H), 0.88 (t, J = 7.2, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 137.7 (C), 120.5 (CH), 81.3 (CH), 35.7 (CH2), 28.2 (CH2), 22.9 (CH2), 17.9 (CH3), 17.70 (CH3), 17.69 (CH3), 17.5 (CH3), 14.3 (CH3), 13.1 (CH3), 12.78 (CH), 12.77 (CH), 10.8 (CH3); 29Si NMR (79.5 MHz, CDCl3) δ 12.0; IR (ATR) 2938, 2867, 1467, 903, 725 cm−1. Anal. Calcd. for C15H32OSi: C, 70.24; H, 12.58. Found: C, 69.97; H, 12.40.
Diisopropyl((2-(4-methoxyphenyl)hex-5-en-2-yl)oxy)silane (28).
Diisopropylsilyl ether 28 was prepared using the representative procedure for the silylation of alcohols using 2-(4-methoxyphenyl)hex-5-en-2-ol (0.230 g, 1.11 mmol), 1H-imidazole (0.152 g, 2.22 mmol), and diisopropylchlorosilane (0.209 mL, 1.22 mmol) in THF (3.4 mL). The reaction time was 40 min. Purification by flash chromatography (hexanes → hexanes:EtOAc = 99:1) afforded diisopropylsilyl ether 28 (0.225 g, 63%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.33 (d, J = 8.8, 2H), 6.85 (d, J = 8.8, 2H), 5.73 (m, 1H), 4.90 (m, 2H), 4.63 (s, 1H), 3.81 (s, 3H), 2.01 (m, 1H), 1.86 (m, 2H), 1.77 (m, 1H), 1.63 (s, 3H), 1.12–0.92 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.0 (C), 140.0 (C), 139.1 (CH), 126.3 (CH), 113.9 (CH2), 113.1 (CH), 76.7 (C), 55.2 (CH3), 44.4 (CH2), 29.4 (CH3), 28.6 (CH2), 18.0 (CH3), 17.7 (CH3), 17.6 (CH3), 13.4 (CH), 13.2 (CH); IR (ATR) 2942, 2097, 1611, 1509, 1462, 1247, 999, 907 cm−1; HRMS (ESI) m/z calcd for C19H33O2Si (M + H)+ 321.2244, found 321.2245.
Representative Procedure for the Co-Catalyzed Silylperoxidation.
3,3-Diisopropyl-5-(4-methoxyphenyl)-5,7-dimethyl-1,2,4,3-trioxasilepane (2).
A flask containing DCE (1,2-dichloroethane, 10 mL) was sparged with oxygen for 15 min. To a separate flask was added Co(thd)2 (0.013 g, 0.032 mmol) followed by diisopropylsilyl ether 1 (0.097 g, 0.316 mmol). The oxygenated DCE (2.9 mL) was added to the reaction flask and the reaction vessel was sparged with oxygen for 2 min. A solution of tert-butyl hydroperoxide in CH2Cl2 (1.0 M, 0.016 mL, 0.016 mmol) was added followed by triethylsilane (0.051 mL, 0.316 mmol) and the reaction mixture was stirred at room temperature under a balloon of oxygen for 50 min. The reaction mixture was filtered through a 6-cm plug of silica, eluted with CH2Cl2 (45 mL), and concentrated in vacuo. Purification by flash chromatography using Brockmann grade III neutral alumina (pentanes:benzene = 80:20) afforded 3-sila-1,2,4-trioxepane 2 (0.046 g, 38%) as a colorless oil. Characterization was performed on an inseparable 67:33 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 7.41 (d, J = 8.9, 1.2H), 7.36 (d, J = 8.9, 2H), 6.88 (m, 3.2H), 4.60 (m, 0.5H), 3.96 (m, 1H), 3.81 (s, 1.8H), 3.80 (s, 3H), 2.50 (dd, J = 15.6, 10.4, 1H), 2.15 (dd, J = 15.4, 10.6, 0.6H), 2.09 (dd, J = 15.6, 3.4, 1H), 1.96 (dd, J = 15.4, 3.1, 0.6H), 1.72 (s, 1.8H), 1.54 (s, 3H), 1.28–1.25 (m, 6H), 1.20–1.17 (m, 3.4H), 1.14–1.09 (m, 18H); 13C{1H} NMR (150 MHz, CDCl3) δ 158.33 (C), 158.27 (C), 142.6 (C), 139.4 (C), 126.2 (CH), 125.7 (CH), 113.6 (CH), 113.5 (CH), 79.4 (CH), 79.1 (CH), 78.4 (C), 55.49 (CH3), 55.45 (CH3), 46.7 (CH2), 34.9 (CH3), 28.3 (CH3), 19.92 (CH3), 19.88 (CH3), 18.03 (CH3), 17.97 (CH3), 17.87 (CH3), 17.71 (CH3), 17.66 (CH3), 17.59 (CH3), 17.58 (CH3), 17.51 (CH3), 13.4 (CH), 13.2 (CH), 12.73 (CH), 12.68 (CH); IR (ATR) 2945, 2867, 1611, 1248, 1033, 907, 731 cm−1; HRMS (ESI) m/z calcd for C18H29O3Si (M + H − H2O)+ 321.1880, found 321.1891. Anal. Calcd. for C18H30O4Si: C, 63.87; H, 8.93. Found: C, 63.95; H, 8.79.
3,3-Diisopropyl-5,7-dimethyl-5-phenyl-1,2,4,3-trioxasilepane (6).
3-Sila-1,2,4-trioxepane 6 was prepared using the representative procedure for the Co-catalyzed silylperoxidation using diisopropylsilyl ether 5 (0.072 g, 0.259 mmol), Co(acac)2 (0.013 g, 0.052 mmol), tert-butyl hydroperoxide (0.013 mL, 0.013 mmol), and triethylsilane (0.082 mL, 0.518 mmol) in DCE (2.4 mL). The reaction time was 1.5 h. Purification by flash chromatography (pentanes:benzene = 80:20) afforded 3-sila-1,2,4-trioxepane 6 (0.017 g, 21%) as a colorless oil. Characterization was performed on an inseparable 59:41 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 7.54–7.41 (m, 3.5H), 7.34 (m, 3.4H), 7.23 (m, 1.5H), 4.63 (m, 0.7H), 3.94 (m, 1H), 2.53 (m, 1H), 2.15 (m, 1.6H), 1.99 (m, 0.7H), 1.75 (s, 1.8H), 1.57 (s, 3H), 1.28 (m, 6H), 1.22–1.08 (m, 22.9H); 13C{1H} NMR (100 MHz, CDCl3) δ 150.2 (C), 147.3 (C), 128.32 (CH), 128.27 (CH), 126.7 (CH), 126.6 (CH), 125.0 (CH), 124.5 (CH), 79.4 (CH), 79.1 (CH), 78.6 (C), 46.58 (CH2), 34.9 (CH3), 28.2 (CH3), 19.9 (CH3), 18.03 (CH3), 17.98 (CH3), 17.87 (CH3), 17.7 (CH3), 17.6 (CH3), 17.5 (CH3), 13.4 (CH), 13.2 (CH), 12.7 (CH); IR (ATR) 2944, 2867, 1464, 1121, 1035, 884, 699 cm−1; HRMS (APCI) m/z calcd for C16H25O2Si (M + H − H2O)+ 277.1618, found 277.1631.
3,3-Diethyl-10-isopropyl-8-(4-methoxyphenyl)-6,8,11-trimethyl-4,5,9-trioxa-3,10-disiladodecane (4).
Linear silyl peroxide 4 was prepared using the representative procedure for the Co-catalyzed silylperoxidation using diisopropylsilyl ether 4 (0.200 g, 0.653 mmol), Co(thd)2 (0.028 g, 0.065 mmol), tert-butyl hydroperoxide (0.033 mL, 0.033 mmol), and triethylsilane (1.04 mL, 6.53 mmol) in DCE (3.0 mL). The reaction time was 2 h. Purification by flash chromatography using Brockmann grade III neutral alumina (pentanes:benzene = 80:20), then silica gel (hexanes:EtOAc = 97:3) yielded linear silyl peroxide 4 as a colorless oil (0.086 g, 29%). Characterization was performed on an inseparable 67:33 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 7.34 (d, J = 8.8, 3.5H), 6.84 (m, 3.5H), 4.34 (s, 1H), 4.30 (0.5H), 4.19 (m, 0.5H), 3.80 (s, 4.5H), 3.75 (m, 1H), 2.26 (dd, J = 14.3, 5.1, 1H), 2.17 (dd, J = 14.4, 4.4, 0.5H), 1.86 (dd, J = 14.3, 5.7, 1H), 1.82 (dd, J = 14.3, 6.2, 0.5H), 1.69 (s, 1.5H), 1.68 (s, 3H), 1.10–1.04 (m, 14.3H), 1.00–0.90 (m, 30.5H), 0.63 (m, 3.6H), 0.57 (m, 6H); 13C{1H} NMR (150 MHz, CDCl3) δ 158.42 (C), 158.40 (C), 140.6 (C), 139.5 (C), 126.9 (CH), 126.7 (CH), 113.3 (CH), 78.9 (CH), 78.6 (CH), 76.5 (C), 75.9 (C), 55.4 (CH3), 50.6 (CH2), 50.1 (CH2), 29.8 (CH3), 28.6 (CH3), 20.19 (CH3), 20.16 (CH3), 18.1 (CH3), 17.94 (CH3), 17.90 (CH3), 17.86 (CH3), 13.54 (CH3), 13.50 (CH3), 13.4 (CH3), 13.3 (CH3), 7.0 (CH3), 6.9 (CH3), 4.0 (CH2), 3.9 (CH2); IR (ATR) 2954, 2099, 1510, 1248, 906, 729 cm−1; HRMS (ESI) m/z calcd for C24H45O3Si2 (M + H − H2O)+ 437.2902, found 437.2907.
4-((Diisopropyl(1-phenylethoxy)silyl)peroxy)-2-phenylpentan-2-ol (9 and 10).
Linear silyl peroxides 9 and 10 were prepared using the representative procedure for the Co-catalyzed silylperoxidation using alcohol 7 (0.050 g, 0.308 mmol), Co(thd)2 (0.013 g, 0.031 mmol), tert-butyl hydroperoxide (0.015 mL, 0.015 mmol), and diisopropylsilane 8 (0.146 g, 0.616 mmol) in DCE (2.8 mL). The reaction time was 19 h. Purification by flash chromatography using Brockmann grade III neutral alumina (hexanes:EtOAc = 97:3) yielded linear silyl peroxide 9 a colorless oil (0.015 g, 11%) and linear silyl peroxide 10 as a colorless oil (0.009 g, 7%). Linear Silyl Peroxide 9: Characterization was performed on an inseparable 53:47 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 7.41 (m, 4H), 7.36–7.27 (m, 12H), 7.25–7.20 (m, 4H), 5.05 (q, J = 6.4, 1H), 5.02 (q, J = 6.4, 0.9H), 4.17 (s, 0.9H), 4.13 (s, 1H), 3.93 (m, 1H), 3.86 (m, 0.8H), 2.21–2.08 (m, 4H), 1.48 (m, 6H), 1.44 (m, 6H), 1.14–0.88 (m, 36.4H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.09 (C), 148.07 (C), 146.41 (C), 146.36 (C), 128.4 (CH), 128.29 (CH), 128.28 (CH), 127.04 (CH), 127.01 (CH), 126.5 (CH), 125.4 (CH), 125.3 (CH), 125.22 (CH), 125.21 (CH), 80.2 (CH), 80.1 (CH), 73.89 (C), 73.86 (C), 71.5 (CH), 71.4 (CH), 49.4 (CH2), 49.3 (CH2), 32.6 (CH3), 27.43 (CH3), 27.42 (CH3), 20.11 (CH3), 20.06 (CH3), 17.61 (CH3), 17.56 (CH3), 17.54 (CH3), 17.51 (CH3), 17.50 (CH3), 17.4 (CH3), 11.90 (CH), 11.85 (CH), 11.75 (CH); IR (ATR) 3466, 2868, 1493, 906, 699 cm−1; HRMS (ESI) m/z calcd for C25H40NO3Si (M + NH4 − H2O)+ 430.2772, found 430.2774. Linear Silyl Peroxide 10: Characterization was performed on a 50:50 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 7.56 (m, 3.8H), 7.39–7.29 (m, 11.6H), 7.22 (m, 3.8H), 5.11 (q, J = 6.3, 2H), 4.29 (m, 2H), 3.28 (s, 0.9H), 3.25 (s, 0.9H), 2.17 (m, 2H), 1.86 (m, 1.9H), 1.58 (s, 5.4H), 1.48 (m, 5.4H), 1.16–0.92 (m, 32.4H); 13C{1H} NMR (150 MHz, CDCl3) δ 148.8 (C), 146.5 (C), 128.3 (CH), 127.1 (CH), 126.7 (CH), 125.4 (CH), 124.9 (CH), 79.7 (CH), 73.4 (C), 71.4 (CH), 49.6 (CH2), 29.8 (CH3), 27.5 (CH3), 20.1 (CH3), 17.6 (CH3), 17.4 (CH3), 11.9 (CH), 11.8 (CH); IR (ATR) 3476, 2867, 1447, 1371, 1095, 907, 731, 699 cm−1; HRMS (ESI) m/z calcd for C25H38NaO4Si (M + Na)+ 453.2432, found 453.2433.
3,3-Diethyl-11-isopropyl-9-(4-methoxyphenyl)-6,9,12-trimethyl-4,5,10-trioxa-3,11-disilatridecane (29).
Linear silyl peroxide 29 was prepared using the representative procedure for the Co-catalyzed silylperoxidation using diisopropylsilyl ether 28 (0.040 g, 0.155 mmol), Co(thd)2 (0.007 g, 0.016 mmol), tert-butyl hydroperoxide (0.008 mL, 0.008 mmol), and triethylsilane (0.025 mL, 0.156 mmol) in DCE (1.4 mL). The reaction time was 20 min. Purification by flash chromatography using Brockmann grade III neutral alumina (pentane → hexanes:EtOAc = 98:2) yielded linear silyl peroxide 29 as a colorless oil (0.028 g, 48%). Characterization was performed on an inseparable 50:50 mixture of diastereomers: 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 8.9, 4H), 6.84 (d, J = 8.9, 4H), 4.36 (s, 1.9H), 3.89 (m, 2H), 3.80 (s, 6H), 1.90 (m, 2H), 1.78 (m, 2H), 1.61 (s, 6H), 1.35 (m, 2H), 1.13–0.93 (m, 54H), 0.66 (q, J = 7.9, 12H); 13C{1H} NMR (125 MHz, CDCl3) δ 158.2 (C), 140.2 (C), 140.1 (C), 126.57 (CH), 126.56 (CH), 113.1 (CH), 81.8 (CH), 81.7 (CH), 76.9 (C), 55.4 (CH3), 41.2 (CH2), 41.0 (CH2), 29.9 (CH3), 29.8 (CH3), 29.5 (CH2), 29.3 (CH2), 18.7 (CH3), 18.6 (CH3), 18.17 (CH3), 18.16 (CH3), 18.0 (CH3), 17.9 (CH3), 17.83 (CH3), 17.80 (CH3), 13.7 (CH), 13.6 (CH), 13.37 (CH), 13.35 (CH), 6.9 (CH3), 4.0 (CH2); IR (ATR) 2953, 2097, 1510, 1246, 906, 729 cm−1; HRMS (ESI) m/z calcd for C24H45O3Si2 (M + H − H2O)+ 437.2902, found 437.2907.
Representative Procedure for the Co-Catalyzed Synthesis of Diisopropylsilylene Acetals.
2,2-Diisopropyl-4-(4-methoxyphenyl)-4,6-dimethyl-1,3,2-dioxasilinane (3).
A flask containing DCE (1,2-dichloroethane, 10 mL) was sparged with oxygen for 15 min. To a separate flask was added Co(thd)2 (0.043 g, 0.100 mmol) followed by diisopropylsilyl ether 1 (0.306 g, 1.00 mmol). The oxygenated DCE (9.1 mL) was added to the reaction flask and the reaction vessel was sparged with oxygen for 2 min. A solution of tert-butyl hydroperoxide in CH2Cl2 (1.0 M, 0.050 mL, 0.050 mmol) was added followed by triethylsilane (0.160 mL, 1.00 mmol) and the reaction mixture was stirred at room temperature under a balloon of oxygen for 30 min. The reaction mixture was filtered through a 6-cm plug of silica, eluted with CH2Cl2 (45 mL), and concentrated in vacuo. To the crude reaction mixture was added toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction mixture was stirred at 40 °C for 3 h. Hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol) was added and the resulting reaction mixture was stirred at room temperature for 15 min. The reaction mixture was diluted with hexanes (9.1 mL) and poured through a glass frit. The filter cake was washed with hexanes (3 × 9 mL). The filtrate was dried over Na2SO4 and concentrated in vacuo. Purification by flash chromatography using Brockmann grade III neutral alumina (pentanes:benzene = 80:20) afforded diisopropylsilylene acetal 3 (0.192 g, 60%) as a colorless oil. Characterization was performed on an inseparable 51:49 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.8, 3.8H), 6.87 (d, J = 8.8, 3.8H), 4.44 (m, 1H), 3.92 (m, 1H), 3.83 (s, 2.5H), 3.80 (s, 3H), 2.30 (dd, J = 14.7, 1.2, 0.9H), 1.90 (dd, J = 14.4, 2.0, 1H), 1.87 (dd, J = 14.8, 10.9, 0.9H), 1.75 (dd, J = 14.6, 11.3, 1H), 1.59 (s, 3H), 1.46 (s, 2.5H), 1.24 (d, J = 6.1, 3H), 1.18 (d, J = 6.2, 2.7H), 1.11–0.91 (m, 27.5H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.3 (C), 158.2 (C), 142.9 (C), 140.0 (C), 126.6 (CH), 125.4 (CH), 113.52 (CH), 113.46 (CH), 76.6 (C), 75.3 (C), 66.3 (CH), 66.2 (CH), 55.5 (CH3), 55.4 (CH3), 50.0 (CH2), 48.9 (CH2), 35.7 (CH3), 29.9 (CH3), 25.0 (CH3), 24.8 (CH3), 17.6 (CH3), 17.4 (CH3), 17.32 (CH3), 17.26 (CH3), 17.23 (CH3), 17.18 (CH3), 17.12 (CH3), 17.11 (CH3), 14.2 (CH2), 13.9 (CH2), 13.41 (CH2), 13.38 (CH2); IR (ATR) 2943, 2865, 1611, 1511, 1248, 983 cm−1; HRMS (ESI) m/z calcd for C18H34NO3Si (M + NH4)+ 340.2302, found 340.2308. Anal. Calcd. for C18H30O3Si: C, 67.03; H, 9.38. Found: C, 67.33; H, 9.46.
Diisopropylsilylene Acetal 3 Prepared Using Catalytic Phenylsilane.
Diisopropylsilylene acetal 3 was prepared using the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 1 (0.165 g, 0.539 mmol), Co(thd)2 (0.034 g, 0.081 mmol), tert-butyl hydroperoxide (0.027 mL, 0.027 mmol), and a solution of phenylsilane in PhCF3 (0.20 M, 0.54 mL, 0.11 mmol) in DCE (4.9 mL). The reaction time was 15 min. Reduction of the peroxide unit was performed using toluene (1.0 mL) and triphenylphosphine (0.283 g, 1.08 mmol). The reaction time was 1 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 0.54 mL, 4.7 mmol). The reaction time was 15 min. Purification by flash chromatography using Brockmann grade III neutral alumina (hexanes → hexanes:EtOAc = 95:5) afforded diisopropylsilylene acetal 3 (0.126 g, 73%) as a colorless oil. Characterization was performed on an inseparable 50:50 mixture of diastereomers. The spectroscopic data are consistent with the data reported above for diisopropylsilylene acetal 3.
2,2-Diisopropyl-4-(4-methoxyphenyl)-6-methyl-1,3,2-dioxasilinane (16) and 1-(4-methoxyphenyl)butane-1,3-diol (20).
Diisopropylsilylene acetal 16 and 1,3-diol 20 were prepared using the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 12 (0.292 g, 1.00 mmol), Co(thd)2 (0.043 g, 0.100 mmol), tert-butyl hydroperoxide (0.050 mL, 0.050 mmol), and triethylsilane (0.160 mL, 1.00 mmol) in DCE (9.1 mL). The reaction mixture was heated to 40 °C for 1 h. Reduction of the peroxide unit was performed using toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction time was 2 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol). Purification by flash chromatography using Brockmann grade III neutral alumina (pentanes:benzene = 95:5 → hexanes:EtOAc 50:50 → hexanes:EtOAc 40:60) afforded diisopropylsilylene acetal 16 (0.047 g, 15%) as a colorless oil and 1,3-diol 20 (0.041 g, 21%) as a colorless oil. Diisopropylsilylene Acetal 16: 1H NMR (400 MHz, CDCl3) δ 7.29 (d, J = 8.7, 2H), 6.89 (d, J = 8.7, 2H), 5.07 (dd, J = 11.3, 2.1, 1H), 4.36 (m, 1H), 3.81 (s, 3H), 1.83 (m, 1H), 1.63 (m, 1H), 1.25 (d, J = 6.3, 3H), 1.13–1.03 (m, 14H); 13C{1H} NMR (100 MHz, CDCl3) δ 159.0 (C), 137.4 (C), 126.5 (CH), 113.9 (CH), 75.4 (CH), 70.3 (CH), 55.5 (CH3), 47.6 (CH2), 24.9 (CH3), 17.34 (CH3), 17.31 (CH3), 17.03 (CH3), 17.00 (CH3), 14.0 (CH), 12.4 (CH); IR (ATR) 2864, 1613, 1513, 1245, 1136, 980, 892 cm−1; HRMS (ESI) m/z calcd for C17H27O2Si (M + H − H2O)+ 291.1775, found 291.1771. 1,3-Diol 20: Characterization was performed on an inseparable 67:33 mixture of diastereomers: 1H NMR (500 MHz, CDCl3) δ 7.27 (m, 2.9H), 6.87 (m, 2.9H), 4.98 (m, 1H), 4.86 (0.5H), 4.07 (m, 1.5H), 3.79 (m, 4.3H), 3.31 (s, 0.5H), 3.28 (s, 0.5H), 3.06 (s, 1H), 2.58 (s, 1H), 1.91–1.69 (m, 3H), 1.21 (m, 4.4H); 13C{1H} NMR (125 MHz, CDCl3) δ 159.3 (C), 159.1 (C), 136.9 (C), 136.8 (C), 127.1 (CH), 127.0 (CH), 114.1 (CH), 114.0 (CH), 75.1 (CH), 71.6 (CH), 68.9 (CH), 65.6 (CH), 55.5 (CH3), 47.2 (CH2), 46.3 (CH2), 24.3 (CH3), 23.4 (CH3); IR (ATR) 3348, 2966, 1611, 1512, 1244, 904, 726, 648 cm−1; HRMS (ESI) m/z calcd for C11H16NaO3 (M + Na)+ 219.0992, found 219.1001.
4-Cyclohexyl-2,2-diisopropyl-4,6-dimethyl-1,3,2-dioxasilinane (17).
Diisopropylsilylene acetal 17 was prepared using the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 13 (0.282 g, 1.00 mmol), Co(acac)2 (0.103 g, 0.400 mmol), tert-butyl hydroperoxide (0.050 mL, 0.050 mmol), and triethylsilane (0.319 mL, 2.00 mmol) in DCE (9.1 mL). The reaction mixture was heated to 40 °C for 2 h. Reduction of the peroxide unit was performed using toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction time was 1 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol). Purification by flash chromatography (pentanes:benzene = 95:5 → pentanes:benzene = 80:20) afforded diisopropylsilylene acetal 17 (0.184 g, 62%) as a colorless oil. Characterization was performed on an inseparable 51:49 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 4.26 (m, 1.9H), 2.09 (m, 1H), 1.97–1.40 (m, 14.3H), 1.37–1.08 (m, 16.8H), 1.06 (s, 3H), 1.05–0.84 (m, 30.9H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.5 (C), 76.1 (C), 65.9 (CH), 65.3 (CH), 51.7 (CH), 46.4 (CH), 45.98 (CH), 45.97 (CH2), 29.3 (CH2), 27.8 (CH2), 27.2 (CH2), 27.1 (CH2), 27.02 (CH2), 26.96 (CH2), 26.91 (CH2), 26.89 (CH2), 26.88 (CH2), 25.3 (CH3), 25.23 (CH3), 25.16 (CH3), 23.3 (CH3), 17.5 (CH3), 17.4 (CH3), 17.33 (CH3), 17.28 (CH3), 17.25 (CH3), 17.0 (CH3), 14.0 (CH), 13.9 (CH), 13.5 (CH), 13.3 (CH); IR (ATR) 2927, 2864, 1464, 1148, 980, 882 cm−1; HRMS (ESI) m/z calcd for C17H33OSi (M + H − H2O)+ 281.2295, found 281.2303.
2,2-Diisopropyl-4-(4-methoxyphenyl)-4,6,6-trimethyl-1,3,2-dioxasilinane (18).
Diisopropylsilylene acetal 18 was prepared using the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 14 (0.320 g, 1.00 mmol), Co(acac)2 (0.103 g, 0.400 mmol), tert-butyl hydroperoxide (0.050 mL, 0.050 mmol), and triethylsilane (0.319 mL, 2.00 mmol) in DCE (9.1 mL). The reaction mixture was heated to 40 °C for 3 h. Reduction of the peroxide unit was performed using toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction time was 2.5 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol). Purification by flash chromatography (pentanes:benzene = 80:20) afforded diisopropylsilylene acetal 18 (0.130 g, 39%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.8, 2H), 6.85 (d, J = 8.8, 2H), 3.80 (s, 3H), 2.30 (d, J = 14.9, 1H), 2.12 (d, J = 14.9, 1H), 1.49 (s, 3H), 1.30 (s, 3H), 1.12–0.96 (m, 14H), 0.89 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.1 (C), 142.2 (C), 125.7 (CH), 113.4 (CH), 75.8 (C), 72.6 (C), 55.4 (CH3), 51.6 (CH2), 36.3 (CH3), 33.5 (CH3), 30.8 (CH3), 17.7 (CH3), 17.60 (CH3), 17.57 (CH3), 17.4 (CH3), 14.3 (CH), 14.2 (CH); IR (ATR) 2967, 2865, 1611, 1510, 1243, 1036, 992 cm−1; HRMS (APCI) m/z calcd for C19H32NaO3Si (M + Na)+ 359.2013, found 359.2017. Anal. Calcd. for C19H32O3Si: C, 67.81; H, 9.58. Found: C, 68.07; H, 9.72.
2,2-Diisopropyl-4-(4-methoxyphenyl)-4,5,5,6-tetramethyl-1,3,2-dioxasilinane (19).
Diisopropylsilylene acetal 19 was prepared using the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 15 (0.290 g, 0.867 mmol), Co(acac)2 (0.089 g, 0.347 mmol), tert-butyl hydroperoxide (0.043 mL, 0.043 mmol), and triethylsilane (0.277 mL, 1.73 mmol) in DCE (7.9 mL). The reaction mixture was heated to 40 °C for 5 h. Reduction of the peroxide unit was performed using toluene (1.7 mL) and triphenylphosphine (0.455 g, 1.73 mmol). The reaction time was 45 min. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 0.87 mL, 7.6 mmol). Purification by flash chromatography (pentanes:benzene = 80:20) afforded diisopropylsilylene acetal 19 (0.098 g, 32%) as a colorless oil. Characterization was performed on an inseparable 70:30 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 7.45–7.36 (m, 2.8H), 6.89–6.83 (m, 2.8H), 4.43 (q, J = 6.3, 1H), 4.11 (q, J = 6.7, 0.4H), 3.81 (m, 4.1H), 1.71 (s, 3H), 1.66 (s, 1.3H), 1.29–1.03 (m, 27H), 0.79 (s, 3H), 0.68 (s. 3H), 0.59 (s, 1.3H); 13C{1H} NMR (100 MHz, CDCl3) δ 158.24 (C), 158.16 (C), 139.7 (C), 138.5 (C), 128.7 (CH), 128.4 (CH), 112.51 (CH), 112.50 (CH), 83.3 (C), 82.5 (C), 73.6 (CH), 72.5 (CH), 55.40 (CH3), 55.39 (CH3), 43.7 (C), 43.6 (C), 27.4 (CH3), 25.9 (CH3), 24.8 (CH3), 22.4 (CH3), 19.5 (CH3), 19.1 (CH3), 18.2 (CH3), 18.1 (CH3), 18.0 (CH3), 17.9 (CH3), 17.8 (CH3), 17.73 (CH3), 17.68 (CH3), 17.66 (CH3), 16.1 (CH3), 15.1 (CH), 15.0 (CH), 14.7 (CH), 13.5 (CH); IR (ATR) 2944, 2865, 1610, 1510, 1245, 1118, 1036, 825, 684 cm−1. Anal. Calcd. for C20H34O3Si: C, 68.52; H, 9.78. Found: C, 68.80; H, 9.86.
2-Cyclohexylpentane-2,4-diol (21).
1,3-Diol 21 was prepared using a modification of the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 13 (0.282 g, 1.00 mmol), Co(thd)2 (0.043 g, 0.100 mmol), tert-butyl hydroperoxide (0.050 mL, 0.050 mmol), and triethylsilane (0.160 mL, 1.00 mmol) in DCE (9.1 mL). The reaction time was 1 h. Reduction of the peroxide unit was performed using toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction time was 1 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol).
To unpurified diisopropylsilylene acetal 17 was added THF (5.0 mL) followed by a solution of tetrabutylammonium fluoride in THF (1.0 M, 2.4 mL, 2.4 mmol). The reaction mixture was stirred for 1 h. Distilled H2O (5.0 mL) was added, then the mixture was extracted with diethyl ether (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes:EtOAc 70:30 → hexanes:EtOAc 65:35) afforded 1,3-diol 21 (0.147 g, 79%) as a colorless oil. Characterization was performed on an inseparable 50:50 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 4.19 (m, 2H), 1.96–1.43 (m, 16H), 1.32–0.92 (m, 24H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.1 (C), 75.9 (C), 65.6 (CH), 65.2 (CH), 50.7 (CH), 47.2 (CH2), 45.5 (CH), 45.2 (CH2), 29.0 (CH2), 27.5 (CH2), 27.14 (CH2), 26.97 (CH2), 26.92 (CH2), 26.91 (CH2), 26.89 (CH2), 26.79 (CH2), 26.76 (CH2), 26.74 (CH2), 25.0 (CH3), 24.7 (CH3), 24.6 (CH3), 23.6 (CH3); IR (ATR) 3329, 2924, 2852, 1450, 1136, 1076 cm−1; HRMS (ESI) m/z calcd for C11H22NaO2 (M + Na)+ 209.1512, found 209.1502.
(1R*,2S*)-2-((R*)-1-Hydroxyethyl)cyclohexan-1-ol (24).
1,3-Diol 24 was prepared using a modification of the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 22 (0.145 g, 0.596 mmol), Co(thd)2 (0.025 g, 0.060 mmol), tert-butyl hydroperoxide (0.030 mL, 0.030 mmol), and triethylsilane (0.091 mL, 0.596 mmol) in DCE (5.4 mL). The reaction time was 1 h. Reduction of the peroxide unit was performed using toluene (1.2 mL) and triphenylphosphine (0.312 g, 1.19 mmol). The reaction time was 2.5 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 0.60 mL, 5.2 mmol).
To unpurified diisopropylsilylene acetal 23 was added THF (3.0 mL) followed by a solution of tetrabutylammonium fluoride in THF (1.0 M, 1.4 mL, 1.4 mmol). The reaction time was for 30 min. Distilled H2O (3.0 mL) was added then, the mixture was extracted with diethyl ether (3 × 3 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes:EtOAc = 50:50) afforded 1,3-diol 24 (0.062 g, 73%) as a colorless oil. Characterization was performed on an inseparable 57:43 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 3.85 (m, 1H), 3.79 (s, 1H), 3.68 (m, 0.8H), 3.58 (m, 1H), 3.44 (m, 0.8H), 3.19 (s, 1H), 3.00 (s, 1H), 1.87 (m, 2H), 1.66–1.44 (m, 6.2H), 1.26–1.03 (m, 11H), 0.94–0.68 (m, 1.8H); 13C{1H} NMR (100 MHz, CDCl3) δ 76.7 (CH), 74.3 (CH), 71.8 (CH), 71.4 (CH), 50.9 (CH), 49.9 (CH), 35.9 (CH2), 35.8 (CH2), 27.6 (CH2), 27.3 (CH2), 25.8 (CH2), 25.5 (CH2), 24.9 (CH2), 24.7 (CH2), 21.9 (CH3), 18.4 (CH3); IR (ATR) 3329, 2930, 2857, 1449, 1035, 907 cm−1; HRMS (ESI) m/z calcd for C8H18NO (M + NH4 − H2O)+ 144.1383, found 144.1388.
3-Methyloctane-3,4-diol (27).
1,2-Diol 27 was prepared using a modification of the representative procedure for the Co-catalyzed synthesis of diisopropylsilylene acetals using diisopropylsilyl ether 25 (0.257 g, 1.00 mmol), Co(thd)2 (0.085 g, 0.200 mmol), tert-butyl hydroperoxide (0.050 mL, 0.050 mmol), and triethylsilane (0.160 mL, 1.00 mmol) in DCE (9.1 mL). The reaction time was 1.5 h. Reduction of the peroxide unit was performed using toluene (2.0 mL) and triphenylphosphine (0.524 g, 2.00 mmol). The reaction time was 4 h. Oxidation of the remaining triphenylphosphine was performed using hydrogen peroxide (27% w/w aqueous solution, 1.0 mL, 8.7 mmol). Characterization was performed on an analytical sample of diisopropylsilylene acetal 26: 29Si NMR (79.5 MHz, CDCl3) δ 15.4.
To unpurified diisopropylsilylene acetal 26 was added THF (5.0 mL) followed by a solution of tetrabutylammonium fluoride in THF (1.0 M, 2.4 mL, 2.4 mmol). The reaction mixture was stirred for 10 min. Distilled H2O (5.0 mL) was added, then the mixture was extracted with diethyl ether (3 × 5 mL). The combined organic layers were dried over MgSO4 and concentrated in vacuo. Purification by flash chromatography (hexanes = hexanes:EtOAc = 80:20) afforded 1,2-diol 27 (0.030 g, 19%) as a colorless oil. Characterization was performed on an inseparable 56:44 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 3.39 (m, 1.8H), 2.22 (s, 1H), 2.12 (s, 0.8H), 2.02–1.87 (m, 1.8H), 1.79 (s, 0.8H), 1.67–1.27 (m, 15H), 1.13 (s, 2.3H), 1.07 (s, 3H), 0.95–0.88 (m, 10.7H); 13C{1H} NMR (100 MHz, CDCl3) δ 78.6 (CH), 77.0 (CH), 75.2 (C), 75.0 (C), 31.7 (CH2), 31.3 (CH2), 31.1 (CH2), 29.2 (CH2), 29.1 (CH2), 28.6 (CH2), 23.1 (CH3), 22.92 (CH2), 22.91 (CH2), 20.6 (CH3), 14.2 (CH3), 7.79 (CH3), 7.75 (CH3); IR (ATR) 3393, 2957, 1461, 1378, 996, 907, 732 cm−1; HRMS (ESI) m/z calcd for C9H23NO (M + NH4 − H2O)+ 161.1774, found 161.1778.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health (1R01GM118730). NMR spectra acquired with the TCI cryoprobe and Bruker Advance 400 spectrometer were supported by the National Institutes of Health (OD016343) and the National Science Foundation (CHE-01162222), respectively. The authors thank Dr. Chin Lin for his assistance with NMR spectroscopy and mass spectrometry.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Spectral data (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).Shigeru I; Teruaki M Novel Method for the Preparation of Triethylsilyl Peroxides from Olefins by the Reaction with Molecular Oxygen and Triethylsilane Catalyzed by Bis(1,3-diketonato)cobalt(II). Chem. Lett 1989, 18, 573–576. [Google Scholar]
- (2).Terent’ev AO; Borisov DA; Vil VA; Dembitsky VM Synthesis of Five- and Six-Membered Cyclic Organic Peroxides: Key Transformations Into Peroxide Ring-Retaining Products. Beilstein J. Org. Chem 2014, 10, 34–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tokuyasu T; Kunikawa S; Masuyama A; Nojima M Co(III)–Alkyl Complex- and Co(III)–Alkylperoxo Complex-Catalyzed Triethylsilylperoxidation of Alkenes with Molecular Oxygen and Triethylsilane. Org. Lett 2002, 4, 3595–3598. [DOI] [PubMed] [Google Scholar]
- (4).Tokuyasu T; Kunikawa S; McCullough KJ; Masuyama A; Nojima M Synthesis of Cyclic Peroxides by Chemo- and Regioselective Peroxidation of Dienes with Co(II)/O2/Et3SiH. J. Org. Chem 2005, 70, 251–260. [DOI] [PubMed] [Google Scholar]
- (5).Hurlocker B; Miner MR; Woerpel KA Synthesis of Silyl Monoperoxyketals by Regioselective Cobalt-Catalyzed Peroxidation of Silyl Enol Ethers: Application to the Synthesis of 1,2-Dioxolanes. Org. Lett 2014, 16, 4280–4283. [DOI] [PubMed] [Google Scholar]
- (6).Barnych B; Vatèle J-M Exploratory Studies Toward the Synthesis of the Peroxylactone Unit of Plakortolides. Tetrahedron 2012, 68, 3717–3724. [Google Scholar]
- (7).Hilf JA; Witthoft LW; Woerpel KA An SN1-type Reaction To Form the 1,2-Dioxepane Ring: Synthesis of 10,12-Peroxycalamenene. J. Org. Chem 2015, 80, 8262–8267. [DOI] [PubMed] [Google Scholar]
- (8).O’Neill PM; Pugh M; Davies J; Ward SA; Park BK Regioselective Mukaiyama Hydroperoxysilylation of 2-Alkyl- or 2-Aryl-prop-2-en-1-ols: Application to a New Synthesis of 1,2,4-Trioxanes. Tetrahedron Lett. 2001, 42, 4569–4571. [Google Scholar]
- (9).Laurent SAL; Boissier J; Coslédan F; Gornitzka H; Robert A; Meunier B Synthesis of “Trioxaquantel”® Derivatives as Potential New Antischistosomal Drugs. Eur. J. Org. Chem 2008, 2008, 895–913. [Google Scholar]
- (10).O’Neill PM; Hindley S; Pugh MD; Davies J; Bray PG; Park BK; Kapu DS; Ward SA; Stocks PA Co(thd)2: A Superior Catalyst for Aerobic Epoxidation and Hydroperoxysilylation of Unactivated Alkenes: Application to the Synthesis of Spiro-1,2,4-trioxanes. Tetrahedron Lett. 2003, 44, 8135–8138. [Google Scholar]
- (11).Oswald JP; Woerpel KA Cobalt-Catalyzed Oxygenation/Dearomatization of Furans. J. Org. Chem 2018, 83, 9067–9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tokuyasu T; Kunikawa S; Abe M; Masuyama A; Nojima M; Kim H-S; Begum K; Wataya Y Synthesis of Antimalarial Yingzhaosu A Analogues by the Peroxidation of Dienes with Co(II)/O2/Et3SiH. J. Org. Chem 2003, 68, 7361–7367. [DOI] [PubMed] [Google Scholar]
- (13).Ahmed A; Dussault PH Stereoselective Allylation of Chiral Monoperoxyacetals. Tetrahedron 2005, 61, 4657–4670. [Google Scholar]
- (14).Wu J-M; Kunikawa S; Tokuyasu T; Masuyama A; Nojima M; Kim H-S; Wataya Y Co-catalyzed Autoxidation of Alkene in the Presence of Silane. The Effect of the Structure of Silanes on the Efficiency of the Reaction and on the Product Distribution. Tetrahedron 2005, 61, 9961–9968. [Google Scholar]
- (15).Sugamoto K; Matsushita Y.-i.; Matsui T Direct Hydroperoxygenation of Conjugated Olefins Catalyzed by Cobalt(II) Porphyrin. J. Chem. Soc., Perkin Trans. 1 1998, 3989–3998. [Google Scholar]
- (16).Shigeru I; Teruaki M Hydration of Olefins with Molecular Oxygen and Triethylsilane Catalyzed by Bis(trifluoroacetylacetonato)cobalt(II). Chem. Lett 1989, 18, 569–572. [Google Scholar]
- (17).The use of catalytic triethylsilane generally resulted in poor conversion of starting material and longer reaction times.
- (18).Use of either silica gel or Brockmann grade III neutral alumina for column chromatography resulted in decomposition of the 3-sila-1,2,4-trioxepane.
- (19).Trost BM; Caldwell CG The Di-t-butylsilylene Protecting Group for Diols. Tetrahedron Lett. 1981, 22, 4999–5002. [Google Scholar]
- (20).Corey EJ; Hopkins PB Diisopropylsilyl Ditriflate and Di-tert-butysilyl Ditriflate: New Reagents for the Protection of Diols. Tetrahedron Lett. 1982, 23, 4871–4874. [Google Scholar]
- (21).Harris JR; Haynes MT; Thomas AM; Woerpel KA Phosphine-Catalyzed Reductions of Alkyl Silyl Peroxides by Titanium Hydride Reducing Agents: Development of the Method and Mechanistic Investigations. J. Org. Chem 2010, 75, 5083–5091. [DOI] [PubMed] [Google Scholar]
- (22).Terent’ev AO; Platonov MM; Tursina AI; Chernyshev VV; Nikishin GI Ring Contraction of 1,2,4,5,7,8-Hexaoxa-3-silonanes by Selective Reduction of COOSi Fragments. Synthesis of New Silicon-Containing Rings, 1,3,5,6-Tetraoxa-2-silepanes. J. Org. Chem 2009, 74, 1917–1922. [DOI] [PubMed] [Google Scholar]
- (23).Shigeru I; Teruaki M A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 18, 1071–1074. [Google Scholar]
- (24).No products were observed during these studies in which incorporation of tert-butyl hydroperoxide had occurred.
- (25).Saussine L; Brazi E; Robine A; Mimoun H; Fischer J; Weiss R Cobalt(III) Alkylperoxy Complexes. Synthesis, X-ray Structure, and Role in the Catalytic Decomposition of Alkyl Hydroperoxides and in the Hydroxylation of Hydrocarbons. J. Am. Chem. Soc 1985, 107, 3534–3540. [Google Scholar]
- (26).Grubbs RH; Miller SJ; Fu GC Ring-Closing Metathesis and Related Processes in Organic Synthesis. Acc. Chem. Res 1995, 28, 446–452. [Google Scholar]
- (27).Schmalz H-G Catalytic Ring-Closing Metathesis: A New, Powerful Technique for Carbon–Carbon Coupling in Organic Synthesis. Angew. Chem. Int. Ed. Engl 1995, 34, 1833–1836. [Google Scholar]
- (28).When the reaction was performed using non-volatile methyldiphenylsilane (Table 1, entry 6), 68% conversion of the methyldiphenylsilane was detected by 1H NMR spectroscopy.
- (29).Unsaturated dimethylsilyl ethers were also screened, but the products from these reactions decomposed upon silica gel column chromatography.
- (30).The unsaturated silyl ethers in Table 3 generally contain a 4-methoxyphenyl group, which was incorporated to simplify the purification of the silylene acetals upon column chromatography.
- (31).Walker AJ; Bruce NC Combined Biological and Chemical Catalysis in the Preparation of Oxycodone. Tetrahedron 2004, 60, 561–568. [Google Scholar]
- (32).Endoma-Arias MAA; Hudlicky T A Short Synthesis of Nonracemic Iodocyclohexene Carboxylate Fragment for Kibdelone and Congeners. Tetrahedron Lett. 2011, 52, 6632–6634. [Google Scholar]
- (33).Farcet J-B; Himmelbauer M; Mulzer J A Non-Photochemical Approach to the Bicyclo[3.2.0]heptane Core of Bielschowskysin. Org. Lett 2012, 14, 2195–2197. [DOI] [PubMed] [Google Scholar]
- (34).Ahmed MAK; Fjellvåg H; Kjekshus A; Dietzel PDC Mixed Ligand Complexes of Cobalt(II) – Synthesis, Structure, and Properties of Co4(thd)4(OEt)4. Z. Anorg. Allg. Chem 2007, 633, 1371–1381. [Google Scholar]
- (35).Ahmed MAK; Fjellvåg H; Kjekshus A; Dietzel PDC Syntheses, Structures, and Polymorphism of β-Diketonato Complexes – Co(thd)3. Z. Anorg. Allg. Chem 2008, 634, 247–254. [Google Scholar]
- (36).Palmer C; Morra NA; Stevens AC; Bajtos B; Machin BP; Pagenkopf BL Increased Yields and Simplified Purification with a Second-Generation Cobalt Catalyst for the Oxidative Formation of trans-THF Rings. Org. Lett 2009, 11, 5614–5617. [DOI] [PubMed] [Google Scholar]
- (37).The yield by 1H NMR spectroscopy of the corresponding 3-sila-1,2,4-trioxepane for this reaction was 35%.
- (38).Chando KM; Bailey PA; Abramite JA; Sammakia T Regioselective Ring Opening of Di-isopropylsilylenes Derived from 1,3-Diols with Alkyl Lithium Reagents. Org. Lett 2015, 17, 5196–5199. [DOI] [PubMed] [Google Scholar]
- (39).Yu M; Pagenkopf BL The Regioselective Mono-deprotection of 1,3-Dioxa-2,2-(di-tert-butyl)-2-silacyclohexanes with BF3·SMe2. J. Org. Chem 2002, 67, 4553–4558. [DOI] [PubMed] [Google Scholar]
- (40).Schulten J; Pfister M; Illi S; Klüfers P Dibutylsilylene Derivatives of Tartaric Acid. Z. Anorg. Allg. Chem 2014, 640, 63–67. [Google Scholar]
- (41).Schulten J; Klüfers P Dibutylsilylene–pentose Bis-chelates: on the Glycoses’ Binding Sites for Strongly Lewis-acidic Centres. Carbohydr. Res 2011, 346, 1767–1775. [DOI] [PubMed] [Google Scholar]
- (42).Bartolo ND; Woerpel KA Mechanistic Insight into Additions of Allylic Grignard Reagents to Carbonyl Compounds. J. Org. Chem 2018, 83, 10197–10206. [DOI] [PubMed] [Google Scholar]
- (43).Ellern JR, R O; Allen RJ; Chatterjee AB; Martin DF Hexacoordinate Complexes of Bis(2,4-Pentanedionato)Cobalt-(II): [Bis(Acetylacetonato) Cobalt(II)] In Inorganic Syntheses; Jolly W, Ed.; Wiley: Hoboken, NJ, 1968; Vol. 11, pp 82–89. [Google Scholar]
- (44).Vasconcelos RS; Silva LF; Giannis A Synthesis of Tetrahydrofurans by Cyclization of Homoallylic Alcohols with Iodine/Iodine(III). J. Org. Chem 2011, 76, 1499–1502. [DOI] [PubMed] [Google Scholar]
- (45).Blanco Jaimes MC; Ahrens A; Pflästerer D; Rudolph M; Hashmi ASK Synthesis of Highly Substituted γ-Butyrolactones by a Gold-Catalyzed Cascade Reaction of Benzyl Esters. Chem. Eur. J 2015, 21, 427–433. [DOI] [PubMed] [Google Scholar]
- (46).Phillips GA; Palmer C; Stevens AC; Piotrowski ML; Dekruyf DSR; Pagenkopf BL Oxidative Cyclization of Tertiary Pentenol Derivatives Forming 2,5,5-Trisubstituted THF Rings and the Total Synthesis of Cyclocapitelline. Tetrahedron Lett. 2015, 56, 6052–6055. [Google Scholar]
- (47).Tobia D; Rickborn B Kinetics and Stereochemistry of LiNR2-induced 1,2-Elimination of Homoallylic Ethers. J. Org. Chem 1989, 54, 777–782. [Google Scholar]
- (48).Gau A-H; Lin G-L; Uang B-J; Liao F-L; Wang S-L Regio- and Diastereoselective Ene Reaction of 4-Phenyl-1,2,4-triazoline-3,5-dione with Chiral Allylic Alcohols and Their Derivatives. J. Org. Chem 1999, 64, 2194–2201. [Google Scholar]
- (49).Fang H; He Q; Liu G; Huang Z Pincer Ruthenium Catalyzed Intramolecular Silylation of C(sp2)–H Bonds. Synlett 2017, 28, 2468–2472. [Google Scholar]
- (50).Williams FJ; Grote RE; Jarvo ER Rhodium-catalyzed Redox Allylation Reactions of Ketones. Chem. Commun 2012, 48, 1496–1498. [DOI] [PubMed] [Google Scholar]
- (51).Barker TJ; Jarvo ER Diene-Ligated Iridium Complexes as Catalysts for Allylation and Methallylation Reactions of Ketones. Synthesis 2010, 2010, 3259–3262. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.