

Mutations in WDR45 cause neurodegeneration with brain iron accumulation. Seibler et al. report that loss of WDR45 function in cells from patients results in increased cellular iron levels and oxidative stress, accompanied by mitochondrial and lysosomal dysfunction. Activation of autophagy may serve as a potential therapeutic approach.
Keywords: WDR45, iron, autophagy, mitochondria, dopaminergic neurons
Abstract
Beta-propeller protein-associated neurodegeneration is a subtype of monogenic neurodegeneration with brain iron accumulation caused by de novo mutations in WDR45. The WDR45 protein functions as a beta-propeller scaffold and plays a putative role in autophagy through its interaction with phospholipids and autophagy-related proteins. Loss of WDR45 function due to disease-causing mutations has been linked to defects in autophagic flux in patient and animal cells. However, the role of WDR45 in iron homeostasis remains elusive. Here we studied patient-specific WDR45 mutant fibroblasts and induced pluripotent stem cell-derived midbrain neurons. Our data demonstrated that loss of WDR45 increased cellular iron levels and oxidative stress, accompanied by mitochondrial abnormalities, autophagic defects, and diminished lysosomal function. Restoring WDR45 levels partially rescued oxidative stress and the susceptibility to iron treatment, and activation of autophagy reduced the observed iron overload in WDR45 mutant cells. Our data suggest that iron-containing macromolecules and organelles cannot effectively be degraded through the lysosomal pathway due to loss of WDR45 function.
Introduction
De novo mutations in the WD repeat domain 45 (WDR45) gene were described to cause ‘beta-propeller protein-associated neurodegeneration’ (BPAN) (Haack et al., 2012), an X-linked form of neurodegeneration with brain iron accumulation (NBIA). NBIA diseases are a heterogeneous group of monogenic disorders that share the feature of high iron levels in defined areas of the brain, usually including the globus pallidus, but occasionally extending to other areas such as cerebellum or substantia nigra, depending on the type of disease (Hayflick et al., 2013; Rouault, 2013). Patients carrying WDR45 mutations present with global developmental delay and autistic features in childhood and further regression in early adulthood with progressive dystonia, parkinsonism, and dementia. Male and female patients share the same distinctive phenotype due to somatic mosaicism or skewing of the X chromosome inactivation in heterozygous female carriers. Post-mortem brain data of patients with WDR45 mutations show iron deposition preferentially in the substantia nigra (Hayflick et al., 2013). WDR45 encodes a member of the WD repeat protein interacting with phosphoinositides (WIPI) family (WIPI-4), a protein that is characterized as a beta-propeller scaffold playing a putative role in autophagy through its interaction with phospholipids and autophagy-related proteins (Lu et al., 2011). In lymphoblast cells from patients carrying WDR45 mutations, autophagosome formation is impaired at an early stage as shown by the accumulation of LC3-II protein (Saitsu et al., 2013). Wdr45 CNS-specific knockout mice exhibit poor motor coordination, reduced learning and memory, and extensive axon swelling (Zhao et al., 2015). Primary cells from these Wdr45 knockout mice show defects in autophagic flux and accumulation of protein aggregates, in both neurons and axons. Mechanistic overlap among the NBIA proteins results from the involvement of mitochondrial function [e.g. pantothenate kinase 2 (PANK2)] and lipid metabolism [e.g. phospholipase A2 group VI calcium-independent (PLA2G6)] (Arber et al., 2016). WDR45 represents an example of a direct link between an autophagy protein and neurodegeneration, with WDR45 being one of the four members of the human WIPI beta-propeller proteins critical for autophagosome formation (Arber et al., 2016). It remains a matter of debate whether iron accumulation is a primary cause or secondary event in this process, but iron-induced oxidative stress contributes to neurodegeneration by generating reactive oxygen species (Berg and Hochstrasser, 2006; Rouault, 2013). In turn, reactive oxygen species can directly damage DNA or oxidize lipids and proteins. Notably, aggregation of proteins involved in neurodegenerative disorders (e.g. alpha-synuclein and hyper-phosphorylated tau protein) has been shown to be triggered by elevated ferric iron concentrations in vitro (Ward et al., 2014). Understanding the role of iron homeostasis will be of great importance towards better understanding of this heterogeneous group of NBIA diseases.
Here, we examined fibroblasts and induced pluripotent stem cell (iPSC)-derived neurons from a female patient with BPAN. The detailed clinical examination of the patient was followed by a thorough molecular study of cellular processes including mitochondrial and lysosomal function. Elevated iron levels were seen in conjunction with an increased susceptibility to oxidative stress, mitochondrial dysfunction, and alterations in the lysosomal/autophagy pathway suggesting a more fundamental impact of WDR45 on cellular function than the known autophagy-related relevance.
Materials and methods
Subjects
We included one female patient (Patient L8172) with BPAN and five healthy control individuals (Subjects L2131, 49, L3293, L3365, L2135). Skin biopsies for functional assays and EDTA blood samples for DNA testing were taken from each individual. The patient was screened for mutations in the coding exons of ATP13A2, CP, C19ORF12, FA2H, FTL, PANK2, PLA2G6, and WDR45 at the Center for Genomics and Transcriptomics, Tübingen, Germany. The study was approved by the local ethics committee of the University of Lübeck and all participants gave written informed consent.
Cell culture, reprogramming, differentiation, and characterization
Primary dermal skin fibroblasts and neuroblastoma SH-SY5Y cells (Life Technologies) were maintained in Dulbecco’s modified Eagle medium (Life Technologies) supplemented with 10% foetal bovine serum (Life Technologies) and 1% penicillin/streptomycin (Life Technologies).
Patient skin fibroblasts were reprogrammed into iPSCs using Sendai virus to deliver the four reprogramming factors OCT4, SOX2, KLF4, and cMYC (CytoTune-iPS 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific). Two iPSC clones (WDR45-C1 and WDR45-C2) were established according to the manufacturer’s protocol and cultured on Matrigel®-coated dishes (BD Biosciences) in mTeSR1 medium (STEMCELL Technologies). The lines were characterized for the expression of pluripotency markers (OCT4, SOX2, GDF3, NANOG) as described previously (Seibler et al., 2011). Karyotype analysis was performed at Cell Line Genetics using standard protocols for high resolution G-banding. Genome integrity was assessed by Illumina Human CytoSNP-12 v2.1 beadchip array, with genomic DNA extracted using the DNeasy® kit (Qiagen) and analysis with GenomeStudio and KaryoStudio software (Illumina) (Supplementary Fig. 4C). In vitro spontaneous differentiation of iPSCs to form embryoid bodies was evaluated as described previously (Grütz et al., 2017). Control lines from three individuals were generated and characterized previously (Subject L2131 = control Subject 1, Subject HFF = control Subject 2, Subject L2135 = control Subject 3) (Mazzulli et al., 2016; Zanon et al., 2017). The direct differentiation of iPSCs into dopaminergic neurons was conducted as described before (Kriks et al., 2011; Mazzulli et al., 2016; Zanon et al., 2017) and neurons were terminally differentiated for 70 days.
Cloning, virus transduction, gene editing, and compound treatment
The patient’s cells were transduced with lentiviral vectors carrying the coding sequences of human wild-type WDR45. The coding sequence was subcloned from the original plasmid (Addgene) into the CSCW2 vector equipped with a puromycin resistance cassette and virus production was performed according to standard protocols (Park et al., 2008). Fibroblast cells were directly infected with viral supernatant. The virus titre was determined using the HIV type 1 p24 Antigen ELISA (ZeptoMetrix). Forty-eight hours post-infection, the culture medium was supplemented with 2 µg/ml puromycin (Sigma-Aldrich) and cells were selected for 2 weeks.
To assay cells upon autophagy induction, cells were treated with 250 nM torin 1 (Tocris Bioscience) for 24 h. To examine the susceptibility of cells to oxidative stressors, cells were treated with 1 mM MPP + (Sigma-Aldrich) or 100 µM FeCl3 (Sigma-Aldrich) for 24 h. All experiments were repeated three to six times.
To induce a knockout into the WDR45 gene gRNA was designed targeting the sequence 5′- ggtgggcagcatgggcttgg-3′ with the immediately following PAM sequence (tgg) located in exon 4 (Transcript ID: ENST00000634944.1). The gRNA was cloned into the pLKO1-Puro vector (Addgene plasmid #8453). Plasmids expressing Cas9 protein and the resistance gene to the antibiotic blastocidin (pCas9-Blast) were produced. SH-SY5Y cells were transfected with pCas9-Blast and the gRNA construct using an Amaxa Nucleofection II device. Transfected cells were grown subsequently until colony formation was apparent. Twenty colonies were picked, cultured independently and examined for a knockout by Sanger sequencing in the targeting region.
Assessment of X-chromosome inactivation and PCR analysis
Genomic DNA was isolated from blood, fibroblasts, iPSCs, and neurons using the DNeasy® kit (Qiagen). The degree of X chromosome inactivation was analysed as described previously (Allen et al., 1992; Domingo et al., 2014). Total RNA from fibroblast cells was prepared by using the RNeasy® kit (Qiagen) according to the manufacturer’s instructions and then reverse transcribed into cDNA with the Maxima First Strand cDNA Synthesis Kit (Thermo Scientific). Quantitative PCR analysis was performed with SYBR® Green (Roche Diagnostics) on a LightCycler480 (Roche Diagnostics). Expression of WDR45 was analysed by amplification of a fragment spanning exons 8–9 (Forward: 5′-CCAGCCTGGAGAAGCAAC-3′; Reverse: 5′-GCTCCACCAGTTTCTCCTTG-3′) and by amplification of specifically the wild-type or the mutant allele. Therefore, two allele-specific forward primers were designed with the 3′ base of each primer matching only the wild-type (wt) or mutant (mut) allele, respectively (wt-Forward: 5′-TGGGAGTCTGCAACTTGAGG-3′; mut-Forward: 5′-TGGGAGTCTGCAACTTGAGA-3′). A common reverse primer was designed and used together with one of the forward primers in separate reactions (Reverse: 5′-CAGAGGAAGGAGGAGTCGTG-3′). Actb served as reference gene.
Quantification of cellular iron content
Cellular iron content was assessed using the Iron Assay Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, cells were homogenized in Iron Assay buffer and centrifuged at 10 000 g for 10 min at 4°C to remove insoluble material. Sample volume was adjusted to a final volume of 100 μl with Iron Assay buffer. To measure the amount of total iron, 5 μl of Iron Reducer was added to each sample well to reduce Fe3+ to Fe2+. After mixing, samples were incubated for 30 min at room temperature and protected from light. Finally, 100 μl of Iron Probe was added to each well, samples were mixed well and incubated for 60 min at toom temperature protected from light. Samples were loaded into a 96-well fluoro plate (F16 Black Maxisorb Plate, Nunc #475515) and absorbance at 593 nm was recorded using a spectramax i3 plate reader (Molecular Devices). A standard was used for normalization.
Immunofluorescence staining and image analysis
For immunocytochemical analysis, cells were fixed in 4% formaldehyde for 15 min, permeabilized, and blocked with 0.1% Triton™ X-100 in 4% normal goat serum in PBS for 1 h. Immunofluorescence staining was performed with primary antibodies against GRP75 (1:400; Abcam), LAMP1 antibody (1:1000; Santa Cruz), tyrosine hydroxylase (1:400; Calbiochem), Microtubule-associated protein 2 (1:400; Millipore), hepcidin (1:100; Santa Cruz), and respective secondary fluorescence antibodies (1:400; Life technologies). Samples for lipofuscin analysis were mounted after fixation in mounting media containing DAPI for nuclear stain (without incubation with primary or secondary antibodies). Images of autofluorescent signal were taken using the 488 channels as shown previously (Valdez et al., 2017). Mitochondrial network interconnectivity was analysed in cells stained for mitochondrial GRP75. The images were taken as z-stacks by using a fluorescence microscope with an ApoTome (Zeiss). The form factor was calculated from the images as a measure for mitochondrial interconnectivity by using ImageJ (NIH software) as described previously (Grünewald et al., 2010). Images of LAMP1 and hepcidin staining were analysed and captured at equal exposure times. Fluorescence intensity of LAMP1 signal was calculated for each field of view by using the following equation: corrected total cell fluorescence = integrated density − (area of selected cell × mean fluorescence of background readings). At least three fields of view were analysed per coverslip. Fluorescence intensity of hepcidin was calculated from z-stack images of single TH-positive neurons.
Assessment of mitochondrial membrane potential and ATP production
The mitochondrial membrane potential was analysed using the green fluorescent JC-1 probe (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine; Life technologies) as reported (Grünewald et al., 2009). ATP production was determined based on the mitochondrial oxygen consumption rate as described previously using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience) (Cooper et al., 2012). Briefly, fibroblasts were plated into XF24 plates. Four wells were left empty for background correction. On the day of the experiment, culture media was replaced with fresh media before plates were preincubated in a CO2-free incubator at 37°C for 1 h for equilibration before processing in the Extracellular Flux Analyzer. For baseline measurements, a 20-min equilibration step was followed by three cycles of 2-min mix, 3-min wait, and 2-min measurement time. For analysis of drug response, three cycles of 2-min mix, 3-min wait, and 2-min measurement were used per condition. After three baseline measurements, oligomycin (1 μM), CCCP (1 μM), and antimycin A (1 μM) were added sequentially and three measurements performed per condition. For normalization of cell number, cells were harvested after the experiment in RIPA buffer and protein concentration was measured.
Western blot analysis
Cell pellets were extracted using SDS extraction buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% DOC, 1% NP-40, and 0.1% SDS) or 1% Triton™ X-100 lysis buffer (containing 10% glycerol, 150 mM NaCl, 25 mM HEPES pH 7.4, 1 mM EDTA, 1.5 mM MgCl2, proteinase inhibitor cocktail) and gels were blotted onto nitrocellulose membranes. Antibodies used for western blotting were anti-GAPDH (1:100 000; Cell Signaling), anti-β-actin (1:1 000 000; Sigma), anti-LC3 (1:1000; Novus Biologicals), anti-L-Ferritin (1:1000; Santa Cruz), anti-H-Ferritin (1:1000; Santa Cruz), anti-Mitochondrial Ferritin (Origene; 1:1000), anti-LAMP1 (1:1000; Santa Cruz), anti-Lamp2 (1:1000; Santa Cruz), and anti-SOD2 (1:1000; Santa Cruz).
Quantification of oxidized proteins and viability assay
Protein carbonyl levels were assessed with the OxyBlot kit (Millipore), according to the manufacturer’s recommendations. The carbonyl groups in the protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNP-hydrazone) through a reaction with 2,4-dinitrophenylhydrazine (DNPH). The DNP-derivatized protein samples were analysed by western blotting using an antibody against DNP. Signals intensities on western blots were measured by Image Lab software (Bio-Rad).
To assess cell viability, we quantitatively measured lactate dehydrogenase (LDH) using the CytoTox 96 Cytotoxicity Assay (Promega). Fibroblasts were examined untreated and upon treatment with 10 mM and 15 mM FeSO4 (Sigma-Aldrich) for 15 h. Released LDH in culture supernatants was determined with the enzymatic assay, which results in the conversion of a tetrazolium salt (iodonitrotetrazolium) into a red formazan product. The amount of colour formed is proportional to the number of lysed cells and was measured at 490 nm in a plate reader.
Assessment of lysosomal proteolysis
A long-lived protein degradation assay was performed by radioactive pulse-chase using tritium-labelled leucine (Perkin-Elmer, #NET460A001MC) as previously described (Kaushik and Cuervo, 2009). Briefly, proteins were labelled with radioactive leucine for 36 h (pulse period), followed by a chasing period of 28 h. Short-lived proteins were excluded from the analysis by replacing the media after 1 h of chasing period with fresh chasing media. For lysosomal inhibition, 100 mM of leupeptin was added to the initial medium (pulse period) and 100 mM of leupeptin and 5 mM of NH4Cl were added to the chasing medium. Aliquots of culture media were sampled after 8, 20 and 28 h during chasing period and precipitated with 20% v/v trichloroacetic acid with 0.5 mg/ml BSA for a minimum of 8 h at 4°C followed by centrifugation at 20 000g, 20 min, 4°C. Pellets were resuspended in 0.1 N NaOH/0.1% Na-deoxycholate. After the last time point, cells were scrapped and harvested in 0.1 N NaOH /0.1% Na-deoxycholate. Radioactive counts of cell lysates and secreted proteins were measured using a liquid scintillation analyser (TriCarb 2800TR, Perkin Elmer). The percentage of labelled acid-soluble amino acids was used as an indication of proteolysis. The percentage of secreted proteins was determined by dividing the radioactive signal obtained from the media by the total radioactive counts obtained from the cell lysate. Lysosomal proteolysis was calculated as the difference between control and inhibited conditions.
Glucocerebrosidase enzyme activity assay
The glucocerebrosidase enzyme activity assay was performed using the artificial enzyme substrate 4-MU-glucopyranoside. Cells were washed in PBS, harvested in 1% Triton lysis buffer, and activity was measured from whole-cell lysates as described previously (Burbulla et al., 2017). Five micrograms of cell lysate was added to 10 μl of 10% BSA [in activity assay buffer: 0.25% (v/v) Triton™ X-100 (Sigma-Aldrich)] and to 20 μl of 5 mM artificial enzyme substrate (in activity assay buffer). The mixture was added up to 100 µl total with activity assay buffer, 0.25% (w/v) taurocholic acid (Sigma-Aldrich), 1 mM EDTA, in citrate/phosphate buffer (pH 5.4). Samples [±1 μM conduritol-b-epoxide (CBE)] were mixed and incubated at 37°C for 30 min. To stop the reaction 100 μl of stop solution (1 M glycine, pH 12.5) was added to each sample. 4-MU fluorescence was determined using a plate reader (excitation: 365 nm, emission: 445 nm; top read). Enzyme activity was quantified by subtracting CBE-treated lysates from non-CBE-treated lysates to obtain the activity from glucocerebrosidase.
Statistical analysis
Differences were analysed statistically using paired and unpaired t-tests or ANOVA with a suitable post hoc test. The error bars indicate standard error of the mean of n ≥ 3 independent experiments (*P < 0.05; **P < 0.01, ***P < 0.001).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.
Results
Clinical presentation and genetic analysis of a patient harbouring a mutation in WDR45
The BPAN patient presented with global developmental delay of all motor and cognitive milestones. She started walking at the age of 3 years, had impaired comprehension, and learned to speak only single words. Fine motor skills were impaired and gait was unstable with recurrent falls. At the age of 1 year, the patient had febrile seizures with recurrent epileptic discharges in EEG recordings until the age of 16 years. At the age of 20 years, neurological examination revealed mental retardation, autistic features, and hand-clapping stereotypies. The patient had vertical supranuclear gaze palsy, generalized axial more than appendicular bradykinesia, and generalized dystonia with involvement of the arms, legs and trunk, and mild spasticity of the legs (Supplementary Video 1). Her gait was slow with small steps but not shuffling. She walked with both feet slightly plantar-flexed and the right foot also everted. When walking, her trunk was flexed by about 20° and slightly tilted to the left.
MRI showed iron deposition in the globus pallidus, the substantia nigra and to a lesser degree in the thalamus bilaterally (Fig. 1A–C). We detected heterozygous mutations in the WDR45 (c.519+1_519+3delGTG; NM_007075.3) and the PLA2G6 (c.91 G>A; NM_003560.2) genes. The same WDR45 mutation has been reported recently in a 31-year-old female with BPAN (Ichinose et al., 2014). Reverse transcriptase-PCR and sequencing of our patient’s cDNA revealed loss of the essential exon 8 splice site leading to aberrant splicing. By use of a cryptic splice donor site within intron 8, 19 bp of intronic sequence were inserted, generating a frameshift and premature stop codon (p.D174Sfs*28) (Fig. 1D). The mutant allele sequence was detected predominantly at the cDNA level, likely due to skewed X chromosome inactivation. Accordingly, X-inactivation analysis showed a skewed pattern for genomic DNA derived from EDTA blood (88:12) and skin fibroblasts (81:19). In addition, we confirmed decreased gene expression levels for WDR45 and specifically for the WDR45 wild-type allele by quantitative RT-PCR (Fig. 1E). While there is strong evidence that loss of WDR45 function is causative for the disease in this patient, an additional heterozygous missense mutation (p.D31N) was detected in PLA2G6. However, mutations in PLA2G6 cause recessively inherited neurodegeneration and should not lead to disease-causing phenotypes in the presence of a PLA2G6 functional wild-type allele. We thus predicted that deficiency of WDR45 contributed to disease in our patient.
Figure 1.
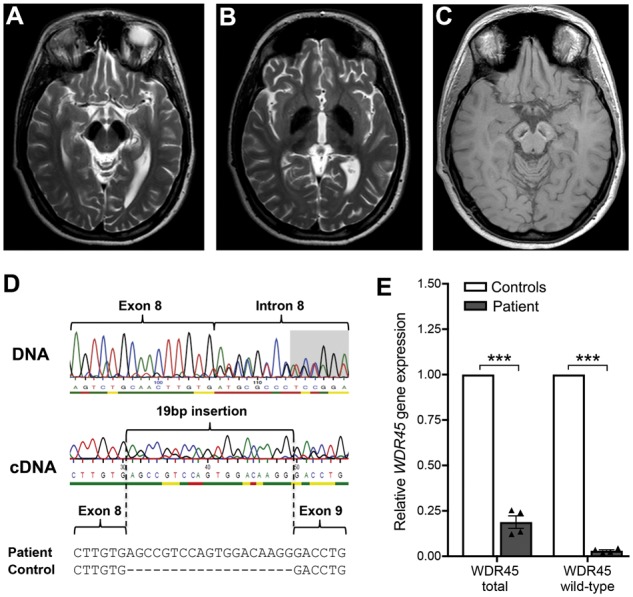
Cranial MRI and genetic analysis of a patient harbouring a mutation in WDR45. (A–C) MRI shows iron deposition. The substantia nigra (A), the globus pallidus and to a lesser degree the thalamus (B) are hypointense bilaterally on axial T2 sequences indicating high levels of iron. On T1-weighted MRI (C), the substantia nigra is hypointense centrally and surrounded by a hyperintense rim, which is a characteristic feature for BPAN. (D) The patient carries a heterozygous mutation in the WDR45 gene (c.519+1_519+3delGTG; NM_007075.3). RT-PCR and sequencing reveals loss of the essential exon 8 splice site leading to aberrant splicing in which 19 bp of intronic sequence are inserted by use of a cryptic splice donor site within intron 8, generating a frameshift and premature stop codon. (E) Skewed X chromosome inactivation results in decreased gene expression levels for WDR45 and specifically for the WDR45 wild-type allele in patient fibroblast cells compared to healthy controls shown by quantitative RT-PCR.
WDR45 mutant fibroblasts exhibit elevated iron and reduced levels of L-ferritin
Since brain iron accumulation is the key feature shared among the heterogeneous group of NBIAs, we examined total iron cellular content in WDR45 mutant fibroblasts. We observed an increase in the levels of basal iron content when comparing patient-derived WDR45 mutant cells with fibroblasts from two healthy controls (Fig. 2A). Ferritin is the major intracellular iron storage protein responsible for storing iron in a soluble and non-toxic state. Western blot analysis revealed reduced L-ferritin and H-ferritin protein levels in WDR45 mutant fibroblasts (Fig. 2B and Supplementary Fig. 1A). Lentiviral overexpression of wild-type WDR45 in the mutant cells rescued L-ferritin levels, suggesting a link between expression of WDR45 and L-ferritin (Fig. 2C and Supplementary Fig. 1B).
Figure 2.

Elevated levels of iron in WDR45 mutant fibroblasts and altered levels of L-ferritin. (A) Patient-derived WDR45 mutant cells demonstrate an increase in the levels of basal iron content when compared to fibroblasts from two healthy controls (ctrl). (B) Western blot analysis shows L-ferritin protein levels in the WDR45 mutant and control fibroblasts with GAPDH as loading control. (C) Lentiviral overexpression of wild-type WDR45 or control vector in the fibroblasts rescued L-ferritin protein levels.
Altered mitochondrial function and increased oxidative stress
Mitochondria represent one of the main cellular storage organelles for iron (Arber et al., 2016). Cells tightly regulate their mitochondrial iron levels because iron shortage affects numerous processes that use iron as co-factor, including the electron transport chain. Conversely, an excess of redox-active iron promotes the generation of radicals (Urrutia et al., 2014). For this reason, we examined mitochondrial function and morphology in WDR45 mutant fibroblasts. Mitochondrial network analysis revealed an increased number of fragmented mitochondria in mutant fibroblasts (Fig. 3A). Further, we observed a decreased mitochondrial membrane potential, reduced production of ATP, and elevated levels of superoxide dismutase 2 (SOD2) in mutant cells (Fig. 3B–D), under basal conditions. SOD2 functions to clear mitochondrial reactive oxygen species and increased levels of SOD2 indicate ongoing mitochondrial stress. We found no additional effect on SOD2 levels upon treatment either with the mitochondrial stressor 1-methyl-4-phenylpyridinium (MPP+), a toxin interfering with oxidative phosphorylation in mitochondria by inhibiting complex I, or with FeCl3 (Suttkus et al., 2014; Imam et al., 2015). Using OxyBlot to measure the total amount of oxidized proteins, we observed no differences between lines under basal conditions, but increased oxidation in mutant fibroblasts after treatment with FeCl3 (Fig. 3E) showing specific susceptibility of mutant cells to iron. Importantly, these effects were rescued by lentiviral overexpression of wild-type WDR45 (Fig. 3D and E). Iron storage in the mitochondria is partly regulated by mitochondrial ferritin, which functions in the sequestration of excess iron and responding to oxidative stress (Guan et al., 2017). In accordance to our data for L- and H-ferritin, we observed reduced protein levels of mitochondrial ferritin precursor in WDR45 mutant fibroblasts (Fig. 3F).
Figure 3.

Altered mitochondrial function and increased oxidative stress in WDR45 mutant fibroblasts. (A) The mitochondrial network was investigated under basal conditions by fluorescence microscopy in fixed cells immunostained with anti-GRP75 (green). The form factor as a measure for mitochondrial interconnectivity was calculated for two control (ctrl) fibroblast lines (n = 72) and one patient line (n = 47). The median, the interquartile range, and the minimum and the maximum value of the investigated individuals are shown. (B) Mitochondrial membrane potential was analysed by using a fluorescent dye and normalized to protein concentration (n = 4 independent experiments). (C) ATP production was determined based on the mitochondrial oxygen consumption rate using an extracellular flux analyser (n = 4 independent experiments). (D) Western blot analysis was performed on samples under basal conditions and upon treatment with 1-methyl-4-phenylpyridinium (MPP+) or ferric chloride (FeCl3). The western blot was probed with antibodies against superoxide dismutase 2 (SOD2) and β-actin (loading control). Statistical significance was analysed by selected unpaired t-tests. (E) Oxidation of proteins in WDR45 mutant fibroblasts and controls was determined by OxyBlot using an antibody against DNP. Statistical significance was analysed by selected unpaired t-tests. (F) Western blot analysis for mitochondrial ferritin (FtMt) precursor protein at 30 kDa. The antibody did not detect the processed protein at 22 kDa (n = 2 independent experiments).
To further validate the findings on mitochondrial dysfunction, we generated a neuroblastoma SH-SY5Y cell line with reduced expression of WDR45 by introducing a knockout (KO) into one allele of WDR45 with CRISPR/Cas9 gene-editing technology (Supplementary Fig. 2A). We compared SH-SY5Y-wt and SH-SY5Y-KO cells, respectively, and found mitochondrial fragmentation under basal conditions similar to that observed in fibroblasts. In addition, SH-SY5Y-KO cells showed elevated levels of SOD2 protein under basal conditions and increased amounts of oxidized proteins upon treatment with FeCl3 (Supplementary Fig. 2B–D). Our data from mutant fibroblasts and SH-SY5Y cells suggest the presence of mitochondrial damage and oxidative stress upon loss of WDR45 function.
Altered autophagy and diminished lysosomal function
Previous studies have shown defective autophagic flux in WDR45 mutant patient-derived lymphoblast cells (Saitsu et al., 2013) and Wdr45 knockout mice (Zhao et al., 2015). Western blotting of protein from WDR45 mutant fibroblast cultures showed equal levels of LC3-II in control, WDR45 mutant and wild-type WDR45 overexpressing cells under basal conditions (Fig. 4A). However, while induction of autophagy with the mTOR inhibitor torin 1 led to an elevation of LC3-II levels in mutant and control cell lines, the level of this increase was markedly reduced in mutant fibroblasts compared to wild-type WDR45 overexpressing cells. This suggests a defect in the induction of autophagy caused by WDR45 deficiency. Since the formation of LC3-II correlates with autophagosome numbers, we treated cells with the lysosomal degradation inhibitors leupeptin and ammonium chloride to measure autophagosome formation. Immunoblotting revealed that LC3-II levels were decreased in WDR45 mutant cells upon torin 1 treatment alone and in the presence of inhibitors, suggesting an overall reduced number of autophagosomes (Fig. 4B). However, comparing the ratio of inhibitor-treated and non-treated wild-type and mutant cells, our data indicate that the process of autophagosome synthesis is still functioning in mutant cells, but occurs at a lower basal level. Importantly, overexpressing WDR45 wild-type protein restored LC3-II levels in patient cells. Since elevated lysosomal iron potentially leads to loss of lysosomal integrity (Appelqvist et al., 2013), we examined whether lysosomes might be affected by the loss of WDR45 function. Levels of the lysosomal proteins LAMP1 and LAMP2 were dramatically lowered in mutant fibroblasts compared to control cells (Fig. 4C). This finding was confirmed by immunostaining for LAMP1 (Fig. 4D) further supporting the hypothesis of a generally reduced lysosomal degradation rate. To determine if a functional relationship exists between WDR45 deficiency and the lysosomal system, we measured the degradation rate of long-lived proteins using radioactive pulse-chase experiments in living cells (Kaushik and Cuervo, 2009; Mazzulli et al., 2011). We found reduced lysosomal proteolysis rates in WDR45 mutant compared to control fibroblasts (Fig. 4E) suggesting that the autophagic flux is reduced and that iron-containing macromolecules and organelles are not properly degraded. Consistent with this notion, lysosomal glucocerebrosidase activity was diminished in WDR45 mutant cells (Fig. 4F), an enzyme that has been implicated in Parkinson’s and Gaucher’s disease (Mazzulli et al., 2011). Additionally, we examined if WDR45 mutant fibroblasts also showed accumulation of lipofuscin, an autofluorescent electron-dense aggregate of undigested lysosomal material. In line with our data of reduced lysosomal function, we found that WDR45 mutant fibroblasts showed an increase in levels of autofluorescent puncta resembling lipofuscin (Valdez et al., 2017) compared to control fibroblasts (Supplementary Fig. 3A). To finally examine whether WDR45 deficiency also affects cell viability, we performed a cytotoxicity assay that quantitatively measures LDH, a stable cytosolic enzyme that is released upon cell lysis. To induce toxicity, we exposed the cells to different concentrations of the iron(II) salt FeSO4. After treatment with FeSO4, LDH levels were significantly higher in mutant compared to control fibroblasts. Overexpression of WDR45 efficiently rescued this increased susceptibility to iron stress (Supplementary Fig. 3B).
Figure 4.

Altered autophagy and lysosomal function in WDR45 mutant fibroblasts. (A) Western blot analysis from control (ctrl), WDR45 mutant, and WDR45 mutant fibroblasts overexpressing wild-type WDR45 using lentiviral vectors. Samples were collected from cells cultured under basal conditions and treated with the mTOR inhibitor torin 1. The western blot was probed against the autophagosome marker LC3-II (membrane-bound form), a cleavage product of LC3-I, and GAPDH (loading control). (B) Control and WDR45 mutant cells were treated with torin 1 alone and torin 1 plus the lysosomal inhibitors leupeptin (Leu) and ammonium chloride (NH4Cl). The western blot was probed against LC3-II and GAPDH (loading control). (C) Levels of the lysosomal proteins LAMP1 and LAMP2 were detected by western blotting. GAPDH was used as loading control. Statistical significance was analysed by one-way ANOVA (LAMP1) and selected unpaired t-test (LAMP2). (D) Immunofluorescence staining of control and WDR45 mutant fibroblasts against LAMP1 (red) and nuclear DAPI (blue). (E) Using radioactive pulse-chase experiments in living cells, lysosomal proteolysis rates were examined in mutant compared to control fibroblasts. To distinguish between proteolysis that occurs within the lysosomal compartment or other degradation pathways, the response to lysosomal inhibitors was measured. (F) Lysosomal glucocerebrosidase activity was measured in whole-cell lysates from WDR45 mutant and control fibroblasts.
Activation of autophagy reduces iron overload in WDR45 mutant neurons
To validate our findings in a neuronal model, we generated two iPSC lines from the BPAN patient’s fibroblasts. In both lines (iPSC-WDR45-C1 and iPSC-WDR45-C2), we confirmed the expression of pluripotency markers, the potential to differentiate into cell types of the three germ layers, and a normal karyotype (Supplementary Fig. 4). These patient-derived WDR45 mutant iPSCs as well as three control iPSC lines from healthy subjects generated previously (Mazzulli et al., 2016; Zanon et al., 2017) were differentiated into midbrain dopaminergic neurons and the expression of neuronal and dopaminergic markers confirmed (Fig. 5A and D). To investigate the impact of the reprogramming and differentiation procedure on the skewed methylation status of the X chromosome, DNA from different WDR45 mutant cell types was extracted. Skewed X chromosome inactivation was detected in all cell types, with the ratio indicating an almost complete silencing of one X chromosomes in mutant neurons (Fig. 5B). As expected, we found about 50% of total WDR45 gene expression levels in the patient compared to control neurons due to the nearly complete methylation of one chromosome (Fig. 5C). Wild-type-specific PCR revealed a dramatic reduction in the expression of wild-type WDR45 suggesting that only the mutant allele is expressed and accounts for the reduced total WDR45 gene expression. In summary, this demonstrates that the skewed X chromosome inactivation is maintained throughout the reprogramming and differentiation process.
Figure 5.

Cellular dysfunction in iPSC-derived dopaminergic neurons harbouring mutant WDR45. (A) Immunofluorescence staining shows expression of microtubule-associated protein 2 (MAP2), tyrosine hydroxylase (TH), and nuclear DAPI. (B) X chromosome inactivation analysis in DNA extracted from blood, fibroblasts, iPSCs, and neurons of patient-derived WDR45 mutant cells. (C) Skewed X chromosome inactivation results in decreased gene expression levels for total WDR45 and specifically for the WDR45 wild-type allele in patient neurons compared to healthy controls shown by quantitative RT-PCR. (D–F) Western blot analysis from control (ctrl) and WDR45 mutant neurons. Samples were collected from cells cultured under basal conditions and probed against SOD2, LAMP1, TH, neuron-specific enolase (NSE), neuronal class III β-tubulin, mitochondrial ferritin (FtMt) precursor protein, and β-actin and GAPDH (loading controls), respectively. (G) Total iron content was analysed in WDR45 mutant neurons and controls under basal conditions and upon treatment with torin 1.
We validated effects identified in mutant fibroblasts including increased expression of SOD2, decreased levels of LAMP1 and mitochondrial ferritin, and elevated iron levels (Fig. 5D–G) indicating that mitochondrial stress and lysosomal dysfunction are also present in neurons. Notably, western blot analysis in neurons revealed no difference in L-ferritin protein levels between control and WDR45 mutant neurons under basal conditions (Supplementary Fig. 5A). However, upon wild-type WDR45 overexpression, L-ferritin levels increased in the mutant neurons similar to our findings in fibroblasts (Supplementary Fig. 5B). Importantly, we confirmed in WDR45 mutant neurons decreased levels of mitochondrial ferritin precursor protein (Fig. 5F).
Since torin 1 treatment led to an activation of autophagy even in WDR45 mutant fibroblasts (Fig. 4A and B), we investigated whether this induction of autophagy might be sufficient to affect iron levels in mutant neurons. Interestingly, treatment with torin 1 completely rescued the elevated iron content in mutant neurons (Fig. 5G) and led to significantly reduced L-ferritin levels (Supplementary Fig. 5A) indicating a link between deficient autophagy, impaired proteolysis of L-ferritin-bound material, and high iron levels caused by WDR45 deficiency.
Discussion
High iron levels in defined areas of the brain are the pathological hallmark of NBIAs. However, the mechanism leading to localized iron overload and its role for neurodegeneration is still unknown and cannot be explained by the ubiquitous expression of the identified NBIA genes (Arber et al., 2016). Among the identified disease genes, only two are directly involved in iron metabolism, while the remaining NBIA genes encode proteins with a wide variety of functions (Rouault, 2013). Fibroblasts from NBIA patients with mutations in L-ferritin (Curtis et al., 2001) and PANK2 (Campanella et al., 2012) have been reported with increased iron levels and defective iron-handling properties, respectively. Likewise, two patient fibroblast lines harbouring WDR45 mutations have been shown to display deficits in iron homeostasis (Ingrassia et al., 2017). After starvation, these fibroblasts showed elevated iron levels when compared to controls. Our analysis of fibroblasts from a BPAN patient revealed increased iron levels under basal conditions accompanied by a decreased amount of L-ferritin, H-ferritin, and mitochondrial ferritin (Figs 2A, B, 3F and Supplementary Fig. 1A), the main iron storage proteins, which play a central role in the regulation of cellular iron metabolism and iron detoxification (Harrison and Arosio, 1996). Notably, similar data have been reported for PANK2 mutant patient fibroblasts (Campanella et al., 2012) indicating that low ferritin levels might be a shared pattern in different NBIA diseases. However, the regulation of L-ferritin and the impact of deficient autophagy might be different in non-dividing neurons (Supplementary Fig. 5A). Importantly, overexpression of wild-type WDR45 increased protein levels of L-ferritin in fibroblasts (Fig. 2C) and neurons (Supplementary Fig. 5B) showing that expression of WDR45 has a direct effect on L-ferritin levels. We found a strong reduction of mitochondrial ferritin precursor levels also in WDR45 mutant neurons (Fig. 5F). Previous studies have shown that mitochondrial ferritin is expressed mainly in neurons and that it protects dopaminergic neurons from oxidative stressors (Guan et al., 2017). Recent findings demonstrated that hepcidin, an iron-regulating hormone, reduced brain iron in iron-overloaded rats (Du et al., 2015). This peptide is predominantly expressed in the liver, but its expression can also be induced in the brain (Wang et al., 2008). Accordingly, we found hepcidin expression also in TH-positive neurons, located primarily to vesicles, but there was no difference in expression between control and WDR45 mutant neurons (Supplementary Fig. 5C).
Iron, the most prevalent metal in the body, reacts with hydrogen peroxide and catalyses the generation of highly reactive hydroxyl radicals, thereby increasing oxidative stress. In accordance with this, we detected an increase in oxidative stress-related markers and an elevated susceptibility to iron-induced stress across all our WDR45 mutant models (Figs 3D, E, 5D and Supplementary Fig. 2 and 3B). Oxidative stress is linked to mitochondrial dysfunction, as mitochondria are both generators and targets of reactive species. Our data showed fragmented morphology, decreased membrane potential, and decreased ATP synthesis rate in WDR45 mutant cells (Fig. 3A–C and Supplementary Fig. 2) indicating mitochondrial damage (Barsoum et al., 2006; Chen and Chan, 2009).
Based on the fact that increased amounts of iron are known to accumulate in lysosomes either as aggregated proteins, damaged mitochondria or due to the accumulation of iron-rich non-degradable materials, such as lipofuscin (Kurz et al., 2008), we hypothesized that the observed iron overload, together with oxidative stress in WDR45 mutant cells, might affect lysosomal function. We found impaired lysosomal proteolysis and reduced glucocerebrosidase activity in WDR45 mutant fibroblasts (Fig. 4E and F). Further, lysosomal LAMP1 protein levels were dramatically decreased in patient fibroblasts and neurons (Figs 4C and 5E) indicating severe dysfunction of the lysosomes. In line with these data, we found increased levels of autofluorescent puncta resembling lipofuscin in WDR45 mutant fibroblasts (Supplementary Fig. 3A).
As WDR45 has been implicated in autophagic dysfunction in patient lymphoblast cells (Saitsu et al., 2013), we examined WDR45 mutant fibroblasts for LC3-II levels under basal conditions and upon mTOR complex inhibition. In contrast to lymphoblasts, WDR45 patient fibroblasts revealed similar LC3-II levels compared to control cells under basal conditions. Upon torin 1 treatment, LC3-II levels increased in control and patient cells as well as patient cells overexpressing wild-type WDR45. However, this increase in LC3-II was less pronounced in mutant cells compared with controls (Fig. 4A and B) suggesting that induction of autophagy via inhibition of mTOR is diminished in WDR45 mutant cells.
Members of the human WIPI beta-propeller proteins associate with autophagosomal membranes based on specific phosphatidylinositol 3-phosphate binding (Vergne and Deretic, 2010; Proikas-Cezanne et al., 2015). It has been suggested that WIPI1, WIPI2, and WIPI4 (WDR45) proteins function as essential and non-redundant phosphatidylinositol 3-phosphate effectors downstream of phosphatidylinositol 3-phosphate production at the onset of autophagy (Proikas-Cezanne et al., 2015). However, the exact function of WDR45 in this process is not yet defined. In accordance with the hypothesis of an underlying autophagic defect as the contributing cause of BPAN, treatment with torin 1 reduced the elevated iron levels in WDR45 mutant neurons (Fig. 5G). We hypothesize that the activation of a reparative autophagy process enabling the degradation of iron binding proteins or iron storage organelles, such as ferritin and mitochondria, contributes to restoration of iron homeostasis. In fact, we observed the proteolysis of L-ferritin in WDR45 mutant neurons upon induction of autophagy (Supplementary Fig. 5A).
In summary, our data show that an overload of iron in WDR45 mutant fibroblasts and midbrain neurons leads to a number of pathological alterations including mitochondrial abnormalities, oxidative stress, autophagic defects, and diminished lysosomal function. Activation of autophagy reduces the amount of iron suggesting that degradation of iron-containing material through the lysosomal pathway may be beneficial. Further studies are needed to explore whether activation of autophagy might be a therapeutic approach in some forms of NBIAs.
Supplementary Material
Acknowledgements
We are indebted to the BPAN patient and healthy control individuals who have participated in this study. We are grateful to N. Andrews for helpful discussions and suggestions.
Glossary
Abbreviation
- BPAN
beta-propeller protein-associated neurodegeneration
- NBIA
neurodegeneration with brain iron accumulation
- iPSC
induced pluripotent stem cell
Funding
The authors declare no competing financial interests. This study was supported by the Hermann and Lilly Schilling Foundation (to C.K.), R01 NS076054 and R01 NS096240 (to D.K.), and the Federal Ministry of Education and Research (DysTract) (to P.S. and C.K.). P.S. is supported by the German Research Foundation (FOR2488).
Competing interests
The authors report no competing interests.
References
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; 51: 1229–39. [PMC free article] [PubMed] [Google Scholar]
- Appelqvist H, Wäster P, Kågedal K, Öllinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5: 214–26. [DOI] [PubMed] [Google Scholar]
- Arber CE, Li A, Houlden H, Wray S. Review: insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol 2016; 42: 220–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 2006; 25: 3900–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Hochstrasser H. Iron metabolism in Parkinsonian syndromes. Mov Disord 2006; 21: 1299–310. [DOI] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017; 357: 1255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella A, Privitera D, Guaraldo M, Rovelli E, Barzaghi C, Garavaglia B et al. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum Mol Genet 2012; 21: 4049–59. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet 2009; 18: R169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 2012; 4: 141ra90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet 2001; 28: 350–4. [DOI] [PubMed] [Google Scholar]
- Domingo A, Lee LV, Brüggemann N, Freimann K, Kaiser FJ, Jamora RD et al. Woman with x-linked recessive dystonia-parkinsonism: clue to the epidemiology of parkinsonism in Filipino women? JAMA Neurol 2014; 71: 1177–80. [DOI] [PubMed] [Google Scholar]
- Du F, Qian ZM, Luo Q, Yung WH, Ke Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol Neurobiol 2015; 52: 101–14. [DOI] [PubMed] [Google Scholar]
- Grünewald A, Gegg ME, Taanman JW, King RH, Kock N, Klein C et al. Differential effects of PINK1 nonsense and missense mutations on mitochondrial function and morphology. Exp Neurol 2009; 219: 266–73. [DOI] [PubMed] [Google Scholar]
- Grünewald A, Voges L, Rakovic A, Kasten M, Vandebona H, Hemmelmann C et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS One 2010; 5: e12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grütz K, Seibler P, Weissbach A, Lohmann K, Carlisle FA, Blake DJ et al. Faithful SGCE imprinting in iPSC-derived cortical neurons: an endogenous cellular model of myoclonus-dystonia. Sci Rep 2017; 7: 41156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Yang H, Yang M, Yanagisawa D, Bellier JP, Mori M et al. Mitochondrial ferritin protects SH-SY5Y cells against H 2 O 2 -induced oxidative stress and modulates α-synuclein expression. Exp Neurol 2017; 291: 51–61. [DOI] [PubMed] [Google Scholar]
- Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet 2012; 91: 1144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1996; 1275: 161–203. [DOI] [PubMed] [Google Scholar]
- Hayflick SJ, Kruer MC, Gregory A, Haack TB, Kurian MA, Houlden HH et al. β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain 2013; 136: 1708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose Y, Miwa M, Onohara A, Obi K, Shindo K, Saitsu H et al. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN). Neurol Clin Pract 2014; 4: 175–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam SZ, Lantz-McPeak SM, Cuevas E, Rosas-Hernandez H, Liachenko S, Zhang Y et al. Iron oxide nanoparticles induce dopaminergic damage: in vitro pathways and in vivo imaging reveals mechanism of neuronal damage. Mol Neurobiol 2015; 52: 913–26. [DOI] [PubMed] [Google Scholar]
- Ingrassia R, Memo M, Garavaglia B. Ferrous iron up-regulation in fibroblasts of patients with Beta Propeller Protein-Associated Neurodegeneration (BPAN). Front Genet 2017; 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Cuervo AM. Methods to monitor chaperone-mediated autophagy. Methods Enzymol 2009; 452: 297–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 2011; 480: 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T, Terman A, Gustafsson B, Brunk UT. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol 2008; 129: 389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell 2011; 21: 343–57. [DOI] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146: 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA 2016; 113: 1931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc 2008; 3: 1180–6. [DOI] [PubMed] [Google Scholar]
- Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci 2015; 128: 207–17. [DOI] [PubMed] [Google Scholar]
- Rouault TA. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat Rev Neurosci 2013; 14: 551–64. [DOI] [PubMed] [Google Scholar]
- Saitsu H, Nishimura T, Muramatsu K, Kodera H, Kumada S, Sugai K et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet 2013; 45: 445–9, 449e1. [DOI] [PubMed] [Google Scholar]
- Seibler P, Graziotto J, Jeong H, Simunovic F, Klein C, Krainc D. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 2011; 31: 5970–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttkus A, Rohn S, Weigel S, Glöckner P, Arendt T, Morawski M. Aggrecan, link protein and tenascin-R are essential components of the perineuronal net to protect neurons against iron-induced oxidative stress. Cell Death Dis 2014; 5: e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urrutia PJ, Mena NP, Núñez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol 2014; 5: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez C, Wong YC, Schwake M, Bu G, Wszolek ZK, Krainc D. Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 2017; 26: 4861–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett 2010; 584: 1313–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Du F, Qian ZM, Ge XH, Zhu L, Yung WH et al. Lipopolysaccharide induces a significant increase in expression of iron regulatory hormone hepcidin in the cortex and substantia nigra in rat brain. Endocrinology 2008; 149: 3920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 2014; 13: 1045–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanon A, Kalvakuri S, Rakovic A, Foco L, Guida M, Schwienbacher C et al. SLP-2 interacts with Parkin in mitochondria and prevents mitochondrial dysfunction in Parkin-deficient human iPSC-derived neurons and Drosophila. Hum Mol Genet 2017; 26: 2412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Sun L, Miao G, Ji C, Zhao H, Sun H et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015; 11: 881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material.