

Abstract
Gpr126/Adgrg6, an adhesion family G protein-coupled receptor (aGPCR), is required for the development of myelinating Schwann cells in the peripheral nervous system. Myelin supports and insulates vertebrate axons to permit rapid signal propagation throughout the nervous system. In mammals and zebrafish, mutations in Gpr126 arrest Schwann cells at early developmental stages. We exploited the optical and pharmacological tractability of larval zebrafish to uncover drugs that mediate myelination by activating Gpr126 or functioning in parallel. Using a fluorescent marker of mature myelinating glia (Tg[mbp:EGFP-CAAX]), we screened hypomorphic gpr126 mutant larvae for restoration of myelin basic protein (mbp) expression along peripheral nerves following small molecule treatment. Our screens identified five compounds sufficient to promote mbp expression in gpr126 hypomorphs. Using an allelic series of gpr126 mutants, we parsed the ability of small molecules to restore mbp, suggesting differences in drug efficacy dependent on Schwann cell developmental state. Finally, we identify apomorphine hydrochloride as a direct small molecule activator of Gpr126 using combined in vivo/in vitro assays and show that aporphine class compounds promote Schwann cell development in vivo. Our results demonstrate the utility of in vivo screening for aGPCR modulators and identify small molecules that interact with the gpr126-mediated myelination program.
Keywords: adhesion GPCR, Gpr126/Adgrg6, Schwann cells, myelin, zebrafish
INTRODUCTION
Adhesion G protein-coupled receptors (aGPCRs) are the second largest class of GPCRs and facilitate proper function across different organ systems1-3. Within the peripheral nervous system, Gpr126/Adgrg6 is necessary for myelination, the process by which Schwann cells iteratively wrap axons to permit rapid conduction of action potentials4-6. Gpr126 functions cell-autonomously in myelinating Schwann cells to elevate cAMP, which drives Schwann cell wrapping in both development and regeneration7, 8. The necessity of Gpr126 in myelination is conserved between zebrafish and mice9-11, illustrating the importance of Gpr126 in nervous system function across vertebrates.
Like other aGPCRs, Gpr126 has a large extracellular N-terminal fragment (NTF) which is cleaved from the 7-pass transmembrane C-terminal fragment (CTF) by the GPCR autoproteolysis-inducing (GAIN) domain at the GPCR proteolysis site (GPS)2, 3, 12, 13. Gpr126-CTF is sufficient for cAMP elevation via Gs protein/adenylyl cyclase pathway to activate protein kinase A, which promotes wrapping of Schwann cells around axons7, 14. Gpr126-NTF has multiple domains that classically promote cell-cell and cell-matrix adhesion (Figure 1A). Recently, we identified laminin-211 as a novel binding partner for Gpr126-NTF that modulates Gpr126 activation states in Schwann cell development15. Other ligands, collagen IV and the cellular prion protein PrPc, are proposed to interact with the CUB/PTX domains in Gpr126-NTF16, 17. Interacting ligands likely regulate Gpr126-CTF signaling by mediating availability of the Stachel, a tethered agonist at the N terminus of Gpr126-CTF that is both necessary and sufficient for Gpr126 activation18. Furthermore, Gpr126-NTF is independently sufficient to mediate radial sorting, the process by which Schwann cells select and encompass a single axon segment in a 1:1 relationship. Based on our in vivo studies and in vitro assays, we predict that laminin-211 stabilizes Gpr126-NTF-CTF interaction to prevent Stachel-mediated activation during Gpr126-NTF-mediated radial sorting. During maturation of the Schwann cell basal lamina, laminin-211 polymerization facilitates mechanical modulation of the Gpr126-NTF or isomerization of the tethered agonist to promote Stachel-mediated signaling and Schwann cell myelination15, 18.
Figure 1. A small molecule suppressor screen in gpr126 hypomorphs for compounds that promote Schwann cell differentiation.

(A) Schematic of zebrafish Gpr126 protein and mutant alleles. (B) Schematic of zebrafish Schwann cell development (green) around axons (gray) in cross-section. Radial sorting begins around 36 hours post-fertilization (hpf); wrapping is observed by 60 hpf. (C) Schematic of 5-6 days post-fertilization (dpf) larval zebrafish with central nervous system (CNS) myelin (blue) and myelinated nerves in the peripheral nervous system (PNS) (green). PLLn = posterior lateral line nerve. Boxed region shown in panels D-E. (D-E) Tg(mbp:EGFP-CAAX) expression (henceforth mbp:gfp) in wild-type (WT) and gpr126st63 larvae at 5 dpf. Brackets denote spinal cord, arrows indicate PLLn, arrowheads mark emerging motor axons. Note strong mbp:gfp expression in the PLLn of WT (D-D’) but reduced expression in gpr126st63 PLLn (E-E’). (F) Logic of gpr126st63 suppressor screen. (G) Workflow for primary small molecule screening of gpr126st63; mbp:gfp larvae.
Because GPCRs promote intracellular signaling in response to extracellular stimuli, they are superb candidate targets for pharmacological manipulation and therapeutics19, 20. Indeed, GPCRs have served as targets in screening compounds for myelination in the central nervous system (CNS)21, 22. Given its central role in multiple stages of Schwann cell development, we reasoned that Gpr126 is an informative target for identification of small molecules mediating myelination. Furthermore, because the Gpr126-mediated myelination program is conserved in zebrafish, we could perform in vivo pharmacological screening for small molecules that modulate Gpr126. Zebrafish larvae are a premiere model system for in vivo drug screening for myelin development due to their optical transparency, large numbers of embryos from a single mating event, and ability to readily absorb small molecules within their culture water23-25. We previously demonstrated pharmacological activation of the Gpr126-mediated peripheral myelination program by addition of the adenylyl cyclase activator forskolin (FSK). A short pulse of FSK during development of gpr126 mutant Schwann cells is sufficient to promote mbp expression and, in radially sorted Schwann cells, wrapping and myelination6, 15. Furthermore, Schwann cell development can be modulated in parallel to Gpr126 signaling; for instance, the ErbB2/3 receptor inhibitor AG1478 is sufficient to disrupt Schwann cell migration and differentiation in zebrafish larvae26. The unique utility of the system is highlighted by several screens and recently developed tools designed to identify compounds that modulate myelination in zebrafish, including those that target gpr12627-33.
In the present study, we describe an unbiased small molecule drug screen in gpr126 hypomorphs that revealed mediators of Schwann cell maturation. For rapid screening, we utilized larvae carrying a fluorescent transgenic marker of myelinating glial cells that reliably labels differentiated glial cells in the central and peripheral nervous systems34, 35. Our screen uncovered five compounds capable of suppressing the hypomorphic gpr126 phenotype and restoring mbp expression in the periphery to different degrees. Using an established allelic series of gpr126 zebrafish mutants6, 15, 18, we characterized the efficacy of drugs that suppress the hypomorphic gpr126st63 phenotype and restore mbp expression at different stages of Schwann cell development as well as different Gpr126 signaling modalities (e.g. Stachel dependent or independent). From these assays, we identified and characterized apomorphine hydrochloride, an aporphine class morphine derivative, as a direct Gpr126 agonist sufficient to elevate cAMP in vitro and promote mbp expression in vivo. Characterization of other aporphines revealed that a related compound, glaucine, has similar ability to interact with Gpr126 and promote myelination. Our screen therefore has identified novel regulators of the Schwann cell myelination program as well as new agonistic ligands for Gpr126.
MATERIALS AND METHODS
For all assays, detailed methods are available in the supplementary material.
Zebrafish strains and husbandry
All animal experiments were performed in compliance with institutional animal protocols. Established wild-type AB*, gpr126st49 and gpr126st63 mutants6, 36, gpr126stl47mutants15, gpr126stl215 mutants18, and Tg[mbp:EGFP-CAAX] (mbp:gfp)34 were used in this study. Zebrafish embryos were collected, raised, and staged using standard techniques37, 38.
Primary and secondary screening in gpr126st63; mbp:gfp
Schematics are provided in Figure 1G and 2G. Primary screen: gpr126st63; mbp:gfp larvae were treated with 10 μM Pharmakon 1600 library (MicroSource Discovery Systems, Inc.) from 48-72 hpf. 25 μM forskolin (FSK, Sigma-Aldrich) treated at 48-53 hpf served as positive control6; negative controls were untreated or 1% DMSO. mbp:gfp in the CNS, PLLn, and motor nerves was scored at 5-6 days post-fertilization (dpf). See detailed methods for scoring criteria including hit threshold. Secondary screen: gpr126st63; mbp:gfp were treated 48-72 hpf with hit compounds from the Pharmakon library and scored as before. Compounds that met hit criteria were validated with whole-mount in situ hybridization (Table 1).
Figure 2. In vivo small molecule screening of gpr126 hypomorphs reveals five suppressor compounds.

(A) mbp:gfp expression in control and (B) 25 μM forskolin (FSK) pulsed gpr126st63 5 dpf larvae. Brackets denote spinal cord, arrows indicate PLLn, dotted lines indicate melanocytes obscuring PLLn. (C) Quantification of mbp:gfp as a percentage of larvae with each mbp PLLn phenotype at 5-6 dpf. *** p<0.001, Fisher’s Exact Test, “non-st63-like” vs “st63-like.” (D) Transformation of data from panel C. Dots indicate individual larva and are jittered to show all samples. Bars indicate mean ± SD. *** p<0.001, Student’s t-test. (E) Quantification of mbp:gfp(+) motor axons at 5-6 dpf. NS = no significant difference. (F) PLLn score and mbp:gfp(+) motor axons for 1462 Pharmakon compounds. Each square = one compound. Dots are jittered and transparent to show all samples. (G) Workflow of screens for gpr126st63 suppressor compounds. (H) Rolipram restores PLLn mbp:gfp but not (I) motor axons in gpr126st63. * p<0.05, Fisher’s Exact Test, “non-st63-like” vs “st63-like”.
Table 1:
mbp expression in larvae treated with small molecules used in primary, secondary, and validation screens
| Primary Screening (mbp:gfp) | Secondary Screening (mbp:gfp) | Validation with ISH | |||||
|---|---|---|---|---|---|---|---|
| average mbp:gfp PLLn score |
average mbp:gfp motor nerves |
n= | # increased PLLn mbp:gfp (score > 2) out of n |
average mbp:gfp motor nerves |
# increased PLLn mbp:gfp (score > 2) out of n |
Hit for allelic series test? |
|
| control (DMSO, untreated) | 0.7 | 1 | 113 | 0/3 | 1.3 | 0/2 | n/a |
| forskolin pulse | 1.6 | 0.7 | 22 | 3/3 | 2.3 | 3/3 | n/a |
| GPCR binding | |||||||
| apomorphine hydrochloride | 1.7 | 0.7 | 3 | 1/2 | 0.5 | ND* | yes* |
| telmisartan | 1 | 6 | 3 | 1/3 | 4 | 1/2 | yes |
| naloxone hydrochloride | 1.7 | 8 | 3 | 0/3 | 3.3 | 2/3 | yes |
| tolazoline hydrochloride | 1.7 | 0 | 3 | 0/3 | 3.4 | 0/3 | |
| oxelaidin citrate | 1.5 | 0 | 2 | 1/3 | 1.7 | 0/3 | |
| norgestrel | 2 | 0.7 | 3 | 1/3 | 0 | 0/2 | |
| cAMP elevator | |||||||
| rolipram | 2.3 | 1 | 3 | 1/3 | 3 | 1/3 | PDE inhibitor |
| steroids/hormones | |||||||
| triamcinolone acetonide | 3 | 0 | 3 | 0/3 | 3.7 | 0/3 | |
| levothyroxine | 2 | 10 | 1 | 1/3 | 4.7 | 0/2 | |
| analgesic | |||||||
| glafenine | 1.3 | 4.7 | 3 | 1/3 | 1 | 0/3 | |
| other nervous system modulator | |||||||
| duloxetine hydrochloride | 1.5 | 3 | 2 | 1/3 | 3 | 0/3 | |
| isaxonine | 0.7 | 4.3 | 3 | 1/3 | 1 | 0/3 | |
| aceglutamide | 1.7 | 1.7 | 3 | 0/2 | 4.5 | 0/2 | |
| antibacterial | |||||||
| ceforanide | 1.3 | 3.3 | 3 | 1/3 | 1.7 | 0/3 | |
| betamipron | 0.7 | 4.7 | 3 | 0/3 | 3.3 | 0/3 | |
| chlortetracycline hydrochloride | 0.7 | 3.3 | 3 | 1/3 | 2.7 | 0/3 | |
| antifungal/antihelminthic | |||||||
| acedapsone | 1.7 | 1.3 | 3 | 1/3 | 2.3 | 0/1 | |
| oxfendazole | 2.7 | 5.7 | 3 | 1/3 | 2.3 | 0/3 | |
| undecylenic acid | 1 | 4.3 | 3 | 1/3 | 2 | 1/2 | yes |
| artemether | 1.7 | 1 | 3 | 5/12** | 11 | 0/10** | |
| antineoplastic | |||||||
| azacitidine | 1.3 | 3.3 | 3 | 0/3 | 4 | 0/3 | |
| other | |||||||
| sodium gluconate | 1 | 3.3 | 3 | 0/3 | 3.3 | 0/3 | |
| tetroquinone | 1.3 | 5.3 | 3 | 0/3 | 6 | 0/3 | |
| allantoin | 1.5 | 2.5 | 2 | 0/3 | 3.3 | 0/1 | |
| benurestat | 1 | 4.5 | 2 | 1/3 | 1 | 0/2 | |
Small molecule treatment for in vivo dose-response and allelic series experiments
See detailed methods for chemical source and dilution information. Embryos were collected from heterozygous in-crosses and raised in 0.003% PTU for whole-mount in situ hybridization. 25-50 μM FSK served as positive control and 1% DMSO served as negative control. Other drugs and concentrations were added as indicated. Whole-mount in situ hybridization was performed using standard protocols39, 40. The mbp riboprobe was previously established26. An established PLLn scoring rubric was applied for measuring mbp expression18. For dose-response experiments with apormorphine and other aporphines, survival was recorded as the proportion of larvae alive immediately before and after drug treatment. Aporphine screening was performed in a smaller volume (2 mL), resulting in higher toxicity for apomorphine; see detailed methods. Scoring was performed with the observer blinded to genotype and treatment. Data are pooled across at least two technical replicates for each drug and allele and analyzed with GraphPad Prism.
In vitro functional assays
Full-length and Δ6bp mutant zebrafish gpr126 constructs were cloned as described previously18 and heterologously expressed in COS-7 cells. For cAMP accumulation assays, cells were transfected with 100 ng of plasmid using Lipofectamine™ 2000 (Thermo Fisher Scientific, Darmstadt, Germany) and incubated with concentrations of test substances. To measure cAMP concentration, the AlphaScreen cAMP assay kit (PerkinElmer Life Sciences) was used according to the manufacturer’s protocol.
Imaging and analysis for aporphine compound screening
gpr126; mbp:gfp larvae were visualized at 5 dpf using a Vertebrate Automated Sorting Technology (VAST) BioImager (Union Biometrica, Holliston, MA) coupled to a spinning disc confocal microscope (SDCM) at 16X magnification (Carl Zeiss Microscopy GmbH, Jena, Germany). Maximum intensity projections were generated from z-stacks to quantify PLLn mbp:gfp in region of interest. See detailed methods for more information. Images were quantified with Fiji41 and data were analyzed with GraphPad Prism.
RESULTS
A suppressor screen for mbp:gfp expression in gpr126 hypomorphs
Because Gpr126 has differential and necessary roles in multiple stages of Schwann cell development, we hypothesized that we could identify small molecule modulators of peripheral myelination by screening for suppressors of gpr126st63 hypomorphs. Several established gpr126 zebrafish alleles result in complete abrogation of mbp expression (gpr126stl47, gpr126st49, and gpr126stl215) due to loss of Stachel-mediated CTF signaling6, 15, 18 (Figure 1A). We instead utilized the gpr126st63 allele, which contains a lesion exchanging a conserved cysteine for tyrosine (Figure 1A) that results in reduced membrane expression, radial sorting, mbp expression, and myelination15, 18. The remainder of the gene sequence is intact in gpr126st63 and should encode a functional NTF, complete Stachel sequence, and partially functional CTF sufficient to elevate cAMP to produce myelinated axons, though fewer than in wild-type. We reasoned that screening for gpr126st63 suppressor molecules would allow us to identify modulators of Schwann cell development mediated by different domains of Gpr126.
Gpr126-NTF is necessary and sufficient for radial sorting, which begins in zebrafish around 36 hpf in the posterior lateral line nerve (PLLn) (Figure 1B-C). Following radial sorting, Gpr126-CTF signals to promote mbp expression and wrapping around 60 hpf in the PLLn. Soon after, Schwann cells populate motor axons emanating from the spinal cord (Figure 1C). We predicted that small molecule treatment from 48-72 hpf would encompass this window and capture potential suppressor molecules that interact with gpr126 during sorting and wrapping stages of Schwann cell development in different peripheral nerves.
To increase throughput, we observed differentiation of Schwann cells using a well-established transgenic zebrafish strain carrying membrane-tagged EGFP under the control of the myelin basic protein (mbp) promoter (Tg[mbp:EGFP-CAAX], henceforth referred to as mbp:gfp)34, 35. In wild-type larvae, mbp:gfp is strongly expressed in the spinal cord, PLLn, and initial segments of motor nerves by 5 dpf. In contrast, gpr126st63 have substantially reduced PLLn mbp:gfp while maintaining strong fluorescence in the spinal cord (Figure 1D-E). We reasoned that small molecule treatment could suppress the gpr126st63; mbp:gfp phenotype, restoring fluorescence in the PLLn (Figure 1F). Because motor nerve myelination commences at 4-5 dpf, it is conceivable that drugs activating Gpr126 could also promote mbp:gfp expression and myelination of motor nerves. In contrast, small molecules that prevent myelination would enhance the gpr126st63 phenotype, resulting in reduced peripheral mbp:gfp.
Given these tools and rationale, we designed a screen to test compounds in the Pharmakon 1600 library for restoration of mbp:gfp in gpr126st63 hypomorphs (Figure 1G). This library contains 1,600 compounds with wide structural variety that are approved for therapeutic use in humans. To screen for gpr126st63 suppressors, we generated large numbers of gpr126st63; mbp:gfp embryos and arrayed the progeny into 96-well plates with three larvae per well, the maximum number that could survive the well volume. At 48 hpf, coincident with Schwann cell radial sorting, we added 10 μM library drug to each well. Larvae were incubated with drug in embryo medium for 24 hours from 48-72 hpf, encompassing the critical window for cAMP elevation and initiation of the Schwann cell terminal differentiation program. At 5-6 dpf, we scored mbp:gfp expression as follows: First, we ensured bright fluorescence mbp:gfp in the CNS to control for transgene expression. Then, we scored relative mbp:gfp fluorescence of the PLLn categorically as previously defined18. We also counted the number of motor nerves with mbp:gfp ventral to the horizontal myoseptum. Because central nervous system glia can myelinate the initial segment of motor axons exiting the spinal cord42, 43 and because myelination is typically incomplete at 5-6 dpf, these could represent drugs promoting Schwann cell myelination of motor nerves. We scored samples manually because bright CNS mbp:gfp directly adjacent to the PNS precludes automated scoring. Manual qualitative scoring (with a dissecting stereoscope and established rubric) was ultimately higher throughput and more consistent than quantitative scoring, which introduced methodological variability.
With these parameters, we were able to screen 1,462 drugs for suppression of the gpr126st63 hypomorphic phenotype, or 91.4% of the Pharmakon 1600 library. Of the 138 drugs not scored, 134 were lethal to all treated larvae, representing 8.4% of the total drugs screened (Supplementary Table 1). This moderate lethality rate suggests that our initial screen dose of 10 μM was appropriate to balance high hit rate with general health of screened larvae. The remaining four unscreened drugs had larvae with no transgenic fluorescence, including no expression of co-selection marker. These larvae likely exhibited transgene silencing independent of drug treatment, a common issue44 that we observed stochastically throughout other drug treatment groups.
Identification of small molecules that restore mbp:gfp in gpr126st63 hypomorphs
To set a threshold for significant suppression of gpr126st63, we analyzed mbp:gfp expression in control (either DMSO-treated or untreated) or 25 μM FSK-treated larvae. Control gpr126 larvae had weak or no fluorescence along the PLLn, the location of which is marked by adjacent pigment cells along the side of the larvae (Figure 2A). In contrast, larvae treated with a moderate dose of 25 μM FSK (comparable to 10 μM dose of drugs in library) exhibit restored PLLn mbp:gfp due to activation of the Schwann cell differentiation program downstream of Gpr126 (Figure 2B). We scored the PLLn of negative and positive control-treated larvae by binning into categories of none, weak, some, or strong mbp:gfp expression and found that the vast majority (95.6%) of control larvae have weak or no fluorescence (40/113 or 35.4% “none”, 68/113 or 60.2% “weak”, Figure 2C). mbp:gfp expression is significantly restored in FSK-treated larvae with over half (13/22, 59.1%) scored as either some or strong mbp:gfp expression. We therefore called “none” and “weak” gpr126st63-like PLLn phenotypes, while “some” and “strong” were considered non-gpr126st63-like PLLn phenotypes (p<0.001, control vs. FSK, Fisher’s exact test). Because we screened a large number of drugs with relatively few larvae (n≤3), we transformed these data to numerical scores to set a threshold for primary screen hits. Conversion of qualitative scores to numerical scores (none = 0, weak = 1, some = 2, strong = 3) demonstrated the significant difference between control and FSK-treated larvae (Figure 2D, 0.69±0.55 in control vs. 1.6±0.85 in FSK-treated, p<0.001, Student’s t-test). Finally, we found that mbp:gfp in motor nerves is not significantly restored with FSK treatment (Figure 2E, 0.96±1.4 in control vs. 0.73±1.1 in FSK, p>0.45).
With these control values, we set a hit threshold for average PLLn score and average number of mbp:gfp(+) motor nerves above the standard deviation for gpr126st63 controls. Primary screen drugs were counted as a “hit” if the average PLLn score across larvae (n≤3) was 1.5 or greater (yellow dotted lines in Figure 2D, F), nearly the mean of FSK rescue. In addition, the presence of at least three mbp:gfp(+) motor nerves signified a primary screen hit (green dotted lines in Figure 2E-F) as this indicated significantly more gfp(+) motor axons than observed in either control treatment. With these thresholds applied, we found 98 compounds that met one or both criteria: 50 compounds (51%) had a PLLn score ≥1.5, 39 (40%) had >3 mbp:gfp motor nerves, and 9 (9%) met both criteria (Figure 2F).
Because of the high hit rate (98/1462, 6.7% of compounds tested), we performed a secondary screen with only hit compounds from the primary screen. We reasoned that additional rounds of screening hit compounds would ultimately remove false positives without the need for repeatedly screening the entire library. In this second round of testing, we found that treatment with 25 compounds increased PLLn score in at least one larvae and/or had an average of >3 myelinated motor nerves (Figure 2G, Table 1). We assigned functional terms to these secondary screen hits based on predicted function in PubChem (pubchem.ncbi.nlm.nih.gov/). Six hits in the secondary screen interact with GPCRs (Table 1), which represents the largest functional class in our secondary screen. Interestingly, two compounds are derivatives of morphine, naloxone hydrochloride and apomorphine hydrochloride.
Rolipram restores mbp:gfp in PLLn of gpr126st63 hypomorphs
As a proof-of-principle for our primary and secondary screens, we found elevated mbp:gfp expression in gpr126st63 larvae treated with rolipram, a selective phosphodiesterase-4 inhibitor. Rolipram treatment is sufficient to elevate Schwann cells’ cAMP levels to promote myelination in mouse models45. Using a new stock of rolipram (i.e., not from the Pharmakon library), we confirmed that PLLn mbp:gfp is partially restored in 10 μM rolipram-treated gpr126st63 larvae (Figure 2H, 3.6% “some” score in control vs. 25.9% “some” in rolipram-treated, p<0.05, Fisher’s exact test). PLLn mbp:gfp is further restored in a dose-dependent manner with 50 μM rolipram treatment (22.9% “some” and 2.9% “strong” in 50 μM rolipram, p<0.05, Fisher’s exact test vs. control). However, similar to FSK, rolipram treatment is insufficient to significantly elevate mbp:gfp in gpr126st63 motor nerves at either 10 μM (1.19±1.44 mbp:gfp(+) motor nerves) or 50 μM (0.94±1.1) rolipram relative to control (0.67±0.98, p>0.05 vs. 10 μM and vs. 50 μM, Student’s t-test, Figure 2I). The identification of rolipram in our screens highlights our ability to identify small molecules that regulate cAMP in Schwann cells in an unbiased manner. Additionally, the difference in restoration of mbp:gfp in PLLn vs. motor nerve with rolipram treatment supports our hypothesis that cAMP elevation can promote differentiation in the PLLn, but is not sufficient to drive Schwann cell development in motor axons.
Validation of secondary screen hits in gpr126st63 hypomorphs
Based on the results of our secondary screen, we attempted to validate these hits using a different assay to visualize mbp expression and an independent gpr126st63 strain. We used Pharmakon 1600 library drug hits from the secondary screen to treat gpr126st63 larvae without mbp:gfp to ensure drug efficacy between different strains of hypomorphic mutants. In addition, we co-treated larvae with phenylthiourea (PTU) to abolish pigment formation and allow visualization of mbp via established whole-mount in situ hybridization methods39, 40. In our validation assay, four drugs were sufficient to elevate PLLn mbp in at least one of three gpr126st63 hypomorphs: telmisartan, naloxone hydrochloride, rolipram, and undecylenic acid (Table 1, Figure 3). Another small molecule hit, apomorphine hydrochloride, did not survive the validation assay, but rescue was observed in subsequent assays using new drug stock (see Figure 4). Because the mechanistic role of rolipram in Schwann cell development is established, apomorphine, telmisartan, naloxone, and undecylenic acid were selected for further study.
Figure 3. An allelic series of gpr126 parses direct versus indirect compound function.

(A) Molecular structure for screen hits (PubChem). (B) Schematic of gpr126 alleles, predicted proteins, phenotypes, and predicted drug functions based on mbp restoration. (C-U) Whole-mount in situ hybridization for mbp in wild-type (WT) or gpr126. Black arrows indicate PLLn. Fraction of gpr126 larvae with increased mbp is noted in upper right of each image. (C-G) mbp expression in control larvae. Note absence of PLLn mbp in gpr126stl47, gpr126st49, and gpr126stl215 (D-F) and reduction in gpr126st63 (G). (H-L) PLLn mbp expression is partially restored in gpr126st49 (J), gpr126stl215 (K), and gpr126st63 (L) in 10 μM undecylenic acid. (M-Q) PLLn mbp is weakly increased in one larva for gpr126st49 (O) and gpr126stl215 (P) but strongly increased in gpr126st63 (Q) with 10 μM naloxone. (R-U) mbp PLLn expression is increased across all alleles in 5 μM telmisartan-treated larvae.
Figure 4. Apomorphine hydrochloride suppresses gpr126 hypomorphic phenotype at high doses.

(A-C, G-J) Whole-mount in situ hybridization for mbp in PLLn of gpr126st63 (A-C) or gpr126stl215 (G-J) with control, apomorphine, or forskolin (FSK) treatment at 5 dpf. Black arrows indicate PLLn. (D, K) Quantification expressed as a percentage of larvae with each mbp PLLn phenotype at 5 dpf. * p<0.05, *** p<0.001, Fisher’s Exact Test, “wild-type” vs. “mutant” (gpr126st63 or gpr126stl215) categories for treated vs. control. (E-F) Survival curve following 24-hour treatment with indicated concentrations of apomorphine. Dots indicate mean ± SD % alive across technical replicates. *** p<0.001, 0 vs.100 μM apomorphine. Sample numbers in Detailed Methods.
Prediction of direct vs. indirect gpr126 interaction with an allelic series of gpr126 mutants
We next considered the structure and function of hit compounds to predict whether they might function by binding/ activating Gpr126 directly (Figure 3A). Undecylenic acid is a hydrophobic fatty acid with antifungal properties; however, its mechanism of action is unknown46, 47. The other three hits are able to bind GPCRs, though they show diverse molecular structures (Figure 3A) and have demonstrated ability to modulate GPCRs in distinct ways. Naloxone hydrochloride is a noncompetitive opioid receptor antagonist that blocks the effects of opiates in drug overdose patients48, 49. Naloxone is a morphine derivative, as is another screen hit, apomorphine hydrochloride. However, apomorphine is structurally distinct from naloxone and is instead a dopamine receptor agonist sufficient to bind both D1 and D2-type receptors which have differential effects on cAMP production50, 51. Telmisartan also modulates cAMP as an antagonist for the angiotensin II type 1 receptor52, 53. Given the variety of screen hits, we elected to test whether they act directly on Gpr126 or in parallel/downstream using an allelic series of gpr126 mutants.
Because gpr126st63 encodes a full-length sub-functional protein, we used three other established gpr126 alleles with distinct phenotypes based on the lesion effect on Gpr126 structure. The strongest loss-of-function allele, gpr126stl47, contains a Δ5+3 base pair lesion in exon 2 that produces an early STOP codon and truncates the protein in the CUB domain (Figure 1A). In gpr126stl47 mutants, Schwann cells neither radially sort nor myelinate axons15 (Figure 3B). In contrast, both gpr126st49 and gpr126stl215 mutants have intact coding sequences for Gpr126-NTF and consequently have normal radial sorting (Figure 1A, 3B). However, gpr126st49 contains a nonsynonymous point mutation that produces a STOP codon within the GAIN domain, and therefore is predicted to lack a functional CTF to promote Schwann cell wrapping of axons. gpr126stl215 mutants have an intact CTF signaling domain with the exception of a Δ6 base pair lesion in the Stachel sequence that precisely deletes two amino acids necessary for Stachel-mediated signaling and myelination18 (Figure 3B). In all three gpr126 alleles, PLLn mbp expression is completely absent due to a failure of Gpr126 signaling, in contrast to strong mbp expression in wild-type (WT) and reduced expression in gpr126st63 (Figure 3C-G). Finally, gpr126st63 has an intact NTF to promote radial sorting, though decreased receptor trafficking results in less available surface NTF and reduced radial sorting15, 18. We therefore tested for restoration of PLLn mbp expression across alleles, which would allow us to infer the mechanism of drug action on Gpr126 as schematized in Figure 3B.
Undecylenic acid, naloxone hydrochloride, and telmisartan modulate mbp expression in parallel to Gpr126-CTF
We found that undecylenic acid is sufficient to drive mbp expression in radially sorted PLLn Schwann cells (Figure 3H-L). In gpr126stl47 mutants, PLLn mbp is absent in control and 10 μM undecylenic acid-treated larvae (Figure 3D, I). However, we observed partial restoration of mbp in a subset of gpr126st49 and gpr126stl215 mutants treated with 10 μM undecylenic acid (Figure 3J-K), though not to the level of mbp expression observed in gpr126st63 (Figure 3I). Because gpr126st49 lacks a CTF signaling domain, we concluded that undecylenic acid acts in parallel or potentially downstream of Gpr126 to promote Schwann cell mbp expression.
Similar results were obtained with naloxone hydrochloride (Figure 3M-Q). We observed no PLLn mbp in 10 μM naloxone-treated gpr126stl47 mutants (Figure 3N), but very slight suppression of gpr126st49 and gpr126stl215 (Figure 3O-P). mbp was observed in only one larva for each allele treated with naloxone; however, mbp is never observed in the PLLn of untreated mutants6, 15, 18 and therefore suggests a small but weak level of mutant phenotype suppression. This is in contrast to gpr126st63 mutants with strong PLLn mbp following naloxone treatment (Figure 3Q). We therefore conclude that naloxone is a weak suppressor in Stachel-absent mutants and requires Schwann cell radial sorting to suppress gpr126 mutant phenotypes.
In contrast to undecylenic acid and naloxone, we observed PLLn mbp expression in all alleles following telmisartan treatment (Figure 3R-U). mbp was weakly expressed in a subset of 5 μM telmisartan-treated gpr126stl47 (Figure 3R), gpr126st49 (Figure 3S), and gpr126stl215 (Figure 3T) mutant larvae. Because all three alleles lack a Stachel-containing CTF, we predict that telmisartan functions either in parallel or downstream of the gpr126-mediated myelination program in Schwann cells. We note that even though gpr126stl47 Schwann cells fail to undergo radial sorting, we have nonetheless observed mbp expression in immature gpr126stl47 Schwann cells treated with FSK15. Therefore, we predict that telmisartan also activates Schwann cell differentiation in the absence of both Gpr126 and radial sorting.
Apomorphine hydrochloride rescues mbp expression in gpr126st63 hypomorphs
We next tested the fourth hit from the validation assay, apomorphine hydrochloride, for its efficacy in gpr126 suppression. Because apomorphine only weakly rescues gpr126st63 mutants at 10 μM, we tested a high dose of apomorphine (100 μM) and found it significantly restores mbp expression along the PLLn in 5 dpf larvae relative to DMSO-treated controls (Figure 4A-B, D), though not to the same degree as a high dose of FSK (50 μM pulse) (Figure 4C-D, 1/34 or 2.9% non-gpr126st63 phenotype in control vs. 25/25, 100% in FSK-treated, p<0.001 st63-like vs. non-st63-like, Fisher’s exact test). Higher doses of FSK (100 μM, analogous to apomorphine) result in larval death. To identify an optimal dose of apomorphine for significant restoration of mbp, we performed a dose-response experiment with increasing concentrations of apomorphine in gpr126st63 larvae. We observed a dose-dependent increase of mbp expression in 10, 20, and 100 μM apomorphine-treated larvae, though the effect was only significant at 100 μM (Figure 4D, 4/11, 36.4% non-gpr126st63 phenotype, p<0.05, Fisher’s exact test). However, this dose had an adverse and often lethal effect on larval development. We counted the number of larvae before and after drug treatment to measure this effect. Nearly all DMSO-treated control gpr126st63 larvae (95.6±4.2%) survive over the 24-hour treatment, and this survival rate is only slightly decreased in 10 and 20 μM (93% and 91%, respectively, Figure 4E). Only 77.8% of larvae survive overnight treatment with 50 μM apomorphine, and the most efficacious dose for mbp expression, 100 μM, has a survival rate of only 26.8±13% (p<0.001, Student’s t-test). A shorter pulse of 100μM apomorphine (48-52 hpf, similar to FSK treatment) permitted larvae survival but was insufficient to restore mbp in gpr126st63 (data not shown). Altogether, these data show that apomorphine partially suppresses the gpr126st63 phenotype but with very similar effective and toxic concentration doses.
We also treated the allelic series of gpr126 mutants with apomorphine to test its ability to promote mbp expression. Because of the high rate of lethality with an effective 100 μM dose of apomorphine, we were unable to efficiently conduct assays across all gpr126 allelic mutants. However, we recovered enough treated Stachel-dead gpr126stl215 larvae to assay despite high lethality rate (30.8+36.9% with 100 μM apomorphine vs. 95.7+5.8% in controls, 97.1% in 10 μM, 93.7% in 25 μM and 92.4% in 50 μM, Figure 4F). Treatment with apomorphine resulted in reduced mbp expression in wild-type larvae relative to control (Figure 4G-H, K, 4/6, 66% non-strong phenotype) likely due to compromised larval health. In control-treated gpr126stl215, we observed complete loss of mbp expression in the PLLn (Figure 4I, K, 0/20, 0%), in line with previous studies demonstrating that the Stachel tethered agonist is absolutely required to promote endogenous Gpr126 signaling in Schwann cells18. Critically, treatment with apomorphine partially restored mbp in the PLLn even though mbp was reduced overall. Like with gpr126st63, we found that sub-threshold doses of apomorphine were insufficient to restore mbp (0/11 10 μM, 0/6 25 μM, 0/7 50 μM), while 100 μM partially restored mbp in the PLLn (Figure 4J, 3/10, 30% non-gpr126stl215 phenotype, p<0.05, Fisher’s exact test vs. gpr126st63 phenotype). These data suggest that the Stachel sequence is dispensable for rescue of mbp expression by apomorphine. We conclude that apomorphine is sufficient to suppress both gpr126st63 and gpr126stl215, though with reduced potency relative to other screen hits at a sublethal dose.
Apomorphine directly agonizes Gpr126 in vitro
To extend and verify the in vivo data, we performed in vitro assays to test for direct Gpr126 activation. Full-length wild-type (WT) Gpr126 expressed in COS-7 cells is sufficient to elevate cAMP compared to vector control (Figure 5A-B). Because 0.1% DMSO has an effect on cAMP levels in vitro, we compared substances that were solubilized in water (Figure 5A) separately from substances solubilized in DMSO (Figure 5A). Upon treatment with 100 μM apomorphine, naloxone, telmisartan, and undecylenic, significant elevation of cAMP was observed only with apomorphine (Figure 5A-B, p<0.05, 1-way ANOVA vs. basal activation). These data suggest that apomorphine is likely a direct activator of Gpr126, whereas the other compounds increase mbp in Schwann cells in a parallel or downstream pathway.
Figure 5. Apomorphine hydrochloride is a direct activating ligand for Gpr126.
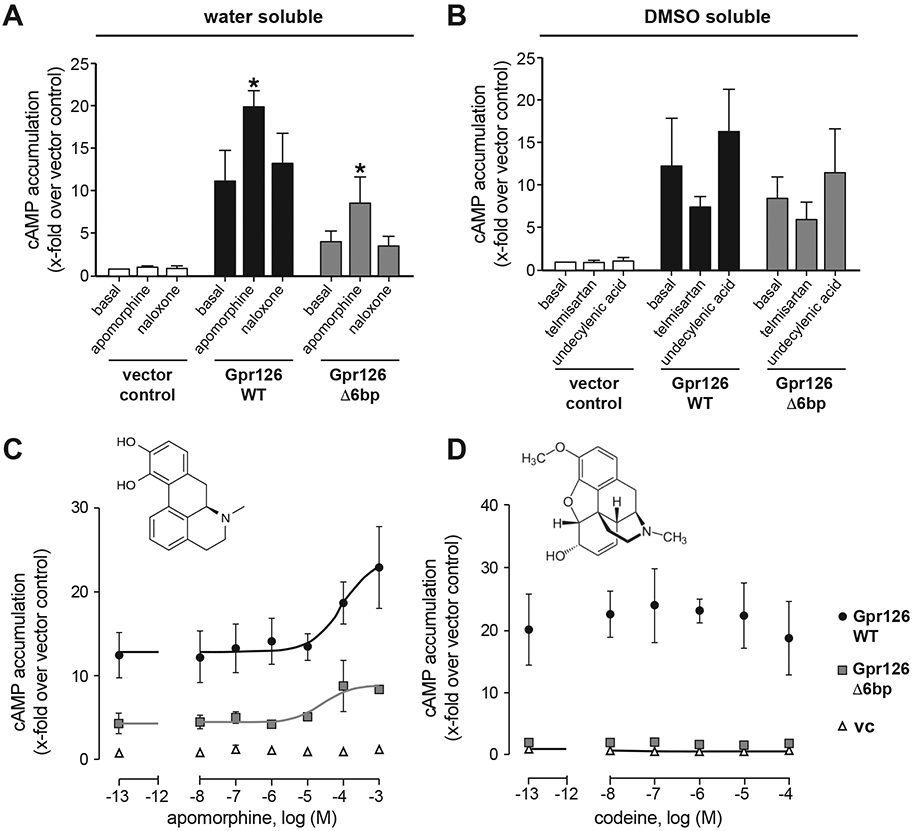
(A-B) COS-7 cells were transiently transfected with empty vector, WT and mutant (Δ6bp lesion as in gpr126stl215) zebrafish gpr126 plasmid, and cAMP accumulation was measured after stimulation with given substances at 100 μM. Results are given as x-folds over vector control (vc: 1.0 ± 1.1 nM) (A) and vector control with 0.1 % DMSO (basal vc: 2.2 ± 3.1 nM) (B) for water and DMSO soluble substances, respectively. Bars show means ± SEM of three independent assays each performed in triplicates. * p<0.05, one-way ANOVA with Dunn’s multiple testing for each substance induced activation vs. basal levels. (C-D) Concentration-response curve of apomorphine (EC50 WT: 21.5 μM Δ6bp mutant: 33.4 μM) (C) and codeine (no dose response detectable) (D). Results are given as x-folds over vector control (vc: 2.9 ± 0.6 nM) of four independent assays each performed in triplicates. Insets show structure of each molecule (PubChem).
Modulation of Gpr126 activity by interacting partners (e.g. Laminin-211) in Schwann cells requires intact Stachel sequence for activation, cAMP accumulation, and mbp expression15, 18, as illustrated by the gpr126stl215 allele (Figure 1A, 3B). We, therefore, expressed an analogous Δ6 base pair Stachel deletion Gpr126 construct (Gpr126 Δ6bp) in COS-7 cells, which shows no basal cAMP elevation, to test whether compound-mediated activation requires a functional tethered agonist. As expected, based on the WT Gpr126 data, we saw no cAMP accumulation in naloxone-, telmisartan-, or undecylenic acid-treated cells carrying Gpr126 Δ6bp. However, treatment with apomorphine significantly elevates cAMP relative to basal levels (Figure 5A, p<0.05, 1-way ANOVA vs. basal activation), suggesting that apomorphine agonizes Gpr126 activation independent from the Stachel sequence.
We also tested whether the effect of apomorphine is specific by analyzing another morphine derivative, codeine, for activation of Gpr126 and Gpr126 Δ6bp. Concentration-response curves revealed an EC50 value of 94.4 μM for apomorphine on WT Gpr126 and 31.8 μM for Gpr126 Δ6bp (Figure 5C). Codeine, however, had no effect on cAMP levels in either Gpr126 construct (Figure 5D). We conclude that undecylenic acid, naloxone, and telmisartan activate the Schwann cell myelination program indirectly, while apomorphine directly and specifically activates Gpr126 independent of Stachel sequence to promote mbp expression.
The aporphine alkaloid glaucine suppresses the gpr126st63 hypomorphic phenotype
Apomorphine suppressed the gpr126 phenotype in vivo (Figure 4) and directly agonized Gpr126 in vitro (Figure 5). Because apomorphine shares an aporphine core scaffold with alkaloid compounds, we hypothesized that other aporphine alkaloids could suppress gpr126st63 hypomorphic phenotype with reduced toxicity. We therefore selected a small set of aporphines for screening: (R)-nuciferine, (S)-glaucine, (S)-boldine, (S)-magnoflorine, and (R)-crebanine (Figure 6A). We first performed small-volume assays (2 mL) with these compounds to measure toxicity compared to apomorphine. Most aporphines exhibited a favorable survival rate (>80%) when incubated from 47-54 hpf with the exception of crebanine and apomorphine (at 10 μM) (Figure 6B). The increased toxicity of apomorphine in this assay is attributed to the smaller incubation volume, which exacerbated the toxicity of apomorphine with increased larval density.
Figure 6. Glaucine suppresses gpr126st63 hypomorphic phenotype likely by direct interaction with Gpr126.

(A) Structures of aporphine compound series. Core scaffold highlighted in blue. (B) Toxicity screening of aporphines from 47-54 hpf. Survival was assessed at 54 hpf. All compounds were at 50 μM unless otherwise noted. Control, 1% DMSO. n≥10 in all cases. (C-G, I-K) PLLn mbp:gfp expression in 5 dpf gpr126st63 (C-G) or gpr126st49 (I-K) following 50 μM compound treatment at 47-54 hpf. Indicated region is the most anterior portion of the PLLn. Scale bar, 25 μm. Control, 1% DMSO. Note increased PLLn mbp:gfp expression in gpr126st63 treated with glaucine (E) compared to control (C) but absence of mbp:gfp in glaucine-treated gpr126st49 (J). (H) Quantification of PLLn mbp expression within ROI of gpr126st63 larvae in C-G. Bars indicate means ± SD. ****p<0.0001, *p<0.05, one-way ANOVA with Dunnett’s multiple testing for each compound against control.
The four remaining aporphines were assessed for their ability to suppress the hypomorphic phenotype in gpr126st63; mbp:gfp (Figure 6C-G). Glaucine showed significant increase in mbp expression along the anterior PLLn compared to control, while the other aporphines had no effect or, for nucifierine, weak inhibition of mbp:gfp (Figure 6H). mbp:gfp expression was not significantly different with a longer incubation with glaucine (47-71 hpf, data not shown), suggesting that a short incubation window from 47-54 hpf is sufficient to suppress the gpr126st63 hypomorphic phenotype for this compound.
To determine whether glaucine might interact directly with Gpr126 like apomorphine, we assayed the ability of glaucine to suppress gpr126st49 phenotype, which encodes Gpr126-NTF but has a premature stop precluding expression of the Gpr126-CTF signaling domain (Figure 3). We observe that glaucine fails to suppress the gpr126st49 phenotype (Figure 6I, J), with no PLLn mbp:gfp in gpr126st49 larvae treated with glaucine (50 μM) from 47-54 hpf. As a positive control, we added 50 μM forskolin, a known small-molecule suppressor of the gpr126st49 phenotype, and saw partial restoration of mbp:gfp expression (Figure 6K). Therefore, we predict glaucine, like apomorphine, acts directly with Gpr126 to promote cAMP elevation and Schwann cell differentiation in vivo.
DISCUSSION
Gpr126 mediates multiple stages of Schwann cell development by virtue of its NTF-CTF interactions and binding of endogenous ligands6, 10, 15, 16, 18. In the present study, we designed a medium-throughput screen in zebrafish to identify small molecules that interact with the gpr126-mediated myelination program. By observing mbp:gfp in gpr126 hypomorphs treated with 1,600 small molecules in the Pharmakon library, we discovered four novel compounds that suppress the gpr126 hypomorph phenotype and partially restore Schwann cell differentiation. Structure-function characterization using an allelic series of gpr126 mutants demonstrates that modulation of the myelination program is dependent upon Schwann cell developmental state. In addition, we discovered that apomorphine hydrochloride, a morphine derivative and dopamine receptor agonist, directly activates Gpr126 in a Stachel-independent manner.
Small molecule screens for regulators of adhesion family GPCRs
Autocatalytic processing of aGPCRs into NTF and CTF and production of the tethered Stachel agonist permits subfunctionalization of receptor domains2, 15, 54, 55. aGPCRs have complex potential to mediate intracellular signal transduction; however, few studies have identified compounds that can perturb aGPCR signaling. Recently, an in vitro screen specifically targeted Gpr56/Adgrg1 and identified dihydromunduletone as a selective antagonist that also antagonizes Gpr114/Adgrg556. Both Gpr56 and Gpr114 are members of the same structural group as Gpr126, Class VIII, which have alike and promiscuous Stachel sequences57, 58. Similarly, a small-scale in vitro screen across a variety of aGPCRs identified beclomethasone diproprionate as an agonist for Adgrg3/Gpr97, another Class VIII aGPCR59. Although in vivo screening in our study is lower throughput than in vitro assays, it offers an attractive complementary approach to find bona fide small molecules interacting with aGPCRs to promote signaling and cellular function.
Myelinating glia in the CNS express multiple druggable targets, including aGPCRs Gpr56/Adgrg1 and Gpr98/Adgrv1 in oligodendrocyte lineage cells60. In mouse and zebrafish gpr56 mutants, oligodendrocyte precursor cells precociously exit the earlier developmental state and result in fewer myelin sheaths61, 62. In contrast, Gpr98 appears to stabilize the mature state of oligodendrocytes, as loss of Gpr98 results in decreased expression of myelin associated glycoprotein (MAG), a mature oligodendrocyte marker63. Therefore, small molecule modulators of Gpr56 and Gpr98 have the potential to restore proper myelination in disease or injury states in the CNS. Our screen serves as a proof-of-principle for identification of therapeutic compounds that target aGPCRs to restore myelination.
Small molecule screens for regulators of glial cell development and myelination
Our screen builds upon previous in vitro64, 65 and in vivo25, 31, 32 screens to identify small molecules promoting glial cell development. Much of the focus has been on restoration of CNS myelin via oligodendrocytes. In contrast, relatively few screens have studied peripheral myelination in vivo. One limitation is the difficulty in screening fluorescence in the periphery of zebrafish (e.g., PLLn or motor nerves) adjacent to the strong transgenic fluorescence in the CNS. The development of more powerful high-throughput analyses of fluorescence and development of new markers may overcome this technical limitation28, 31. Furthermore, we observed that PLLn mbp was more easily restored relative to motor nerves in both fluorescent and in situ hybridization assays, suggesting cAMP elevation alone is not sufficient to restore mbp expression in gpr126 motor axons with treatment up to 72 hpf. This disparity could be due to differences in the nerve structure or function or differences in the populations of Schwann cell precursors along these nerves. Alternatively, we observe few Schwann cells ventral to the horizontal myoseptum at our timepoints (data not shown), thus we may not observe substantial rescue because too few Schwann cells are present.
Another gpr126 suppressor screen was recently performed using different hypomorphic and loss-of-function alleles and took advantage gpr126 function in the ear33. Rather than a fluorescent marker, this screen used whole-mount in situ hybridization in both ear and PLLn. Of 3120 molecules tested, this screen yielded 41 molecules rescuing mbp in the PLLn of gpr126 hypomorphs, with 19 predicted to interact directly with Gpr126 based on suppression of a strong gpr126 allele that lacks CTF function (see rationale in Figure 3). Importantly, this screen was conducted at 60-90 hpf versus our primary screen at 48-72 hpf, which reflects difference in ear developmental stages compared to earlier Gpr126 function in PLLn Schwann cells. Furthermore, while our assay investigated the PLLn in the primary screen, this study tested PLLn mbp in a secondary assay following a primary screen for otic versican b (vcanb). These differences in timing and tissue-specificity likely underlies differences in hits. Our screen primarily uncovered GPCR interactors with morphine-like structures, whereas the other screen recovered calcium channel agonist and antagonists, particularly with dihydropyridine structure. These contrasting hits could distinguish tissue-specific activation mechanisms for Gpr126, as compounds that did not rescue otic gpr126 function would not have been screened in the PLLn. Similarly, we did not see otic vesicle swelling suppression with any of the drugs in our screen with treatment at 48-72 hpf (data not shown). Future studies can parse hits from these screens on Gpr126 function at differing developmental stages, or for interaction with gpr56/adgrg1, which is also necessary for proper peripheral myelin development and maintenance66.
Models for drug action based on influence of Schwann cell developmental state
Gpr126-NTF is required for radial sorting in Schwann cells, though the mechanism by which it intracellularly transduces this cue is unclear15. Our allelic series analyses suggest that undecylenic acid and naloxone are sufficient to promote mbp expression specifically in Schwann cells that are predicted to express Gpr126-NTF and naloxone cannot activate the differentiation program in immature Schwann cells until radial sorting is underway (Figure 3). Undecylenic acid is an antifungal drug without defined receptors47 and thus the nature of activation, and whether it is Schwann cell-autonomous, remains unclear. The mechanism of action may involve a receptor either exclusively expressed in Schwann cells following radial sorting, or one acting non-cell-autonomously as an instructive cue for myelination by pre-myelinating Schwann cells (e.g., as an axonal ligand).
In contrast, naloxone is a well-studied opioid receptor antagonist routinely used to block the effects of opiate overdoses. Opioid receptors are expressed in terminals and processes of sensory and nociceptive neurons67 and promote myelination in cultured oligodendrocytes65, but their expression in Schwann cells is not established. Thus, one model is a non-cell-autonomous effect on Schwann cells mediated by the axons they myelinate. However, the low level of rescue in gpr126st49 and gpr126stl215 hints at potential indirect and direct Gpr126 activation by naloxone specifically in pre-myelinating Schwann cells. We previously found that Laminin-211 suppresses cAMP accumulation in static Gpr126-expressing cultures, but nonetheless acts to elevate cAMP in dynamic in vitro assays and in vivo15. Similarly, we do not observe cAMP accumulation in static naloxone-treated Gpr126-expressing cells in vitro (Figure 5). Therefore, naloxone may act in two ways: indirectly via opioid receptors on adjacent axons, and by cell-autonomous activation of Gpr126 in dynamic and later-developing states.
The angiotensin II type 1 (AT) receptor antagonist, telmisartan, was sufficient to promote mbp expression across all gpr126 alleles, even gpr126stl47 which lacks a functional Gpr126-NTF and thus fails to radially sort peripheral axons. AT receptors are upregulated in functional recovery of injured peripheral nerves68, and telmisartan treatment improves peripheral nerve regeneration69. However, the expression of AT receptors in Schwann cells is not established and the cell autonomy of telmisartan’s action is unclear. Notably, AT1 receptors are coupled to Gq/11 and Gi and would therefore inhibit the production of cAMP70. Telmisartan as an AT receptor antagonist should then increase or at least stabilize cAMP levels. However, we observe a reduction of basal GPR126-induced cAMP levels, which indicates additional AT receptor-independent effect of telmisartan. We have previously observed that cAMP elevation by FSK is sufficient to promote mbp expression in gpr126stl47 mutants in a similar fashion; however, FSK-treated gpr126stl47 mutants still fail to complete radial sorting and express terminal differentiation markers in the absence of myelin15. Further investigation into telmisartan’s mechanism of action can include both the target receptor as well as whether receptor activation mediates radial sorting and wrapping in addition to mbp expression.
Apomorphine directly activates Gpr126-mediated cAMP accumulation
In addition to compounds that act in parallel to or potentially downstream of gpr126, our study revealed a novel direct activator of Gpr126, apomorphine hydrochloride. Due to high lethality with efficacious doses, we could not dissect the ability of apomorphine to rescue mbp expression across all gpr126 alleles. However, our in vivo assays demonstrate that apomorphine promotes mbp expression in Stachel-dead gpr126stl215 mutants, suggesting a Stachel-independent mode of action (Figure 4). While we cannot formally exclude a Gpr126-independent mechanism in vivo, we favor a model in which apomorphine directly agonizes Gpr126-CTF in Schwann cells to promote mbp expression, rather than via apomorphine’s established role agonizing Gi-coupled D2 dopamine receptors71. In the latter case, activation of D2 receptors in Schwann cells would result in reduction of cAMP, which would normally prevent Schwann cell differentiation and mbp expression. Thus, we conclude that apomorphine directly activates Gpr126 in vivo, a model which is corroborated by our heterologous in vitro assays showing apomorphine directly activates both wild-type and Stachel-dead Gpr126 (Figure 5). Our previous work showed Stachel-mediated activation is facilitated by dynamic assays and suggested physical dissociation of Gpr126-NTF could liberate the cryptic tethered agonist13, 15, 18 . In the present study, the elevation of cAMP in static cultures suggests a bypass of mechanical activation of Gpr126 and direct activation of Gs signaling upon apomorphine binding to the Gpr126-CTF. To our knowledge, our study is the first to demonstrate activation of Gpr126 signaling in a Stachel-independent fashion.
Intriguingly, two hits in our screen, apomorphine and naloxone, are both morphine derivatives with different structures and pharmacologic effects. Importantly, apomorphine does not bind opioid receptors, whereas naloxone is an opioid receptor antagonist. We decided to include codeine, an agonist of opioid receptors to test specificity and found only apomorphine is sufficient to elevate cAMP in Gpr126-expressing cells (Figure 5). To investigate if this effect is specific to apomorphine in vivo, we treated gpr126st63 hypomorphs and gpr126st49 NTF-only mutants with related alkaloids. We found that one of these aporphines, glaucine, suppresses the gpr126st63 hypomorphic phenotype. Glaucine, like apomorphine, interacts with dopamine receptors, but can also modulate serotonergic receptors and intriguingly interacts with L-type Ca2+ channels in vitro72-74, mirroring the findings of another gpr126 suppressor screen33. However, glaucine did not suppress gpr126st49, suggesting that it also functions specifically through gpr126 as a direct agonist like apomorphine (Figure 6).
Taken together, our screen demonstrates the feasibility of identifying pharmacological modulators of aGPCRs that interact directly with the receptor or act in parallel pathways. Because the functions of aGPCRs span many tissue types, we emphasize the ability to perform similar in vivo small molecule screens for other aGPCRs using the larval zebrafish model. Finally, our study revealed five novel small molecule regulators of Schwann cell development that act at different points in the differentiation pathway. These compounds represent an exciting opportunity to dissect the basic biology of Schwann cell development and develop therapeutics for neurological disorders by targeting Gpr126 and other GPCRs.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Steve Johnson, Lila Solnica-Krezel, John Hofferberth, and members of the Monk and Petersen laboratories for valuable discussions and feedback. We thank Becky Gallagher and the Animal Care staff at Kenyon College as well as Charleen Johnson and the Washington University Zebrafish Consortium staff for excellent zebrafish care. This work was supported by Kenyon College Summer Scholars fellowships to EB; National Science Foundation Graduate Research Fellowship DGE-1745038 to RLC, the junior research grant by the Medical Faculty of the University of Leipzig to CW, a Collins Medical Trust grant to RKM, the German Research Foundation (Research Unit FOR 2149, project numbers: 266022790/266061011; CRC 1052 B06, project number: 209933838) to TS and IL, the BMBF (IFB AdipositasDiseases Leipzig AD2-7102) and the European Social Fund and the Free State of Saxony to IL; NIH R01 NS079445 and NIH R01 HD080601 to KRM; and an Integrative Research in Pharmacology fellowship from the American Society for Pharmacology and Experimental Therapeutics (ASPET-IRP) to SCP. SCP accepts responsibility for data analysis integrity.
Footnotes
COMPETING INTERESTS
The authors declare no conflicts of interest.
REFERENCES
- 1.Arac D, Aust G, Calebiro D, et al. 2012. Dissecting signaling and functions of adhesion G protein-coupled receptors. Annals of the New York Academy of Sciences. 1276: 1–25. [DOI] [PubMed] [Google Scholar]
- 2.Langenhan T, Aust G & Hamann J. 2013. Sticky signaling--adhesion class G protein-coupled receptors take the stage. Science signaling. 6: re3. [DOI] [PubMed] [Google Scholar]
- 3.Bjarnadottir TK, Fredriksson R & Schioth HB. 2007. The adhesion GPCRs: a unique family of G protein-coupled receptors with important roles in both central and peripheral tissues. Cellular and molecular life sciences : CMLS. 64: 2104–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jessen KR & Mirsky R. 2005. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 6: 671–682. [DOI] [PubMed] [Google Scholar]
- 5.Woodhoo A & Sommer L. 2008. Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia. 56: 1481–1490. [DOI] [PubMed] [Google Scholar]
- 6.Monk KR, Naylor SG, Glenn TD, et al. 2009. A G protein-coupled receptor is essential for Schwann cells to initiate myelination. Science. 325: 1402–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mogha A, Benesh AE, Patra C, et al. 2013. Gpr126 functions in Schwann cells to control differentiation and myelination via G-protein activation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 33: 17976–17985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mogha A, Harty BL, Carlin D, et al. 2016. Gpr126/Adgrg6 Has Schwann Cell Autonomous and Nonautonomous Functions in Peripheral Nerve Injury and Repair. J Neurosci. 36: 12351–12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackerman SD & Monk KR. 2016. The scales and tales of myelination: using zebrafish and mouse to study myelinating glia. Brain Res. 1641: 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monk KR, Oshima K, Jors S, et al. 2011. Gpr126 is essential for peripheral nerve development and myelination in mammals. Development. 138: 2673–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyons DA & Talbot WS. 2014. Glial cell development and function in zebrafish. Cold Spring Harb Perspect Biol. 7: a020586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liebscher I, Ackley B, Araç D, et al. 2014. New functions and signaling mechanisms for the class of adhesion G protein-coupled receptors. Ann N Y Acad Sci. 1333: 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arac D, Boucard AA, Bolliger MF, et al. 2012. A novel evolutionarily conserved domain of cell-adhesion GPCRs mediates autoproteolysis. EMBO J. 31: 1364–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glenn TD & Talbot WS. 2013. Analysis of Gpr126 function defines distinct mechanisms controlling the initiation and maturation of myelin. Development. 140: 3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen SC, Luo R, Liebscher I, et al. 2015. The Adhesion GPCR GPR126 Has Distinct, Domain-Dependent Functions in Schwann Cell Development Mediated by Interaction with Laminin-211. Neuron. 85: 755–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Küffer A, Lakkaraju AK, Mogha A, et al. 2016. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 536: 464–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paavola KJ, Sidik H, Zuchero JB, et al. 2014. Type IV collagen is an activating ligand for the adhesion G protein-coupled receptor GPR126. Science signaling. 7: ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liebscher I, Schon J, Petersen SC, et al. 2014. A tethered agonist within the ectodomain activates the adhesion G protein-coupled receptors GPR126 and GPR133. Cell reports. 9: 2018–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson KA 2015. New paradigms in GPCR drug discovery. Biochem Pharmacol. 98: 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao Y & McCammon JA. 2016. G-protein coupled receptors: advances in simulation and drug discovery. Curr Opin Struct Biol. 41: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hennen S, Wang H, Peters L, et al. 2013. Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci Signal. 6: ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu C, Dong L, Zhou H, et al. 2018. G-Protein-Coupled Receptor Gpr17 Regulates Oligodendrocyte Differentiation in Response to Lysolecithin-Induced Demyelination. Sci Rep. 8: 4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zon LI & Peterson RT. 2005. In vivo drug discovery in the zebrafish. Nature reviews. Drug discovery. 4: 35–44. [DOI] [PubMed] [Google Scholar]
- 24.D’Rozario M, Monk KR & Petersen SC. 2017. Analysis of myelinated axon formation in zebrafish. Methods Cell Biol. 138: 383–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buckley CE, Goldsmith P & Franklin RJ. 2008. Zebrafish myelination: a transparent model for remyelination? Dis Model Mech. 1: 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons DA, Pogoda HM, Voas MG, et al. 2005. erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr Biol. 15: 513–524. [DOI] [PubMed] [Google Scholar]
- 27.Preston MA & Macklin WB. 2015. Zebrafish as a model to investigate CNS myelination. Glia. 63: 177–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Preston MA, Finseth LT, Bourne JN, et al. 2019. A novel myelin protein zero transgenic zebrafish designed for rapid readout of in vivo myelination. Glia. [DOI] [PMC free article] [PubMed]
- 29.Buckley CE, Marguerie A, Roach AG, et al. 2010. Drug reprofiling using zebrafish identifies novel compounds with potential pro-myelination effects. Neuropharmacology. 59: 149–159. [DOI] [PubMed] [Google Scholar]
- 30.Cole KLH, Early JJ & Lyons DA. 2017. Drug discovery for remyelination and treatment of MS. Glia. 65: 1565–1589. [DOI] [PubMed] [Google Scholar]
- 31.Early JJ, Cole KL, Williamson JM, et al. 2018. An automated high-resolution in vivo screen in zebrafish to identify chemical regulators of myelination. Elife. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kazakova N, Li H, Mora A, et al. 2006. A screen for mutations in zebrafish that affect myelin gene expression in Schwann cells and oligodendrocytes. Dev Biol. 297: 1–13. [DOI] [PubMed] [Google Scholar]
- 33.Diamantopolou E, Baxendale S, de la Vega de Léon A, et al. 2019. Identification of compounds that rescue otic and myelination defects in the zebrafish adgrg6 (gpr126) mutant. eLife. 520056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almeida RG, Czopka T, Ffrench-Constant C, et al. 2011. Individual axons regulate the myelinating potential of single oligodendrocytes in vivo. Development. 138: 4443–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czopka T & Lyons DA. 2011. Dissecting mechanisms of myelinated axon formation using zebrafish. Methods in cell biology. 105: 25–62. [DOI] [PubMed] [Google Scholar]
- 36.Pogoda HM, Sternheim N, Lyons DA, et al. 2006. A genetic screen identifies genes essential for development of myelinated axons in zebrafish. Dev Biol. 298: 118–131. [DOI] [PubMed] [Google Scholar]
- 37.Westerfield M 2000. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio) Institute of Neuroscience. University of Oregon. [Google Scholar]
- 38.Kimmel CB, Ballard WW, Kimmel SR, et al. 1995. Stages of embryonic development of the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 203: 253–310. [DOI] [PubMed] [Google Scholar]
- 39.Cunningham RL & Monk KR. 2018. Whole Mount In Situ Hybridization and Immunohistochemistry for Zebrafish Larvae. Methods Mol Biol. 1739: 371–384. [DOI] [PubMed] [Google Scholar]
- 40.Thisse C & Thisse B. 2008. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 3: 59–69. [DOI] [PubMed] [Google Scholar]
- 41.Schindelin J, Arganda-Carreras I, Frise E, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods. 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kucenas S, Wang WD, Knapik EW, et al. 2009. A selective glial barrier at motor axon exit points prevents oligodendrocyte migration from the spinal cord. J Neurosci. 29: 15187–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith CJ, Morris AD, Welsh TG, et al. 2014. Contact-mediated inhibition between oligodendrocyte progenitor cells and motor exit point glia establishes the spinal cord transition zone. PLoS Biol. 12: e1001961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goll MG, Anderson R, Stainier DY, et al. 2009. Transcriptional silencing and reactivation in transgenic zebrafish. Genetics. 182: 747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo L, Moon C, Niehaus K, et al. 2012. Rac1 controls Schwann cell myelination through cAMP and NF2/merlin. J Neurosci. 32: 17251–17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petrović M, Bonvin D, Hofmann H, et al. 2018. Fungicidal PMMA-Undecylenic Acid Composites. Int J Mol Sci. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi D, Zhao Y, Yan H, et al. 2016. Antifungal effects of undecylenic acid on the biofilm formation of Candida albicans. Int J Clin Pharmacol Ther. 54: 343–353. [DOI] [PubMed] [Google Scholar]
- 48.Kerensky T & Walley AY. 2017. Opioid overdose prevention and naloxone rescue kits: what we know and what we don’t know. Addiction science & clinical practice. 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snyder SH & Childers SR. 1979. Opiate receptors and opioid peptides. Annual review of neuroscience. 2: 35–64. [DOI] [PubMed] [Google Scholar]
- 50.Beaulieu J-M & Gainetdinov RR. 2011. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological reviews. 63: 182–217. [DOI] [PubMed] [Google Scholar]
- 51.Chipkin RE, McQUADE RD & Iorio LC. 1987. D1 and D2 dopamine binding site up-regulation and apomorphine-induced stereotypy. Pharmacology Biochemistry and Behavior. 28: 477–482. [DOI] [PubMed] [Google Scholar]
- 52.Benson SC, Pershadsingh HA, Ho CI, et al. 2004. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 43: 993–1002. [DOI] [PubMed] [Google Scholar]
- 53.Michel MC, Foster C, Brunner HR, et al. 2013. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacological reviews. 65: 809–848. [DOI] [PubMed] [Google Scholar]
- 54.Promel S, Frickenhaus M, Hughes S, et al. 2012. The GPS Motif Is a Molecular Switch for Bimodal Activities of Adhesion Class G Protein-Coupled Receptors. Cell reports. 2: 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patra C, van Amerongen MJ, Ghosh S, et al. 2013. Organ-specific function of adhesion G protein-coupled receptor GPR126 is domain-dependent. Proceedings of the National Academy of Sciences of the United States of America. 110: 16898–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoveken HM, Bahr LL, Anders MW, et al. 2016. Dihydromunduletone Is a Small-Molecule Selective Adhesion G Protein-Coupled Receptor Antagonist. Mol Pharmacol. 90: 214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harty BL, Krishnan A, Sanchez NE, et al. 2015. Defining the gene repertoire and spatiotemporal expression profiles of adhesion G protein-coupled receptors in zebrafish. BMC Genomics. 16: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demberg LM, Winkler J, Wilde C, et al. 2017. Activation of Adhesion G Protein-coupled Receptors: AGONIST SPECIFICITY OF STACHEL SEQUENCE-DERIVED PEPTIDES. J Biol Chem. 292: 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gupte J, Swaminath G, Danao J, et al. 2012. Signaling property study of adhesion G-protein-coupled receptors. FEBS Lett. 586: 1214–1219. [DOI] [PubMed] [Google Scholar]
- 60.Mogha A, D’Rozario M & Monk KR. 2016. G protein-coupled receptors in myelinating glia. Trends in pharmacological sciences. 37: 977–987. [DOI] [PubMed] [Google Scholar]
- 61.Ackerman SD, Garcia C, Piao X, et al. 2015. The adhesion GPCR Gpr56 regulates oligodendrocyte development via interactions with Gα12/13 and RhoA. Nat Commun. 6: 6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giera S, Deng Y, Luo R, et al. 2015. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat Commun. 6: 6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin D, Lin S-T, Fu Y-H, et al. 2013. Very large G protein-coupled receptor 1 regulates myelin-associated glycoprotein via Gαs/Gαq-mediated protein kinases A/C. Proceedings of the National Academy of Sciences. 110: 19101–19106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mei F, Fancy SP, Shen YA, et al. 2014. Micropillar arrays as a high-throughput screening platform for therapeutics in multiple sclerosis. Nature medicine. 20: 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mei F, Mayoral SR, Nobuta H, et al. 2016. Identification of the Kappa-Opioid Receptor as a Therapeutic Target for Oligodendrocyte Remyelination. J Neurosci. 36: 7925–7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ackerman SD, Luo R, Poitelon Y, et al. 2018. GPR56/ADGRG1 regulates development and maintenance of peripheral myelin. J Exp Med. 215: 941–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mambretti EM, Kistner K, Mayer S, et al. 2016. Functional and structural characterization of axonal opioid receptors as targets for analgesia. Mol Pain. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gallinat S, Yu M, Dorst A, et al. 1998. Sciatic nerve transection evokes lasting up-regulation of angiotensin AT2 and AT1 receptor mRNA in adult rat dorsal root ganglia and sciatic nerves. Brain Res Mol Brain Res. 57: 111–122. [DOI] [PubMed] [Google Scholar]
- 69.Yuksel TN, Halici Z, Demir R, et al. 2015. Investigation of the effect of telmisartan on experimentally induced peripheral nerve injury in rats. Int J Neurosci. 125: 464–473. [DOI] [PubMed] [Google Scholar]
- 70.Higuchi S, Ohtsu H, Suzuki H, et al. 2007. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clinical science. 112: 417–428. [DOI] [PubMed] [Google Scholar]
- 71.Usiello A, Baik JH, Rougé-Pont F, et al. 2000. Distinct functions of the two isoforms of dopamine D2 receptors. Nature. 408: 199–203. [DOI] [PubMed] [Google Scholar]
- 72.Asencio M, Hurtado-Guzmán C, López JJ, et al. 2005. Structure–affinity relationships of halogenated predicentrine and glaucine derivatives at D1 and D2 dopaminergic receptors: halogenation and D1 receptor selectivity. Bioorganic & medicinal chemistry. 13: 3699–3704. [DOI] [PubMed] [Google Scholar]
- 73.Heng HL, Chee CF, Thy CK, et al. 2019. In vitro functional evaluation of isolaureline, dicentrine and glaucine enantiomers at 5‐HT2 and α1 receptors. Chemical biology & drug design. 93: 132–138. [DOI] [PubMed] [Google Scholar]
- 74.Cortijo J, Villagrasa V, Pons R, et al. 1999. Bronchodilator and anti‐inflammatory activities of glaucine: In vitro studies in human airway smooth muscle and polymorphonuclear leukocytes. British journal of pharmacology. 127: 1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.