

Abstract
Caveolae position CaV3.2 (T‐type Ca2+ channel encoded by the α‐3.2 subunit) sufficiently close to RyR (ryanodine receptors) for extracellular Ca2+ influx to trigger Ca2+ sparks and large‐conductance Ca2+‐activated K+ channel feedback in vascular smooth muscle. We hypothesize that this mechanism of Ca2+ spark generation is affected by age. Using smooth muscle cells (VSMCs) from mouse mesenteric arteries, we found that both Cav3.2 channel inhibition by Ni2+ (50 µM) and caveolae disruption by methyl‐ß‐cyclodextrin or genetic abolition of Eps15 homology domain‐containing protein (EHD2) inhibited Ca2+ sparks in cells from young (4 months) but not old (12 months) mice. In accordance, expression of Cav3.2 channel was higher in mesenteric arteries from young than old mice. Similar effects were observed for caveolae density. Using SMAKO Cav1.2−/− mice, caffeine (RyR activator) and thapsigargin (Ca2+ transport ATPase inhibitor), we found that sufficient SR Ca2+ load is a prerequisite for the CaV3.2‐RyR axis to generate Ca2+ sparks. We identified a fraction of Ca2+ sparks in aged VSMCs, which is sensitive to the TRP channel blocker Gd3+ (100 µM), but insensitive to CaV1.2 and CaV3.2 channel blockade. Our data demonstrate that the VSMC CaV3.2‐RyR axis is down‐regulated by aging. This defective CaV3.2‐RyR coupling is counterbalanced by a Gd3+ sensitive Ca2+ pathway providing compensatory Ca2+ influx for triggering Ca2+ sparks in aged VSMCs.
Keywords: aging, calcium sparks, caveolae, ryanodine receptors, T‐type calcium channels, vascular smooth muscle
Cav3.2‐RyR axis in smooth muscle cell is down‐regulated by age‐related ultrastructural alterations of caveolae and reduced Cav3.2 expression. This defective Cav3.2‐RyR coupling is counterbalanced by a Gd3+ sensitive Ca2+ pathway providing compensatory Ca2+ influx for triggering Ca2+ sparks in aged VSMCs.
1. INTRODUCTION
In resistance arteries, voltage‐dependent Ca2+ channels activate ryanodine receptors (RyRs) to cause elementary Ca2+ release events (Ca2+ sparks) from the sarcoplasmic reticulum (SR) (Essin et al., 2007; Jaggar et al., 1998; Nelson et al., 1995; Wang et al., 2004). Ca2+ release from the SR in the form of Ca2+ sparks opens numerous large‐conductance Ca2+‐sensitive K+ (BKCa) channels, causing spontaneous transient outward K+ currents (STOCs) (Knot, Standen, & Nelson, 1998; Nelson et al., 1995). As a result, Ca2+ spark–BKCa channel coupling induces vascular smooth muscle cell (VSMCs) hyperpolarization and attenuation of arterial constriction (Brenner et al., 2000; Löhn et al., 2001; Pérez, Bonev, Patlak, & Nelson, 1999). In previous studies, we demonstrated that L‐type CaV1.2 channels play the predominant role (~75%) in Ca2+ sparks generation in mesenteric arterial VSMCs, and T‐type CaV3.2 channels, localized in caveolae, represent an additional source (~25%) (Fan, Kaßmann, Hashad, Welsh, & Gollasch, 2018; Hashad et al., 2018). In the latter pathway, caveolae position CaV3.2 channels sufficiently close to RyRs (<40 nm) of the sarcoplasmic reticulum (SR) for extracellular Ca2+ influx to trigger Ca2+ sparks and large‐conductance Ca2+‐activated K+ channel feedback in vascular smooth muscle (Fan et al., 2018; Harraz et al., 2014; Hashad et al., 2018; Löhn et al., 2000). These conclusions were mainly derived from experiments using CaV3.2 channel (Cacna1h −/−) and caveolin‐1 (Cav1 −/−) knockout mice. Although genetic caveolin‐1 deletion leads to a complete lack of caveolae from the VSMC plasma membrane, data interpretation is limited because Cav1 deletion may affect SR Ca2+ load and is known to increase the density of BKCa channels in VSMCs (Cheng & Jaggar, 2006). Caveolins affect also trafficking of other K+ channels (Kv1.5) to cholesterol‐rich membrane microdomains (McEwen, Li, Jackson, Jenkins, & Martens, 2008).
Little is known about the effects of aging on the T‐type CaV3.2‐RyR axis to generate Ca2+ sparks. While L‐type Ca2+ current densities are preserved in VSMCs, aging has been reported to cause decrements in Ca2+ signaling in response to either ryanodine receptor stimulation by caffeine or inositol trisphosphate (InsP3) receptor activation with phenylephrine in mesenteric arteries of mice (del Corsso et al., 2006). Loss of CaV3.2 channels attenuates a protective function to excess myogenic tone in response to intravasal pressure (Mikkelsen, Björling, & Jensen, 2016). Advanced age can also alter the composition of lipid rafts and caveolae, which could affect a variety of signaling molecules (Bergdahl & Sward, 2004; Parton & Simons, 2007) to contribute to the pathophysiology of Alzheimer's, Parkinson's, diabetes, and cardiovascular diseases (Boersma et al., 2001; Headrick et al., 2003; Ohno‐Iwashita, Shimada, Hayashi, & Inomata, 2010; Simons & Ehehalt, 2002). Aging has been also found to alter the number and morphology of caveolae in smooth muscle cells (Bakircioglu et al., 2001; Lowalekar, Cristofaro, Radisavljevic, Yalla, & Sullivan, 2012; Ratajczak et al., 2003). We hypothesize that aging affects the T‐type CaV3.2‐RyR axis to generate Ca2+ sparks in vascular smooth muscle. To test this hypothesis, we used methyl‐ß‐cyclodextrin, smooth muscle‐specific (SMAKO) CaV1.2−/− mice and a novel Eps15 homology domain‐containing protein (EHD2) knockout mouse model, which leads to destabilization of caveolae at the plasma membrane (Lian, Matthaeus, Kassmann, Daumke, & Gollasch, 2019). We also evaluated the role of luminal SR calcium on T‐type CaV3.2‐RyR coupling. Clarification of this hypothesis is important for understanding age‐dependent effects in cardiovascular disease and may provide new therapeutic avenues in the elderly.
2. RESULTS
2.1. Age effects on T‐type CaV3.2‐RyR axis
The T‐type Cav3.2 channel blocker Ni2+ decreased Ca2+ spark frequency and fraction of cells with sparks in young VSMCs (see also (Fan et al., 2018; Hashad et al., 2018)), while it failed to decrease Ca2+ spark events in old VSMCs (Figure 1). These data suggest that Cav3.2 channels contribute to generation of Ca2+ sparks in young but not in old VSMC. To address whether the reduced function of T‐type Cav3.2 channels in generating Ca2+ sparks in old VSMCs could rely on reduced protein expression, we analyzed Cav3.2 protein expression in mesenteric arteries from young mice versus old mice. In Western blot analyses, we found that Cav3.2 expression decreased with age (Figure 1g,h).
Figure 1.
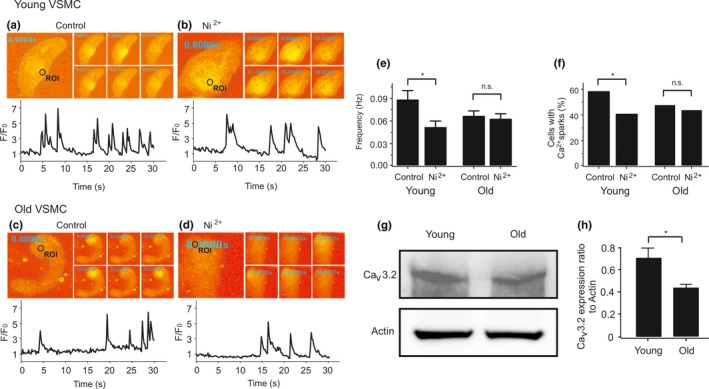
Age attenuates the role of CaV3.2 channels in Ca2+ spark generation and decreases CaV3.2 protein expression in VSMC. (a), Ca2+ fluorescence images of a Fluo‐4‐AM–loaded VSMC from a young mouse and time course of Ca2+ fluorescence changes in the cellular ROI (upper panel). Cell boundary is marked with dashed line. (b), same as (a) but in the presence of Ni2+ (50 µM). (c), same as (a) but in a VSMC from an old mouse. (d), same as (c) but in the presence of Ni2+ (50 µM). (e, f), summary of the results. Ca2+ spark frequency (e) and fraction of cells producing Ca2+ sparks (f) in VSMCs from young mice (n = 102), in VSMCs from young mice cells incubated with Ni2+ (n = 85), in VSMCs from aged mice (n = 129), and in VSMCs from aged mice cells incubated with Ni2+ (n = 127). Cells were isolated from 4 mice in each group; 25–40 cells were recorded and analyzed from each mouse. VSMC, vascular smooth muscle cell. (g), Western blot analysis of CaV3.2 proteins in mesenteric arteries of young versus old mice. (h), quantification of Western blot results. Mesenteric arteries were taken from 9 mice in each group. *, p < .05. n.s., not significant
2.2. Role of luminal SR calcium on T‐type CaV3.2‐RyR axis
Thapsigargin inhibits the SR Ca2+ transport ATPase (SERCA) and thereby reduces SR [Ca2+] load (Janczewski & Lakatta, 1993; Sagara & Inesi, 1991; Thastrup, 1990). We studied the effects of thapsigargin on [Ca2+]SR load and its role on T‐type CaV3.2‐RyR axis. Caffeine (10 mM)‐induced peak fluorescence was measured to monitor maximal RyR Ca2+ release from SR stores. Our data showed that thapsigargin decreased concentration‐dependently caffeine‐induced cytosolic [Ca2+] peaks (Figure S1a–d). The results confirm that thapsigargin causes luminal SR calcium depletion. We next studied the individual contributions of Cav1.2 versus Cav3.2 channels to generate Ca2+ sparks under these different [Ca2+]SR loads. We found that [Ca2+]SR depletion by thapsigargin reduced Ca2+ spark frequency and the percentage of cells firing Ca2+ sparks in Cav1.2+/+ VSMCs (Figure 2a,c) (see also (Essin et al., 2007)). In contrast, thapsigargin had no or little effects on Ca2+ spark frequency and the percentage of cells firing Ca2+ sparks in Cav1.2−/− (SMAKO) VSMCs (Figure 2b,c). These data are consistent with the idea that L‐type Cav1.2 channels couple indirectly to RyRs, that is, by influencing luminal SR calcium load to generate Ca2+ sparks (Essin et al., 2007). The data also show that SR Ca2+ load is controlled by SERCA (Nelson et al., 1995). We next studied how Cav1.2 channel ablation and reduced [Ca2+]SR load affect the CaV3.2‐RyR axis, that is, direct coupling between CaV3.2 channels and RyRs to generate Ca2+ sparks (Fan et al., 2018; Hashad et al., 2018; Löhn et al., 2000). Consistent with our previous results (Essin et al., 2007), we found that [Ca2+]SR was lower in Cav1.2−/− (SMAKO) VSMCs compared to Cav1.2+/+ control cells. As illustrated in Figure 2d–f, caffeine‐induced cytosolic [Ca2+] peaks were larger in Cav1.2+/+ cells compared to SMAKO Cav1.2−/− VSMCs, consistent with the idea that L‐type CaV1.2 channels are critical for SR Ca2+ load and peak [Ca2+] release. We compared the role of Ca2+ uptake into SR in these cells. 15 min after the first caffeine pulse, subsequent application of caffeine induced a strong [Ca2+] peak in Cav1.2+/+ control compared to Cav1.2−/− (SMAKO) cells (Figure 2d–f). We also compared the effects of caffeine on mesenteric arteries in the absence and presence of Ni2+. Ni2+ did not alter caffeine‐induced constrictions (Figure S1i–k). These data indicate that SR Ca2+ load mainly depends on Ca2+ influx through L‐type CaV1.2 channels (see also (Essin et al., 2007)). We confirmed these results by measuring BKCa channel currents activated by Ca2+ sparks (STOCs) in VSMCs (Figure S1e–h). STOCs were measured in presence of Cd2+ and/or Ni2+ after depletion of the [Ca2+]SR by thapsigargin. The holding potential was set to −40 mV, a physiological membrane potential that should drive T‐type Ca2+ channel‐mediated Ca2+ sparks, enabling the activation of BKCa channels (Fan et al., 2018; Harraz et al., 2014; Hashad et al., 2018). Figure S1 shows that thapsigargin removed ~60% of STOCs in VSMCs (Figure S1e–g). The Cav1.2 channel blocker Cd2+ blocked all STOCs in thapsigargin‐treated cells (Figure S1f), while Ni2+ had no effects (Figure S1e,h). Together, the results indicate that (a) Ca2+ influx through L‐type CaV1.2 channels is the main source of filling the SR with Ca2+ and (b) proper function of the T‐type CaV3.2‐RyR axis requires sufficient high [Ca2+]SR load.
Figure 2.
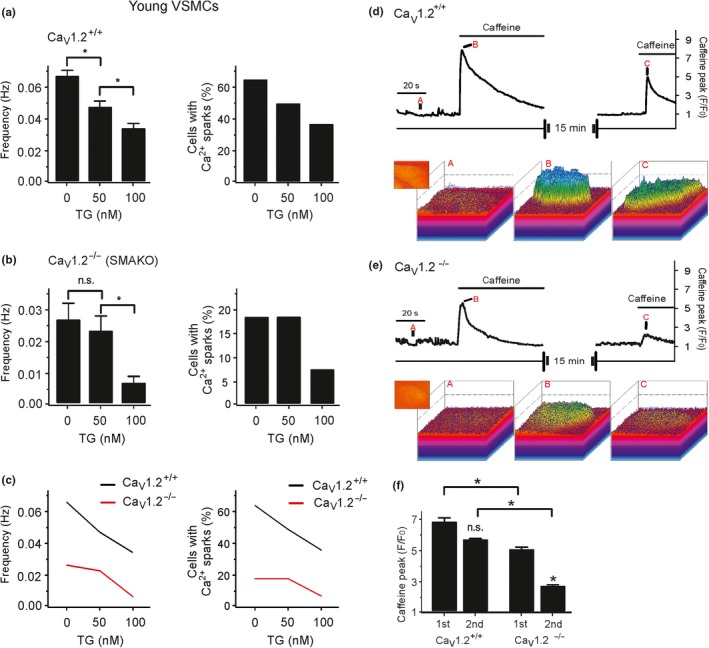
Role of luminal SR calcium on T‐type CaV3.2‐RyR axis. Effects of different concentrations of thapsigargin on Ca2+ spark frequency (a, left) and fraction of cells producing Ca2+ sparks (a, right) in Cav1.2+/+ VSMCs from young mice. Effects of different concentrations of thapsigargin on Ca2+ spark frequency (b, left) and fraction of cells producing Ca2+ sparks (b, right) in VSMCs from Cav1.2−/− (SMAKO) mice. (c), overlay of the data for Ca2+ spark frequency (left) and fraction of cells producing Ca2+ sparks (right). Cells were isolated from 4 mice in each group; 30–35 cells were recorded and analyzed from each mouse. (d), time course of Ca2+ fluorescence changes in the cellular ROI in a wild‐type (CaV1.2+/+) Fluo‐4‐AM–loaded VSMC induced by 10 mM caffeine (upper panel) and Ca2+ fluorescence plots (lower panel). (e), the same as (d), but in CaV1.2−/− VSMC. (f), summary of the 10 mM caffeine‐induced Ca2+ peaks in wild‐type versus CaV1.2−/− VSMCs. n = 7 cells from 3 mice, 2–3 cells were recorded and analyzed from each mouse. *, p < .05. n.s., not significant
2.3. Aging and alterations of VSMC caveolae
Defective CaV3.2‐RyR axis in old VSMCs could result from alterations in the ultrastructure of caveolae, where Cav3.2 channels reside to drive RyR‐mediated Ca2+ sparks (Fan et al., 2018; Harraz et al., 2014; Hashad et al., 2018). We first explored the contribution of caveolae to Ca2+ spark generation in VSMCs using methyl‐ß‐cyclodextrin (10 mM), a cholesterol‐depleting drug, which is known to disturb caveolae and inhibit a significant fraction of Ca2+ sparks in VSMCs (Löhn et al., 2000). In accordance with our previous data (Fan et al., 2018; Löhn et al., 2000), we found that methyl‐ß‐cyclodextrin decreased the frequency of Ca2+ spark and the fraction of cells with sparks by ~30% in young VSMCs. However, methyl‐ß‐cyclodextrin did not alter Ca2+ spark generation in old VSMCs (Figure 3a–f). Ni2+ (50 µM) did not further reduce Ca2+ sparks in methyl‐β‐cyclodextrin treated VSMCs neither from young nor old mice. Next, we evaluated the ultrastructure of caveolae in young versus old VSMCs. Although caveolae were present in cells of both groups, the density of caveolae was reduced in old VSMCs compared to young VSMCs (Figure 3g–i). We next confirmed our results by using a novel EHD2 genetic knockout (KO) mouse model. Since EHD2 localizes to the caveolar neck region of all caveolae, genetic abolition of EHD2 increases ubiquitously detachment of caveolae from the plasma membrane (Matthaeus et al., 2019). In line with these findings, we found detachment of caveolae in EHD2 del/del VSMCs compared to control VSMCs (Figure 4a). These changes were accompanied by reduced expression of Cav3.2 channels in EHD2 KO (del/del) VSMCs compared to control cells. Furthermore, Ca2+ spark frequency and the percentage of cells firing Ca2+ sparks were diminished in VSMCs from EHD2 del/del mice (Figure 4). Together, ultrastructural alterations of caveolae, reduced expression of Cav3.2 channels or both could underlie the observed attenuation of the vascular T‐type CaV3.2‐RyR axis to generate Ca2+ sparks in aged vascular smooth muscle.
Figure 3.
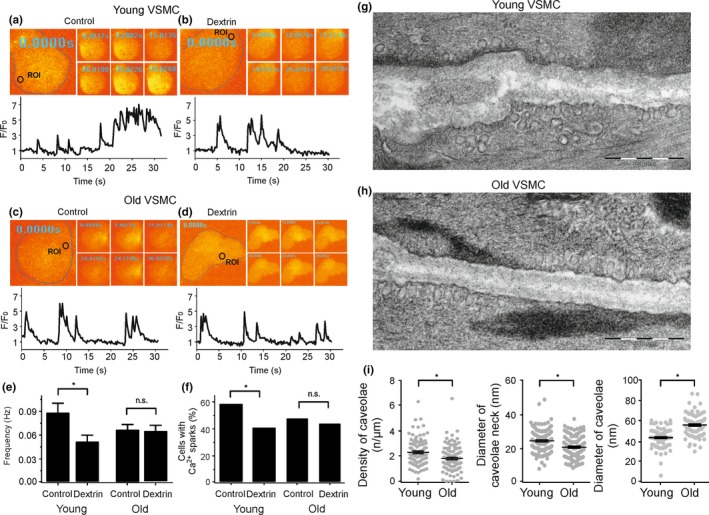
Defective CaV3.2‐RyR axis in aged VSMC result from alterations in the ultrastructure of caveolae. (a), Ca2+ fluorescence images of a Fluo‐4‐AM–loaded VSMC from a young mouse and time course of Ca2+ fluorescence changes in the cellular ROI (upper panel). Cell boundary is marked with dashed line. (b), same as (a) but with a cell incubated with methyl‐ß‐cyclodextrin (10 mM, 90 min at room temperature) to disrupt caveolae. (c), same as (a) but with VSMCs from old mice. (d), same as (c) but with a cell incubated with methyl‐ß‐cyclodextrin. (e, f), summary of the results. Ca2+ spark frequency (e) and fraction of cells producing Ca2+ sparks (f) in VSMCs from young mice (n = 98), in VSMCs from young mice cells incubated with methyl‐ß‐cyclodextrin (n = 111), in VSMCs from old mice (n = 121), and in VSMCs from old mice cells incubated with methyl‐ß‐cyclodextrin (n = 128). Cells were isolated from 4 mice in each group; 25–40 cells were recorded and analyzed from each mouse. (g), Electron microscopy image of a young VSMC. (h), same as (g) but from old VSMC. (i), summary of the results. Caveolae density, diameter of caveolae neck, caveolae size in VSMCs from young versus old mice (10–20 cells from each mouse, 4 mice in each group). *, p < .05. n.s., not significant
Figure 4.
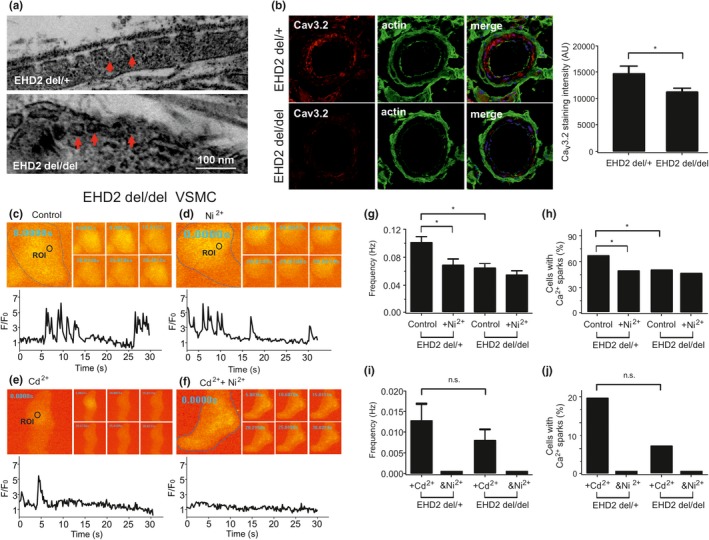
EHD2 knockout (EHD2 del/del) alters the ultrastructure of caveolae and decrease CaV3.2 expression, resulting in CaV3.2‐RyR axis malfunction. (a), Electron microscopy image of a EHD2 del/+ VSMC and a EHD2 del/del VSMC. (b, left), CaV3.2 immuno‐staining in BAT cryostat sections from EHD2 del/+ and del/del mice. (b, right), summary of the results, n (del/+)=46/5 mice and n (del/del)=53/5 mice. (c), Ca2+ fluorescence images of a Fluo‐4‐AM–loaded VSMC from EHD2 del/del mouse and time course of Ca2+ fluorescence changes in the cellular ROI (upper panel). Cell boundary is marked with dashed line. (d), same as (c) but in the presence of Ni2+ (50 µM). (e), same as (c) but in the presence of Cd2+ (200 µM). (f), same as (e) but in the presence of Ni2+ (50 µM). (g, h), summary of the results. Ca2+ spark frequency (g) and fraction of cells producing Ca2+ sparks (h) in VSMCs from EHD2 del/+ mice (n = 99), in VSMCs from EHD2 del/+ mice cells incubated with Ni2+ (n = 96), in VSMCs from EHD2 del/del mice (n = 144), and in VSMCs from EHD2 del/del mice cells incubated with Ni2+ (n = 125). Cells were isolated from 4 mice in each group; 25–40 cells were recorded and analyzed from each mouse. (I, j), summary of the results. Ca2+ spark frequency (i) and fraction of cells producing Ca2+ sparks (j) in VSMCs from EHD2 del/+ mice incubated with Cd2+ (n = 56), in VSMCs from EHD2 del/+ mice cells incubated with Ni2++Cd2+ (n = 56), in VSMCs from EHD2 del/del mice incubated with Cd2+ (n = 75), and in VSMCs from EHD2 del/del mice cells incubated with Ni2++Cd2+ (n = 68). Cells were isolated from 4 mice in each group; 15–20 cells were recorded and analyzed from each mouse. *, p < .05. n.s., not significant
2.4. Residual Ca2+ sparks in aged VSMCs
We noticed that there was a fraction of Ca2+ sparks in old VSMCs, which was insensitive to CaV1.2 and CaV3.2 channel blockade by Cd2+ and Ni2+, respectively (Figure 5). Surprisingly, Gd3+, a permissive TRP channel blocker, inhibited these remaining Ca2+ sparks (Figure 5). In contrast, Gd3+ (100 µM) had no effects on Ca2+ sparks in young VSMCs (Figure S1l,m). Together, the data suggest that defective CaV3.2‐RyR coupling in old VSMCs is counterbalanced by putative Gd3+ sensitive (TRP) cation channels providing sufficient Ca2+ influx to generate Ca2+ sparks.
Figure 5.
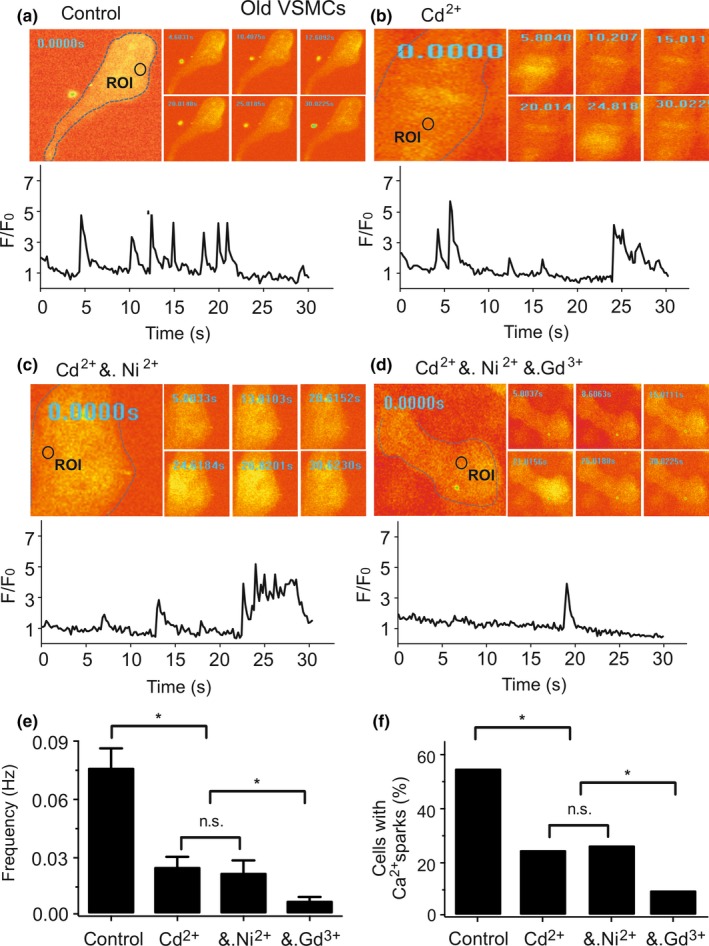
Gd3+ sensitive (TRP) cation channels generate Ca2+ sparks in old VSMCs. (a), Ca2+ fluorescence images of a Fluo‐4‐AM–loaded VSMC from an old mouse and time course of Ca2+ fluorescence changes in the cellular ROI (upper panel). Cell boundary is marked with dashed line. (b), same as (a) but with a cell incubated with Cd2+ (200 µM). (c), same as (a) but with Ni2+ (50 µM) following Cd2+ treatment. (d), same as (a) but with Gd3+ (100 µM) following Cd2++ Ni2+ treatment. (e, f), summary of the results. Ca2+ spark frequency (e) and fraction of cells producing Ca2+ sparks (f) in cells (n = 66), in cells incubated with Cd2+ (n = 69), in cells incubated with Cd2++ Ni2+ (n = 61), and in cells incubated with Cd2++ Ni2++ Gd3+ (n = 86). Cells were isolated from 4 old mice in each group; 15–30 cells were recorded and analyzed from each mouse. *, p < .05. n.s., not significant
2.5. Age‐dependent regulation of myogenic tone by Cav3.2 channels
To ascertain the importance of the CaV3.2‐RyR relationship to regulate arterial tone, we performed video microscopic measurements on isolated arteries. In young wild‐type mesenteric arteries, the Cav3.2 blocker Ni2+ 50 µM increased myogenic tone from 9.2% ± 1.2% to 13.04% ± 0.8% at 60 mmHg, from 11.6% ± 1.2% to 19.7% ± 0.5% at 80 mmHg, and from 17.7% ± 2% to 27.8% ± 1.3% at 100 mmHg (Figure 6), whereas Ni2+ 50 µM did not affect myogenic constriction in old vessels. Despite these differences, 60 mM K+‐induced vasoconstrictions were similar between young (54.2% ± 1.2%) and old (60.7% ± 2.1%) pressurized arteries (Figure 6c).
Figure 6.
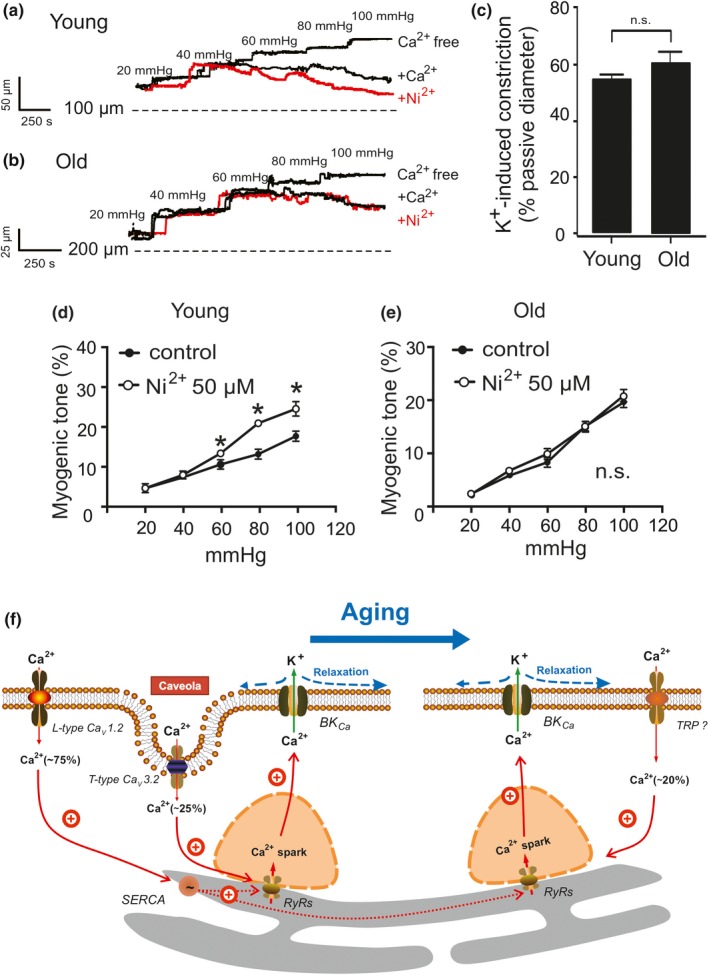
T‐type CaV3.2 blockade does not constrict mesenteric arteries from old mice. (a, b), representative traces and summary data show the effect of Ni2+ (50 µM) on mesenteric arteries pressurized to 60–100 mmHg from young and old mice, respectively. (c), vasoconstriction evoked by 60 mM K+ was similar in young and old pressurized (15 mmHg) arteries. (d, e), summary of myogenic tone measurements in pressurized mesenteric arteries from young and old mice (n = 5 arteries from 5 mice, one artery was recorded and analyzed from each mouse). Experiments were performed in the absence and presence of 50 µM Ni2+. *, p < .05. n.s., not significant. (f), schematic illustration of major Ca2+ influx pathways regulating Ca2+ sparks in VSMCs during aging. Ca2+ sparks, which result from opening of clustered RyRs in the SR, activate large‐conductance Ca2+‐activated K+ (BKCa) channels to produce a negative feedback effect on vasoconstriction. L‐type Cav1.2 channels contribute to global cytosolic [Ca2+], which in turn influences luminal SR calcium (via SERCA) and thus generates the majority (75%) of Ca2+ sparks. Caveolae position CaV3.2 channels sufficiently close to RyRs for extracellular Ca2+ influx to trigger (~25%) Ca2+ sparks. In aged mice, this CaV3.2‐RyR pathway loses importance. Instead, a gadolinium‐sensitive Ca2+ influx pathway is upregulated to trigger (20%) Ca2+ sparks. This pathway may compromise nonselective TRP channels. RyRs, ryanodine receptors; SERCA, sarcoplasmic/endoplasmic calcium pump; SR, sarcoplasmic reticulum; VSMC, mesenteric artery vascular smooth muscle cell
3. DISCUSSION
In this study, we analyzed the effects of aging on the Cav3.2 channels‐RyR axis on Ca2+ sparks generation in VSMCs. We employed pharmacological tools, smooth muscle‐specific Cav1.2 channel (SMAKO) and EHD2 genetic knockout mice. Our studies demonstrate that caveolar Cav3.2 channels‐RyR axis is impaired in aged VSMCs. We observed age‐related ultrastructural alterations of caveolae, which together with decreased Cav3.2 expression, may underlie incomplete caveolae‐Cav3.2‐RyR coupling for extracellular Ca2+ influx to trigger Ca2+ sparks and BKCa feedback in aged vascular smooth muscle.
3.1. Local and tight caveolar CaV3.2‐RyR coupling
L‐type Cav1.2 channels provide the predominant Ca2+ pathway for Ca2+ spark generation in VSMCs (Brenner et al., 2000; Filosa et al., 2006; Gollasch et al., 1998; Nelson et al., 1995; Pluger et al., 2000; Sausbier et al., 2005). This pathway increases Ca2+ load in the SR ([Ca2+]SR) can activate RyRs from the SR luminal side of the receptor to produce Ca2+ sparks (Figure 6f) (Ching, Williams, & Sitsapesan, 2000; Essin et al., 2007). T‐type Cav3.2 channels, which are located in pits structures of caveolae, constitute an additional Ca2+ influx pathway to trigger Ca2+ sparks (Figure 6f) (Abd El‐Rahman et al., 2013; Braunstein et al., 2009; Chen et al., 2003; Fan et al., 2018; Hashad et al., 2018). Our recent data show that RyR2 is the predominant RyR isoform responsible for Ca2+ sparks in VSMCs (Kassmann et al., 2019). The results from the present study are in line with these conceptual views. We first used low concentrations of the SR Ca2+‐ATPase inhibitor thapsigargin to decrease the SR calcium content (Janczewski & Lakatta, 1993; Lewartowski & Wolska, 1993; Nelson et al., 1995; Sagara, Fernandez‐Belda, Meis, & Inesi, 1992) and found that [Ca2+]SR depletion reduced Ca2+ spark frequency. In contrast, thapsigargin did not affect Ca2+ spark frequency in the absence of Cav1.2 channels. These data indicate that SR calcium filling through SERCA is critical for CaV1.2‐mediated Ca2+ sparks, but not for CaV3.2‐RyR axis. They support that local and tight coupling between the CaV1.2 channels and RyRs is not required to initiate Ca2+ sparks as previously suggested by our group (Essin et al., 2007). Indeed, the data indicate that Cav1.2 channels contribute to global cytosolic [Ca2+], which in turn influences luminal SR calcium and thus Ca2+ sparks (Figure 6f) (Essin et al., 2007). We also found that Cav3.2 channel blockade by Ni2+ had no effects on Ca2+ sparks and STOCs after treatment of cells with thapsigargin, that is, in conditions of suboptimal filled [Ca2+]SR stores. These data indicate that proper function of the caveolar T‐type CaV3.2‐RyR axis requires sufficient high [Ca2+]SR load. Second, we also explored the function of Cav1.2 and Cav3.2 channels for luminal SR Ca2+ load. We used high concentrations of caffeine (10 mM), a well‐known activator RyRs, to induce SR calcium release. Caffeine evoked smaller Ca2+ transients through SR Ca2+ release in SMAKO Cav1.2−/− VSMCs, in which T‐type Cav3.2 channels play a minor role in providing Ca2+ influx to induce Ca2+ sparks. These findings support the view that Ca2+ influx through L‐type Cav1.2 channels, but not T‐type Cav3.2 channels, represents the main source for luminal SR calcium load (Essin et al., 2007; Fan et al., 2018). To confirm this conclusion, we studied Ca2+ uptake into luminal SR by 2 pulse‐protocol of caffeine applications. We found that 10 mM caffeine evoked weak caffeine‐induced peaks in SMAKO Cav1.2−/− cells compared to control cells fifteen minutes after the 1st‐pulse caffeine application. We failed to observe Ca2+ sparks in SMAKO Cav1.2−/− cells before the 2nd‐pulse caffeine application, whereas cells with functional Cav1.2 channels enabled generation of Ca2+ sparks within the fifteen minutes interval. The poor recovery of the luminal SR calcium in SMAKO Cav1.2−/− VSMCs suggests that T‐type Cav3.2 channels play a minor role in [Ca2+]SR filling. The results were also confirmed by our electrophysiological experiments.
3.2. Effects of aging on T‐type CaV3.2‐RyR axis
In order to explore the effects of aging on caveolar T‐type Cav3.2 channel‐mediated Ca2+ sparks, we treated VSMCs from young and old mice with Ni2+ and methyl‐ß‐cyclodextrin. Consistent with our previous findings (Fan et al., 2018; Hashad et al., 2018), both compounds inhibited Ca2+ sparks in young VSMCs. In contrast, neither Ni2+ nor methyl‐ß‐cyclodextrin inhibited Ca2+ sparks in old VSMCs. These results indicate that the T‐type CaV3.2‐RyR axis loses its function to generate Ca2+ sparks in aged VSMCs to drive negative feedback control of myogenic tone in resistance arteries (Figure 6a‐e). The data are consistent with other data showing that CaV3.2 channels lose their protective role against excess myogenic tone and the loss of CaV3.2 channels induces a loss of flow‐mediated vasodilation with advanced age (Mikkelsen et al., 2016). Since RyR2 is the predominant RyR isoform responsible for Ca2+ sparks in VSMCs (Kassmann et al., 2019) and CaV1.2‐RyR2 axis works efficient in old VSMCs (Figure 5), RyRs reorganization should not be a key reason for altered calcium sparks in aged VSMCs. Thus, we propose that the observed malfunction of T‐type CaV3.2‐RyR axis in aging results from reduced CaV3.2 expression and ultrastructural alterations in caveolar microdomains responsible for CaV3.2‐RyR coupling. In accordance, we found that caveolae density was decreased and caveolae necks were narrowed in old VSMCs. T‐type CaV3.2‐RyR axis provides an important vascular Ca2+ influx pathway for triggering Ca2+ sparks in young VSMCs that deserves further attention since CaV3.2 T‐type calcium channels contribute to cardiovascular diseases (Chiang et al., 2009; David et al., 2010). Defective T‐type CaV3.2‐RyR axis may contribute to age‐related cardiovascular complications involving increased myogenic tone and blood pressure with advanced age.
3.3. Role of EHD2 on T‐type CaV3.2‐RyR axis
EHD2 is a dynamin‐related ATPase located at the neck of caveolae, which constitutes a structural component of caveolae involved in controlling the stability and turnover of this organelle (Ludwig et al., 2013; Morén et al., 2012; Stoeber et al., 2016). Knockout or down‐regulation of EHD2 in vivo results in decreased surface association and increased mobility of caveolae, whereas EHD2 overexpression stabilizes caveolae at the plasma membrane (Matthaeus et al., 2019; Morén et al., 2012; Shvets, Bitsikas, Howard, Hansen, & Nichols, 2015; Stoeber et al., 2016). Here we used EHD del/del mice to disturb the stability of caveolae to explore the effect of caveolar microdomains on CaV3.2‐RyR axis. Loss of EHD2 decreased the plasma membrane localization of caveolae and CaV3.2 channel expression, thus impaired the ability of T‐type CaV3.2 on Ca2+ sparks generation in the mesenteric SMC. It aligns with our above results and provides firm evidence that CaV3.2 channels in caveolar microdomains contribute to Ca2+ sparks in VSMCs of young but not old mice.
3.4. Possible role of TRP channels
We found that complete blockade of both CaV1.2 and CaV3.2 channels (by Cd2+ and Ni2+) abolished all Ca2+ sparks in young VSCMs (see also Fan et al., 2018) but only ~70% of Ca2+ sparks in old VSMCs. The findings suggest appearance of an additional Ca2+ influx pathway evoking Ca2+ sparks only in aged VSMCs. We found that gadolinium, a permissive TRP channel blocker (Hashad et al., 2017; Riehle et al., 2016), inhibited these remaining Ca2+ sparks. In order to rule out possible effects of gadolinium on Cav1.2 channel and/or Cav3.2 channel‐mediated Ca2+ sparks, we tested the effects of gadolinium on Ca2+ sparks in young VSMCs (in the absence of Cd2+ and Ni2+) and found that gadolinium had no effects on these Ca2+ sparks. Although gadolinium has been identified as nonspecific blocker (Berrier, Coulombe, Szabo, Zoratti, & Ghazi, 1992; Gottlieb, Suchyna, Ostrow, & Sachs, 2004; Trollinger, Rivkah Isseroff, & Nuccitelli, 2002), it is likely that a Ca2+ permeable conductance (TRP channels) has been upregulated to compensate for loss of T‐type CaV3.2 channels driving Ca2+ sparks in aged VSMCs (Figure 6f). Besides, TRP channels might trigger calcium sparks through reloading the SR with calcium since methyl‐ß‐cyclodextrin treatment failed to alter calcium events in old VSMCs (Figure 3e,f). Further works are required to ascertain which TRP cation channel(s) or pathways are responsible for generation of these Ca2+ sparks. Identification of the underlying pathways might be important for understanding age‐dependent factors contributing to cardiovascular disease and providing novel therapeutic approaches.
3.5. Summary
Our data provide further evidence that CaV3.2 channels colocalize in microdomains with RyRs to initiate Ca2+ sparks and activate BKCa channels to drive a feedback response on vascular tone. Here we demonstrate that caveolar CaV3.2 channels are impaired in triggering Ca2+ sparks in aged VSMCs. This defective caveolae‐RyR coupling may be caused by age‐related ultrastructural alterations of caveolae and reduced CaV3.2 expression in VSMCs. Furthermore, we found that proper function of the T‐type CaV3.2‐RyR axis requires sufficiently high SR Ca2+ load, which is regulated via Ca2+ influx through L‐type CaV1.2 channels. T‐type CaV3.2‐RyR axis malfunction may provide a straightforward explanation on how aging affects blood pressure (Chiossi et al., 2016; Hilgers et al., 2017; Wirth et al., 2016). Targeting defective CaV3.2‐RyR coupling may provide new therapeutic avenues for treatment of cardiovascular disease in the elderly.
4. EXPERIMENTAL PROCEDURES
4.1. Mice
In this study, young (12–14 weeks) versus old (48–56 weeks) male mice were used. The generation and usage of mice deficient in the smooth muscle Cav1.2 Ca2+ channel (SMAKO, smooth muscle α1c‐subunit Ca2+ channel knockout) has been described previously (Moosmang et al., 2003). Briefly, a conditional lox P‐flanked allele (L2) of the Cav1.2 gene (i.e., exons 14 and 15) was generated by homologous recombination in R1 embryonic stem cells (Seisenberger et al., 2000). In addition, mice carried a knock‐in allele (SM‐CreER T2 (ki)) (Kuhbandner et al., 2000), which expresses the tamoxifen‐dependent Cre recombinase, CreER T2, from the endogenous SM22 α gene locus, which is selectively expressed in smooth muscle of adult mice. Thus, tamoxifen treatment results in conversion of the lox P‐flanked Cav1.2 allele (L2) into a Cav1.2 knockout allele (L1) specifically in SMCs (Moosmang et al., 2003) (Essin et al., 2007). Mice were maintained at the breeding facility of the Max Delbrück Center for Molecular Medicine Berlin (MDC) in individually ventilated cages under standardized conditions that included a 12‐hr dark‐light cycle and free access to standard chow (0.25% sodium; SSNIFF Spezialitäten, Soest, Germany) and drinking water. SMAKO mice (Cav1.2 flox/flox; SM22α‐CreT2 or Cav1.2 flox/flox; SM22α‐CreT2/T2) and corresponding control mice (Cav1.2 +/+; SM22α‐CreT2/T2, Cav1.2 +/+; SM22α‐CreT2, Cav1.2 +/+ or Cav1.2 flox/flox) (12–14 weeks each) were i.p. injected with tamoxifen (30 µg/g body weight/day) for five consecutive days and sacrificed within 2–4 days after the injections. EHD2 del/del or EHD2 del/+ littermates (as control) mice (20–35 weeks each) were used as previously described (Matthaeus et al., 2019). All mice were deeply anesthetized by inhalation of isoflurane until cessation of breathing and then killed by cervical dislocation, and the mesentery arteries were removed. Experiments were performed on the same day with arteries from litter‐matched young versus aged mice, EHD2 control versus EHD2 del/del, and control versus SMAKO mice. All animal protocols were approved by the local animal care committee (LAGeSo, Berlin, Germany) and the animal welfare officers of the MDC (No. X9011/16, G0154/14). There are no ethical concerns.
4.2. Isolation of arterial vascular smooth muscle cells
Arterial VSMCs from mesenteric arteries were isolated as previously described (Gollasch et al., 1998; Kassmann et al., 2019; Pluger et al., 2000; Schleifenbaum et al., 2014). Briefly, arteries were removed and quickly transferred to cold (4°C) oxygenated (95% O2‐5% CO2) physiological salt solution (PSS) of the following composition (mM): 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 MgSO4, 1.6 CaCl2, and 11.1 glucose. The arteries were cleaned, cut into pieces, and placed into a Ca2+‐free Hank's solution (mM): 55 NaCl, 80 sodium glutamate, 5.6 KCl, 2 MgCl2, 1 mg/ml bovine serum albumin (BSA, Sigma, Taufkirchen), 10 glucose, and 10 HEPES (pH 7.4 with NaOH) containing 0.5 mg/ml papain (Sigma) and 1.0 mg/ml DTT for 37 min at 37°C. The segments were then placed in Hank's solution containing 1 mg/ml collagenase (Sigma, type F and H, ratio 30% and 70%) and 0.1 mM CaCl2 for 17 min at 37°C. Following several washes in Ca2+‐free Hank's solution (containing 1 mg/ml BSA), single cells were dispersed from artery segments by gentle triturating. Cells were then stored in the same solution at 4°C.
4.3. Ca2+ imaging measurements
Ca2+ sparks were measured as previously described (Essin et al., 2007; Fan et al., 2018). Isolated VSMCs were placed onto glass coverslips and incubated with the Ca2+ indicator fluo‐4 a.m. (10 µM) and pluronic acid (0.005%, w/v) for 60 min at room temperature in Ca2+‐free Hanks’ solution (Fan et al., 2018; Kassmann et al., 2019). After loading, cells were washed with bath solution for 10 min at room temperature. Isolated cells and intact arterial segments were imaged in a bath solution containing (mM): 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 HEPES (pH 7.4, NaOH). Images were recorded using a Nipkow disc‐based UltraView LCI confocal scanner (Perkin Elmer, Waltham, MA, USA) linked to a fast digital camera (Hamamatsu Photonics Model C4742‐95‐12ERG, 1,344 × 1,024 active pixel resolution, 6.45 µm square pixels). The confocal system was mounted on an inverted Nikon Eclipse Ti microscope with a x40 oil‐immersion objective (NA 1.3, Nikon). Images were obtained by illumination with an argon laser at 488 nm and recording all emitted light above 515 nm. Ca2+ spark analyses were performed off‐line using the UltraView Imaging Suite software (Perkin Elmer). The entire area of each image was analyzed to detect Ca2+ sparks. Ca2+ sparks were defined as local fractional fluorescence increase (F/F 0) above the noise level of 1.5. The frequency was calculated as the number of detected sparks divided by the total scan time. Caffeine‐induced peak was measured as previously described (Fernandez‐Sanz et al., 2014). After the VSMCs loaded with Ca2+ indicator fluo‐4 a.m. (10 µM, 60 min at room temperature), images were obtained following a single pulse of 10 mM caffeine. Maximal amplitude of caffeine‐induced peak fluorescence was normalized by the initial fluorescence value (F/F0) and considered as an index of total SR Ca2+ load.
4.4. Electrophysiology
Currents were measured in the whole‐cell perforated‐patch mode of the patch‐clamp technique (Essin et al., 2007; Gollasch, Ried, Bychkov, Luft, & Haller, 1996; Kassmann et al., 2019). Patch pipettes (resistance, 1.5–3.5 MΩ) were filled with a solution containing (in mM): 110 K‐Asp, 30 KCl, 10 NaCl, 1 MgCl2, and 0.05 EGTA (pH 7.2). The patch pipette solution was supplemented with 200 µg/ml Amphotericin B, dissolved in dimethyl sulfoxide (DMSO), to measure K+ currents in the whole‐cell perforated‐patch mode. The external bath solution contained (in mM): 134 NaCl, 6 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES (pH 7.4); holding potential was −60 mV. Whole‐cell currents were recorded using an Axopatch 200B amplifier (Axon Instruments/Molecular Devices) or an EPC 7 amplifier (List) at room temperature. Data were digitized at 5 kHz, using a Digidata 1440A digitizer (Axon CNS, Molecular Devices) and pClamp software versions 10.1 and 10.2. STOC analysis was performed off‐line using IGOR Pro (WaveMetrics) and Microsoft Excel software (Microsoft Corporation). A STOC was identified as a signal with at least three times the BKCa single‐channel current amplitude (Kassmann et al., 2019).
4.5. Ultrastructure and quantitative assessment of caveolae
Quantitative assessment of caveolae was carried out as previously described (Lowalekar et al., 2012). Isolated VSMCs from mesenteric arteries were dehydrated in a graded series of ethanol and embedded in the PolyBed® 812 resin (Polysciences Europe GmbH), ultrathin sections (60–80 nm) were cut (Leica microsystems), and uranyl acetate and lead citrate staining was performed. Samples were examined at 80 kV with a Zeiss EM 910 electron microscope (Zeiss), and image acquisition was performed with a Quemesa CDD camera and the iTEM software (Emsis GmbH). The density of caveolae was calculated as number of caveolae per µm. The diameter of caveolae neck (nm) and caveolae size (nm) were determined by using the parallel dimension function of CorelDRAW. Values from all electron microscopy images (n = 18 cells in each group) were averaged for each group.
4.6. Western blot analysis
Mesenteric arteries were isolated from mice and placed into cold physiological saline solution (PSS) previously oxygenated for 30 min (95% O2, 5% CO2). Vessels were cleaned of perivascular fat, and all tissues were immediately placed on dry ice and kept at −80°C until use. Samples were homogenized in RIPA buffer (Cell Signaling Technology) containing protease inhibitors (Sigma‐Aldrich). Tubes containing homogenates were freeze‐thawed three times at −80°C and 37°C, respectively, and then centrifuged at 11,200 g for 20 min at 4°C. After determining protein concentration, samples prepared in Laemmli buffer (50 mM Tris pH 6.8, 10% SDS, 10% glycerol, 5% mercaptoethanol, and 2 mg/ml bromophenol blue) were boiled for 2 min, separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) on 7% polyacrylamide gels and transferred onto polyvinylidene fluoride membranes. Membranes were blocked in 5% nonfat dry milk in phosphate‐buffered saline (PBS) containing 0.1% Tween 20 and then incubated overnight at 4°C with primary anti‐CaV3.2 antibody (Mouse. NBP1‐22444, 1:1,000 final dilution; Novus Biologicals). After washing, membranes were incubated with anti‐mouse IgG‐peroxidase‐linked secondary antibody (1:5,000 final dilution; GE Healthcare) for 1 hr at room temperature. Blots were washed and incubated in enhanced chemiluminescence reagents (ECL Prime, Amersham Bioscience), after which bands were detected using a ChemiDoc XRS+ Imaging System (Bio‐Rad). An anti‐Actin antibody (Mouse. sc‐8432, 1:500 final dilution; Santa Cruz) was used as a loading control, and Precision Plus Protein Prestained Standard (Bio‐Rad) was used as a molecular weight marker.
4.7. Immunohistostaining of mesenteric arteries for confocal imaging
EHD2 del/+ and EHD2 del/del mice were anesthetized with 2% ketamine/10% rompun, perfused by 30 ml PBS and 50 ml 4% PFA (Roth, diluted in PBS), and afterward, vessels were dissected, and tissue pieces were further fixed for 4 hr in 4% PFA, transferred to 15% sucrose (in PBS, Merck) for 4 hr and incubated in 30% sucrose overnight. After embedding in TissueTek (Sakura), the tissue is frozen at −80°C and 8‐µm sections were obtained in a Leica cryostat at −30°C. For immunostainings, the cryostat sections were incubated with blocking buffer (1% donkey serum/1% Triton X‐100/PBS), the first antibody was applied overnight at 4°C, and after washing with PBS/1% Tween, the secondary antibody and DAPI were applied for 2 hr. Afterward, the sections were embedded in ImmoMount (Thermo Scientific #9990402). The stained sections were analyzed with Zeiss LSM700 microscope provided with Zeiss 40x objective, and images were analyzed by ImageJ/Fij. Antibodies: anti‐beta‐actin‐mouse (Sigma #A2228), anti‐Cav3.2‐rabbit (Alomone Labs #ACC‐025), anti‐mouse‐Alexa488 (Invitrogen #R37114), anti‐rabbit‐Cy3 (Dianova #711‐165–152), and DAPI (Sigma #D9542).
4.8. Vessel myography
Vessel myography was performed as previously described (Schleifenbaum et al., 2014) (Kassmann et al., 2019). Mesenteric arteries (third or fourth order) were mounted on glass cannula and superfused continuously with physiological saline solution (95% O2 −5% CO2; pH, 7.4; 37°C) containing (mM): 119 NaCl, 4.7 KCl, 25 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, and 11.1 glucose. The intravascular pressure was incrementally elevated from 20 to 100 mmHg using a pressure servo control system (Living System Instrumentation), and the inner diameter of the vessel was measured (Nikon Diaphot). The recording system was connected to a personal computer for data acquisition and analysis (HaSoTec). Arteries were equilibrated at 15 mmHg for 60 min and contractile responsiveness assessed by applying 60 mM KCl before starting experiments.
4.9. Materials
Fluo‐4‐AM was purchased from Molecular Probes (Eugene). Thapsigargin was purchased from Alomone Laboratories. All salts and other drugs were obtained from Sigma‐Aldrich or Merck. In cases where DMSO was used as a solvent, the maximal DMSO concentration after application did not exceed 0.5% (Kassmann et al., 2019; Tsvetkov et al., 2016).
4.10. Statistics
Data are presented as means ± SEM. Statistically significant differences in mean values were determined by Student's unpaired t test or one‐way analysis of variance (ANOVA) or Mann–Whitney U test. p‐values < .05 were considered statistically significant; “n” represents the number of cells.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
G.F., M.K., Y.C., D.T, C.M., S.K., C.Z., S.Z., and Y.X. were responsible for data collection, analysis, and interpretation. M.K. and M.G. were responsible for the conception and design of the experiments. G.F. and M.G. drafted the manuscript. All authors were responsible for interpretation of the data, contributed to the drafting, and revised the manuscript critically for important intellectual content. All authors have approved the final version of the manuscript and agreed to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Supporting information
Fig S1
ACKNOWLEDGMENTS
M.G. is supported by grants from the Deutsche Forschungsgemeinschaft (DFG) and Deutscher Akademischer Austauschdienst (DAAD). G.F. is supported by the CSC (China Scholarship Council). Y.X is supported by the Health Commission of Hunan and by the Science and Technology Department of Hunan. We acknowledge support from the Open Access Publication Fund of Charité–Universitätsmedizin Berlin.
Fan G, Kaßmann M, Cui Y, et al. Age attenuates the T‐type CaV3.2‐RyR axis in vascular smooth muscle. Aging Cell. 2020;19:e13134 10.1111/acel.13134
Gang Fan and Mario Kaßmann contributed equally to this work.
DATA AVAILABILITY STATEMENT
I confirm that my article contains a Data Availability Statement even if no new data was generated (list of sample statements) unless my article type does not require one.
REFERENCES
- Abd El‐Rahman, R. R. , Harraz, O. F. , Brett, S. E. , Anfinogenova, Y. , Mufti, R. E. , Goldman, D. , & Welsh, D. G. (2013). Identification of L‐ and T‐type Ca2+ channels in rat cerebral arteries: Role in myogenic tone development. American Journal of Physiology Heart and Circulatory Physiology, 304, H58–H71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakircioglu, M. E. , Sievert, K.‐D. , Nunes, L. , Lau, A. , Lin, C.‐S. , & Lue, T. F. (2001). Decreased trabecular smooth muscle and caveolin‐1 expression in the penile tissue of aged rats. The Journal of Urology, 166, 734–738. [PubMed] [Google Scholar]
- Bergdahl, A. , & Sward, K. (2004). Caveolae‐associated signalling in smooth muscle. Canadian Journal of Physiology and Pharmacology, 82, 289–299. [DOI] [PubMed] [Google Scholar]
- Berrier, C. , Coulombe, A. , Szabo, I. , Zoratti, M. , & Ghazi, A. (1992). Gadolinium ion inhibits loss of metabolites induced by osmotic shock and large stretch‐activated channels in bacteria. European Journal of Biochemistry, 206, 559–565. [DOI] [PubMed] [Google Scholar]
- Boersma, E. , Poldermans, D. , Bax, J. J. , Steyerberg, E. W. , Thomson, I. R. , Banga, J. D. , … Group DS (2001). Predictors of cardiac events after major vascular surgery: Role of clinical characteristics, dobutamine echocardiography, and β‐blocker therapy. JAMA, 285, 1865–1873. [DOI] [PubMed] [Google Scholar]
- Braunstein, T. H. , Inoue, R. , Cribbs, L. , Oike, M. , Ito, Y. , Holstein‐Rathlou, N. H. , & Jensen, L. J. (2009). The role of L‐ and T‐type calcium channels in local and remote calcium responses in rat mesenteric terminal arterioles. Journal of Vascular Research, 46, 138–151. [DOI] [PubMed] [Google Scholar]
- Brenner, R. , Peréz, G. J. , Bonev, A. D. , Eckman, D. M. , Kosek, J. C. , Wiler, S. W. , … Aldrich, R. W. (2000). Vasoregulation by the β1 subunit of the calcium‐activated potassium channel. Nature, 407, 870. [DOI] [PubMed] [Google Scholar]
- Chen, C. C. , Lamping, K. G. , Nuno, D. W. , Barresi, R. , Prouty, S. J. , Lavoie, J. L. , … Campbell, K. P. (2003). Abnormal coronary function in mice deficient in alpha1H T‐type Ca2+ channels. Science, 302, 1416–1418. [DOI] [PubMed] [Google Scholar]
- Cheng, X. , & Jaggar, J. H. (2006). Genetic ablation of caveolin‐1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. American Journal of Physiology. Heart and Circulatory Physiology, 290, H2309–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, C. S. , Huang, C. H. , Chieng, H. , Chang, Y. T. , Chang, D. , Chen, J. J. , … Chen, C. C. (2009). The Ca(v)3.2 T‐type Ca(2+) channel is required for pressure overload‐induced cardiac hypertrophy in mice. Circulation Research, 104, 522–530. [DOI] [PubMed] [Google Scholar]
- Ching, L. L. , Williams, A. J. , & Sitsapesan, R. (2000). Evidence for Ca2+ activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circulation Research, 87, 201–206. [DOI] [PubMed] [Google Scholar]
- Chiossi, G. , Costantine, M. M. , Tamayo, E. , Hankins, G. D. , Saade, G. R. , & Longo, M. (2016). Fetal programming of blood pressure in a transgenic mouse model of altered intrauterine environment. The Journal of Physiology, 594, 7015–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, L. S. , Garcia, E. , Cain, S. M. , Thau, E. , Tyson, J. R. , & Snutch, T. P. (2010). Splice‐variant changes of the Ca(V)3.2 T‐type calcium channel mediate voltage‐dependent facilitation and associate with cardiac hypertrophy and development. Channels (Austin), 4, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Corsso, C. , Ostrovskaya, O. , McAllister, C. E. , Murray, K. , Hatton, W. J. , Gurney, A. M. , … Wilson, S. M. (2006). Effects of aging on Ca2+ signaling in murine mesenteric arterial smooth muscle cells. Mechanisms of Ageing and Development, 127, 315–323. [DOI] [PubMed] [Google Scholar]
- Essin, K. , Welling, A. , Hofmann, F. , Luft, F. C. , Gollasch, M. , & Moosmang, S. (2007). Indirect coupling between Cav1. 2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. The Journal of Physiology, 584, 205–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, G. , Kaßmann, M. , Hashad, A. M. , Welsh, D. G. , & Gollasch, M. (2018). Differential targeting and signalling of voltage‐gated T‐type Cav3. 2 and L‐type Cav1. 2 channels to ryanodine receptors in mesenteric arteries. The Journal of Physiology, 596:4863–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Sanz, C. , Ruiz‐Meana, M. , Miro‐Casas, E. , Nuñez, E. , Castellano, J. , Loureiro, M. , … Garcia‐Dorado, D. (2014). Defective sarcoplasmic reticulum–mitochondria calcium exchange in aged mouse myocardium. Cell Death & Disease, 5, e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filosa, J. A. , Bonev, A. D. , Straub, S. V. , Meredith, A. L. , Wilkerson, M. K. , Aldrich, R. W. , & Nelson, M. T. (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nature Neuroscience, 9, 1397–1403. [DOI] [PubMed] [Google Scholar]
- Gollasch, M. , Ried, C. , Bychkov, R. , Luft, F. C. , & Haller, H. (1996). K+ currents in human coronary artery vascular smooth muscle cells. Circulation Research, 78, 676–688. [DOI] [PubMed] [Google Scholar]
- Gollasch, M. , Wellman, G. C. , Knot, H. J. , Jaggar, J. H. , Damon, D. H. , Bonev, A. D. , & Nelson, M. T. (1998). Ontogeny of local sarcoplasmic reticulum Ca2+ signals in cerebral arteries: Ca2+ sparks as elementary physiological events. Circulation Research, 83, 1104–1114. [DOI] [PubMed] [Google Scholar]
- Gottlieb, P. A. , Suchyna, T. M. , Ostrow, L. W. , & Sachs, F. (2004). Mechanosensitive ion channels as drug targets. Current Drug Targets‐CNS & Neurological Disorders, 3, 287–295. [DOI] [PubMed] [Google Scholar]
- Harraz, O. F. , Abd El‐Rahman, R. R. , Bigdely‐Shamloo, K. , Wilson, S. M. , Brett, S. E. , Romero, M. , … Welsh, D. G. (2014). Ca(V)3.2 channels and the induction of negative feedback in cerebral arteries. Circulation Research, 115, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashad, A. M. , Harraz, O. F. , Brett, S. E. , Romero, M. , Kassmann, M. , Puglisi, J. L. , … Welsh, D. G. (2018). Caveolae link CaV3. 2 channels to BKCa‐mediated feedback in vascular smooth muscle. Arteriosclerosis, Thrombosis, and Vascular Biology, 38:2371–2381. [DOI] [PubMed] [Google Scholar]
- Hashad, A. M. , Mazumdar, N. , Romero, M. , Nygren, A. , Bigdely‐Shamloo, K. , Harraz, O. F. , … Welsh, D. G. (2017). Interplay among distinct Ca2+ conductances drives Ca2+ sparks/spontaneous transient outward currents in rat cerebral arteries. The Journal of Physiology, 595, 1111–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headrick, J. P. , Willems, L. , Ashton, K. J. , Holmgren, K. , Peart, J. , & Matherne, G. P. (2003). Ischaemic tolerance in aged mouse myocardium: The role of adenosine and effects of A1 adenosine receptor overexpression. The Journal of Physiology, 549, 823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers, R. H. , Kundumani‐Sridharan, V. , Subramani, J. , Chen, L. C. , Cuello, L. G. , Rusch, N. J. , & Das, K. C. (2017). Thioredoxin reverses age‐related hypertension by chronically improving vascular redox and restoring eNOS function. Science Translational Medicine, 9:eaaf6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar, J. H. , Wellman, G. C. , Heppner, T. J. , Porter, V. A. , Perez, G. J. , Gollasch, M. , … Nelson, M. T. (1998). Ca2+ channels, ryanodine receptors and Ca(2+)‐activated K+ channels: A functional unit for regulating arterial tone. Acta Physiologica Scandinavica, 164, 577–587. [DOI] [PubMed] [Google Scholar]
- Janczewski, A. M. , & Lakatta, E. G. (1993). Thapsigargin inhibits Ca2+ uptake, and Ca2+ depletes sarcoplasmic reticulum in intact cardiac myocytes. American Journal of Physiology‐Heart and Circulatory Physiology, 265, H517–H522. [DOI] [PubMed] [Google Scholar]
- Kassmann, M. , Szijarto, I. A. , Garcia‐Prieto, C. F. , Fan, G. , Schleifenbaum, J. , Anistan, Y. M. , … Gollasch, M. (2019). Role of ryanodine type 2 receptors in elementary Ca2+ signaling in arteries and vascular adaptive responses. Journal of the American Heart Association, 8, e010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot, H. J. , Standen, N. B. , & Nelson, M. T. (1998). Ryanodine receptors regulate arterial diameter and wall [Ca2+] in cerebral arteries of rat via Ca2+‐dependent K+ channels. The Journal of Physiology, 508, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhbandner, S. , Brummer, S. , Metzger, D. , Chambon, P. , Hofmann, F. , & Feil, R. (2000). Temporally controlled somatic mutagenesis in smooth muscle. Genesis, 28, 15–22. [DOI] [PubMed] [Google Scholar]
- Lewartowski, B. , & Wolska, B. M. (1993). The effect of thapsigargin on sarcoplasmic reticulum Ca2+ content and contractions in single myocytes of guinea‐pig heart. Journal of Molecular and Cellular Cardiology, 25, 23–29. [DOI] [PubMed] [Google Scholar]
- Lian, X. , Matthaeus, C. , Kassmann, M. , Daumke, O. , & Gollasch, M. (2019). Pathophysiological Role of Caveolae in Hypertension. Front Med (Lausanne), 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhn, M. , Fürstenau, M. , Sagach, V. , Elger, M. , Schulze, W. , Luft, F. C. , … Gollasch, M. (2000). Ignition of calcium sparks in arterial and cardiac muscle through caveolae. Circulation Research, 87, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Löhn, M. , Lauterbach, B. , Haller, H. , Pongs, O. , Luft, F. C. , & Gollasch, M. (2001). β1‐Subunit of BK channels regulates arterial wall [Ca2+] and diameter in mouse cerebral arteries. Journal of Applied Physiology, 91, 1350–1354. [DOI] [PubMed] [Google Scholar]
- Lowalekar, S. K. , Cristofaro, V. , Radisavljevic, Z. M. , Yalla, S. V. , & Sullivan, M. P. (2012). Loss of bladder smooth muscle caveolae in the aging bladder. Neurourology and Urodynamics, 31, 586–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, A. , Howard, G. , Mendoza‐Topaz, C. , Deerinck, T. , Mackey, M. , Sandin, S. , … Nichols, B. J. (2013). Molecular composition and ultrastructure of the caveolar coat complex. PLoS Biology, 11, e1001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthaeus, C. , Lahmann, I. , Kunz, S. , Jonas, W. , Melo, A. A. , Lehmann, M. , … Mueller, D. N. (2019). EHD2‐mediated restriction of caveolar dynamics regulates cellular lipid uptake. bioRxiv, 511709. [DOI] [PMC free article] [PubMed]
- McEwen, D. P. , Li, Q. , Jackson, S. , Jenkins, P. M. , & Martens, J. R. (2008). Caveolin regulates kv1.5 trafficking to cholesterol‐rich membrane microdomains. Molecular Pharmacology, 73, 678–685. [DOI] [PubMed] [Google Scholar]
- Mikkelsen, M. F. , Björling, K. , & Jensen, L. J. (2016). Age‐dependent impact of CaV3.2 T‐type calcium channel deletion on myogenic tone and flow‐mediated vasodilatation in small arteries. The Journal of Physiology, 594, 5881–5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang, S. , Schulla, V. , Welling, A. , Feil, R. , Feil, S. , Wegener, J. W. , … Klugbauer, N. (2003). Dominant role of smooth muscle L‐type calcium channel Cav1. 2 for blood pressure regulation. The EMBO Journal, 22, 6027–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morén, B. , Shah, C. , Howes, M. T. , Schieber, N. L. , McMahon, H. T. , Parton, R. G. , … Lundmark, R. (2012). EHD2 regulates caveolar dynamics via ATP‐driven targeting and oligomerization. Molecular Biology of the Cell, 23, 1316–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M. T. , Cheng, H. , Rubart, M. , Santana, L. F. , Bonev, A. D. , Knot, H. J. , & Lederer, W. J. (1995). Relaxation of arterial smooth muscle by calcium sparks. Science, 270, 633–637. [DOI] [PubMed] [Google Scholar]
- Ohno‐Iwashita, Y. , Shimada, Y. , Hayashi, M. , & Inomata, M. (2010). Plasma membrane microdomains in aging and disease. Geriatrics & Gerontology International, 10, S41–S52. [DOI] [PubMed] [Google Scholar]
- Parton, R. G. , & Simons, K. (2007). The multiple faces of caveolae. Nature Reviews Molecular Cell Biology, 8, 185–194. [DOI] [PubMed] [Google Scholar]
- Pérez, G. J. , Bonev, A. D. , Patlak, J. B. , & Nelson, M. T. (1999). Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. The Journal of General Physiology, 113, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluger, S. , Faulhaber, J. , Furstenau, M. , Lohn, M. , Waldschutz, R. , Gollasch, M. , … Pongs, O. (2000). Mice with disrupted BK channel beta 1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circulation Research, 87, E53–E60. [DOI] [PubMed] [Google Scholar]
- Ratajczak, P. , Damy, T. , Heymes, C. , Oliviero, P. , Marotte, F. , Robidel, E. , … Samuel, J. L. (2003). Caveolin‐1 and ‐3 dissociations from caveolae to cytosol in the heart during aging and after myocardial infarction in rat. Cardiovascular Research, 57, 358–369. [DOI] [PubMed] [Google Scholar]
- Riehle, M. , Büscher, A. K. , Gohlke, B.‐O. , Kaßmann, M. , Kolatsi‐Joannou, M. , Bräsen, J. H. , … Hoyer, P. F. (2016). TRPC6 G757D loss‐of‐function mutation associates with FSGS. Journal of the American Society of Nephrology, 27:2771–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara, Y. , Fernandez‐Belda, F. , De Meis, L. , & Inesi, G. (1992). Characterization of the inhibition of intracellular Ca2+ transport ATPases by thapsigargin. Journal of Biological Chemistry, 267, 12606–12613. [PubMed] [Google Scholar]
- Sagara, Y. , & Inesi, G. (1991). Inhibition of the sarcoplasmic‐reticulum Ca2+ transport Atpase by thapsigargin at subnanomolar concentrations. Journal of Biological Chemistry, 266, 13503–13506. [PubMed] [Google Scholar]
- Sausbier, M. , Arntz, C. , Bucurenciu, I. , Zhao, H. , Zhou, X. B. , Sausbier, U. , … Ruth, P. (2005). Elevated blood pressure linked to primary hyperaldosteronism and impaired vasodilation in BK channel‐deficient mice. Circulation, 112, 60–68. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum, J. , Kassmann, M. , Szijártó, I. A. , Hercule, H. C. , Tano, J.‐Y. , Weinert, S. , … Alenina, N. (2014). Stretch–activation of angiotensin II type 1a receptors contributes to the myogenic response of mouse mesenteric and renal arteries. Circulation Research, 115, 263–272. [DOI] [PubMed] [Google Scholar]
- Seisenberger, C. , Specht, V. , Welling, A. , Platzer, J. , Pfeifer, A. , Kühbandner, S. , … Hofmann, F. (2000). Functional embryonic cardiomyocytes after disruption of the L‐type α1C (Ca v 1.2) calcium channel gene in the mouse. Journal of Biological Chemistry, 275, 39193–39199. [DOI] [PubMed] [Google Scholar]
- Shvets, E. , Bitsikas, V. , Howard, G. , Hansen, C. G. , & Nichols, B. J. (2015). Dynamic caveolae exclude bulk membrane proteins and are required for sorting of excess glycosphingolipids. Nature Communications, 6, 6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, K. , & Ehehalt, R. (2002). Cholesterol, lipid rafts, and disease. The Journal of Clinical Investigation, 110, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeber, M. , Schellenberger, P. , Siebert, C. A. , Leyrat, C. , Helenius, A. , & Grünewald, K. (2016). Model for the architecture of caveolae based on a flexible, net‐like assembly of Cavin1 and Caveolin discs. Proceedings of the National Academy of Sciences of the United States of America, 113, E8069–E8078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup, O. (1990). Role of Ca‐2+‐Atpases in regulation of cellular Ca‐2+ signaling, as studied with the selective microsomal Ca‐2+‐Atpase inhibitor, thapsigargin. Agents and Actions, 29, 8–15. [DOI] [PubMed] [Google Scholar]
- Trollinger, D. R. , Rivkah Isseroff, R. , & Nuccitelli, R. (2002). Calcium channel blockers inhibit galvanotaxis in human keratinocytes. Journal of Cellular Physiology, 193, 1–9. [DOI] [PubMed] [Google Scholar]
- Tsvetkov, D. , Shymanets, A. , Huang, Y. , Bucher, K. , Piekorz, R. , Hirsch, E. , … Nurnberg, B. (2016). Better understanding of phosphoinositide 3‐kinase (PI3K) pathways in vasculature: Towards precision therapy targeting angiogenesis and tumor blood supply. Biochemistry‐Moscow+. 81, 691–699. [DOI] [PubMed] [Google Scholar]
- Wang, S.‐Q. , Wei, C. , Zhao, G. , Brochet, D. X. P. , Shen, J. , Song, L.‐S. , … Cheng, H. (2004). Imaging microdomain Ca2+ in muscle cells. Circulation Research, 94, 1011–1022. [DOI] [PubMed] [Google Scholar]
- Wirth, A. , Wang, S. , Takefuji, M. , Tang, C. , Althoff, T. F. , Schweda, F. , … Offermanns, S. (2016). Age‐dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by G‐protein signalling. Cardiovascular Research, 109, 131–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Data Availability Statement
I confirm that my article contains a Data Availability Statement even if no new data was generated (list of sample statements) unless my article type does not require one.