

Abstract
A general and efficient synthesis of α-haloglycine esters from commercially available feedstock chemicals, in a single step, is reported. The reactivity of these α-haloglycine esters with various nucleophiles was studied as surrogates of α-iminoesters upon activation with hydrogen-bond donor catalysts. DFT calculations on the α-haloglycine structures (X = F, Cl, Br) accompanied by an X-ray characterization of the α-bromoglycine ester support the existence of a “generalized” anomeric effect created by hyperconjugation. This peculiar hyperconjugative effect is proposed to be responsible for the enhanced halogen nucleofugality leading to a facile halogen abstraction by hydrogen-bond donor catalysts. This reactivity was exploited with thiourea catalysts on several catalytic transformations (aza–Friedel-Crafts and Mannich reactions) for the synthesis of several types of non-proteinogenic α-amino esters.
Keywords: Anomeric effect, Homogeneous catalysis, Hyperconjugation, Non-proteinogenic amino acids, Synthetic methods
Introduction
Non-proteinogenic α-amino acid residues are essential motifs of proteins, non-ribosomal peptides, natural products, and other marketed drugs.[1] Due to their exceptional array of structural and functional diversity, building blocks derived from non-proteinogenic α-amino acids are also found in numerous chiral auxiliaries, organocatalysts, ligands, and bioactive peptides, thus imparting them with a crucial role in modern organic chemistry.[2] This is why developing the most versatile and scalable synthesis of non-proteinogenic α-amino acids has attracted significant attention in the past decades. Conceptually, several synthons have been proposed and studied to achieve the amino acids α-stereocenter functionalization via the corresponding glycine-like radical, anion or carbocation.[3] Most recent efforts have focused either on a Schiff base approach (α-anion), or the activation of an iminoglycine A by BrØnsted or Lewis acids to unveil the glycinyl iminium B reactivity. Numerous functionalizations of α-iminoglycine esters A through Strecker,[4] Mannich,[5] aza-Friedel–Crafts[6] or Petasis[7] reactions have been reported to synthesize an exquisite variety of highly functionalized and optically active α-amino esters (Scheme 1). However, the innate instability of these iminoglycines A is a major limitation which often translates into cumbersome preparation techniques.[8] To counter this obstacle, α-amido sulfones[9] and α-haloglycine esters[10] have been tentatively exploited as in situ imine precursors.
Scheme 1.
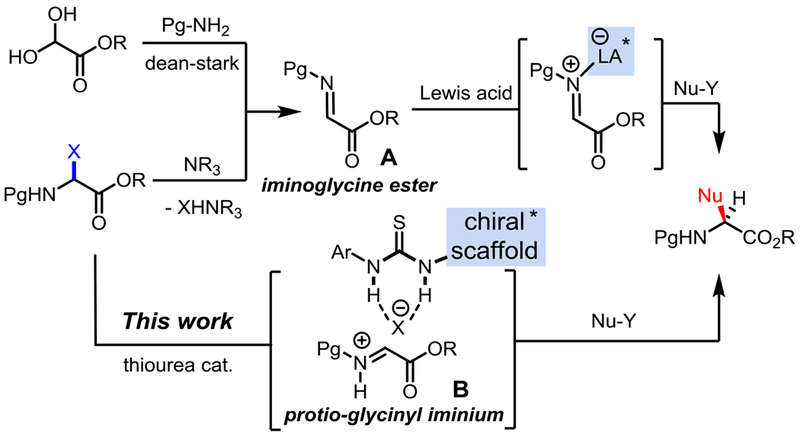
Asymmetric synthetic strategies toward α-amino esters from iminoglycine ester A, and an N-acyliminium equivalent B.
For a long time, however α-haloglycine esters have been disregarded in the synthesis of α-amino esters because they were dubiously thought to be overly reactive, and moisture sensitive.[11] Not surprisingly, there have been only a few reports using α-haloglycine derivatives for the synthesis of non-proteinogenic α-amino acids or esters,[12] heterocycles[13] and peptides.[14] Important to our strategy, the works of Williams[15] and Davies[11b] validated that α-haloglycine bearing a chiral auxiliary moiety can be functionalized with nucleophiles to prepare a wide range of α-amino acids in a diastereoselective fashion. Until recently, the chemistry of α-haloglycines was largely unexplored because the asymmetric maneuvers for halogen abstraction by a chiral catalysts were limited. In 2014, Jacobsen reported the first asymmetric Mannich synthesis of α-amino esters via the halogen abstraction of a α-chloroglycine residue by thiourea anion-binding chiral catalysts.[16] Since this report, we have been particularly drawn to examine the role of halogens from α-haloglycine esters 4a-c (X = F, Cl, and Br) in the abstraction mechanism leading to glycinyl iminiums B (Scheme 1 and Scheme 2). Herein, we are reporting for the first time the synthesis and characterization of three N-carbamoyl α-haloglycine 4a-c, as well as their innate reactivity which is proposed to rise from a very unique “generalized” anomeric effect. Finally, we are demonstrating the efficiency of several hydrogen-bond donor catalysts in promoting noteworthy applications via halogen abstractions for the synthesis of two different classes of α-amino esters.
Scheme 2.
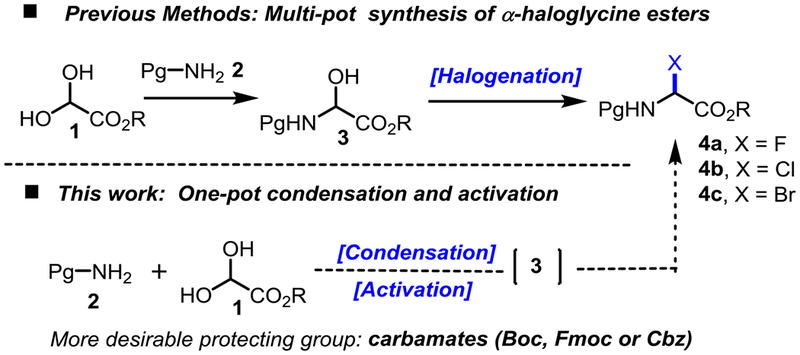
Synthesis of α-haloglycine esters.
Results and Discussion
Synthesis of α-haloglycines.
While α-fluoroglycine ester 4a has never been reported, α-haloglycine esters 4b (X = Cl) and 4c (X = Br) are typically synthesized in two steps which entail the isolation and purification of the hemiaminal 3 followed by a halogenation step (Scheme 2).[17]
We recently reported that α-chloroglycine ester 4b can be prepared in a single step and further functionalized in the same reaction vessel to afford a general strategy to several classes of α-amino esters.[16,18] We proposed that the key to a multicomponent synthesis of α-haloglycine is the simultaneous activation of a glyoxylic ester 1 to facilitate the condensation with a selected primary carbamate 2 while enabling a facile deoxyhalogenation of the hemiaminal intermediate 3 (Scheme 3). Encouraged by our initial findings that the AcOH (cat.)/AcCl system promotes the synthesis of α-chloroglycine 4b in a single step (Table 1, entry 1),[18] several other halogenation reagents have been evaluated to optimize the synthesis of 4a-c (Scheme 3 and Table 1).[19] As we initially reported, water needs to be fully extruded to avoid the reaction reversibility via hydrolysis, and other oxidative side reactions.[20] Therefore, for each deoxyhalogenation reagent or “promoter” tested (Scheme 3), the stoichiometry in promoters was carefully calculated to account for the initial dehydration of glyoxylate 1 (step 1) and to remove a second molecule of water during the deoxyhalogenation of hemiaminal 3 (step 3). For each successful promoter evaluated in Table 1, a reaction profile was established through the reactions’ advancement monitoring by 1H NMR.[21] As shown in our proposed mechanism (Scheme 3), all the promoters release stoichiometric amounts of acid (H-X) during the initial dehydration of glyoxylate hydrate 1 thus facilitating further the condensation producing hemiaminal 3 (step 2). A second molecule of water is then extruded during deoxyhalogenation (step 3) to access α-haloglycines 4a-c. The preliminary kinetic studies[21] confirmed what could be intuitively assumed, that N-carbamoyl iminium formation 5→6 is the rate-limiting step of the cascade reaction.[22] We also observed a correlation between the Lewis acid strength of the promoters (nucleofugality of leaving groups transiently formed in 5) and the overall rate of halogenation, as it would be expected from a typical SN1 reaction via the N-carbamoyl iminium 6.[23] Thus, the net reactivity increase observed may be rationalized by considering the promoter strength from TMSCl, AcCl, SiCl4 ≈ (CO)2Cl2 to the most reactive SOCl2 (Table 1, entries 1–6). To sum-up the results presented in Table 1, thionyl chloride (SOCl2: entry 2), and silicon tetrachloride (SiCl4: entry 3) were found to be the most efficient promoters to synthesize 4b in quantitative yields (r.t. or 35 °C). Along with the formation of HCl, by-products of these reactions are easily removed by evaporation or filtration respectively (SO2 or SiO2). Similarly, several promoters were evaluated to prepare α-bromoglycine ester 4c (entries 7–9). As expected, bromination reagents are more reactive, and SOBr2 was found to be extremely efficient to produce 4c at −20 °C in a quantitative manner and only 20 min. Finally, numerous attempts to synthesize α-fluoroglycine ester 4a have been unsuccessful with only small amounts of hemiaminal 3 observed in some cases (entries 10–11). Therefore the synthesis of substrate 4a was optimized via a stepwise process in one-pot: First, the initial condensation leading to hemiaminal 3 was achieved using catalytic amounts of AcOH at 40 °C which was followed by an in situ deoxyfluorination promoted by DAST (entry 12).[24] The addition of molecular sieves (4Å MS) or Ag2O as acid scavengers severely slows down reactions (2–3 folds), suggesting that the condensation–deoxyhalogenation reactions toward 4a-c are likely catalyzed by acid (HX).
Scheme 3.
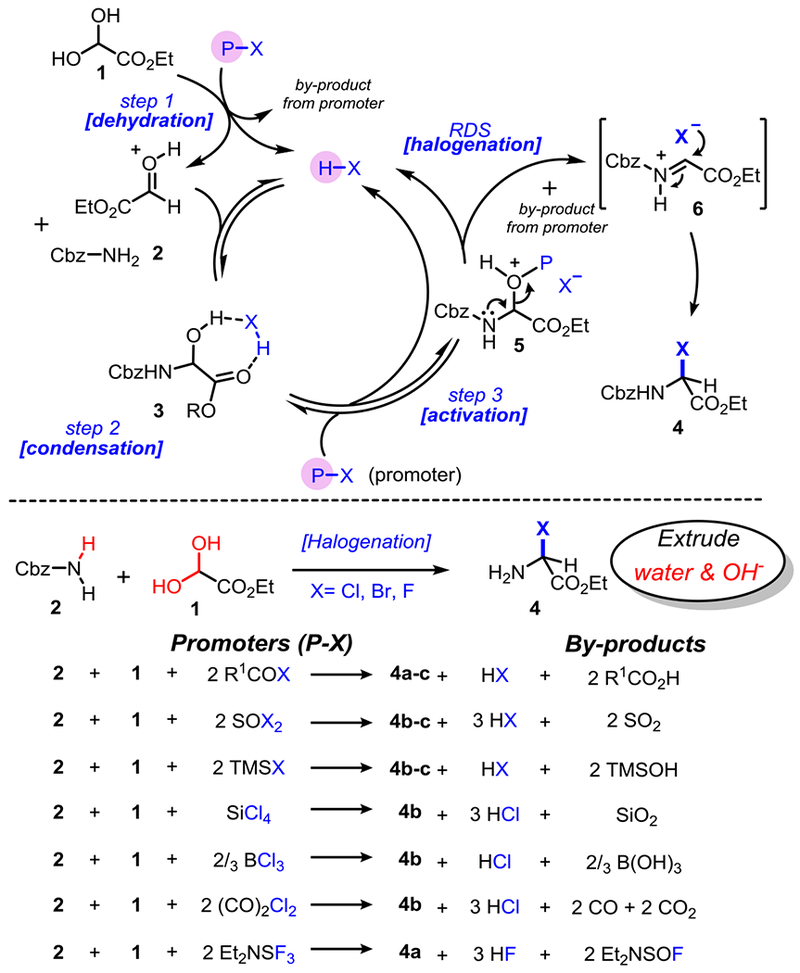
Proposed mechanism and reagents for the one-pot synthesis of α-haloglycine esters 4.
Table 1.
Optimization studies to access α-haloglycines 4a-c in one-pot.[a]
 | ||||
|---|---|---|---|---|
| Entry | Promoter (eq.) | Temp | Time | Yield (%)[b] |
| 1 | AcCI (3.0) | RT | 48 h | 4b (62) |
| AcOH (0.1) | 60 °C | 11 h | 4b (100) | |
| 2 | SOCI2 (3.0) | RT | 15 h | 4b (100) |
| 60 °C | 6 h | 4b (100) | ||
| 3 | SiCI4 (1.5) | 35 °C | 24 h | 4b (100) |
| 4[c] | BCI3 (1.0) | 0 °C | 24 h | 4b (0) |
| 5 | TMSCl (3.0) | RT | 60 h | 4b (89) |
| 6 | (CO)2Cl2 (3.0) | RT | 18 h | 4b (94) |
| 7 | AcBr (3.0) | RT | 6.5 h | 4c (100) |
| AcOH (0.1) | ||||
| 8 | SOBr2 (3.0) | −20 °C | 15 mins | 4c (100) |
| 9 | TMSBr (3.0) | RT | 4 h | 4c (100) |
| 10[d] | BzF (3.0) | 0 °C to 60 °C | 72 h | 4a (0) |
| BzOH (0.1) | ||||
| 11 | Et2NSF3 (3.0) | 0 °C to RT | 72 h | 4a (0) |
| 12[e] | Et2NSF3 (3.0) | 40 °C then −78 °C | 15 h then 2 h | 4a (100) |
| AcOH(0.1) | ||||
1H NMR recorded in CD3CN, aH: 4a (F), δ = 5.95 ppm (dd, J = 9.5, 53.7 Hz); 4b (Cl), δ = 6.18 ppm (d, J = 10.4 Hz); 4c (Br), δ = 6.39 ppm (d, J = 10.8 Hz).
Yields determined by 1H NMR on crude reaction mixtures using mesitylene as internal standard.
Complete decompostion of starting material Cbz-carbamate 2 was observed in presence of BCl3.
Hemiaminal 3 was formed (≈ 25 % conv.).
Experiment run in in a a stepwise manner in CH2Cl2 for the condensation-deoxyfluorination.
To gain mechanistic insight into the condensation-deoxyhalogenation cascade, the reaction with AcOH(cat.)/AcCl at 60 °C in CDCl3 (Table 1, entry 1) was monitored in situ by 1H NMR (Figure 1). Interestingly, while the starting material Cbz-carbamate 2 disappearance followed a typical exponential decay, the plot for the conversion of α-chloroglycine ester 4b over time best fit a sigmoid-like curve, suggesting a complex reaction mechanism (Figure 1A). Indeed, the slow formation of product 4b in the initial 100 minutes corresponds to the time-frame of glyoxylate dehydration (step 1) and a plausible oxonium formation which further initiates the condensation to the hemiaminal intermediate 3 (step 2). During this initial dehydration, the first equivalent of HCl and AcOH are released which appeared to further catalyze the reaction. The maximum conversion in 3 can be seen after 150 minutes suggesting that the rate-determining step is the deoxyhalogenation via the intermediacy of iminium 6. From there on, the formation of AcOH and 4b (monitored by 1H NMR) followed similar kinetics (sigmoid curve) with an acceleration phase followed by a deceleration characteristic of an autocatalysis-like mechanism (Figure 1A).[25] The rate in AcOH formation (d[AcOH]/dt) was plotted against time which confirmed that the acceleration phase proceeded from 100 to 250 minutes at which time the rate of formation in 4b is maximal before decelerating toward the end of the reaction. Given that similar kinetic profiles (autocatalysis-like) were not observed in other reactions (Table 1, entries 2–9) suggested that the rate-determining step could be shifted based on the promoter strength.[21] As such, in the reactions of condensation–deoxybromination to synthesize 4b (Table 1, entries 7–9), hemiaminal 3 was not observed by 1H NMR throughout the entire reaction course.
Figure 1.
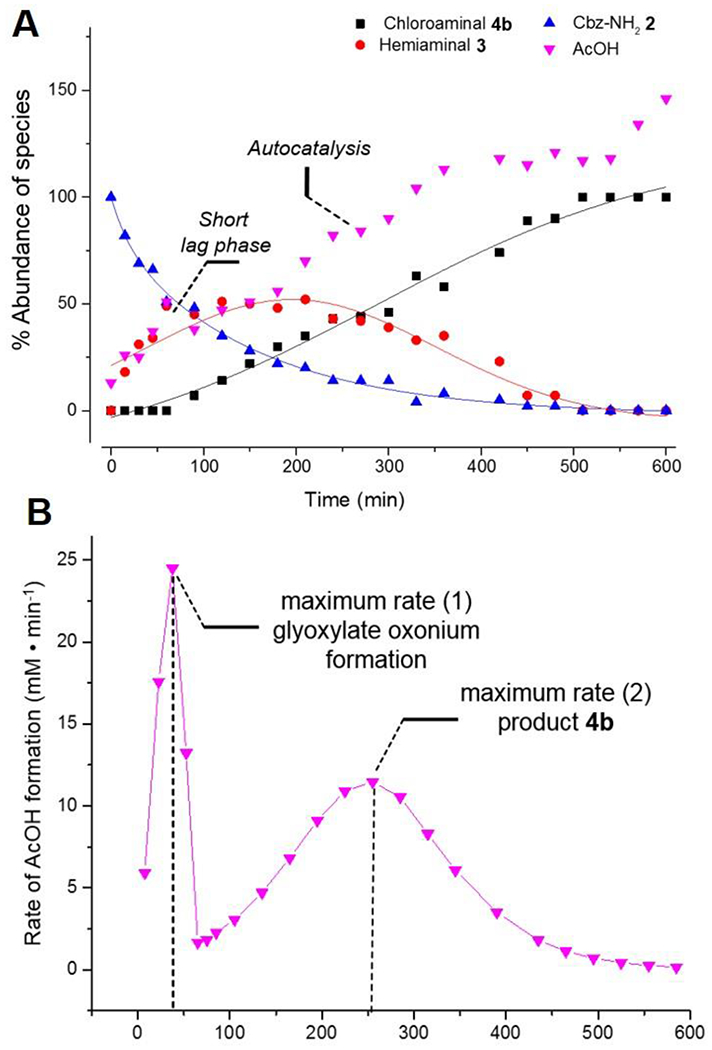
Panel A: kinetic profile for the synthesis of α-chloroglycine 4b at 60 °C (Table 1, entry 1) with best-fitted curves. Panel B: rate of AcOH formation with time suggesting an autocatalysis-like mechanism.
Reactivity of α-haloglycines.
Our next goal was to study the reactivity of α-haloglycines 4a-c under the control of hydrogen-bond donor thiourea catalysts TA,B (Figure 2).
Figure 2.
Thiourea catalysts TA,B used in this study.
A comparative reactivity was examined via a competitive cross functionalization experiment in an aza-Friedel–Crafts reaction (Table 2). Using an equimolar mixture of α-haloglycines (4a-F, 4b-Cl and 4c-Br) with N-methylindole as π-nucleophile, the innate reactivity of α-haloglycines (Condition A: entry 1) was investigated and compared under the presence of 10 mol-% of the achiral Schreiner’s thiourea TA (Condition B: entry 2). Under both reactions’ conditions, α-fluoroglycine 4a remained mostly untouched ([4a] deviation over time is due to substrate degradation[26]) which suggested that 4a is unreactive under these conditions. More interestingly, in the initial hour of the uncatalyzed reaction (entry 1), α-chloroglycine 4b reacted the fastest (57 % consumed) while α-bromoglycine 4c slowly initiated a reaction (14% consumed). After this mark, from 1.5 to 3.5 h, α-chloroglycine 4b was surprisingly regenerated (blue box in Table 2), which might be the result from an anion exchange (Br− to Cl−). Indeed, after the first hour of reaction, ≈ 60 % of α-chloroglycine was consumed therefore the concentration of chloride in the reaction media is significant. It is likely that the degree of conversion in chloride enhanced an external ion return mechanism 4c → 4b (as [Cl−] increased), which also contributed to retard the glycinyl iminium 6 trapping by the indole nucleophile, therefore, promoting the regeneration of α-chloroglycine 4b (through ionic-pair exchange).[27] When similar reaction conditions were applied to evaluate the anion-binding catalysis with thiourea TA (10 mol-%), α-chloroglycine 4b was not consumed faster (entry 2). In contrary, α-bromoglycine 4c appeared to interact with the hydrogen-bond donor catalyst TA which translated in an overall rate acceleration in product formation. Indeed, during the first hour of reaction, the consumption of 4c is largely increased (49% vs. 14%: entry 2 vs. 1). In this case, the counteranion exchange occurred much later, after 6 hours of reaction. Overall, under the reaction conditions tested at −20 °C, both α-haloglycines 4b and 4c have shown an innate propensity to heterolysis, forming the glycinyl iminium 6 in either contact or solvent-separated ion pairs.[18b]
Table 2.
Cross-functionalization experiments to examine the competitive reactivity of α-haloglycine esters 4a–c.[a–d]
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Time (h) | 0 | 0.25 | 0.5 | 1 | 1.5 | 2.5 | 3.5 | 6 | 8 | 10 | |
| 1 Condition A Uncat. | α-haloglycine % consumption | 4a | 0 | 12 | 15 | 18 | 12 | 30 | 32 | 33 | 30 | 36 |
| 4b | 0 | 37 | 40 | 57 | 53 | 47 | 40[d] | 47 | 50 | 43 | ||
| 4c | 0 | 8 | 14 | 14 | 24 | 32 | 43 | 54 | 59 | 76 | ||
| % Yield (7) | 0 | 9 | 11 | 12 | 14 | 19 | 20 | 28 | 29 | 37 | ||
| 2 Condition B TA (10 mol%) | α-haloglycine % consumption | 4a | 0 | 18 | 15 | 18 | 15 | 15 | 21 | 21 | 15 | 27 |
| 4b | 0 | 33 | 50 | 43 | 50 | 47 | 53 | 67 | 40 | 37[d] | ||
| 4c | 0 | 27 | 49 | 49 | 57 | 59 | 70 | 70 | 73 | 86 | ||
| % Yield (7) | 0 | 20 | 23 | 25 | 29 | 32 | 36 | 40 | 43 | 45 | ||
Reactions performed on 0.5 mmol scale, conditions A: 4a–4c (1.0 eq.), conditions B: 4a–4c (1.0 eq.) with thiourea TA (10 mol-%).
The conversions were determined using 1H NMR with mesitylene as internal standard, from reaction aliquots in CHCl3 transferred in NMR tubes and adjusted to a mixture of CHCl3/CD3CN (2:1) at low temperature.
Average measurements are reported from triplicate experiments.
A bromide-chloride exchange is likely occurring through an external ion-return between the two glycinyl iminium ion-pairs 6b:6c (X = Br, Cl).
The direct comparison between catalyzed and uncatalyzed reactions demonstrated that the Schreiner’s thiourea catalyst TA has a pronounced halide-binding effect on α-bromoglycine 4c (Figure 3). In addition, it is proposed that a “generalized” anomeric effect (vide infra) in 4c develops a partial negative-charge on the halogen leaving group, which promotes an early event of halogen-binding by thiourea TA resulting in a more facile C-Br bond cleavage and a greater equilibrium concentration in glycinyl iminium 6, favorable to the ensuing C-C bond-forming step.
Figure 3.
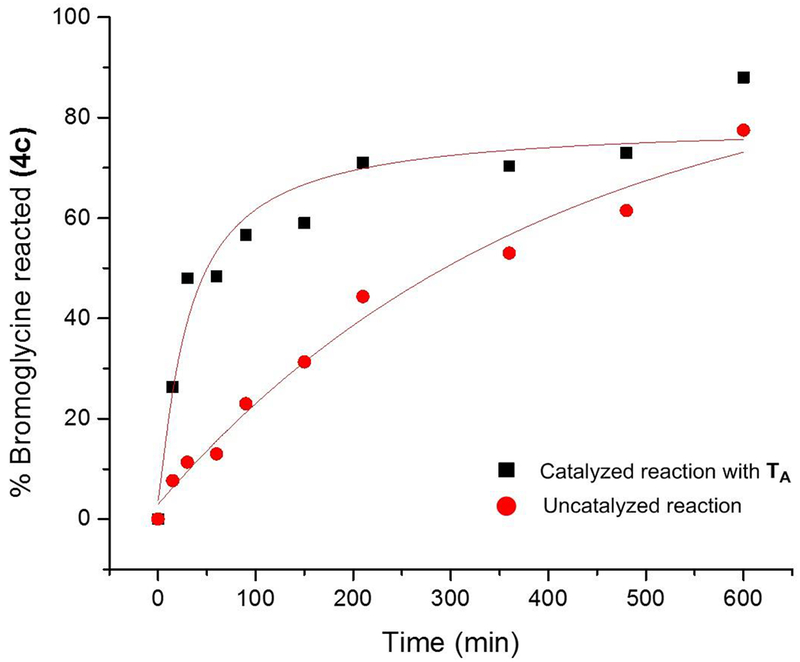
Effect of the hydrogen-bond donor catalyst TA on the arylation of α-bromoglycine 4c with N-methyl indole.
To test this hypothesis, a side by side comparison of α-chloro and α-bromoglycines 4b and 4c reactivity catalyzed by an H-bond donor thiourea was deemed necessary. Thus the reactivity of α-haloglycine esters 4b and 4c as iminoglycine precursors were tested by kinetic profiling in our previously reported Mannich reaction (Scheme 4). While the uncatalyzed reaction between 4b and dibenzoylmethane afforded the expected Mannich product 8 in only 14% yield, thiourea catalyst TA was found to exert a profound effect on the reaction, affording 8 in 90 % yield. As a control, the course of the uncatalyzed reactions of 4b and 4c with dibenzoylmethane were monitored in situ by 1H NMR in CD2Cl2 and shown that product 8 was formed in less than 7 % yield in both cases (initial rate constant kobs of 75 and 0 × 10−5 M−1•min−1 respectively). Similarly to the Friedel–Crafts reaction, the uncatalyzed reaction of 4b seemed to be faster than the reaction of 4c. Furthermore, both reactions proved to be catalyzed by the Takemoto’s tertiary aminothiourea TB leading to about 25 % yield in 8 from α-chloroglycine 4b in the first hour, while only 15 % yield was observed from α-bromoglycine 4c. From these initial reactions profile, it appears that the crucial tertiary amine moiety in catalyst TB might be rapidly quenched by the in situ formation of an HBr salt. Nonetheless, catalyst TB displayed a markedly enhanced reactivity toward 4b, affording good conversion in product 8 as previously reported[16] (conv. > 35 % at 2 h).[21]
Scheme 4.
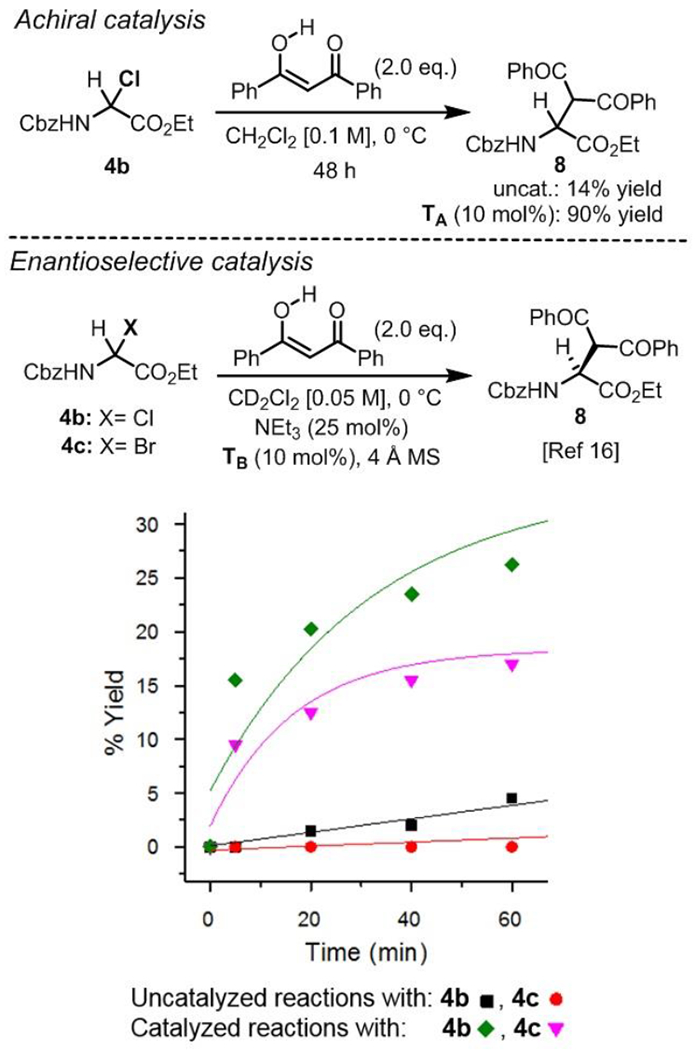
Effect of the hydrogen-bond donor catalyst TB on the direct Mannich reaction of α-haloglycines 4b and 4c.
Having established some interesting trends in reactivity, between 4b and 4c, we further investigated some potential modes of activation for α-fluoroglycine ester 4a (Table 3). Even though the fluoride anion is known to be the strongest hydrogen-bond acceptor halide, the activation of covalently bound organofluorines by hydrogen-bond donors for nucleophilic substitution remains a particularly challenging reaction.[28] Inspired by the original study from Bull and Davies on the reactivity of α-haloglycine and their halogen exchange reactions,[11b] we hypothesized that 4a could be converted into 4b under an appropriate set of conditions. Upon exposure with TMSCl, 4a was chlorinated with high conversions in 2 hours at 0 °C (entries 1–2). When a similar reaction was carried out at −78 °C for 30 min, product 4b was obtained in 20 % yield (entry 3). In comparison, the chlorination of 4a was catalyzed smoothly at −78 °C by thiourea TA to deliver 4b in 60 % yield (entry 4). Remarkably the catalyzed reaction proceeded to full conversion in only one hour to afford 4b in 96% yield after a practical and simple evaporation (entry 5).
Table 3.
Halogen exchange on α-fluoroglycine esters 4a.
 | |||||
|---|---|---|---|---|---|
| Entry[a] | Conditions | Time | Temp | % Conv.[b] | % Yield[b,c] |
| 1 | TMSCl (66 mol%) | 2 h | 0 °C | 72 | 45 |
| 2 | TMSCl (100 mol%) | 2 h | 0 °C | 82 | 82 |
| 3 | TMSCl (130 mol%) | 30 mins | −78 °C | 23 | 20 |
| 4 | TMSCl (130 mol%) TA (10 mol%) | 30 mins | − 78 °C | 60 | 60 |
| 5 | TMSCl (130 mol%) TA (10 mol%) | 1 h | − 78 °C | 100 | 96[c] |
Reactions run on a 0.25 mmol scale.
Conversions and Yields determined by 1H NMR on crude reaction mixtures using mesitylene as internal standard.
Isolated yield.
The overall results of functionalization in both the aza-Friedel–Crafts and in the Mannich reactions suggest that α-chloroglycine 4b reacts innately faster than the α-bromo-analogue 4c leading to an unexpected order of reactivity (Cl > Br >> F).[34d]
Structural Studies.
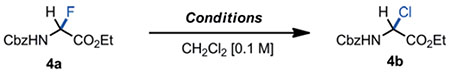
The fact that α-haloglycines 4a-b engaged so easily in heterolysis at cryogenic temperatures, prompted us to draw similarities with the reactivity of pyranosyl halides[29] and the well-established anomeric effect in carbohydrate chemistry[30] Functionalizations of Csp3–F bonds are sparse with the exception of the facile C-F bond cleavage at the anomeric center of glycopyranosyl fluorides to achieve glycosylation reactions.[31] This reactivity is facilitated by the anomeric effect embedded at the C1-position of fluorinated carbohydrates through favorable dipole-dipole repulsion and hyper-conjugation n(O)→σ*(C-F).[32] Hyperconjugative effects in the series of pyranosyl halides have been extensively studied and characterized by abnormal bond lengths. X-ray crystallographic data have shown that O-C(1) bonds are typically shortend, and C(1)-Xax elongated.[33] Even though hyperconjugation is widely accepted as a major stereoelectronic effect that contributes to organic reactivity,[34] examples of hyperconjugative effect on acyclic molecules - so-called “generalized” anomeric effect - are limited.[35] Having access to the α-haloglycine esters 4a-c in pure form, spectroscopic data were obtained to correlate the ease of these substrates toward heterolysis with a potential hyperconjugative effect. Crude material 4c was crystallized in toluene and the X-ray crystal structure of 4c provided several interesting structural features (Figure 4A). For instance, the bond angles around the central Cα are larger than expected for a sp3-hybridized carbon. Also, all dihedral angles observed in the crystal structure correlated well with our conformational analysis of 4b obtained by density functional theory computations (DFT) at the B3LYP 6-311++G (3df,3pd) level of theory (including solvent corrections)[21] Torsion angle Φ, which is a key dihedral angle in the conformational analysis of peptides[36] was found to be unusually large which is characteristic of a fully extended conformation like in trans-amides (ΦX-ray = 150.5°; Φcalc = 159.3°). Also, torsion angle θ between the H-N-C(2) and N-C(2)-H planes which can be determined experimentally from the vicinal scalar coupling constant 3JNH-Hα in 1H NMR was well estimated by the DFT calculations (θX-ray = 161.9°; θcalc = 160.8°).
Figure 4.
Panel A: X-ray structure of α-bromoglycine ester 4c. Ellipsoids are shown at the 50 % probability level. Panel B: model for hyperconjugation for the major conformer of 4c in solution.
Indeed, this peculiar large torsional angle in 4c matches the abnormally large vicinal coupling constant in CDCl3 (JNH-Hα = 10.2 Hz), thus supporting the argument that the low-lying conformation in the crystal lattice reflects closely the major conformer of 4c in solution (Figure 4B)[37,38] Similarly, the JNH-Hα coupling constants for 4a, 4b, and 4c are abnormally large in both apolar and polar protic solvents CDCl3 and CD3CN (J > 9.2 Hz), suggesting that the three α-haloglycines experience a comparable solvent exposure and have a similar gauche-conformation.[39] In such gauche-conformation, 4c is also stabilized by a weak intramolecular hydrogen bond NH•••O=C (2.491 Å) leading to the alignment of the C-Br bond with the carbonyl π* antibonding orbital of the ester.[37] Furthermore the quasi-orthogonality of the α dihedral angle C(3)-N-C(2)-Br (α = −92.6°) supports the fact that the nitrogen lone pair not only engaged in conjugation with the carbonyl π* orbital (Cbz carbamate) but more importantly aligned impeccably in an antiperiplanar manner to the σ*C-Br antibonding orbital to allow a spatial electronic donation through hyperconjugation (n(N) → σ*(C-Br)). This proposed phenomenon of hyperconjugation in 4c is further characterized by abnormal bond lengths: (C2)–N and C(2)–Br (see X-ray crystal structure and the DFT minimized models).[21] With a 140.6 pm length, the (C2)–N bond is substantially shorter than the reported average length for Csp3–Nsp2 of 145.4 pm in acyclic amides,[40] suggesting that the C(2) carbon of 4c has a partial sp2-character. Finally, the C(2)–Br bond length of 200.6 pm is the longest ever reported Csp3–Br bond in the CCDC database (Cambridge Crystallographic Data Centre), +4 pm longer than the average Csp3–Br bond lengths reported in the literature (196.6 pm) which is consistent with a bond elongation magnitude caused by hyperconjugative effects in pyranosyl bromides (avg +2 pm).[21,41] Taken altogether, these spectroscopic and structural results suggest that a “generalized” anomeric effect might take place in the α-haloglycines studied 4a-c due to an hyperconjugation that stabilizes gauche conformations in which the C–X bonds are unusually elongated.
CCDC 1878828 (for 4c) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Conclusions
In conclusion, some highly practical and efficient preparations of α-haloglycine esters 4a-c in one-pot have been developed to generate useful precursors of non-proteinogenic α-amino esters. X-ray characterization and other spectroscopic data combined with the DFT calculations support the existence of an anomeric effect in these acyclic α-haloglycines 4a-c, which is the first reported example of hyperconjugation initiated by a nitrogen heteroatom bearing an electron-withdrawing carbamoyl protecting group (n(N)→σ*(C–Br)). This peculiar hyperconjugative effect is proposed to be responsible for the innate reactivity of the α-haloglycines studied herein by developing a partial negative-charge on the halogen leaving group, thus enhancing nucleofugality and binding by hydrogen-bond donor catalysts. Interestingly, α-chloroglycine 4b reacts faster than the bromoanalog 4c leading to an innate order of reactivity Cl > Br >>F. The unique “generalized” anomeric effect was therefore advantageously exploited to enable several types of transformations (halogen exchange, and C-C bond formations) catalyzed by two thiourea catalysts TA-B. This work resulted in the synthesis of two kinds of non-proteinogenic α-amino esters 7 and 8 as proof of principle. While a catalyzed halogen exchange was achieved with the Schreiner thiourea on α-fluoroglycine 4a, the catalyzed Friedel–Crafts reaction was found to be optimum not from α-chloroglycine 4b as it was initially reported,[18] but from α-bromoglycine 4c. In the other end, the kinetic study for the direct asymmetric Mannich reaction catalyzed by the Takemoto bifunctional thiourea was found to proceed more smoothly from α-chloroglycine 4b. Given the importance of haloacetals in glycosylation chemistry and other C-C bond formation in small molecules,[42] as well as the role of stereoelectronic factors in glycosylation mechanisms,[43] we anticipate that the characterization of the hyperconjugative effect herein and its implication for halogen binding will stimulate new catalytic coupling reactions to be developed with H-bond donor catalysts.[44]
Experimental Section
Instrumentation and methods:
All reactions were performed in flame-dried glassware under a positive pressure of argon. Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise noted. Analytical TLC was performed on 0.25 mm glass-backed 60 Å F-254 TLC plates (Silicycle, Inc.). The plates were visualized by exposure to UV light (254 nm). Infrared spectra were recorded on a Nicolet IS5 FT-IR spectrophotometer. 1H NMR spectra were recorded on a Varian Mercury400 and a Bruker Biospin GmbH (400 MHz) spectrometers and are reported in ppm using solvent as an internal standard (CDCl3 at 7.26 ppm, or CD3CN at 1.96 ppm). 1H NMR spectra were performed using standard parameters, and data are reported as b = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet; coupling constant(s) in Hz, integration. 13C NMR spectra were recorded on a Varian Mercury400 (100 MHz) spectrometer. Chemical shifts are reported in ppm, with solvent resonance employed as the internal standard (CDCl3 at 77.2 ppm, or CD3CN at 1.3 and 118.3 ppm).
Reagents and solvents:
All reagents used in this paper were acquired from Alfa Aesar or Sigma Aldrich. All bulk solvents were acquired from Fischer Scientific. Freshly distilled solvents were used in the reactions presented herein. Chloroform was dried with CaCl2 overnight prior to distillation and transferred under argon to a dark glass bottle with 3 Å molecular sieves for storage. Tetrahydrofuran was purified by refluxing with sodium metal and benzophenone and transferred under argon to a dark glass bottle with 3 Å molecular sieves for storage. Dichloromethane was dried with CaCl2 over-night, prior to distillation and transferred under argon to a dark glass bottle with 4 Å molecular sieves for storage. Full procedures can be found in Purification of Laboratory Chemicals by Armarego, W. L. F.; and Chai C. L. L., Elsevier (Sixth Edition).
Products 7 and 8 have been reported and fully characterized by us in ref.[18,16] respectively.
N-Cbz-α-fluoroglycine ethyl ester 4a:
In a polypropylene vial, a mixture of benzyl carbamate 2 (76 mg, 0.50 mmol, 1.0 eq.), ethyl glyoxylate hydrate in 50 % toluene (w/v) 1 (120 μL, 0.50 mmol, 1.0 equiv.) and acetic acid (3 μL, 0.05 mmol, 0.10 eq.) were stirred in anhydrous dichloromethane (5.0 mL) for 15 h under argon at 40 °C. The reaction was monitored by the 1H NMR for the full conversion to the corresponding hemiaminal. The reaction mixture was then cooled to −78 °C and diethylaminosulfurtrifluoride (DAST: 132 μL, 1.0 mmol, 2.0 eq.) was added dropwise over 5 min to the reaction mixture. At the end of the addition, the reaction mixture was allowed to slowly warm up to room temperature over another 2 h. After consumption of all starting materials (observed by the 1H NMR), the reaction mixture was quenched with ice-cold water (10 mL) and extracted with CH2Cl2 (3 × 5.0 mL). The combined organic layers were then dried with sodium sulfate, filtered and concentrated under vacuum in a glass scintillation vial to obtain the desired product 4a as a pale yellow liquid (144 mg containing residual CH2Cl2, 0.50 mmol, >95 % yield overall) which is finally transferred in a polypropylene vial for storage. [Note: Complete removal of the residual CH2Cl2 in a scintillation vial leads to rapid decomposition of compound 4a]. Rf = 0.25 (EtOAc/hexanes, 30:70; UV active, stains green-yellow with vanillin). Caution!! α-Fluoroglycine 4a is not stable, neither on silica nor on neutral alumina; It decomposes rapidly into 3, but 4a can be stored in a polypropylene vial under argon in a freezer at −78 °C for 4 days with a minimum decomposition <10 %. IR (Neat) νmax = 697, 736, 779, 857, 970, 1021, 1205, 1335, 1374, 1399, 1455, 1520, 1732, 2982, 3320 cm−1. 1H NMR (400 MHz, CD3CN): δ (ppm) 7.44–7.33 (m, 5H), 7.08 (bs, 1H, NH), 5.96 (dd, J = 53.7, 9.5 Hz, 1H), 5.15 (s, 2H), 4.24 (q, J = 7.1 Hz, 2H), 1.26 (t, J = 7.2 Hz, 3H). 19F NMR (377 MHz, CDCl3) δ (ppm) −140.2 (d, J = 54.0 Hz). (Standard hexafluorobenzene δ = −164.9 ppm). 13C NMR (100 MHz, CD3CN): δ (ppm) 166.1 (C; d, J = 33.3 Hz), 137.2 (C), 129.5(2 CH), 129.3(CH), 129.1 (2 CH), 88.34 (C-F: d, J = 205.1 Hz), 68.2 (CH2), 63.3 (CH2), 14.2 (CH3). HRMS (ESI): Compound 4a hydrolyzed into the corresponding hemiaminal 3 in the mass spectrometer with m/z Calcd. for [C12H15NO5+Na]+ = 276.0842, found 276.0848 (+2.2 ppm).
SMILES: FC([H])(C(OCC)=O)NC(OCC1=CC=CC=C1)=O.
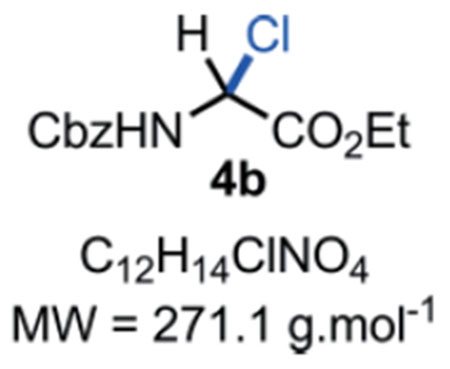
N-Cbz-α-chloroglycine ethyl ester 4b:
In a flame dried scintillation vial, a mixture of benzyl carbamate 2 (76 mg, 0.50 mmol, 1.0 eq.), ethyl glyoxylate hydrate in 50 % toluene (w/v) 1 (143 μL, 0.60 mmol, 1.2 eq.) and thionyl chloride (109 μL, 1.50 mmol, 3.0 eq.) were stirred under argon in anhydrous chloroform (5.0 mL) for 6 hours at 60 °C. The reaction mixture was cooled down to r.t. and directly evaporated under vacuum to obtain the desired product 4b as a white solid (135 mg, 0.50 mmol, quant. yield). Compound 4b can be stored in an amber glass container at r.t. for 14 days in a desiccator without major decomposition <5–10%). Rf = Caution!! Compound 4b is not stable on silica gel; it hydrolyses back to the corresponding hemiaminal 3 which can be observed by TLC at the Rf mentioned above. M.P. = 117–119 °C. IR (Neat) νmax = 660, 694, 753, 778, 976, 1023, 1056, 1204, 1240, 1337, 1529, 1700, 1742 cm−1. 1H NMR (400 MHz, CD3CN): δ (ppm) 7.48–7.22 (m, 5H), 7.03 (bs, NH), 6.18 (d, J = 10.4 Hz, 1H), 5.16 (s, 2H), 4.26 (q, J = 7.1, 2H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 165.9 (C), 153.9 (C), 135.3 (C), 128.7 (2 CH), 128.6 (CH), 128.4 (2 CH), 68.1 (CH), 63.4 (CH2), 63.2 (CH2), 13.9 (CH3). HRMS (ESI): Compound 4b hydrolysed into the corresponding hemiaminal 3 in the mass spectrometer with m/z Calcd. for [C12H15NO5+Na]+ = 276.0842, found 276.0835 (−2.5 ppm). SMILES: ClC([H])(C(OCC)=O)NC(OCC1=CC=CC=C1)=O.
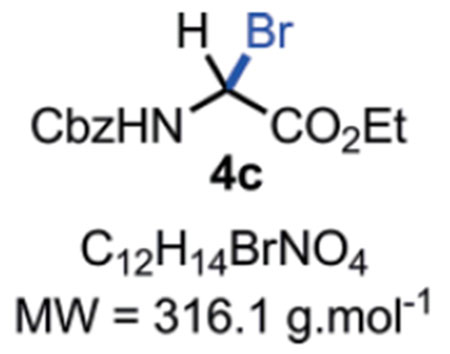
N-Cbz-α-bromoglycine ethyl ester 4c:
N-Protected α-bromoglycine 4c was synthesized using the either following procedures:
In a flame dried scintillation vial, a mixture of benzyl carbamate 2 (76 mg, 0.50 mmol, 1.0 eq.), ethyl glyoxylate hydrate in 50 % toluene (w/v) 1 (143 μL, 0.60 mmol, 1.2 eq.), acetyl bromide (111 μL, 1.50 mmol, 3.0 eq.) and acetic acid (3 μL, 0.05 mmol, 0.1 eq.) were stirred in anhydrous chloroform (5.0 mL) for 6.5 hours under argon at room temperature. The reaction mixture was then evaporated under vacuum to obtain the desired product 4c as a pale brown solid (157 mg, 0.50 mmol, quant. yield).
Alternatively, the title compound can be prepared in a flame dried scintillation vial, with a mixture of benzyl carbamate 2 (76 mg, 0.50 mmol, 1.0 eq.), ethyl glyoxylate hydrate in 50% toluene (w/v) 1 (143 μL, 0.60 mmol, 1.2 eq.) which were stirred in anhydrous chloroform (5.0 mL) under argon at −20 °C. Into the reaction mixture thionyl bromide (116 μL, 1.50 mmol, 3.0 eq.) was added dropwise and stirred for 15 min until full consumption of all starting materials. The reaction mixture was then evaporated under vacuum to obtain the desired product 4c as a pale brown solid (158 mg, 0.50 mmol, quant. yield).
Compound 4c can be stored in a brown glass container at r.t. in a desiccator for 7 days without major decomposition < 5 %). Rf = Caution!! Compound 4c is not stable on silica gel; it hydrolyses back to the corresponding hemiaminal 3 which can be observed by TLC at the Rf mentioned above. M.P. = 63–65 °C. IR (Neat) νmax = 661, 752, 762, 776, 814, 844, 861, 910, 950, 992, 1112, 1144, 1230, 1274, 1333, 1367, 1381, 1396, 1525, 1700, 1736, 3032, 3292 cm−1. 1H NMR (400 MHz, CD3CN): δ (ppm) 7.47–7.27 (m, 5H), 7.08 (bs, NH), 6.38 (d, J = 10.8 Hz, 1H), 5.17 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 166.2 (C), 153.5 (C), 135.3 (C), 128.7 (2 CH), 128.7 (2 CH), 128.4 (CH), 68.2 (CH), 63.2 (CH2), 53.4 (CH2), 13.9 (CH3). HRMS (ESI): Compound 4c hydrolysed into the corresponding hemiaminal 3 in the mass spectrometer with m/z Calcd. for [C12H15NO5+Na]+ = 276.0842, found 276.0855 (+4.7 ppm).
SMILES: BrC([H])(C(OCC)=O)NC(OCC1=CC=CC=C1)=O.
Supplementary Material
Acknowledgments
We are very grateful for the financial support from the NIH (NIGMS Grant: R15GM116025 to S.S.S and S.P.R.). The authors thank Timothy Foo and Dr. Andrew Terentis from Florida Atlantic University for guiding Shyam Samanta to perform the preliminary DFT calculations. The authors also thank Dr. Maren Pink, director of the Molecular Structure Center at the Indiana University Bloomington for the high-resolution crystal structure analysis (X-ray).
Footnotes
Supporting information and ORCID(s) from the author(s) for this article are available on the WWW under https://doi.org/10.1002/ejoc.201901033.
References
- [1].a) Fenteany G, Standaert R, Lane W, Choi S, Corey E, Schreiber S, Science 1995, 268, 726–731; [DOI] [PubMed] [Google Scholar]; b) Cane DE, Walsh CT, Khosla C, Science 1998, 282, 63–68; [DOI] [PubMed] [Google Scholar]; c) Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M, Drug Discovery Today 2010, 15, 40–56; [DOI] [PubMed] [Google Scholar]; d) Walsh CT, ACS Chem. Biol. 2014, 9, 1653–1661. [DOI] [PubMed] [Google Scholar]
- [2].a) Soloshonok VA, Izawa K, Asymmetric Synthesis and Application of α-Amino Acids; Oxford University Press: Washington, DC, 2009; [Google Scholar]; b) Blaskovich MA, Handbook on Syntheses of Amino Acids: General Routes for the Syntheses of Amino Acids; Oxford University Press: Oxford; New York, 2010. [Google Scholar]
- [3].a) Duthaler RO, Tetrahedron 1994, 50, 1539–1650; [Google Scholar]; b) Wirth T, Angew. Chem. Int. Ed. Engl. 1997, 36, 225–227; [Google Scholar]; Angew. Chem 1997, 109, 235; [Google Scholar]; c) Ma J-A, Angew. Chem. Int. Ed. 2003, 42, 4290–4299; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2003, 115, 4426. [Google Scholar]
- [4].For selected examples of Strecker Reaction, see:; a) Krueger CA, Kuntz KW, Dzierba CD, Wirschun WG, Gleason JD, Snapper ML, Hoveyda AH, J. Am. Chem. Soc. 1999, 121, 4284–4285; [Google Scholar]; b) Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN, Nature 2009, 461, 968–970 and references cited herein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For selected examples of Mannich reaction, see: ; a) Hagiwara E, Fujii A, Sodeoka M, J. Am. Chem. Soc. 1998, 120, 2474–2475; [Google Scholar]; b) Ferraris D, Young B, Dudding T, Lectka T, J. Am. Chem. Soc. 1998, 120, 4548–4549; [Google Scholar]; c) Juhl K, Gathergood N, Jørgensen KA, Angew. Chem. Int. Ed. 2001, 40, 2995–2997; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2001, 113, 3083. [Google Scholar]
- [6].For selected examples of Friedel–Crafts reaction, see: ; a) Johannsen M, Chem. Commun. 1999, 2233–2234; [Google Scholar]; b) Saaby S, Fang X, Gathergood N, Jørgensen KA, Angew. Chem. Int. Ed. 2000, 39, 4114–4116; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2000, 112, 4280; [Google Scholar]; c) Uraguchi D, Sorimachi K, Terada M, J. Am. Chem. Soc. 2004, 126, 11804–11805. [DOI] [PubMed] [Google Scholar]
- [7].a) Petasis NA, Goodman A, Zavialov IA, Tetrahedron 1997, 53, 16463–16470; [Google Scholar]; b) Lou S, Schaus SE, J. Am. Chem. Soc. 2008, 130, 6922–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Manabe K, Oyamada H, Sugita K, Kobayashi S, J. Org. Chem. 1999, 64, 8054–8057; [Google Scholar]; b) Porter JR, Traverse JF, Hoveyda AH, Snapper ML, J. Am. Chem. Soc. 2001, 123, 10409–10410; [DOI] [PubMed] [Google Scholar]; c) Schleusner M, Gais H-J, Koep S, Raabe G, J. Am. Chem. Soc. 2002, 124, 7789–7800; [DOI] [PubMed] [Google Scholar]; d) Prenzel AHGP, Deppermann N, Maison W, Org. Lett. 2006, 8, 1681–1684; [DOI] [PubMed] [Google Scholar]; e) Oliver LH, Puls LA, Tobey SL, Tetrahedron Lett. 2008, 49, 4636–4639. [Google Scholar]
- [9].a) Fini F, Sgarzani V, Pettersen D, Herrera RP, Bernardi L, Ricci A, Angew. Chem. Int. Ed. 2005, 44, 7975–7978; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 8189; [Google Scholar]; b) Palomo C, Oiarbide M, Laso A, Lopez R, J. Am. Chem. Soc. 2005, 127, 17622–17623; [DOI] [PubMed] [Google Scholar]; c) Gianelli C, Sambri L, Carlone A, Bartoli G, Melchiorre P, Angew. Chem. Int. Ed. 2008, 47, 8700–8702; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8828; [Google Scholar]; d) Galzerano P, Agostino D, Bencivenni G, Sambri L, Bartoli G, Melchiorre P, Chem. Eur. J. 2010, 16, 6069–6076. [DOI] [PubMed] [Google Scholar]
- [10].a) Gaitanopoulos DE, Weinstock J, J. Heterocycl. Chem. 1985, 22, 957–959; [Google Scholar]; b) Ermert P, Meyer J, Stucki C, Schneebeli J, Obrecht J-P, Tetrahedron Lett. 1988, 29, 1265–1268; [Google Scholar]; c) McFarlane AK, Thomas G, Whiting A, Tetrahedron Lett. 1993, 34, 2379–2382; [Google Scholar]; d) Hafez AM, Taggi AE, Dudding T, Lectka T, J. Am. Chem. Soc. 2001, 123, 10853–10859; [DOI] [PubMed] [Google Scholar]; e) Kobayashi S, Kitagawa H, Matsubara R, J. Comb. Chem. 2001, 3, 401–403; [DOI] [PubMed] [Google Scholar]; f) Dudding T, Hafez AM, Taggi AE, Wagerle TR, Lectka T, Org. Lett. 2002, 4, 387–390; [DOI] [PubMed] [Google Scholar]; g) Nakamura Y, Matsubara R, Kiyohara H, Kobayashi S, Org. Lett 2003, 5, 2481–2484. [DOI] [PubMed] [Google Scholar]
- [11].a) Trost BM, Lee C, J. Am. Chem. Soc. 2001, 123, 12191–12201; [DOI] [PubMed] [Google Scholar]; b) Bull SD, Davies SG, Garner AC, Savory ED, Snow EJ, Smith AD, Tetrahedron: Asymmetry 2004, 15, 3989–4001. [Google Scholar]
- [12].For selected examples of α-chloroglycine in synthesis: ; a) Mori M, Kanda N, Ban Y, J. Chem. Soc., Chem. Commun. 1986, 1375–1376; [Google Scholar]; b) Mooiweer HH, Hiemstra H, Speckamp WN, Tetrahedron 1989, 45, 4627–4636; [Google Scholar]; c) Mooiweer HH, Ettema KWA, Hiemstra H, Speckamp WN, Tetrahedron 1990, 46, 2991–2998; [Google Scholar]; d) Williams RM, Aldous DJ, Aldous SC, J. Org. Chem. 1990, 55, 4657–4663; [Google Scholar]; e) Kobayashi S, Matsubara R, Nakamura Y, Kitagawa H, Sugiura M, J. Am. Chem. Soc. 2003, 125, 2507–2515; [DOI] [PubMed] [Google Scholar]; f) Hafez AM, Dudding T, Wagerle TR, Shah MH, Taggi AE, Lectka T, J. Org. Chem. 2003, 68, 5819–5825; [DOI] [PubMed] [Google Scholar]; g) Xu F, Devine P, Org. Process Res. Dev. 2010, 14, 666–667. [Google Scholar]; For selected examples of α-bromoglycine in synthesis: ; h) Kober R, Steglich W, Liebigs Ann. Chem. 1983, 1983, 599–609; [Google Scholar]; i) Bretschneider T, Miltz W, Munster P, Steglich W, Tetrahedron 1988, 44, 5403–5414; [Google Scholar]; j) Easton CJ, Scharfbillig IM, J. Org. Chem. 1990, 55, 384–386; [Google Scholar]; k) Kohn H, Sawhney KN, LeGall P, Conley JD, Robertson DW, Leander JD, J. Med. Chem. 1990, 33, 919–926; [DOI] [PubMed] [Google Scholar]; l) Martin CL, Overman LE, Rohde JM, J. Am. Chem. Soc. 2010, 132, 4894–4906. [DOI] [PMC free article] [PubMed] [Google Scholar]; Up to date there are no literature precedents highlighting the use of α-fluoroglycine in synthesis .
- [13].For selected examples of α-chloroglycine in heterocycle synthesis, see: ; a) Evans DA, Biller SA, Tetrahedron Lett. 1985, 26, 1911–1914; [Google Scholar]; b) Fujimoto K, Iwano Y, Hirai K, Bull. Chem. Soc. Jpn. 1986, 59, 1887–1896; [Google Scholar]; c) Shah NV, Cama LD, Heterocycles 1987, 25, 221–227; [Google Scholar]; d) Coqueron P-Y, Didier C, Ciufolini MA, Angew. Chem. Int. Ed. 2003, 42, 1411–1414; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2003, 115, 1451; [Google Scholar]; e) Zhang J, Ciufolini MA, Org. Lett. 2009, 11, 2389–2392; [DOI] [PubMed] [Google Scholar]; f) Brenek SJ, Caron S, Chisowa E, Colon-Cruz R, Delude MP, Drexler MT, Handfield RE, Jones BP, Nadkarni DV, Nelson JD, Olivier M, Weekly RM, Bellinger GCA, Brkic Z, Choi N, Desneves J, Lee MAP, Pearce W, Watson JK, Org. Process Res. Dev. 2012, 16, 1338–1347. [Google Scholar]; For selected examples of α-bromoglycine in heterocycle synthesis, see: ; g) Zhang J, Coqueron P-Y, Vors J-P, Ciufolini MA, Org. Lett. 2010, 12, 3942–3945. [DOI] [PubMed] [Google Scholar]; See also ref.[8d,13c] For the sole example of a α-fluoroglycine in an heterocycle, see ref.[11b].
- [14].For α-chloroglycines in peptide synthesis, see: ; a) Apitz G, Jager M, Jaroch S, Kratzel M, Schaffeler L, Steglich W, Tetrahedron 1993, 49, 8223–8232. [Google Scholar]; For α-bromoglycines in peptide synthesis, see: ; b) Repine JT, Kaltenbronn JS, Doherty AM, Hamby JM, Himmelsbach RJ, Kornberg BE, Taylor MD, Lunney EA, Humblet C, J. Med. Chem. 1992, 35, 1032–1042; [DOI] [PubMed] [Google Scholar]; c) Annedi SC, Li W, Samson S, Kotra LP, J. Org. Chem. 2003, 68, 1043–1049. [DOI] [PubMed] [Google Scholar]; For α-fluoroglycines in peptide synthesis, see: ; d) Takeuchi Y, Kamezaki M, Kirihara K, Haufe G, Laue KW, Shibata N, Chem. Pharm. Bull. 1998, 46, 1062–1064. [Google Scholar]
- [15].a) Sinclair PJ, Zhai D, Reibenspies J, Williams RM, J. Am. Chem. Soc. 1986, 108, 1103–1104; [Google Scholar]; b) Williams RM, Sinclair PJ, Zhai D, Chen D, J. Am. Chem. Soc. 1988, 110, 1547–1557; [Google Scholar]; c) Williams RM, Hendrix JA, J. Org. Chem. 1990, 55, 3723–3728. [Google Scholar]
- [16].Wasa M, Liu RY, Roche SP, Jacobsen EN, J. Am. Chem. Soc. 2014, 136, 12872–12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].For the synthesis of α-chloroglycines, see ref.[12a–12g,13a–13f,14a–14d] For the synthesis of α-bromoglycines, see ref.[12h–12k,13g,14b–14c] For the synthesis of α-fluoroglycines, see: ; a) Takeuchi Y, Nabetani M, Takagi K, Hagi T, Koizumi T, J. Chem. Soc., Perkin Trans 1 1991, 49–53; [Google Scholar]; b) Takeuchi Y, Kirihara K, Kirk KL, Shibata N, Chem. Commun. 2000, 785–786; [Google Scholar]; c) Wolfer J, Bekele T, Abraham CJ, Dogo-Isonagie C, Lectka T, Angew. Chem. Int. Ed. 2006, 45, 7398–7400; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2006, 118, 7558; [Google Scholar]; d) Fang Y-Q, Bio MM, Hansen KB, Potter MS, Clausen A, J. Am. Chem. Soc. 2010, 132, 15525–15527. [DOI] [PubMed] [Google Scholar]; See also ref.[14d].
- [18].a) Roche SP, Samanta SS, Gosselin MMJ, Chem. Commun. 2014, 50, 2632–2634; [DOI] [PubMed] [Google Scholar]; b) Samanta SS, Roche SP, J. Org. Chem. 2017, 82, 8514–8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stach T, Dräger J, Huy PH, Org. Lett. 2018, 20, 2980–2983. [DOI] [PubMed] [Google Scholar]
- [20].a) Armesto XL, Canle M, García MV, Losada M, Santaballa JA, J. Phys. Org. Chem. 1996, 9, 552–560; [Google Scholar]; b) Armesto XL, Canle M, García LMV, Santaballa JA,Chem. Soc. Rev. 1998, 27, 453–460. [Google Scholar]
- [21].See Supporting Information for complete experimental details.
- [22].A correlation between the leaving group ability (nucleofugality) and the Lewis acid strength of the promoter is characterized by the relative amount of α-hydroxyglycine 3 building-up in each reaction (monitored by 1H NMR). More reactive promoters enhance the leaving group departure leading to the N-carbamoyl-iminium 6 and a faster clearance of 3 by accelerating the deoxyhalogenation rate-limiting step.
- [23].a) Denegri B, Ofial AR, Jurić S, Streiter A, Kronja O, Mayr H, Chem. Eur. J. 2006, 12, 1657–1666; [DOI] [PubMed] [Google Scholar]; b) Streidl N, Denegri B, Kronja O, Mayr H, Acc. Chem. Res. 2010, 43, 1537–1549. [DOI] [PubMed] [Google Scholar]
- [24].a) Posner GH, Haines SR, Tetrahedron Lett. 1985, 26, 5–8; [Google Scholar]; b) Beauve C, Bouchet M, Touillaux R, Fastrez J, Marchand-Brynaert J, Tetrahedron 1999, 55, 13301–13320. [Google Scholar]
- [25].a) Bissette AJ, Fletcher SP, Angew. Chem. Int. Ed. 2013, 52, 12800–12826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13034; [Google Scholar]; b) Champagne PA, Benhassine Y, Desroches J, Paquin J-F, Angew. Chem. Int. Ed. 2014, 53, 13835–13839; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14055; [Google Scholar]; c) Blackmond DG, J. Am. Chem. Soc. 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- [26].α-Fluoroglycine 4a was found to degrade rapidly at –78 °C when kept in a typical borosilicate glass reaction vessel (<10 % recovery after 24 h), but 4a can be preserved intact in a PTFE container for 48 h.
- [27].a) Streidl N, Antipova A, Mayr HJ, Org. Chem 2009, 74, 7328. [DOI] [PubMed] [Google Scholar]; For reviews covering the topic of ion returns, see: ; b) Raber DJ, Harris JM, Schleyer P. v. R., In Ions and Ion Pairs in Organic Reactions (Ed.: Szwarc M); Wiley: New York, 1974; Vol. 2; [Google Scholar]; c) Peters KS, Chem. Rev. 2007, 107, 859–873. [DOI] [PubMed] [Google Scholar]
- [28].a) Restorp P, Berryman OB, Sather AC, Ajami D, Rebek J Jr., Chem. Commun. 2009, 5692–5694; [DOI] [PubMed] [Google Scholar]; b) Champagne PA, Pomarole J, Therien M-E, Benhassine Y, Beaulieu S, Legault CY, Paquin J-F, Org. Lett. 2013, 15, 2210–2213; [DOI] [PubMed] [Google Scholar]; See also ref.[25a].
- [29].a) Holland CV, Horton D, Jewell JS, J. Org. Chem. 1967, 32, 1818–1821; [DOI] [PubMed] [Google Scholar]; b) Anderson CB, Sepp DT, J. Org. Chem. 1967, 32, 607–611; [Google Scholar]; c) Lemieux RU, Hendriks KB, Stick RV, James K, J. Am. Chem. Soc. 1975, 97, 4056–4062; [Google Scholar]; d) Lemieux RU, Driguez H, J. Am. Chem. Soc. 1975, 97, 4063–4069; [DOI] [PubMed] [Google Scholar]; e) Matsumoto T, Maeta H, Suzuki K, Tsuchihashi l. G.-i., Tetrahedron Lett. 1988, 29, 3567–3570; [Google Scholar]; f) Lichtenthaler FW, Ronninger S, Kreis U, Liebigs Ann. Chem. 1990, 1990, 1001–1006; [Google Scholar]; g) Lichtenthaler FW, Schneider-Adams T, J. Org. Chem. 1994, 59, 6728–6734; [Google Scholar]; h) Hadd MJ, Gervay J, Carbohydr. Res. 1999, 320, 61–69. [Google Scholar]
- [30].a) Juaristi E, Cuevas G, Tetrahedron 1992, 48, 5019–5087; [Google Scholar]; b) Toshima K, Tatsuta K, Chem. Rev. 1993, 93, 1503–1531; [Google Scholar]; c) Demchenko AV, Curr. Org. Chem. 2003, 7, 35–79; [Google Scholar]; d) Mydock LK, Demchenko AV, Org. Biomol. Chem. 2010, 8, 497–510. [DOI] [PubMed] [Google Scholar]
- [31].a) Teruaki M, Yoshiyuki M, Shin-ichiro S, Chem. Lett. 1981, 10, 431–432; [Google Scholar]; b) Nicolaou KC, Dolle RE, Papahatjis DP, J. Am. Chem. Soc. 1984, 106, 4189–4192; [Google Scholar]; c) Nicolaou KC, Caulfield T, Kataoka H, Kumazawa T, J. Am. Chem. Soc. 1988, 110, 7910–7912; [Google Scholar]; d) Deshpande PP, Kim HM, Zatorski A, Park T-K, Ragupathi G, Livingston PO, Live D, Danishefsky SJ, J. Am. Chem. Soc. 1998, 120, 1600–1614. [Google Scholar]
- [32].a) Mo Y, Nat. Chem 2010, 2, 666–671; [DOI] [PubMed] [Google Scholar]; b) Lee SS, Greig IR, Vocadlo DJ, McCarter JD, Patrick BO, Withers SG, J. Am. Chem. Soc. 2011, 133, 15826–15829. [DOI] [PubMed] [Google Scholar]
- [33].a) Jeffrey GA, Yates JH, J. Am. Chem. Soc. 1979, 101, 820–825; [Google Scholar]; b) Allen FH, Kirby AJ, J. Am. Chem. Soc. 1984, 106, 6197–6200; [Google Scholar]; c) Briggs AJ, Glenn R, Jones PG, Kirby AJ, Ramaswamy P, J. Am. Chem. Soc. 1984, 106, 6200–6206; [Google Scholar]; d) Hillig KW, Lattimer RP, Kuczkowski RL, J. Am. Chem. Soc. 1982, 104, 988–993. [Google Scholar]
- [34].a) Bader RFW, Slee TS, Cremer D, Kraka E, J. Am. Chem. Soc. 1983, 105, 5061–5068; [Google Scholar]; b) Lambert JB, Zhao Y, Emblidge RW, Salvador LA, Liu X, So J-H, Chelius EC, Acc. Chem. Res. 1999, 32, 183–190; [Google Scholar]; c) Alabugin IV, J. Org. Chem. 2000, 65, 3910–3919; [DOI] [PubMed] [Google Scholar]; d) Alabugin IV, Zeidan TA, J. Am. Chem. Soc. 2002, 124, 3175–3185; [DOI] [PubMed] [Google Scholar]; e) Alabugin IV, Manoharan M, Peabody S, Weinhold F, J. Am. Chem. Soc. 2003, 125, 5973–5987. [DOI] [PubMed] [Google Scholar]
- [35].a) Bingham RC, J. Am. Chem. Soc. 1975, 97, 6743–6746; [Google Scholar]; b) Salzner U, Schleyer P. v. R., J. Am. Chem. Soc. 1993, 115, 10231–10236; [Google Scholar]; c) Christen D, Mack H-G, Rüdiger S, Oberhammer H, J. Am. Chem. Soc. 1996, 118, 3720–3723; [Google Scholar]; d) Cortés F, Tenorio J, Collera O, Cuevas G, J. Org. Chem. 2001, 66, 2918–2924; [DOI] [PubMed] [Google Scholar]; e) Takahashi O, Yamasaki K, Kohno Y, Ueda K, Suezawa H, Nishio M, Carbohydr. Res. 2009, 344, 1225–1229; [DOI] [PubMed] [Google Scholar]; f) Geng S, Ren Y, Wong N-B, Li W-K, J. Phys. Chem. A 2012, 116, 3952–3959; [DOI] [PubMed] [Google Scholar]; g) Wang C, Chen Z, Wu W, Mo Y, Chem. Eur. J. 2013, 19, 1436–1444. [DOI] [PubMed] [Google Scholar]
- [36].a) Bystrov VF, Pro. Nucl. Magn. Reson. Sp. 1976, 10, 41–82; [Google Scholar]; b) De Leeuw FAAM, Altona C, Int. J. Pept. Protein Res. 1982, 20, 120–125. [DOI] [PubMed] [Google Scholar]
- [37].For an excellent review interpreting spectroscopic data induced by anomeric effects (X-ray, torsional angles and NMR), see; Lemieux RU, Pure Appl. Chem. 1971, 27, 527–548. [Google Scholar]
- [38].For an example presenting a typical value of a spin-spin coupling constant in acyclic chloroaminals (3J = 5–6 Hz), see:; Denmark SE, Wynn T, Beutner GL, J. Am. Chem. Soc. 2002, 124, 13405–13407. [DOI] [PubMed] [Google Scholar]
- [39].a) Janetka JW, Raman P, Satyshur K, Flentke GR, Rich DH, J. Am. Chem. Soc. 1997, 119, 441–442; [Google Scholar]; b) Reid RC, Kelso MJ, Scanlon MJ, Fairlie DP, J. Am. Chem. Soc. 2002, 124, 5673–5683. [DOI] [PubMed] [Google Scholar]
- [40].Bond length in 4b have been compared with the average bond lengths determined statistically from the CCDC data bank (Cambridge Crystallographic Data Centre):; Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R, J. Chem. Soc., Perkin Trans 2 1987, S1–S19. [Google Scholar]
- [41].For bond elongation in pyranosyl bromide, see: ; a) Doherty RM, Stewart JM, Benson WR, Maienthal MM, De Camp WH, Carbohydr. Res. 1983, 116, 150–155; [Google Scholar]; b) Praly J-P, Brard L, Descotes G, Toupet L, Tetrahedron 1989, 45, 4141–4152; [Google Scholar]; c) Benz A, Immel S, Lichtenthaler FW, Tetrahedron: Asymmetry 2007, 18, 1108–1114; [Google Scholar]; d) Hugenberg V, Frohlich R, Haufe G, Org. Biomol. Chem. 2010, 8, 5682–5691; [DOI] [PubMed] [Google Scholar]; e) Monch B, Gebert A, Emmerling F, Becker R, Nehls I, Carbohydr. Res. 2012, 352, 186–190. [DOI] [PubMed] [Google Scholar]
- [42].a) Ford DD, Lehnherr D, Kennedy CR, Jacobsen EN, ACS Catal. 2016, 6, 4616–4620; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN, Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Crich D, Acc. Chem. Res. 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- [44].Bendelsmith AJ, Kim SC, Wasa M, Roche SP, Jacobsen EN, J. Am. Chem. Soc. 2019, 141, 11414–11419. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.