

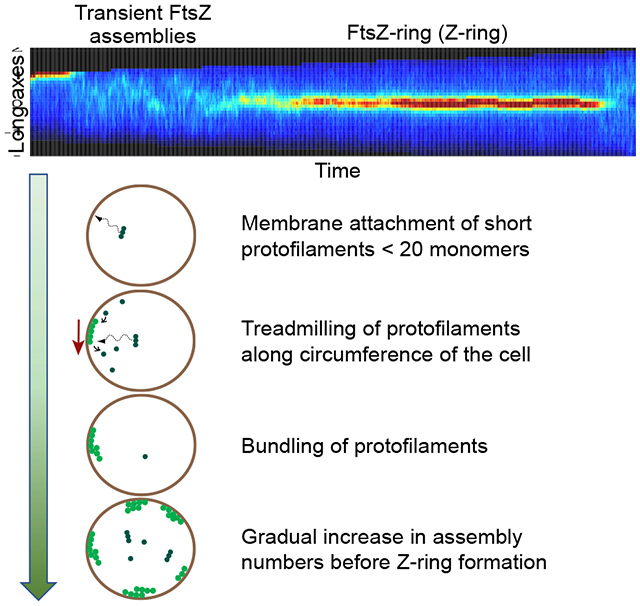
Summary
During the early stages of cytokinesis FtsZ protofilaments form a ring-like structure, the Z-ring, in most bacterial species. This cytoskeletal scaffold recruits downstream proteins essential for septal cell wall synthesis. Despite progress in understanding the dynamic nature of the Z-ring and its role in coordinating septal cell wall synthesis, the early stages of protofilament formation and subsequent assembly into the Z-ring are still not understood. Here we investigate a sequence of assembly steps that lead to the formation of the Z-ring in Escherichia coli using high temporal and spatial resolution imaging. Our data show that formation of the Z-ring is preceded by transient membrane-linked FtsZ assemblies. These assemblies form after attachment of short cytosolic protofilaments, which we estimate to be less than 20 monomers long, to the membrane. The attachments occur at random locations along the length of the cell. The filaments treadmill and show periods of rapid growth and shrinkage. Their dynamic properties imply that protofilaments are bundled in these assemblies. Furthermore, we establish that the size of assemblies is sensitively controlled by the availability of FtsZ molecules and by the presence of ZapA proteins. The latter has been implicated in cross-linking the protofilaments. The likely function of these dynamic FtsZ assemblies is to sample the cell surface for the proper location for the Z-ring.
Graphical Abstract
eTOC Blurb
By using high spatial and temporal resolution imaging, Walker et al. show that Z-ring formation in E. coli is preceded by a period in which FtsZ protofilament assemblies transiently form at the cytosolic face of the inner membrane. These assemblies are dynamic and compete with each other for the available FtsZ pool.
Introduction
In most studied bacterial species cell division starts with the formation of the Z-ring assembly at the inner face of the plasma membrane [1]. The assembly is organized around single subunit thick filaments of FtsZ, referred to as protofilaments. These cytoskeletal filaments are present in most bacterial species and also exist in several clades of archaea and plant plastids [2]. FtsZ shares structural homology and some dynamic properties with tubulin. It was recently shown that FtsZ filaments exhibit a treadmilling behavior [3, 4]. Typical half-lifetimes of monomers in FtsZ protofilaments has been measured to be about ten seconds [5, 6]. These dynamic properties are dependent on GTP hydrolysis at the interface of two FtsZ monomers. FtsZ protofilaments are located in the cytosol in the vicinity of the inner membrane, and in Escherichia coli are attached to it via FtsA and ZipA linkers [1]. In addition to the essential proteins FtsZ, FtsA, and ZipA, the early Z-ring assembly in this organism also includes non-essential Z-ring associated proteins ZapA-D. ZapA, ZapC, and ZapD have been implicated in bundling together individual protofilaments, [7] and ZapA and ZapB in positioning the division ring relative to the replication terminus of the chromosome [8]. This early cytokinetic assembly subsequently recruits about two dozen different proteins involved in the synthesis of the septal cell wall and in the partitioning of chromosomal DNA between daughter cells [9]. In this assembly FtsZ protofilaments act as a control hub that receives signals from different cellular processes including replication [10], metabolism [11], and DNA damage [12].
It is currently unclear how FtsZ protofilaments assemble into the Z-ring. The first assembly step is likely the formation of FtsZ protofilaments in the cytosol. This step is then followed by the attachment of the protofilament to the cell membrane via FtsA and ZipA linkers. Thereafter, it is possible that individual membrane attached filaments form higher order protofilament structures such as bundles or ribbons as has been seen in vitro [13]. However, the intermediary products of these steps are yet to be observed directly in vivo. Also, if the assembly proceeds via these steps then it is not clear if any of these steps are rate-limiting for the Z-ring assembly.
Based on in vitro studies, protofilaments should be abundantly present in the cytosol. These studies have shown that FtsZ protofilament assembly is cooperative with a critical concentration of about 1 μM [13]. At the same time the cytosolic concentration of FtsZ has been determined to be about 4-6 μM; thus much higher than the critical concentration [13]. However, it is unclear to what degree the in vitro studies are able to predict the critical concentration in vivo. The latter can be expected to be higher because of the presence of antagonists in vivo, such as the nucleoid occlusion factor SlmA [14], which prevents polymerization of FtsZ. So far, it has remained unclear what the polymerization state of cytosolic FtsZ is prior to the formation of the Z-ring. No cytosolic FtsZ protofilaments have yet been conclusively detected in experiments where FtsZ is present at physiological levels [15, 16]. Thus, it is possible that the effective critical concentration is higher in the cellular environment than in vitro and that the formation of a protofilament is a rate limiting step for the Z-ring formation in cells.
Attachment of protofilaments to the plasma membrane can also be a possible rate-limiting step. Based on in vitro measurements, FtsZ monomers bind to their membrane linkers with relatively low affinity, having Kd in the 10-50 μM range [17]. However, the binding of FtsZ protofilaments to ZipA was found to be about 100-fold stronger because of the possibility to link an FtsZ protofilament to multiple ZipA proteins [17]. Based on these data, it has been proposed that instead of being rate-limiting, protofilaments are rapidly captured by the cell membrane, via ZipA and FtsA membrane linkers, after their formation [18]. However, in vivo evidence for this idea is yet lacking.
Another possible rate-limiting step in the formation of the Z-ring can be protofilament bundling [19] but existing evidence for protofilament bundling in vivo is not conclusive. While super-resolution imaging has found that the Z-ring consists of FtsZ protofilament clusters/bundles [20, 21], higher resolution TEM imaging has shown just sparse individual filaments scattered around the constriction region of the cell in physiologically relevant levels of FtsZ [15, 16]. Protofilament-protofilament bundling via their lateral contacts has been reported in some in vitro studies but not in conditions that most closely mimic physiological ones [13]. It is more likely that protofilaments are linked together not by direct lateral contacts but cross-linked via the Zap proteins [22, 23]. However, none of the Zaps are individually essential nor are they essential as a group, although their absence leads to a cell division defect [24]. Protofilament cross-linking has also been reported in vitro in the presence of the FtsZ membrane anchor, ZipA [25, 26]. Altogether, there is growing evidence for protofilament-protofilament cross-linking via linker proteins from both in vivo and in vitro studies, but it is still unclear to what degree this process is essential for Z-ring formation.
Here we investigate the above described putative assembly steps that lead to the formation of the Z-ring using live cell microscopy at high spatial and temporal resolution. Our study focuses on slow growth conditions, where there is a distinct period between the birth of the cell and the formation of the Z-ring. In fast growth conditions this period is lacking [27], and therefore the possibility to observe intermediaries is much more limited. We show that in slow growth conditions transient attachments of FtsZ protofilaments to the plasma membrane precede the Z-ring formation. These attachments sample the whole cell surface for possible attachment sites but are more stable at the cell center compared to the poles. We find that the assemblies are nucleated by short protofilaments that are less than 20 monomers long. These protofilaments appear to be abundantly present in the cytosol while longer ones (20-40 monomers), although expected based on in vitro measurements [13, 28–30], are absent. After attachment to the plasma membrane, these FtsZ filaments show frequent periods of rapid growth and decay at rates that exceed, five-fold, the ones expected from a simple treadmilling model that accounts only for monomer turnover indicating involvement of protofilament bundling.
Results
Transient assemblies of FtsZ on the cell membrane precede the formation of the Z-ring
To understand the intermediaries leading to Z-ring formation we followed two types of strains with fluorescent FtsZ fusions using time lapse microscopy. One type expressed either FtsZ55-56mNeonGreen (FtsZ-mNG) [31] or FtsZ55-56sfGFP sandwich fusion (FtsZ-sfGFP) from its native locus [4] while the other type expressed FtsZ-GFP as an extra copy from the plasmid [4] (Table S1). Since the half lifetime of FtsZ monomers in protofilaments is about ten seconds [5, 6], FtsZ assemblies should be tracked at least at comparable rates to observe any possible intermediary structures. To prevent phototoxicity of such rapid and light-intensive measurements, we used grazing angle illumination (also referred to as HILO [32]), which decreases background fluorescence. We grew cells in mother machine channels [33, 34] to guarantee steady state conditions. To be compatible with HILO illumination we used agarose as the material for the channels instead of PDMS (poly-dimethylsiloxane) polymer (Methods: Preparing and using agarose based mother machine chips). The FtsZ-mNG strain was imaged in these channels for three to four consecutive cell cycles while recording about one thousand frames per cell cycle (Figure 1A, Video S1). There was no noticeable effect on the doubling time (Td=170±50min, N=25) stemming from the light exposure conditions when we compared it to measurements where the imaging rate was 40 times lower (Td=160±30 min) [27]. Once the Z-ring has formed, the FtsZ protofilaments or filament bundles could be observed treadmilling around the circumference of the cell, which gives rise to the characteristic oscillatory pattern of the FtsZ distribution along the short axis of the cell (Figure 1B). Although the cell length at birth in this strain is about 10% longer than in the wild type (WT) strain in slow growth conditions [27], the unperturbed doubling times and observations of FtsZ treadmilling indicated that our measurement conditions do not significantly perturb the processes associated with FtsZ protofilament dynamics.
Figure 1. Transient membrane-linked FtsZ assemblies precede Z-ring formation.
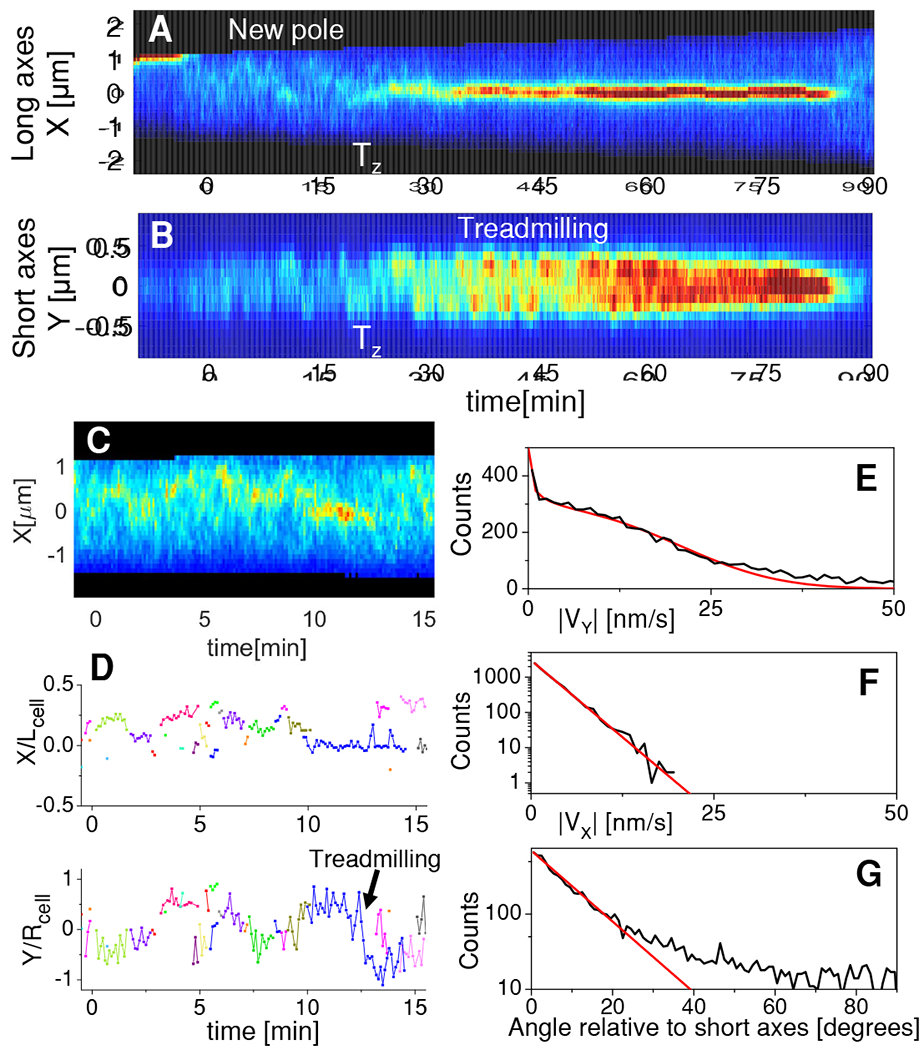
(A) Kymograph of FtsZ-mNG intensity distribution along the long axis of a cell during one cell cycle. Red corresponds to high and blue to low concentrations. Black color shows regions outside the cell. Vertical white dashed lines mark cell birth and white arrows mark the time when a persistent Z-ring forms (Tz). See also Video S1.
(B) Kymograph along the short axes for the same cell.
(C) Zoom in of the kymograph in panel (A) for the time preceding Z-ring formation.
(D) Trajectories of FtsZ assemblies along the long (top) and short axis (bottom) before Z-ring formation. Different colors indicate different assemblies. Arrow points to trajectory showing treadmilling motion. Trajectories are normalized by cell length, Lcell, and radius, Rcell, respectively. All data in panels (A-D) are from the same cell.
(E) Distribution of apparent speeds of assemblies relative to the short axis of the cell (black). Red line is a fitting of the distribution taking into account the variation of apparent speeds on the cell perimeter (Methods: Determination of treadmilling velocities and distribution of treadmilling speeds). The fitting yields true speeds of 19±11 nm/s (mean±std).
(F) Distribution of apparent speeds relative to the long axis of cell (black). Red line is exponential fitting. The characteristic speed from the fitting is 2.5 nm/s.
(G) Angles of velocity vectors relative to the short axis of the cell (black). Red line is exponential fitting for angles 0-20°. The characteristic angle from the fitting is 9.2°. In panels (E-F) the number of cells analyzed is Ncell=20 and the number of clusters analyzed Ncluster=352. All data is from strain JM26. For data related to strain JM60 see Figure S1.
We then examined cells for intermediary FtsZ assemblies preceding Z-ring formation. An earlier study has reported dynamic helical patterns of FtsZ in cells that lacked a Z-ring [35]. Our measurements also revealed dynamic FtsZ assemblies in cells that had not yet formed a Z-ring. Instead of helixes we observed less-extended assemblies, most of them limited by the resolution of the microscope (about 250 nm). These FtsZ foci formed and disappeared throughout the cell (Figures 1 C–D; S1A–B). Since the observed foci showed displacements less than about 300 nm during 6 seconds frame interval, it can be concluded that these foci correspond to membrane attached FtsZ assemblies. Cytosolic FtsZ assemblies would have diffused throughout the cell length (≈2 μm) during this measurement interval.
On the other hand, the membrane attached FtsZ foci were not static. Some of the larger foci were clearly moving along the circumference of the cell (Figure 1D Bottom; pointed by arrow). Such movement can be attributed to treadmilling rather than to diffusion on the membrane because of its directionality. By tracking larger foci, which can be distinguished by segmentation, we determined their apparent movements relative to the short and long axis in a population of cells (Figures 1E–F). Since the image is a projection of a cylindrical cell to a (image) plane, the apparent speed of the foci along the short axis (y-axis) is not uniform even when the FtsZ assembly moves at a uniform speed along the circumference of the cell. Accounting for this variation and assuming the distribution of the true circumferential speeds to be Gaussian yields a distribution of apparent treadmilling speeds (Methods: Determination of treadmilling velocities and distribution of treadmilling speeds), which we compared to experimentally measured distributions (Figure 1E). This comparison yields an estimate of 19±11 nm/s (mean±std) for the true treadmilling speeds of FtsZ transient assemblies at the cell’s circumference (Figure 1E). Similar treadmilling speeds (15±11 nm/s) were also found for the strain expressing FtsZ-GFP from a plasmid (Figures S1 C–D). These values are comparable to those reported previously for treadmilling FtsZ clusters that are part of the Z-ring [4].
Direction of treadmilling was not always circumferential, but also showed a velocity component along the long axis (Figure 1E). For most assemblies, the angles of the velocity vectors relative to the direction of the short axis of the cell followed an exponential distribution with a characteristic angle of 9° (Figure 1G). However, the distribution had a long tail that reached to 90°. So while a few assemblies treadmilled along the long axis of the cell, the majority of them were well aligned relative to the short axis.
Size of transient assemblies is limited by Z-ring components.
The transition from transient mid-cell assemblies to the Z-ring occurred gradually over the period of about 30 minutes (Figures 2A, B). As more FtsZ accumulated to the mid-cell Z-ring, the transient assemblies in other locations in the cell disappeared. Once the mature Z-ring emerged, which contained about 35% of the cellular FtsZ content, no transient assemblies were present from visual inspections of the kymographs and cell images (cf. Figures 1A, S1A). We used spectral analysis to determine if the transient assemblies were completely absent. For that end we collected fluorescent intensity traces from the cell middle and two quarter positions (Figure 2A). Formation and dissociation of distinct FtsZ foci in the cell images leads to variations in the intensity for these traces. At the same time treadmilling, which the direction is predominantly circumferential, did not cause fluctuations in these intensities because the assemblies should have remained within the band-shaped region of interest that we defined for this analysis (Figure 2A, Schematics). To analyze these fluctuations we calculated a population-averaged (N = 20 cells) power spectral density and autocorrelation function before and after Z-ring formation using the intensity traces from different cells (see Methods: Power spectral density and autocorrelation function analysis of FtsZ fluctuations). As a control we imaged the same strain, which was transformed with a plasmid carrying an inducible copy of sulA [36], and determined the intensity traces after upregulation of SulA. SulA binds to FtsZ monomers, thereby lowering the amount of free FtsZ below the critical concentration required for cooperative assembly. As a result, very few, if any, polymeric FtsZ molecules are expected to be present in these cells [36]. Intensities from the SulA control showed a frequency independent (white) noise spectra while the two WT strains showed power spectra that decayed approximately as an inverse of the frequency (Figures 2B, S2A,C). The fluctuation levels in FtsZ-mNG exceeded the levels in the control, more than an order of magnitude, at lower frequencies showing that the observed fluctuations in WT intensity traces originated from higher order FtsZ structures. Furthermore, the long decay of the autocorrelation functions with a decay constant of about 20 seconds (Figures 2C, S2B–D) confirmed that these higher order structures are membrane bound.
Figure 2. Comparison of FtsZ transient assemblies before and after Z-ring formation.
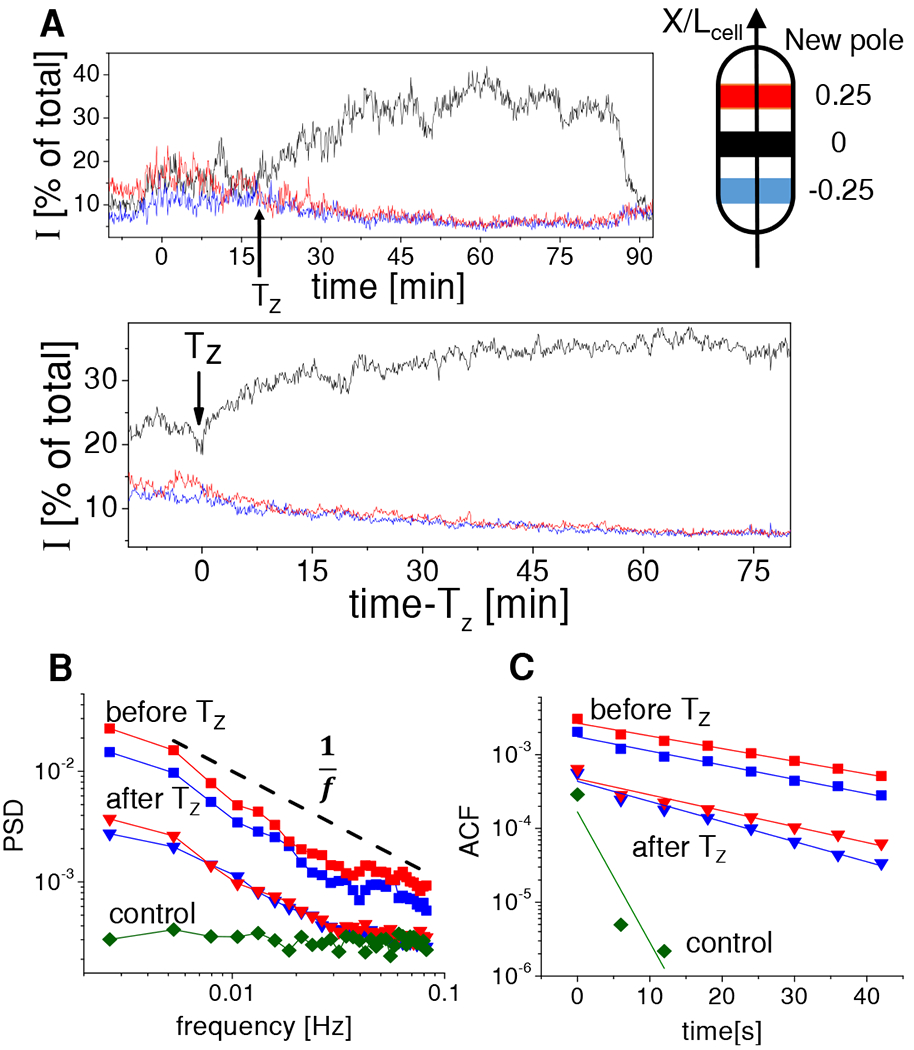
(A) Top: Intensity traces from three separate regions within the cell (diagram shown on right). Each region represents 0.3 mm wide band. Red corresponds to the band centered at a quarter of the cell length from the new cell pole, blue to a quarter of the cell length from the old cell pole, and black to the cell center region. This color coding applies to all panels. Time zero marks cell birth, Tz corresponds to time of Z-ring formation, and the trace ends at cell division. Data from the cell shown in Figure 1. Bottom: population averaged curves (N=20).
(B) Population averaged power spectral density of the old pole and new pole traces before (squares) and after Tz (triangles). PSD from the central traces are shown in Figure S2. Dashed black line shows for reference an inverse frequency dependence. Control data (diamonds) is from cells where FtsZ polymerization is inhibited by upregulation of SulA (N=10). In these cells the PSD is not dependent on cellular location.
(C) Population averaged autocorrelation function of old and new pole traces before and after Tz. Exponential fits to the autocorrelation functions are shown by solid lines. All data is from strain JM26. See also Figure S2.
The peripheral regions of the cell show about ten-fold higher levels of power spectral density before Z-ring formation compared to that after the ring has formed. This quantitative conclusion is consistent with visual observations. The power spectral density analysis also reveals that membrane-bound FtsZ assemblies still appear after the Z-ring has formed, even though they are not visually distinguishable from the images. From the power spectrum and autocorrelation measurements, we estimate that the number of FtsZ monomers present in the foci is a factor of lower after Z-ring formation. These data point out that the sizes of the transient assemblies are controlled by the available amount of FtsZ and/or its membrane linkers. Once the Z-ring forms, the number of early Z-ring components available to form transient assemblies decreases. Nevertheless, the smaller sized assemblies are still able to form throughout the cell body.
Spatial and temporal distribution of large FtsZ assemblies.
In what follows we will characterize the distribution of membrane bound FtsZ assemblies as a function of cell cycle time and location within the cell. Our analysis is pertinent to large size assemblies that contain 2.5% or higher amounts of total cellular FtsZ. These assemblies can be unambiguously distinguished from images using thresholding based methods (Methods: Determination of FtsZ Cluster Characteristics). To quantify position dependence on a coarser scale, we divided each cell into two regions - a mid-cell and a cell periphery where the periphery also included cell poles (Figure 3A, Schematics). Here, the mid-cell region spans 1/3 of the total length of the cell around its center while the rest is referred to as the periphery. One could perhaps have expected FtsZ contents and lifetimes of transient assemblies at the mid-cell to increase as the cell cycle time progressed until the Z-ring formed. Contrarily, we found that the location of the large assemblies along the long axis of the cell (Figure 3A), their size (Figure 3B), and their lifetimes (Figure 3C) were all only very weakly correlated with the cell cycle times during the period that preceded the Z-ring formation (R<0.2; Table S2). The same conclusions were also reached based on data from the FtsZ-GFP strain (Figures S3A–C, Table S2). The exception to these steady behaviors was the number of assemblies, which started to increase before the Z-ring formed (Figure 3A, bottom). Our previous work has shown that prior to the formation of the Z-ring the synthesis rate of FtsZ increases four-fold [27]. This observation was confirmed at the qualitative level in current studies. Thus, the increase in the number of assemblies correlates with the increase in the number of FtsZ within the cell. It follows then that the increase in FtsZ numbers in the cell leads to more assemblies with the same kinetic properties (size and lifetime) rather than to the same number of larger assemblies.
Figure 3. Characterization of spatial and temporal distributions of FtsZ transient assemblies that contain 2.5% or higher amount of FtsZ relative to the cell total.

(A) Top: The positions of assemblies along the long axis of cell as a function of time. Assemblies at the mid-cell are shown by black and in cell peripheries by red markers as explained by the inset. Time axis is scaled by time when the Z-ring forms (Tz). Bottom: Number of assemblies in the mid-cell and two peripheries combined as a function of normalized time. Solid lines in panels (A)-(C) are linear fits. Parameters of these fittings are collected to Table S2.
(B) The fluorescent intensities from assemblies as function of normalized time. The intensities are scaled by the total fluorescent intensity from the cell and averaged over the lifetime of each assembly. The small markers are individual data points and the large markers the binned data.
(C) Lifetimes of assemblies at the mid-cell (black) and in the periphery (red) as a function of normalized time.
(D) Distribution of assemblies (left) and Z-ring positions (right) along the long axes of the cell. Positions are normalized by the cell length. Panels (A-D) correspond to WT cells (strain JM26).
(E-F) Same distributions for ΔzapA (strain BEW2), and ΔminC cells (strain BEW1), respectively.
(G) Comparison of sizes of assemblies in WT, ΔzapA and ΔminC strains. See also Figure S3.
(H) Comparison of lifetimes of the assemblies in these three strains. In last two panels error bars are std error.
The large assemblies were completely missing from the cell poles, and their frequency in both peripheries combined was five times lower than at the mid-cell (Figure 3A, bottom panel). Nevertheless, the spatial distribution of assemblies in the cell was broad having a standard deviation of 0.14 in units of the cell length (Figure 3D, left). The corresponding distribution was about 10 times wider than the spatial distribution of the Z-rings in these cells (Figure 3D, right). Interestingly, the distribution of the transient assemblies was asymmetric relative to the cell center. 72% of all assemblies localized to the cell half closest to the new pole (Figure 3D; p=7·10−17 in Wilcoxon test). The asymmetry of the distribution correlates with the localization of the replication terminus region of the chromosome to the new pole side of the cell during the early stages of the cell cycle [37]. Deletion of a genetic region encoding ZapA, which is necessary for the Ter linkage, lead to a reduction of the asymmetry (Figure 3E, 57%; p=0.007) but statistically significant bias towards the new pole side still remained. The distribution of the transient assemblies and in particular their absence from the cell poles was even more strongly influenced by the Min system, which is involved in the positioning of the Z-ring [38]. In the absence of MinC, which couples Min oscillations and FtsZ, the spatial distribution of assemblies broadened further (standard deviation 0.24) and transient assemblies also appeared in the polar regions (Figure 3F).
Although the large scale assemblies were rarer at the cell peripheries in WT cells, their size, in terms of their FtsZ content, was only weakly correlated with their position in the cell. We found the assemblies in the mid-cell region to be on average only 5% larger in terms of FtsZ numbers (p=0.3 in Wilcoxon test) than those in the cell peripheries (Figures 3G left, S3G). Comparing the sizes of transient assemblies with the size of the Z-ring, we found that the transient assemblies were on average six times smaller than the Z-ring in terms of FtsZ numbers. The latter contained about 33% of the cellular FtsZ (Figure 3G right), which is in agreement with earlier reports [20, 39]. In the absence of ZapA, which has been implicated in cross-linking FtsZ protofilaments, the assemblies were about 30% smaller in both the mid-cell and peripheral regions than in WT cells. Moreover, in the absence of ZapA, the lifetimes of the assemblies were more than two times shorter than in WT type cells (Figure 3H). The effect was more pronounced at the cell peripheries. These data imply that protofilament-protofilament crosslinking via ZapA occurs in the transient assemblies.
The sizes of the assemblies in ΔminC cells were comparable to their sizes in WT cells (Figure 3G). There was no significant difference in assembly sizes at the mid-cell and cell periphery in ΔminC cells (p=0.4 in Wilcoxon test). Also, the lifetimes of the assemblies in ΔminC cells were the same at the mid-cell and peripheries (Figure 3H), and were comparable to the assembly lifetimes at the mid-cell in the WT strain. Unlike in ΔminC cells, the WT transient assemblies’ lifetimes were 30% shorter at the cell peripheries. These findings show that the Min system is responsible for the smaller sizes and shorter lifetimes of the FtsZ assemblies at the cell peripheries in WT cells. The smaller abundance of peripheral assemblies can also be explained, to some extent, by the Min system. The findings are largely consistent with the known function of the Min system, which is to prevent the formation of the Z-ring at polar regions. Altogether, the data presented here indicate that transient assemblies are affected by the Z-ring positioning systems. However, their effects are weaker than one could have expected based on studying the Z-ring. In particular, the spatial distribution of transient assemblies is much broader than the distribution of Z-ring positions.
Cytosolic protofilaments in vivo are shorter than ones observed from in vitro measurements
We next investigated assembly-disassembly kinetics of membrane attached FtsZ assemblies in WT cells. The first step in this assembly must be attachment of an FtsZ oligomer from the cytosol to the plasma membrane via its linkers. Based on in vitro studies, one could expect that the typical protofilament length in the cytosol is about 100-200 nm (≈25 monomers) [13, 28–30], although it has also been proposed that cytosolic filaments could be smaller because of their rapid capture to the membrane via their linkers [18]. We aimed to capture attachment of such protofilaments to the plasma membrane from the cytosol during time lapse movies, which were taken at higher frame rates (1 Hz) than earlier ones. By using 100 ms long excitation pulses, we expected to observe a step-like increase in the intensity at the location of FtsZ focus once the protofilament attached. Note that prior to the attachment, motion blurring of the filament diffusing in the cytosol should have completely smeared out its image. Contrary to our expectations, none of the 1973 analyzed foci showed distinct step-like increases in the beginning of their lifetime that could be classified as protofilament attachment. We did notice that many of these assemblies grow rapidly but with further examination of the images it became clear that prior to the rapid growth an already existing FtsZ assembly was present (Figure 4A). We also did not observe any step-like decreases although in many assemblies the amount of FtsZ-sfGFP decreased rapidly during the end of their lifetimes (Figure 4B). This indicates that at least longer filaments are not able to detach from the membrane which could have occurred if their binding to the membrane, via linkers, was sufficiently weak.
Figure 4. Rate of change in the size of FtsZ transient assemblies.
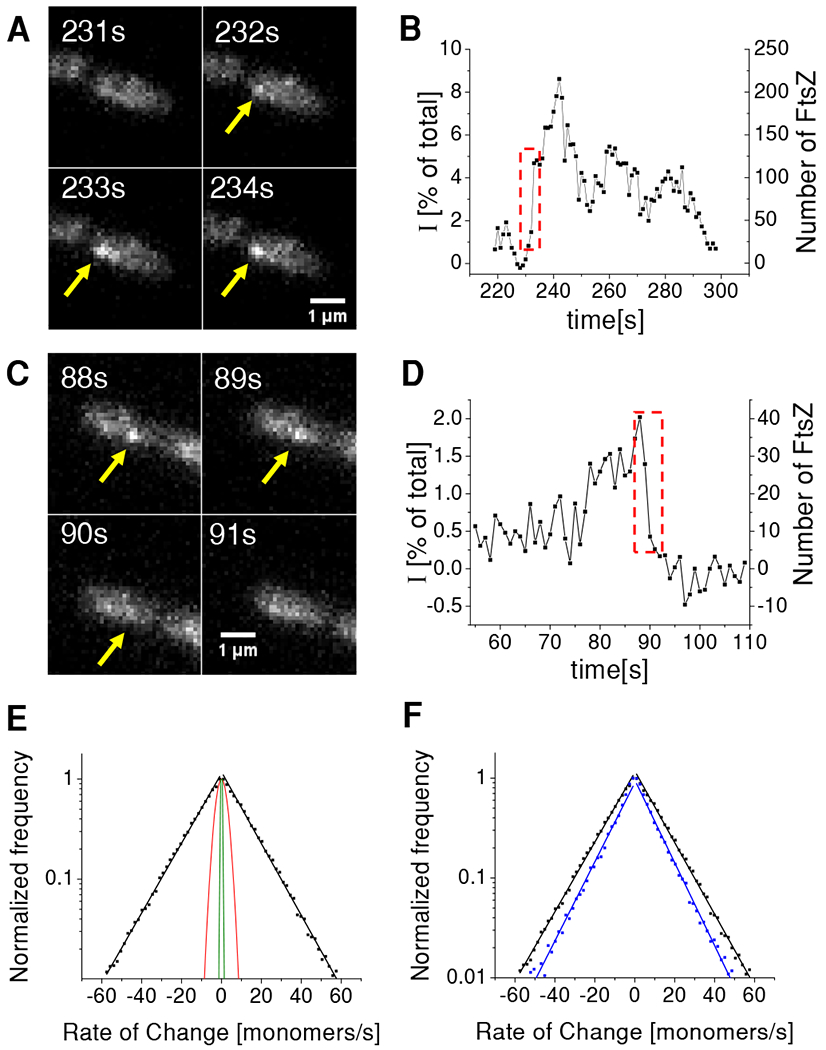
(A) Time-lapse images of a cell with an assembly that rapidly increases in size.
(B) Corresponding intensity time-trace from this assembly. Left axis shows intensity of the assembly relative to the total intensity of FtsZ-sfGFP from the cell. Right axis corresponds to the absolute number of fluorescent FtsZ-sfGFP in the assembly. Dashed box region corresponds to frames in panel (A). For calibration of right axes see Figure S4 and Methods for details.
(C) Time-lapse images of a cell with an assembly that rapidly decreases in size.
(D) Corresponding time-trace from this assembly. Dashed box region corresponds to frames in panel (C).
(E) Distribution of rates for cell population (Ncell=199; Ncluster=1973) plotted on logarithmic scale (black squares). Each side of the distribution was fitted to an exponential (black solid lines). The characteristic rates from the fitting is 15.6 monomeres/s for addition and 15.7 monomers/s for FtsZ removal. Red solid line represents the prediction of a model where assemblies change in size via treadmilling which include only monomer addition and removal (see Methods for details). Green line is the estimated distribution of the measurement noise (see Methods for details).
(F) Comparison of rates from WT (black) and ΔzapA strains (blue). The latter distribution has a characteristic monomer addition rate of 10.1/s and monomer removal rate of 10.3/s. WT data is from strain KC606 and ΔzapA from strain BEW2.
The lack of any step-like increases could have been caused by our limited sensitivity to detect small FtsZ assemblies. To have a better understanding of the measurement sensitivity we determined the number of FtsZ-sfGFP within the cells for these growth conditions (Methods: Determination of total FtsZ numbers in cells). These measurements yielded an estimated average concentration of 4.7±0.7 μM (Figure S4). This number is in the range, 4-6 μM, reported before [36, 40, 41] although concentrations as high as 10 μM have also been measured [13]. The concentration above corresponds to 2300±400 FtsZ molecules for a cell having a population average length of 2.8 μm. By using the above calibration we determined the sizes of the assemblies in terms of FtsZ numbers (right axis, Figures 4B, D). Based on the noise level in the traces we conservatively estimate the sensitivity of the detection to be better than 20 monomers. Based on this estimation we conclude that the transient FtsZ assemblies start from protofilaments that are shorter than 20 monomers.
The above data left open the possibility that longer protofilaments are present in the cytosol, but their attachment to the membrane may be hindered. To find evidence for longer (>20 monomers) cytosolic protofilaments diffusing in the cytosol we employed short 1 ms stroboscopic pulses to excite the fluorophores and to eliminate motion blurring. We first verified that 1 ms long excitation pulses were sufficiently short enough for stroboscopic imaging using a photoactivatable FtsZ-mMaple3 [31] construct in a monomeric state in the cytosol (Methods: Photoactivation Experiment, Videos S2, S3). We then imaged non-photoactivatable FtsZ-sfGFP in normally growing cells using excitation pulses of the same duration. Given the low frame rate (1 Hz) of the measurement and the rapid diffusion of protofilaments in the cytosol, we expected to observe foci that would have shown large displacements (>0.5 μm) from one frame to another. Contrary to this expectation, the measurement showed 748 out of 755 (99.1 %) foci to be static. The observations included 101 cells for a total of about 5·105 frames. The remaining 7 events (0.9%) corresponded to foci that were present for only a single frame. Some of these single-framed events may have corresponded to membrane attached assemblies whose size increased and decreased rapidly within the 1 second interval. Altogether, these measurements lend support to the idea that protofilaments longer than 20 monomers are exceedingly rare, if at all present, in the cytosol.
Rapid kinetics of FtsZ transient assemblies
While we did not find evidence of longer protofilaments in the cytosol and their attachment to the inner membrane of the cell, the recorded time-traces of FtsZ transient assemblies (Figure 4A–D) showed frequent periods of rapid growth and shrinkage. To understand these dynamics further, we determined instantaneous growth and shrinkage rates of the assemblies (Figure 4E). The distributions of both rates were approximately exponential with an average rate of 16 monomers/sec. To understand the effect of the measurement noise for this distribution we imaged a strain carrying inducible SulA in conditions where the cells lacked any visible FtsZ foci or Z-rings. Since these cells did not have any detectable FtsZ foci, we simulated the foci by randomly selecting regions within the SulA-expressing cells based on the area and duration of foci obtained from WT cells. The resulting control distribution was about 25 times narrower (Figure 4E, green line) than the one from non-induced cells. Thus, the measurement noise contributes only to a narrow region at the center of the distribution and does not affect its tails.
The high rates at the tails of the distribution in Figure 4E were unexpected. If the assembly were to consist of only a single protofilament, and only monomers and not oligomers were added and removed from the assemblies then a much narrower rate distribution would be expected based on treadmilling kinetics (Figure 4E, red line; Methods: Simple model for monomer addition and removal during treadmilling). We found the calculated rate distribution to be about 5 times narrower than the experimental one (Figure 4E, red line). The higher rates in the experimental distribution could be explained by assemblies that consist of protofilament bundles instead of single protofilaments. In this case, the monomer addition and removal rates would increase in proportion to the number of protofilament ends in the bundle. Bundling would also mean that additional protofilaments can be added or removed to the assemblies via lateral contacts or via cross-linking. The latter processes could possibly explain the high-rate tails of the distribution in Figure 4E. To investigate the possibility of protofilament cross-linking further, we carried out the same measurements in a strain lacking ZapA. We found the monomer addition and removal rates of transient assemblies in this strain to be about 35% lower compared to WT cells (Figure 4F). This finding together with the decrease in assembly sizes and lifetimes upon zapA deletion (Figures 3G, H) supports the idea that at least some of the observed assemblies consists of cross-linked FtsZ protofilaments.
Discussion
Dynamics of transient assemblies
Our experimental findings are consistent with the following scenario summarized in Figure 5. The life cycle of transient assemblies starts with nucleation and growth of protofilaments in the cytosol. Based on the frequent formation of transient assemblies, it is clear from our data that the effective FtsZ concentration in the cell prior to the Z-ring formation is above the critical concentration. Furthermore, the presence of transient assemblies show that 1) protofilament formation in the cytosol and 2) protofilament attachment to the cell membrane via its linkers are not rate limiting steps for Z-ring formation. However, we found cytosolic protofilaments shorter than one would have expected based on in vitro measurements. We estimate protofilament lengths to be less than 20 monomers in vivo while 25-50 (100-200 nm) [28–30] and even 100 monomer lengths [42] have been reported from in vitro measurements. A variety of factors can contribute to this discrepancy. For example, the filament length distribution depends on the concentrations of ions (K+, Mg2+) and GTP levels. Even though concentrations of these ions and molecules are to some degree known for in vivo conditions, their activities are less well characterized and depend on the cell growth environment and exact genetic make-up. Additionally, FtsZ polymerization antagonists, such as SlmA, can effectively shorten protofilament length. It has also been proposed that cytosolic filaments cannot elongate to their in vitro lengths because of their capture to the membrane via FtsA and ZipA linkers [18]. The quantitative extent by which the latter mechanism limits cytosolic protofilament lengths is not clear from our data, but our measurements do show frequent attachments of protofilaments to the plasma membrane. These attachments occur at random locations along the cell length except possibly at the cell poles where the apparent occurrence of observable assemblies is smaller. The randomness in positioning appears to stem from stochastic collisions of cytosolic protofilaments with the membrane.
Figure 5. Inferred sequence for formation of transient assemblies.
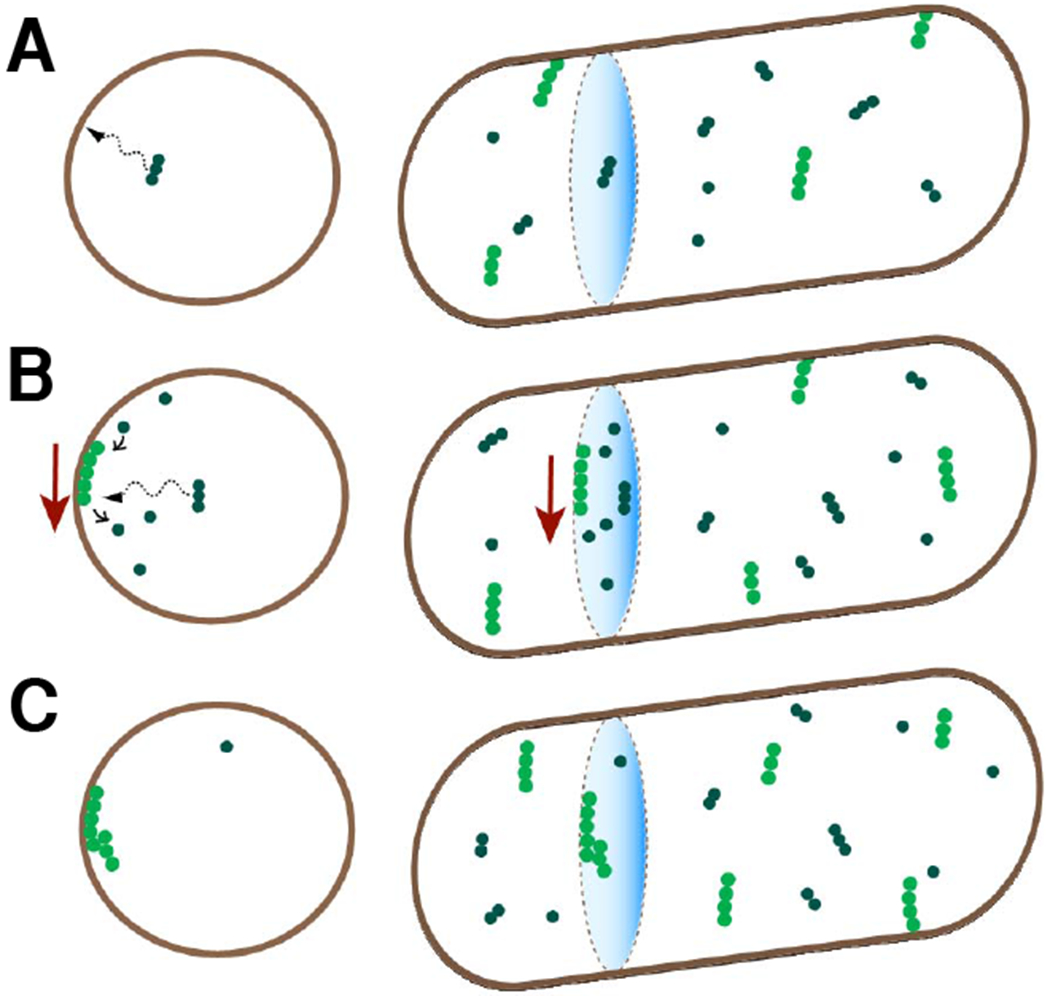
(A) FtsZ protofilaments are formed in the cytosol and are recruited to the membrane by FtsA and ZipA membrane tethers (not shown). The right column is a 3D view of cell contours and FtsZ filaments and the left column is a cross-sectional view. Bright green circles correspond to membrane attached FtsZ and dark green circles correspond to cytosolic FtsZ.
(B) The membrane attached filaments treadmill preferentially in a direction perpendicular to the long axis of the cell.
(C) These filaments bundle/crosslink at early stages after their attachment to the membrane. All filaments in the bundle are oriented in the same direction.
Once attached, some of the filaments give rise to assemblies that contain several hundreds of monomers. Their rapid growth rate implies that at least some of these assemblies must become protofilament bundles. In agreement with earlier reports [40], our data show that ZapA enhances protofilament cross-linking, but the latter can occur in the absence of ZapA.
Competition between different assemblies for the pool of available FtsZ
FtsZ assemblies forming during the early stages of the cell cycle (about the first 20% of the cell cycle) remain unstable; even those that form at the cell middle. Then what causes the transition from transient assemblies to a persistent Z-ring which carries out cell division? There appears to be no well-defined trigger for Z-ring assembly. The transition in slow growth conditions is gradual taking about 20% of the cell cycle (Figure 2A). During this time the dominant mid-cell assembly on average grows in size, but the growth is not uniform and is interspersed with periods of shrinkage. Previous research has identified the amount of FtsZ in the cell as one of the limiting factors for Z-ring formation [27, 43, 44]. The data reported here provides further support to this idea. Specifically, the data show that once the dominant mid-cell assembly forms, which is to become the Z-ring, the numbers of other larger transient assemblies within the cell drops gradually to zero. A more sensitive analysis based on power spectra and autocorrelation functions show smaller FtsZ assemblies still form throughout the inner membrane, but the size of these assemblies is a factor of 3 smaller than the ones present prior to Z-ring formation. Our data show that Z-ring formation leads to about a 35% reduction in the available pool of FtsZ for transient assemblies (cf. Figure 2A, Figure 3) since this amount is sequestered to the Z-ring. This relatively small decrease in the available pool of FtsZ then leads to a significant (3-fold) decrease in FtsZ transient assembly sizes. The exact mechanism giving rise to this very sensitive concentration dependence is not clear. We hypothesize that the out competition of other assemblies by the dominant mid-cell assembly is likely to occur because this assembly is a bundle that contains more protofilaments. Addition of monomers and short oligomers to a larger bundle would be more favorable than to smaller bundles or to single protofilaments. For cellular function, out competition appears to be an important characteristic of protofilament dynamics as it guarantees that not more than one Z-ring forms per cell.
Transient FtsZ assemblies probe for the proper location for the Z-ring
The assembly that outcompetes others and forms the Z-ring is not a random one but the one that has formed in the vicinity of the cell center. Its higher stability can be expected to stem from its more favorable interaction with the spatial positioning systems of the Z-ring (the Min system, the Ter linkage, and SlmA based nucleoid occlusion). Tracking the assemblies (Figure 1) shows that the spatial positioning systems mostly select among the randomly forming assemblies that stay a fixed distance from the cell center. However, some movement of these assemblies along the long axes of the cell does occur because of treadmilling, and likely this motion also plays a role in the positioning of the Z-ring. Surprisingly, transient assemblies are present in the wide band around the cell center, which covers about one half of the cell length. It remains yet unclear by what mechanism this broad distribution of transient assemblies collapses into a narrow distribution of FtsZ in the cell center, which has about a 3% variation relative to the cell center (Figure 3D). This process possibly combines the effects from different spatial positioning systems and the competition effects between different assemblies. A detailed understanding of how this process unfolds remains an important and open question for future studies.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Jaan Männik (JMannik@utk.edu). New strains generated during this study have not been deposited in a public repository but are available from the Lead Contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All strains are derivatives of E. coli K-12. The strains and plasmids are described in Table S1. All cells were grown and imaged in M9 minimal medium (Teknova) supplemented with 2 mM magnesium sulfate (Millipore Sigma) and 0.3% glycerol (Fisher Scientific). When needed antibiotics were added to the culture media at final concentration described in Table S1. No antibiotics were used during cell growth on 2% agarose dishes, and in agarose and PDMS (polydimethylsiloxane) microfluidic devices, except for the plasmid expressing FtsZ-GFP strain (chloramphenicol, 40 μg ml−1). For the strain with photoactivatable FtsZ mMaple3 0.2% glucose and 0.2% arabinose were added to the M9 media. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 400μM to induce SulA expression from the plasmid pA3 in the MB43 strain.
METHOD DETAILS
Preparing and using agarose based mother machine chips.
The process of agarose chip fabrication follows the one described by Moffitt et al. [45], but with some modifications. Unlike this previous work, which used channels with two open ends, we utilized dead end channels with one open end. The latter design is usually referred to as the mother machine [33].
The channels were patterned into 4% agarose to which M9 salts and glycerol were added. A silicon template with protruding channel reliefs were used for patterning. The fabrication procedure for the silicon template has been reported before [34]. Fabrication was carried out on a 4” silicon wafer. After fabrication, the wafer was cut into individual patterns using a diamond knife and cleaned in isopropanol (Fisher Scientific) by sonication. The resulting silicon template was clamped between two Teflon slabs of 5.1 mm in height. One of the slabs contained a rectangular hole with dimensions 9x11 mm. This hole was centered relative to the pattern on the silicon template. Molten agarose was poured into the hole formed by the Teflon slabs. After the agarose solidified, excess agarose that overfilled the hole was cut by a razor blade. The Teflon pieces were unclamped and a rectangular agarose piece was released from the hole.
The agarose piece with patterned channels was then inserted into a custom made jig. The jig design follows the one reported earlier [45]. Briefly, the holder consisted of a PDMS slab and a #1.5 glass coverslip (Fisher Scientific). The two were bonded together after cleaning and O2 plasma treatment. The PDMS slab contained a central through hole of 5.0 mm in height and cross-sectional dimensions of 8x10 mm in which the agarose piece was inserted. The PDMS slab also contained two channels leading to the central hole. The PDMS slab was made in a machined aluminum mold, which followed the design described by Moffit et al. [45].
Once the agarose piece was inserted into the central hole in the jig, the whole assembly was placed on a custom-made aluminum plate that fit onto the microscope stage. A piece from a glass coverslide (Fisher Scientific) was cut by a diamond knife, cleaned in isopropanol by sonication, and dried. This glass slide was then pressed against the agarose piece, which protruded from the jig, by custom-made clamps that attached to the microscope plate. These clamps pushed the coverslide tightly against the PDMS part of the jig to prevent leakage and raising of the agarose piece. During application of the pressure the oversized agarose piece contracted and some of its liquid content flowed out. After this flow had stopped, tubing was connected to the jig and M9 medium, to which 1 mg/ml BSA was added, was circulated through the chip for about 20 minutes using a New Era 1000 syringe pump.
Our loading procedure of cells to these dead-end channels is similar to the one described for PDMS based mother machine channels [33] [34] but differs from the one described for agarose chips before [45]. In the latter case cells were pipetted onto the glass slide and spread when patterned agarose was pressed against the slide. Here to load the cells, extra tubing with a concentrated solution of cells was connected to the jig and cells were injected into the main flow channel by a syringe that was connected to the other side of the tubing. For loading an overnight culture of cells was used (OD600 > 0.4). 1 μg ml−1 of bovine serum albumin (BSA) was added to the overnight cell culture and cells were concentrated about 100x by centrifugation. To allow cells from the main flow channel to fill the dead-end channels, the clamps that held the coverslide on the top of the jig were slightly loosened. Once the mother machine channels were filled with cells, flow of the medium was started in the main channel. The flow was maintained in the main channel overnight at a rate of 8 μl/min. The next day, while maintaining the same flow rate, cells were imaged using a Nikon Ti-E inverted microscope.
Preparing and using PDMS-based mother machine chips.
PDMS (polydimethylsiloxane) based chips were used to image the JM60 strain for long time-lapse measurements. The agarose based chips were less suitable for imaging of this strain because its plasmid expressed Plac::ftsZ-gfp product required IPTG induction. Since IPTG diffuses into agarose, PDMS based chips allowed for better control over the induction level. The induction level was kept as small as possible (10 μg/ml) to avoid over-expression of the non-complementing construct yet allowing it to be visible in the microscope.
The fabrication procedure for PDMS based microfluidic chips has been reported before [34]. Briefly, silicon molds were fabricated combining e-beam and photolithography with reactive ion etching. The mold defined dead-end channels, which were 0.8-0.9 μm wide, 1.1 μm high, and 20 μm long. PDMS (Sylgard 184, Dow Corning) was poured onto a passivated mold and baked for 15 min at 90 °C in a convection oven. Subsequently, the solidified PDMS layer was peeled from the mold and cut into pieces. Access holes to each piece, corresponding to one microfluidic device, were then punched using a biopsy needle. A PDMS piece was then plasma treated together with a clean #1.5 coverslip (Fisher Scientific). The two were bonded together after the treatment.
To load the cells to the chip an overnight culture of cells was used. 1 μg ml−1 of bovine serum albumin (BSA) was added to 1.5 ml of overnight cell culture (OD600 > 0.4) and concentrated 100x by centrifugation. A 2-3 μl of resuspended culture was then pipetted into the main flow channel. The cells were then allowed to populate the dead-end channels for about 1 hr. Once these channels were sufficiently populated, tubing was connected to the devices, and the flow of fresh M9 medium with BSA (1 μg ml−1) started. The flow was maintained at 5 μl min−1 during the entire experiment by an NE-1000 Syringe Pump (New Era Pump Systems, NY). To ensure steady-state growth, the cells were left to grow in the channels for at least 14 hours before imaging started.
Microscopy.
A Nikon Ti-E inverted fluorescence microscope with a 100X NA 1.40 oil immersion phase contrast objective and Perfect Focus system was used for imaging the bacteria. Fluorescence was excited by a 488nm diode laser with a 5 mW power through ND16 and ND2 neutral density filters for 100ms exposure and 15 mW power through no filters for 1 ms exposure experiments. Chroma 41001 filter-cube was used to record mNG, sfGFP, and GFP images. Images were captured by an Andor iXon DU897 camera and recorded using NIS-Elements software.
Photoactivation experiment.
Cells with photoactivatable construct FtsZ-mMaple3 (strain HE5) [31] were grown up in minimal M9 salts medium supplemented with 0.2% glucose, 0.2% arabinose, and 30 μg/ml chloramphenicol for 24 hours. Cells were then diluted and resuspended in PBS (phosphate-buffered saline) for 20 hours to lower the amount of GTP to almost zero making all FtsZ monomeric. During imaging an Optomask (Andor Technologies, Ireland) adapter was used to limit the image to a small region near the readout register of the EMCCD camera. Reading out a smaller number of pixels enabled faster frame rates (22.9 frames per second during 1ms exposure and 9.8 frames per second during 100 ms exposure). FtsZ-mMaple3 was fluorescently activated by a single pulse from a 405 nm diode laser having a power of 3mW and pulse length of 100 ms. After photoactivation, we imaged the cells with 1 ms and 100 ms long excitation pulses using a 561 nm laser. Both types of pulses had approximately the same excitation energy. With 1 ms pulses we were able to observe foci of monomeric photoactivated FtsZ-mMaple3 molecules diffusing in the cytosol (Video S2) while the foci were absent when imaged with 100 ms pulses (Video S3) due to motion blurring. Note that both videos originate from the same cell. Total fluorescent intensity averaged over all frames were comparable in these two excitation conditions. The recorded videos were smoothed with a Gaussian filter having σ = 1 pixel. Once activated, FtsZ-mMaple3 was apparently bleached and re-activated by a 561nm diode laser repeatedly so that the number of fluorescent photo-activated molecules during the imaging period (5-10 minutes) remained approximately constant.
Simple model for monomer addition and removal during treadmilling.
Rate of change in Figures 4E–F shows how transient assembly size, which is expressed in number of monomers, changes in time. This rate is the difference in monomer addition and removal rates to the assembly. Positive rate corresponds to more monomers being added and negative rate more monomers being removed from the assembly. Treadmilling means that these two rates are on average equal. However, the number of added and removed monomers from a protofilament in each measurement interval (1 sec) is not in general equal because of stochastic nature of reactions. This leads to fluctuations in assembly size which is measured experimentally.
We next estimate this rate of change assuming that monomers are added and removed one at a time. The number of monomers added and removed during a fixed time interval follows a Poisson distribution. Both addition and removal have the same average rate (λ) for persistently treadmilling filaments. We estimate this rate based on measured treadmilling speeds, which we determined to be about 20 nm/sec. Since the length of an FtsZ monomer is about 4.3 nm [13], this rate corresponds to adding monomers to one end and of losing monomers from the other end at an average rate of λ=5 monomers/sec. In our measurements the rate is determined as a change in monomer numbers during Δt = 1 sec interval. During this interval say m monomers could be added to (presumably from one side) and n removed from (presumably from the other side of) the filament. The instantaneous rate during this interval would then be (m − n)/Δt. The probability of this particular process is:
The probability that a total of N monomers are added during the time interval Δt is the sum of all possible processes where n = N + m.
This distribution is used to compare the experimentally measured rate distribution in Figure 4E.
QUANTIFICATION AND STATISTICAL ANALYSIS
Matlab, along with the Image Analysis Toolbox and DipImage Toolbox, (http://www.diplib.org/) were used for image analysis. In all analysis of time-lapse recordings, corrections to subpixel shifts between different frames were applied first. These shifts were determined by correlating phase contrast images in adjacent frames. The cells were then segmented based on phase contrast images using a custom Matlab script. FtsZ fluctuations and cluster tracking were carried out using custom Matlab scripts (see below: Power spectral analysis of FtsZ fluctuations, and Determination of FtsZ cluster characteristics for more detailed information). ImageJ (Fiji) was used for montage and video creations.
Power spectral density and autocorrelation function analysis of FtsZ fluctuations.
Using fluorescent intensity traces from cell middle and two quarter positions, power spectral density (PSD) and autocorrelation function (ACF) for FtsZ fluctuations were determined. The traces were first separated into two parts, transition period (t < Tz) and Z-ring period (t > Tz) so that FtsZ fluctuations before and after Z-ring formation could be compared. In each period, i.e. before and after Z-ring formation, the traces were divided into 64 frame segments. For each trace the mean intensity of the trace was then subtracted and divided by the total intensity of the cell.
The intensity in each of these segments was then multiplied by a Hanning window, and then the Fast Fourier transform (F) and Fast Inverse Fourier transform (F−1) were calculated to obtain the PSD and ACF for each trace respectively.
All power spectral densities and autocorrelation functions before and after Z-ring formation for each cell were averaged together. The resulting PSD and ACF were furthermore averaged over the population of cells. These calculations were done in Matlab 2016b.
Determination of FtsZ cluster characteristics.
Images were analyzed with custom Matlab scripts using functions from Matlab Image Analysis and DipImage (http://www.diplib.org/) toolboxes. Each image had the background intensity subtracted first. The resulting image then was convolved with a Gaussian kernel (low-pass filter) with σ = 1 pixel. Cell coordinates were then determined by our own Matlab script or Oufti [46]. Our own script was used for microfluidic measurements while Oufti for agarose dish/pad measurements. Based on cell coordinates a binary mask for each cell was defined. Within this mask a thresholding value was calculated as Ithresh = 2Iaverage ≈ Iaverage + 2.5σI. Here Iaverage is the average pixel intensity and σI is the standard deviation of the image intensity within the cell. The resulting binary image was then dilated by a disk-shaped structuring element with a radius equal to 1 pixel. Individual clusters were subsequently labelled using the label function from the DipImage toolbox. Clusters were connected in the time series by using their proximity and intensity. For cluster connectivity an intensity threshold of 2.5% of the total intensity and distance thresholds of 500 nm for short axis and 200 nm for long axis movement were used. First, all clusters with an intensity lower than the threshold were disregarded for analysis. Then the distances between each cluster from image (i) and image (i + 1) were obtained (i = 1,2, ….,Nimages – 1). Each cluster from image (i) was connected with the corresponding cluster with the minimum distance that stayed within the threshold values for short and long axis movement from image (i + 1). Once the cluster was matched that cluster was taken out of the pool of possible connections between the remaining clusters. Clusters that had no connections within the proximity thresholds, retained their initial labels.
In long time lapse measurements (Figures 1–3) each frame was segmented separately and then cluster characteristics analyzed. The analysis used the measure function from the DipImage tool box to determine the binary centroid coordinates, average intensity, and total intensity of each cluster. The centroid coordinates were calculated relative to the center of the cell. The coordinate axis were oriented along the long (x-axis) and short axis (y-axis) of the cell. Intensity (% of total) in Figures 3A, 3D, 3E, and 3I were found as the time-averaged intensity of the cluster during its lifetime. It was normalized by the total fluorescent intensity from the cell.
During the analysis of the rate of change of FtsZ numbers in the assemblies (Figure 4), cluster sizes and their trajectories were determined first. Then the largest size cluster of pixels in each trajectory was found. The binary mask of this cluster was propagated to other frames in the time-lapse sequence. This also included the frames beyond those in which the program was able to identify this cluster using thresholding. The total intensity from this masked area was determined and plotted as a function of time. The procedure was only applied to clusters that did not significantly displace during the period when the program was able to identify these clusters based on thresholding. The rate of change was determined by the difference of the normalized intensity from a given cluster between two consecutive frames. The frame rate in these measurements was 1 Hz.
Determination of treadmilling velocities and distribution of treadmilling speeds.
Treadmilling velocity components νx and νy in Figures 1 E–F were determined from tracking FtsZ-mNG foci. Here νx is the velocity component along the long and νy the velocity component along the short axes of the cell; both components lay on the image plane. The tracking of the foci is described in Determination of FtsZ cluster characteristics (the previous section). Displacements of foci between subsequent image frames determined velocity vector components. All displacements between subsequent frames during the entire lifetime of the assembly from all cells were used to compile the distributions in Figures 1E–F. The angle relative to the short axes of the cell (Figure 1G) was calculated as the arctan (νx/νy).
The treadmilling velocity on the image plane, , is not the same as the actual 3D velocity of the treadmilling assembly, . While the νx component of both vectors is the same, the νy component is the projection of the azimuthal velocity to the image plane and can be expressed as νy = νφsin(φ), where φ is the azimuthal angle measured from the image plane. Thus, |νy| changes from 0 to |νφ| when the transient assembly moves around the circumference of the cell at a constant velocityνφ.
Of interest is not the projection νy, which we directly determine from the images, but the 3D velocity component νφ. To estimate the distribution of νφ among different assemblies in a cell population, we assume that the azimuthal positions of FtsZ foci, φ, are uniformly distributed around the circumference of the cell over time. First, we consider a situation where all assemblies treadmill at the same velocity νφ. In this case the νy component is distributed as
However, not all assemblies would treadmill at the same velocity νφ. If νφ is spread out according to a distribution G(νφ − νφ,0) around the mean velocity νφ,0 then distribution for νy becomes
In our analysis we assumed that G(νφ − νφ,0) is a normal distribution (a Gaussian). We then compared ρ(νy; νφ,0) at different νφ,0 and σν,φ (std) values to experimentally measured distribution at Figure 1E. In this procedure ρ(νy; νφ,0) was calculated using numerical integration. We found the best agreement for νφ,0 = 19 nm/s and σν,φ = 11 nm/s for the data shown in Figure 1E.
Determination of total FtsZ numbers in cells.
To determine FtsZ numbers in the cells we compared FtsZ- sfGFP fluorescent intensity from cells to fluorescent intensity from a calibrated GFP standard (37 μM, Clontech Laboratories Inc). Both cells and GFP solution were loaded into microfluidic channels made of PDMS that had a height of Hch=0.92 μm, widths of either Wch=0.8 μm or 0.9 μm, and length of 19 μm. The widths and heights of the channels on the silicon mold were determined by SEM and profilometer, respectively, as described in [34]. The shrinkage of the PDMS after its release from the mold is expected to be ε≈2% [47]. This shrinkage was used to correct channel width and heights.
To measure the intensity of the calibrated GFP standard, it was diluted 10 times into an M9 minimal medium (pH=7.0) to a final concentration of 3.7 μM. At this concentration channels with GFP showed a comparable brightness to channels filled with cells. Thereby GFP solution and cells were both imaged in the same illumination and exposure settings. The GFP solution was pipetted into the PDMS mother machine channels and imaged within 10 minutes after loading. Integrated intensity from the channel, IGFP, was used in further calculations. Integration included a 2.5 μm wide region centered on the channel midline. The integration region did not include a 2 μm long segment from the dead end and 4 μm long segment from the open end side of the channel. From this integrated intensity the background fluorescence was subtracted. The background was calculated using the same size region as described above but shifted 5 μm from the center of the channels in a perpendicular direction to the centerline of the channel.
To image cells, the same channels were used (although one could have used different ones without changing noticeably the outcome as we verified). The channels were flushed with growth media + BSA (1 mg/ml) first. This quickly got rid of any GFP in the channels that had been there from the previous measurement. After loading the cells, they were left growing in the channels with M9 glycerol media circulating overnight. The cells were then treated with 300 μg/ml chloramphenicol for 75 minutes to stop protein synthesis and cell growth. This treatment provided that effectively all sfGFP had time to mature [27, 48]. Cells were then imaged exactly in the same conditions as the channels with GFP. The integrated intensity from the cells, Icell, was also calculated the same way. Only channels where cells were well-aligned with the channel centerline and had no gaps between them were used in the analysis.
The measured intensities were corrected for different brightness’s of fluorophores, Br. For WT GFP BrGFP = 16.2 and for sfGFP BrsfGFP = 54.1 were used (http://nic.ucsf.edu/dokuwiki/doku.php?id=fluorescent_proteins). Measured intensities were also corrected for the changes in the power of the Hg lamp that was used as an illumination source. Power of the Hg lamp prior to each measurement (PGFP and Pcells, respectively) was measured by a Thorlabs PM121D power meter. The final formula used in calculations of cellular FtsZ was:
Here Rcell is the cytosolic radius of the cells estimated to be 0.32 μm in these growth conditions for this strain [49]. Additionally, a correction factor, corr, was introduced to the formula to account for cells that were not forming a continuous tube-like volume but consisted of individual cylindrical cells with hemispherical endcaps. Using an average cell length of Lcell = 2.80 μm [27] and cell radius Rcell = 0.32 μm, we estimated this geometrical factor to be corr = 0.08.
The cellular FtsZ concentration from measurements of different channels is shown in Figure S4. In these data, cells from 0.8 and 0.9 μm wide channels are pooled together. The average concentration was found to be 4.7±0.7 μM (mean±std). This is in good agreement with previous estimates reported to be in the range of 4-6 μM [36, 40, 41].
To calculate FtsZ numbers in an average cell in an asynchronous population, an average cell length of < Lcell >= 2.80 μm, and radius of 0.32 μm were used. Cell shape was assumed to be cylindrical with hemispherical endcaps. This corresponds to a cell volume of 0.83 μm3. Then the FtsZ number in an average cell was found to be 2300±400 molecules.
Determination of FtsZ numbers in individual cells.
To determine FtsZ numbers in individual cells in different measurements, we plotted the total intensity of the labeled FtsZ from each cell versus its length. These plots were made for every measurement where different strains or imaging parameters were used. Linear fits to Itotal vs Lcell data were then found. Based on these fits an average intensity at Lcell = 2.80 μm was determined. From that intensity and assuming 2300 molecules per average cell, we calculated the fluorescent intensity corresponding to a single labelled FtsZ molecule under the given experimental condition. This intensity was subsequently used to convert fluorescent intensities into the number of FtsZ molecules in the cell.
Estimation of measurement noise contribution to rate of change distributions.
To estimate noise contribution to the rate of change distributions in Figures 4E–F we imaged cells where SulA protein expression form an extra copy plasmid was induced (plasmid pA3, strain MB43). In these cells transient assemblies were absent due to antagonizing effect of SulA on FtsZ polymerization [36]. The measured rates in these cells thus originated from sources not related to transient assemblies, i.e. from measurement noise. Since the SulA overexpression strain lacked FtsZ foci we generated focal regions in these cells based measured distributions of focal parameters in WT cells. The size, shape and locations of each foci were drawn from distribution measured for WT cells. These parameters defined a region in the image (a binary mask), which was overlaid with the image of a SulA cell (which were picked randomly from the measured cells). It was centered at the same relative position as the focus in the WT cell and it lasted the same duration as the focus in WT cell. Fluorescent intensity as function of time from this region was then collected and analyzed for changes.
DATA AND CODE AVAILABILITY
The datasets and codes supporting current study are available from the Lead Contact on request.
Supplementary Material
Video S1. FtsZ-mNG cells (strain JM26) grown in agarose mother machine microfluidic chip. Cells were imaged at 6 second frame rate in slow growth conditions (M9 glycerol). Related to Figure 1.
Video S2. Time-lapse of photoactivated FtsZ-mMaple molecules (strain HE5). Exposure time is 1 ms and frame rate 22.9 frames/sec. Related to STAR Methods: Photoactivation experiment.
Video S3. Time-lapse of photoactivated FtsZ-mMaple molecules (strain HE5). Exposure time is 100 ms and frame rate 9.83 frames/sec. Related to STAR Methods: Photoactivation experiment.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| SeeTable S1 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Chloramphenicol | Sigma | Cat# C0378-5G |
| Kanamycin | Sigma | Cat# K4000-5G |
| Ampicillin | Sigma | Cat# A0166-5G |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Sigma | Cat# I6758-1G |
| Isopropanol | Fisher Scientific | Cat# A464-4 |
| Bovine Serum Albumin (BSA) | Sigma | Cat# A7906-10G |
| M9 minimal media | Teknova.com | Cat# M1902 |
| UltraPure™ Agarose | Invitrogen | Cat# 16500100 |
| Sylgard 184 | Dow Corning | N/A |
| GFP standard | Clontech Laboratories Inc | Cat# 632373 |
| Critical Commercial Assays | ||
| Gibson Assembly® Master Mix | New England Biolabs, Inc | Cat# E2611 |
| Oligonucleotides | ||
| Primers for cloning mNeonGreen (mNG) into pROD Forward: TGCTGGTTCTGGCGAATTCATGGTCAGCAAAGGTGAAGA | This paper | N/A |
| Primers for cloning mNeonGreen (mNG) into pROD Reverse: GAAGCAGCTCCAGCCTACAACACCCGGGTTACTTGTACA |
This paper | N/A |
| Primers for λ-Red engineering of the C-terminal fusion of mNeonGreen to LacY Forward: CCGCTTTCCCTGCTGCGTCGTCAGGTGAATGAAGTCGCTTCGGCTGGCTCCGCTGCTGG3 |
This paper | N/A |
| Primers for λ-Red engineering of the C-terminal fusion of mNeonGreen to LacY Reverse: GTCGGATAAGGCGCTCGCGCCGCATCCGACATTGATTGCCTTATGAATATCCTCCTTAG |
This paper | N/A |
| Software and Algorithms | ||
| MATLAB R2016b | Mathworks, Inc | RRID:SCR_001622 |
| NIS-Elements | Nikon Instruments Inc. | RRID:SCR_014329 |
| DipImage toolbox | http://www.diplib.org/ | N/A |
| Oufti | http://www.oufti.org/ | RRID:SCR_016244 |
| ImageJ (Fiij) | https://imagej.net/Fiji | RRID:SCR_002285 |
| Other | ||
| Nikon Ti-E inverted microscope | Nikon Instruments Inc. | Cat# MEA53100 |
| Nikon Perfect Focus system | Nikon Instruments Inc. | Cat# MEP59390 |
| LightHub-4 with 405 nm LuxX, 488 nm LuXx and 561 nm Obis lasers | Omicron | Cat# 214172 |
| Andor EMCCD camera | Andor Technologies | iXon897 |
| Power meter | Thorlabs | PM121D |
| Optomask | Andor Technologies | OPTMSK-L |
| O2 plasma asher | March Instruments | Plasmod |
| Syringe pump | New Era Pump Systems | Model: NE-1000 |
| Syringe pump | New Era Pump Systems | Model: NE-2000 |
Highlights.
Z-ring formation in E. coli is preceded by formation of transient FtsZ assemblies
The assemblies show rapid growth and shrinkage in addition to treadmilling
Transient assemblies compete for the available FtsZ pool
FtsZ protofilaments in the cytosol are less than 20 monomers long
Acknowledgements
The authors thank Alex Dajkovic, Harold Erickson, Rodrigo Reyes-Lamothe, and Jie Xiao for strains and plasmids, Da Yang and Scott Retterer for help in microfluidic chip making, Harold Erickson and Sriram Tiruvadi Krishnan for valuable comments. Authors acknowledge technical assistance and material support from the Center for Environmental Biotechnology at the University of Tennessee. A part of this research was conducted at the Center for Nanophase Materials Sciences, which is sponsored at Oak Ridge National Laboratory by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. This work has been supported by NSF research grant MCB-1252890, US-Israel BSF research grant 2017004, and National Institutes of Health award under R01GM127413.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1.Haeusser DP, and Margolin W (2016). Splitsville: structural and functional insights into the dynamic bacterial Z ring. Nat. Rev. Microbiol 14, 305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wagstaff J, and Lowe J (2018). Prokaryotic cytoskeletons: protein filaments organizing small cells. Nat. Rev. Microbiol 16, 187–201. [DOI] [PubMed] [Google Scholar]
- 3.Bisson AW, Hsu YP, Squyres GR, Kuru E, Wu FB, Jukes C, Sun YJ, Dekker C, Holden S, VanNieuwenhze MS, et al. (2017). Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science 355, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang XX, Lyu ZX, Miguel A, McQuillen R, Huang KC, and Xiao J (2017). GTPase activity-coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis. Science 355, 744–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stricker J, Maddox P, Salmon ED, and Erickson HP (2002). Rapid assembly dynamics of the Escherichia coli FtsZ-ring demonstrated by fluorescence recovery after photobleaching. Proc. Natl. Acad. Sci. U. S. A 99, 3171–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DE, Gueiros-Filho FJ, and Erickson HP (2004). Assembly dynamics of FtsZ rings in Bacillus subtilis and Escherichia coli and effects of FtsZ-regulating proteins. J. Bacteriol 186, 5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang KH, Durand-Heredia J, and Janakiraman A (2013). FtsZ ring stability: of bundles, tubules, crosslinks, and curves. J. Bacteriol 195, 1859–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Männik J, and Bailey MW (2015). Spatial coordination between chromosomes and cell division proteins in Escherichia coli. Frontiers in Microbiology 6, doi: 10.3389/fmicb.2015.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Persons L, Lee L, and de Boer PAJ (2015). Roles for both FtsA and the FtsBLQ subcomplex in FtsN-stimulated cell constriction in Escherichia coli. Mol. Microbiol. 95, 945–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey MW, Bissichia P, Warren BT, Sherratt DJ, and Männik J (2014). Evidence for divisome localization mechanisms independent of the Min system and SlmA in Escherichia coli. PLoS Genet. 10, doi: 10.1371/journal.pgen.1004504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill NS, Buske PJ, Shi Y, and Levin PA (2013). A moonlighting enzyme links Escherichia coli cell size with central metabolism. PLoS Genet. 9, doi: 10.1371/journal.pgen.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huisman O, Dari R, and Gottesman S (1984). Cell-division control in Escherichia coli - specific induction of the SOS function SfiA protein is sufficient to block septation. Proc. Natl. Acad. Sci. U. S. A 81, 4490–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erickson HP, Anderson DE, and Osawa M (2010). FtsZ in bacterial cytokinesis: cytoskeleton and force generator all in one. Microbiol. Mol. Biol. Rev 74, 504–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernhardt TG, and de Boer PAJ (2005). SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol. Cell 18, 555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Z, Trimble MJ, Brun YV, and Jensen GJ (2007). The structure of FtsZ filaments in vivo suggests a force-generating role in cell division. EMBO J. 26, 4694–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao Q, Jewett AI, Chang YW, Oikonomou CM, Beeby M, lancu CV, Briegel A, Ghosal D, and Jensen GJ (2017). Short FtsZ filaments can drive asymmetric cell envelope constriction at the onset of bacterial cytokinesis. EMBO J. 36, 1577–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du SS, Park KT, and Lutkenhaus J (2015). Oligomerization of FtsZ converts the FtsZ tail motif (conserved carboxy-terminal peptide) into a multivalent ligand with high avidity for partners ZipA and SlmA. Mol. Microbiol 95, 173–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pichoff S, and Lutkenhaus J (2005). Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol. Microbiol 55, 1722–1734. [DOI] [PubMed] [Google Scholar]
- 19.Lan GH, Daniels BR, Dobrowsky TM, Wirtz D, and Sun SX (2009). Condensation of FtsZ filaments can drive bacterial cell division. Proc. Natl. Acad. Sci. U. S. A 106, 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coltharp C, Buss J, Plumer TM, and Xiao J (2016). Defining the rate-limiting processes of bacterial cytokinesis. Proc. Natl. Acad. Sci. U. S. A 113, E1044–E1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holden SJ, Pengo T, Meibom KL, Fernandez CF, Collier J, and Manley S (2014). High throughput 3D super-resolution microscopy reveals Caulobacter crescentus in vivo Z-ring organization. Proc. Natl. Acad. Sci. U. S. A 111, 4566–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan FH, Yu JY, Yu J, Liu Y, Li Y, Feng XH, Huang KC, Chang ZY, and Ye S (2018). Lateral interactions between protofilaments of the bacterial tubulin homolog FtsZ are essential for cell division. Elife 7, doi: 10.7554/eLife.35578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roseboom W, Nazir MG, Meiresonne NY, Mohammadi T, Verheul J, Buncherd H, Bonvin A, de Koning LJ, de Koster CG, de Jong L, et al. (2018). Mapping the contact sites of the Escherichia coli division-initiating proteins FtsZ and ZapA by BAMG coss-linking and site-directed mutagenesis. Int. J. Mol. Sci 19, 2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durand-Heredia J, Rivkin E, Fan GX, Morales J, and Janakiraman A (2012). Identification of ZapD as a cell division factor that promotes the assembly of FtsZ in Escherichia coli. J. Bacteriol 194, 3189–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.RayChaudhuri D (1999). ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J. 18, 2372–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hale CA, Rhee AC, and de Boer PAJ (2000). ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J. Bacteriol 182, 5153–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Männik J, Walker BE, and Männik J (2018). Cell cycle-dependent regulation of FtsZ in Escherichia coli in slow growth conditions. Mol. Microbiol 110, 1030–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YD, and Erickson HP (2005). Rapid in vitro assembly dynamics and subunit turnover of FtsZ demonstrated by fluorescence resonance energy transfer. J. Biol. Chem 280, 22549–22554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huecas S, Llorca O, Boskovic J, Martin-Benito J, Valpuesta JM, and Andreu JM (2008). Energetics and geometry of FtsZ polymers: Nucleated self-assembly of single protofilaments. Biophys. J 94, 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romberg L, Simon M, and Erickson HP (2001). Polymerization of FtsZ, a bacterial homolog of tubulin - Is assembly cooperative? J. Biol. Chem 276, 11743–11753. [DOI] [PubMed] [Google Scholar]
- 31.Moore DA, Whatley ZN, Joshi CP, Osawa M, and Erickson HP (2017). Probing for binding regions of the FtsZ protein surface through site-directed insertions: discovery of fully functional FtsZ-fluorescent proteins. J. Bacteriol 199, doi: 10.1128/jb.00553-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tokunaga M, Imamoto N, and Sakata-Sogawa K (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 5, 455–455. [DOI] [PubMed] [Google Scholar]
- 33.Wang P, Robert L, Pelletier J, Dang WL, Taddei F, Wright A, and Jun S (2010). Robust growth of Escherichia coli. Curr. Biol. 20, 1099–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang D, Jennings AD, Borrego E, Retterer ST, and Männik J (2018). Analysis of factors limiting bacterial growth in PDMS mother machine devices. Frontiers in Microbiology 9, doi: 10.3389/fmicb.2018.00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thanedar S, and Margolin W (2004). FtsZ exhibits rapid movement and oscillation waves in helix-like patterns in Escherichia coli. Curr. Biol 14, 1167–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dajkovic A, Mukherjee A, and Lutkenhaus J (2008). Investigation of regulation of FtsZ assembly by SulA and development of a model for FtsZ polymerization. J. Bacteriol 190, 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Männik J, Castillo D, Yang D, Siopsis G, and Männik J (2016). The role of MatP, ZapA, and ZapB in chromosomal organization and dynamics in Escherichia coli. Nucleic Acids Res. 44, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowlett VW, and Margolin W (2015). The Min system and other nucleoid-independent regulators of Z ring positioning. Frontiers in Microbiology 6, doi: 10.3389/fmicb.2015.00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vischer NOE, Verheul J, Postma M, van den Berg van Saparoea B, Galli E, Natale P, Gerdes K, Luirink J, Vollmer W, Vicente M, et al. (2015). Cell age dependent concentration of Escherichia coli divisome proteins analyzed with ImageJ and ObjectJ. Front. Microbiol 6, doi: 10.3389/fmicb.2015.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buss J, Coltharp C, Huang T, Pohlmeyer C, Wang S-C, Hatem C, and Xiao J (2013). In vivo organization of the FtsZ-ring by ZapA and ZapB revealed by quantitative super-resolution microscopy. Mol. Microbiol 89, 1099–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rueda S, Vicente M, and Mingorance J (2003). Concentration and assembly of the division ring proteins FtsZ, FtsA, and ZipA during the Escherichia coli cell cycle. J Bacteriol 185, 3344–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Monterroso B, Ahijado-Guzman R, Reija B, Alfonso C, Zorrilla S, Minton AP, and Rivas G (2012). Mg2+-linked self-assembly of FtsZ in the presence of GTP or a GTP analogue involves the concerted formation of a narrow size distribution of oligomeric cpecies. Biochemistry 51, 4541–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Si FW, Le Treut G, Sauls JT, Vadia S, Levin PA, and Jun S (2019). Mechanistic origin of cell-size control and homeostasis in bacteria. Curr. Biol 29, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sekar K, Rusconi R, Sauls JT, Fuhrer T, Noor E, Nguyen J, Fernandez VI, Buffing MF, Berney M, Jun S, et al. (2018). Synthesis and degradation of FtsZ quantitatively predict the first cell division in starved bacteria. Mol. Syst. Biol 14, e8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moffitt JR, Lee JB, and Cluzel P (2012). The single-cell chemostat: an agarose-based, microfluidic device for high-throughput, single-cell studies of bacteria and bacterial communities. Lab Chip 12, 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paintdakhi A, Parry B, Campos M, Irnov I, Elf J, Surovtsev I, and Jacobs-Wagner C (2016). Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol. Microbiol 99, 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madsen MH, Feidenhans’l NA, Hansen PE, Garnaes J, and Dirscherl K (2014). Accounting for PDMS shrinkage when replicating structures. J. Micromech. Microeng 24, doi: 10.1088/0960-1317/24/12/127002. [DOI] [Google Scholar]
- 48.Balleza E, Kim JM, and Cluzel P (2018). Systematic characterization of maturation time of fluorescent proteins in living cells. Nat Methods 15, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang D, Männik J, Retterer ST, and Männik J (2019). The effects of polydisperse crowders on the compaction of the Escherichia coli nucleoid. bioRxiv, doi: 10.1101/803130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. FtsZ-mNG cells (strain JM26) grown in agarose mother machine microfluidic chip. Cells were imaged at 6 second frame rate in slow growth conditions (M9 glycerol). Related to Figure 1.
Video S2. Time-lapse of photoactivated FtsZ-mMaple molecules (strain HE5). Exposure time is 1 ms and frame rate 22.9 frames/sec. Related to STAR Methods: Photoactivation experiment.
Video S3. Time-lapse of photoactivated FtsZ-mMaple molecules (strain HE5). Exposure time is 100 ms and frame rate 9.83 frames/sec. Related to STAR Methods: Photoactivation experiment.
Data Availability Statement
The datasets and codes supporting current study are available from the Lead Contact on request.