

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive, incurable cancer with a 20% 1 year survival rate. While standard-of-care therapy can prolong life in a small fraction of cases, PDAC is inherently resistant to current treatments, and novel therapies are urgently required. Histone deacetylase (HDAC) inhibitors are effective in killing pancreatic cancer cells in in vitro PDAC studies, and although there are a few clinical studies investigating combination therapy including HDAC inhibitors, no HDAC drug or combination therapy with an HDAC drug has been approved for the treatment of PDAC. We developed an inhibitor of HDACs, AES-135, that exhibits nanomolar inhibitory activity against HDAC3, HDAC6, and HDAC11 in biochemical assays. In a three-dimensional coculture model, AES-135 kills low-passage patient-derived tumor spheroids selectively over surrounding cancer-associated fibroblasts and has excellent pharmacokinetic properties in vivo. In an orthotopic murine model of pancreatic cancer, AES-135 prolongs survival significantly, therefore representing a candidate for further preclinical testing.
Graphical Abstract
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers, with only a 20% 1 year survival rate and a 7% 5 year survival rate for all stages combined, and is widely considered incurable.1,2 It is currently the third leading cause of cancer-related mortality in the United States3 and is characterized by a complex tumor microenvironment that is immunosuppressive and contains myeloid-derived suppressor cells (MDSCs) as well as cancer-associated fibroblasts (CAFs), heterogeneity within the tumor, and an innate capacity for metastasis.4–7 Therefore, there is an imminent need for therapies in PDAC, inhibiting novel targets.
Histone/lysine deacetylases control post-translational protein acetylation,8–13 in conjunction with histone acetyltransferases (HATs), which fulfill an antagonistic role,9,14 for a large number of substrates, most notably histones. By regulating histone acetylation/deacetylation, HATs and histone deacetylases (HDACs) play a key indirect role in gene expression.11 Oncogenic HDAC activity has been observed in aggressive human cancers, including pancreatic cancer.1,2,15 To date, four small-molecule HDAC inhibitors have been approved by the Food and Drug Administration (FDA) for hematological cancer treatment (cutaneous T-cell lymphoma, peripheral T-cell lymphoma and multiple myeloma):8,11,13 suberoylanilide hydroxamic acid (SAHA, Vorinostat),16 Romidepsin (depsipeptide-FK228),17 Belinostat (PXD101),18 and Panobinostat (LBH-589).19 Current HDAC clinical trials in PDAC consist of adjuvant therapies using Vorinostat or Panobinostat in combination with radiation, surgery, or standard-of-care chemotherapy.20–27 Three of the four HDAC drugs contain an N-hydroxamic acid, which mimics the hydrogen bonds formed by acetylated lysine substrates, competitively coordinating to the metal ion within the catalytic domain, rendering the HDAC inactive.8 The catalytic domain is the most structurally conserved region in the HDAC family primary sequence, and targeting of this domain by small molecules often results in the inhibition of more than one HDAC. Despite this, clinical efficacy with pan-HDAC inhibitors has been observed in select cancer subtypes, but with adverse side effects including diarrhea and bone marrow toxicity observed in patients.8 HDAC inhibitors, with the exception of Romidepsin,28–30 possess a similar linear structural design, with a metal chelating group (e.g., hydroxamic acid) at one end and a hydrophobic capping group (e.g., a 2-methylindole) at the other, connected by a linear hydrophobic scaffold, for example, a benzene ring or an alkyl chain.13,31 A lack of structural diversity might infer that many of the current clinical candidates are likely to encounter the same pitfalls in clinical trials.32
Herein, we introduce a small family of novel HDAC inhibitors, including lead compound, AES-135, which biochemically inhibits HDACs 3, 6, 8, and 11 with half-maximal inhibitory concentration (IC50) values of 190–1100 nM and exhibits selective in vitro cytotoxicity in low-passage patient-derived pancreatic cancer cells even in the presence of cancer-associated fibroblast (CAF) cells. AES-135 has other favorable in vivo properties such as metabolic stability in mouse hepatocytes and bioavailability in micromolar (μM) concentrations in NSG mice for >10 h (intraperitoneal (IP) injection). AES-135 combines the proven attributes of an N-hydroxamic acid with a new chemotype for exploration as an HDAC inhibitor.
RESULTS AND DISCUSSION
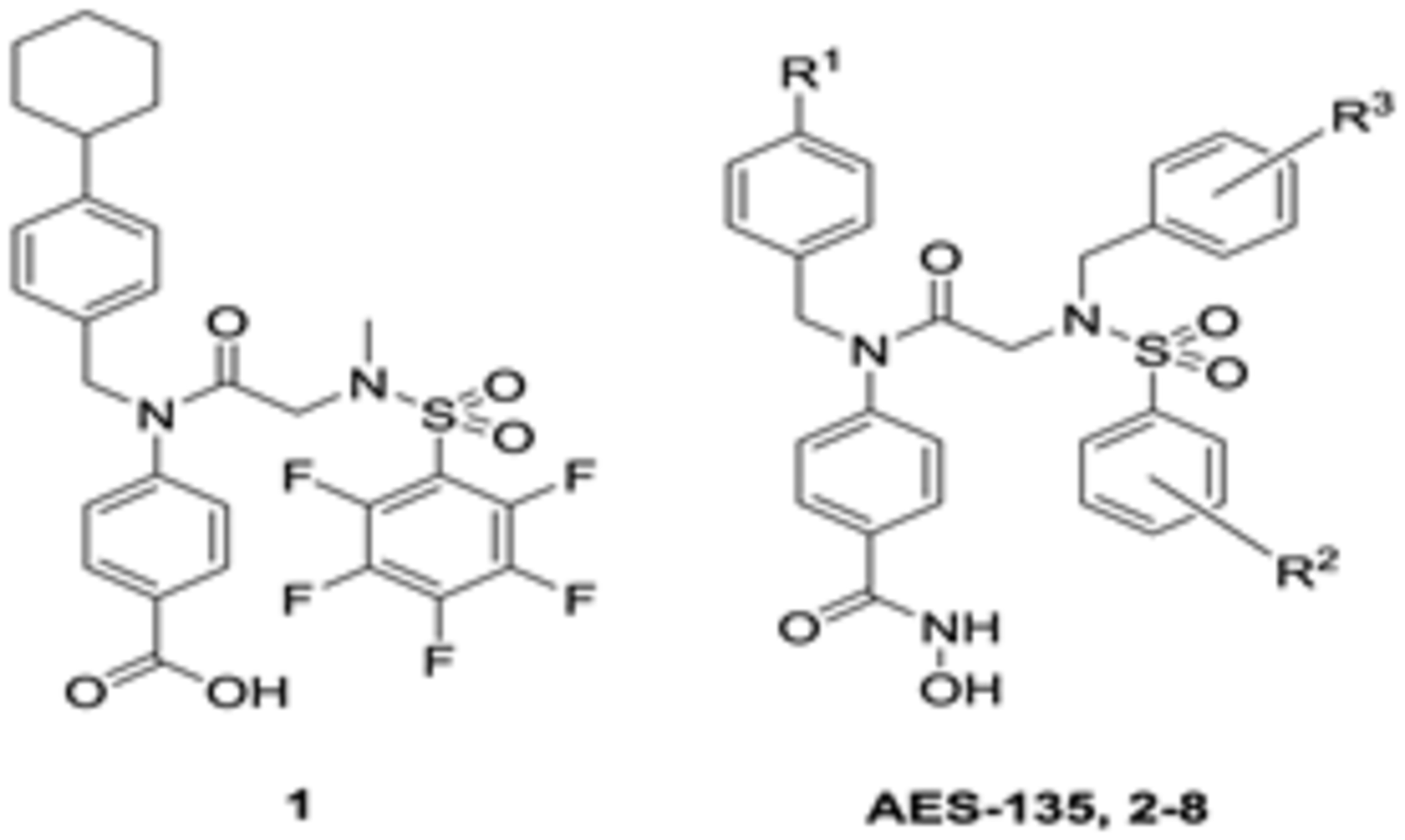
AES-135 was identified as part of a structure–activity relationship (SAR) study designed to repurpose a class of signal transducer and activator of transcription 3 (STAT3)-targeting compounds, including SH-4–54 (1, Table 1), toward HDACs.33–35 Efforts were predominantly focused on replacing the STAT3 SH2 domain-targeting benzoic acid substituent with an isosteric N-hydroxamic acid for HDAC catalytic domain targeting.36 A brief SAR around the general structure of 1 sought to identify direct-binding nanomolar (nM) IC50 HDAC inhibitors. A series of analogs were prepared with substitutions made at positions R1, R2, and R3 (Table 1).
Table 1.
IC50 Values for AES-135 and Its Analogs against HDACs 3, 6, 8, and 11, Evaluated up to 1 μM (Electrophoretic Mobility Shift Assay (EMSA), n = 1)
 | |||||||
|---|---|---|---|---|---|---|---|
| substituents | IC50 (μM) | ||||||
| compound # | R1 | R2 | R3 | HDAC3 | HDAC6 | HDAC8 | HDAC11 |
| AES-13S | t-Bu | 4-F | 2,3,4,5,6-F | 0.654 | 0.190 | >1 | 0.636 |
| 2 | t-Bu | 3,4-F | 2-CF3 | >1 | 0.362 | >1 | 0.254 |
| 3 | CF3 | 3,4-F | 2-CF3 | >1 | 0.188 | >1 | 0.396 |
| 4 | t-Bu | 2,4-F | 2-CF3 | >1 | 0.289 | >1 | 0.288 |
| S | CF3 | 2,4-F | 2-CF3 | >1 | 0.151 | >1 | 0.346 |
| 6 | t-Bu | 4-F | 2-CF3 | >1 | 0.230 | >1 | 0.253 |
| 7 | CF3 | 4-F | 2-CFj | >1 | 0.093 | >1 | 0.304 |
| 8 | CF3 | 4-F | 2,3,4,5,6-F | >1 | 0.086 | >1 | 0.191 |
Compounds were synthesized as outlined in Scheme 1. Briefly, anilines 11 and 12 were prepared in good to excellent yields via reductive amination of benzyl 4-aminobenzoate 10 with different benzaldehydes under standard conditions. Glycine tert-butyl (t-Bu) ester hydrochloride (13) was sulfonylated, and the resulting sulfonamides 14–17 were benzylated under basic conditions. Removal of the t-Bu protecting group with diluted trifluoroacetic acid (TFA) furnished the carboxylic acids 23–27 quantitatively. Anilines and carboxylic acids were coupled using dichlorotriphenylphosphorane (PPh3Cl2) under microwave conditions, and subsequent hydrogenation cleaved the carboxybenzyl group. Chlorination of the benzoic acids using oxalyl chloride, followed by coupling with O-benzylhydroxylamine, generated the hydroxamate esters, and the O-benzyl group was removed by hydrogenation.
Scheme 1.
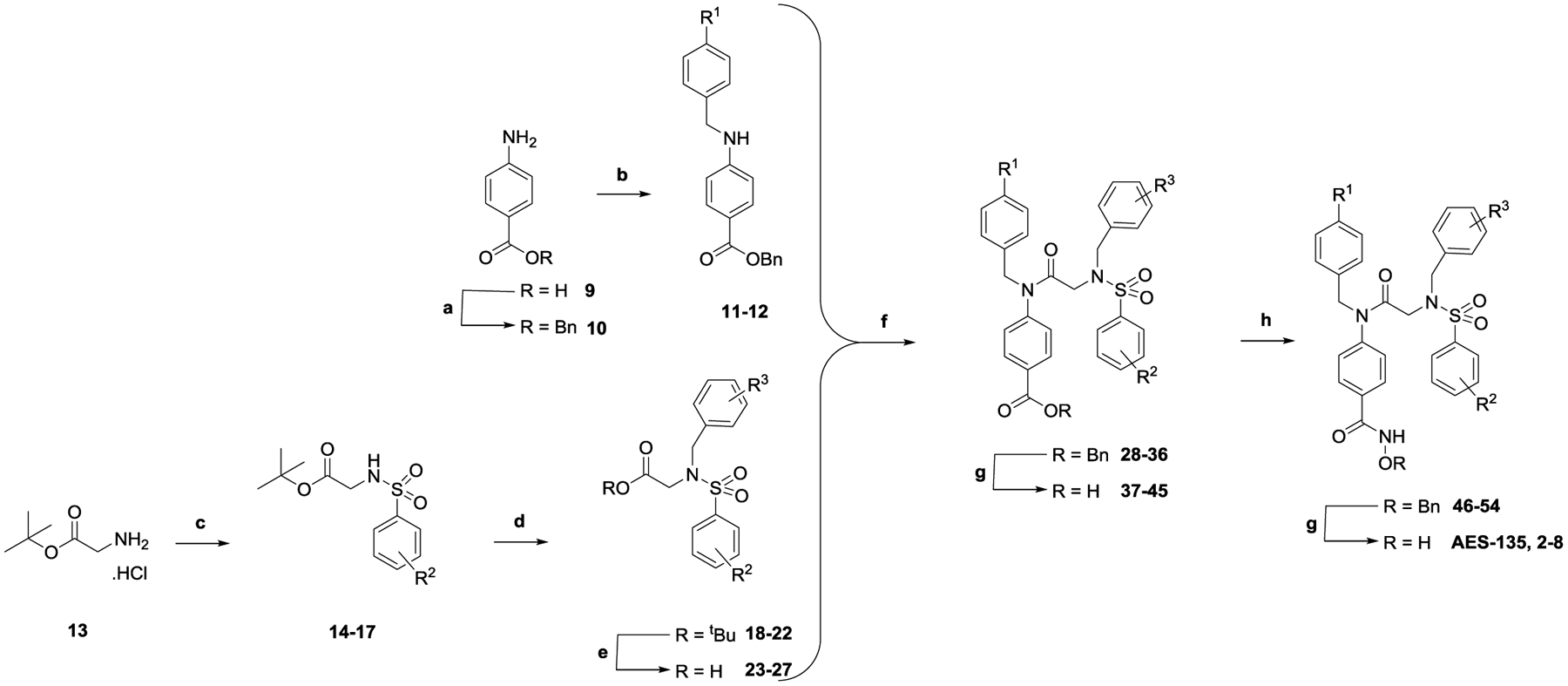
Synthesis of N-Hydroxamic Acid-Based HDAC Inhibitorsa
a(a) BnBr, Cs2CO3, dimethylformamide (DMF), 24 h, room temperature (RT); (b) (i) ArCHO, MgSO4, tetrahydrofuran (THF), 16 h, RT; (ii) NaBH4, trifluoroethanol (TFE), 16 h, RT; (c) ArSO2Cl, iPr2NEt, CH2Cl2, 16 h, 0 °C to RT, N2; (d) BrCH2Ar, Cs2CO3, MeCN, 16 h, 50 °C to RT; (e) CF3CO2H/CHCl3 (1:3), 3 h, RT; (f) 11 or 12, PPh3Cl2, CHCl3, 1 h, 100 °C, N2, microwave; (g) H2, 10% Pd/C, THF/MeOH (2:1), 16 h, RT; (h) (i) (COCl)2, THF, DMF, 2 h, 0 °C, N2; (ii) H2NOBn, iPr2NEt, THF, 16 h, RT, N2.
The R2 position of 1, occupied by a pentafluorobenzenesulfonamide, was substituted with less electron-deficient monoand difluorinated benzene rings to minimize potential nucleophilic addition in vivo.35 The cyclohexyl R1 group reduced solubility and was susceptible to phase I oxidation,35 so this was replaced with less lipophilic t-Bu and trifluoromethyl (CF3) groups.33 Finally, the R3 N-methyl group, previously shown to be sensitive to oxidation in mouse hepatocytes, was substituted with either a pentafluorobenzyl (PFB) or 2-(trifluoromethyl)benzyl appendage.37 The prepared library was evaluated for inhibitory activity against select HDAC representative of groups I (3 and 8), II (6), and IV (11) using an electrophoretic mobility shift assay (EMSA). In this assay, enzymatic deacetylation of a FAM-labeled peptide substrate is measured as a change in the relative fluorescence intensity of the substrate and product following incubation. In the presence of an inhibitor, deacetylation is impeded, altering the fluorescence intensity of the product and substrate (a detailed procedure is provided in the Supporting Information). With the exception of AES-135, compounds in this library demonstrated selective activity for HDAC groups II and IV, with limited activity observed against either HDAC3 or HDAC8 (group I). AES-135 exhibited nanomolar (nM) inhibition of HDACs 3, 6, and 11, with low-μM activity against HDAC8 (IC50 was later confirmed to be 1.10 μM when AES-135 was evaluated up to 10 μM against these targets (Figures S22–S25, Supporting Information)).
To explain the observed results, compounds were modeled in silico using AutoDock Vina/AutoDockTools (ADT) v4.2.6. Specifically, AES-135 and 6 were chosen as representative ligands due to their differing HDAC selectivity profiles despite being structurally similar, differing only at the R3 position (pentafluorobenzyl vs 2-(trifluoromethyl)benzyl, respectively). These compounds were docked against eukaryotic, zinc-dependent hHDAC 3, 6, and 8 (PDB: 4A69, 5EDU, and 1T64) (Figure 1). hHDAC11 analysis was not possible as no crystal structure has been resolved to date. A detailed description of the experiments performed can be found in the Supporting Information.
Figure 1.
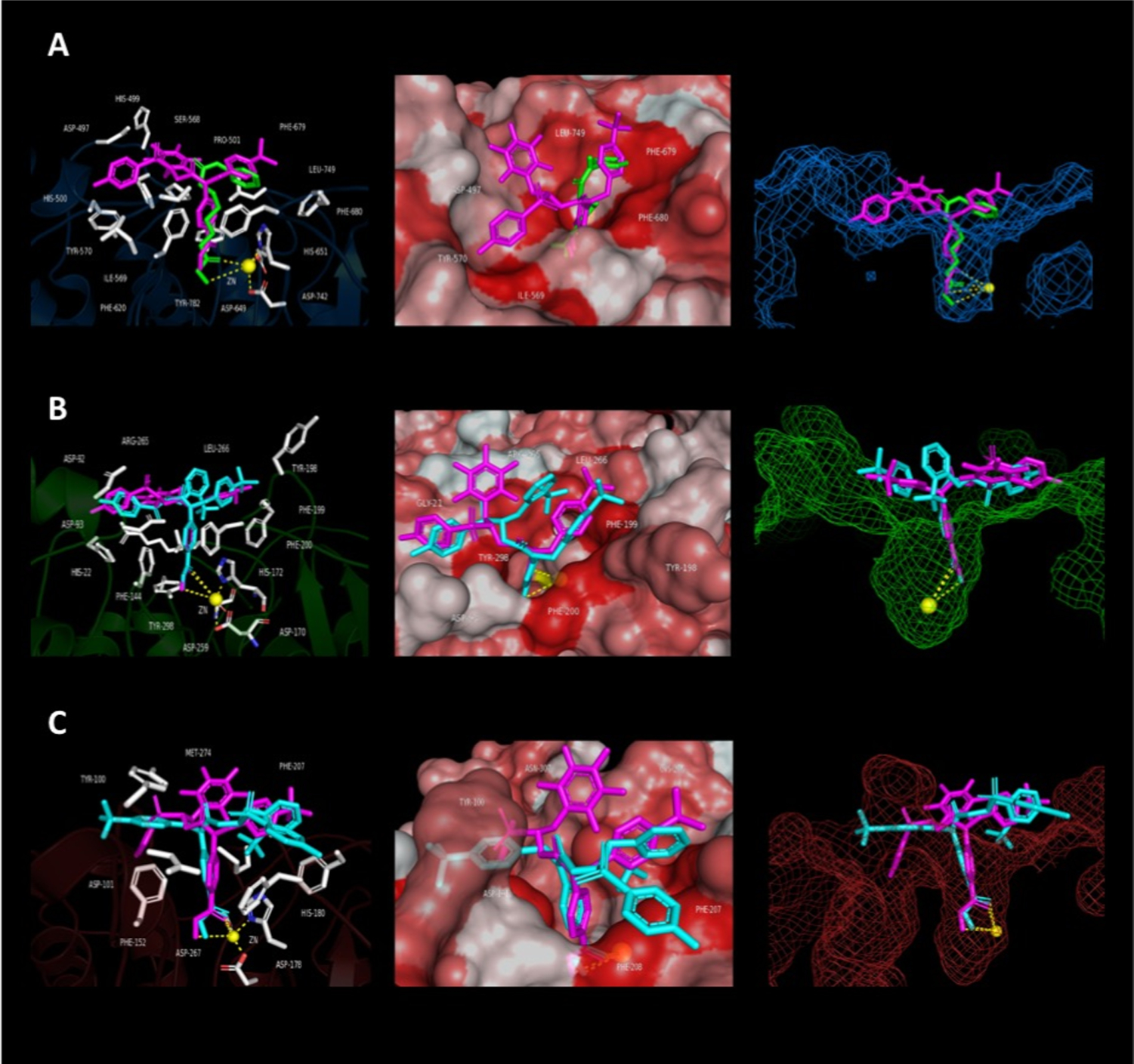
Computational modeling/docking studies of AES-135 against hHDAC 3, 6, and 8 (PDB: 4A69, 5EDU, and 1T64, respectively). (Left column) catalytic active site of enzyme, Zn2+ (yellow sphere), residues within/around the lysine-substrate channel (shown as white ball-and-stick) and catalytic triad residues (shown as colored ball-and-stick). (Centre column) molecular surface view of channel entrance, low hydrophobicity residues (white), and high hydrophobicity residues (red). (Right column) side view of ligands docked within the active site. Panel A: hHDAC6 (blue cartoon), AES-135 (magenta), and SAHA (green). Panel B: hHDAC3 (green cartoon), AES-135 (magenta), and 6 (cyan). Panel C: hHDAC8 (red cartoon), AES-135 (magenta), and 6 (cyan).
All three enzymes have a largely hydrophobic surface proximal to the lysine-substrate channel. In hHDAC6, the lysine tunnel surroundings are largely featureless and flat, whereas hHDAC3 and 8 contain greater surface topology. While the importance of the metal binding group for HDAC targeting is critical, the contribution of the capping group to binding and selectivity among the HDAC isoforms is significant. Increased hydrophobic interaction between the enzyme surface and the capping group is postulated to greatly increase binding affinity.38–40 These interactions were of interest when analyzing the in silico binding conformations of AES-135 and 6 to hHDACs 3, 6, and 8 (Figure 1), relative to the in vitro EMSA data shown in Table 1. The docking of AES-135 to hHDAC3, 6, and 8 returned average free energy of binding values (ΔGB) of −8.31 ± 0.08, −8.98 ± 0.17, and −8.80 ± 0.13 kcal/mol, whereas 6 scored −7.31 ± 0.44, −8.64 ± 0.17, and −7.74 ± 0.12 kcal/mol, respectively (Tables S17–S23, Supporting Information). In silico, AES-135 binds more favorably to all three isoforms than 6, while having greater affinity for hHDAC3/8, yet has more comparable binding to hHDAC6. Comparing binding to hHDAC 3 and 8 (Figure 1B,C), the perfluorinated ring of AES-135 makes significantly more interactions with residues proximal to the channel, with minimal steric clash compared to the 2-(trifluoromethyl) group of 6, which appears to occupy poses that unfavorably clash with the HDAC surface.
To confirm AES-135 as the lead candidate, cytotoxicity profiles were determined against a bank of human cancer cell lines (Tables S1, S2 and Figures S1–S6, Supporting Information). Encouragingly, AES-135 was shown to be the most promising candidate, with low μM potency being observed in multiple brain tumor stem cell (BTSC) glioblastoma lines, MV4–11 and MOLM-13 acute myeloid leukemia (AML) cells, and PC-3 prostate cancer cells. In D425 primary medulloblastoma (MDB) and D458 recurrent MDB cells, nM activity was observed. Patient-derived pancreatic cancer cells were sensitive to single-digit μM/high nM concentrations of AES-135. Of the lines tested, only chronic myelogenous leukemia K562 cells were resistant to AES-135. Encouragingly, in MRC-9 lung cells (noncancerous), AES-135 demonstrated minimal toxicity, identifying a clear therapeutic window.
To assess the stability of the pentafluorobenzyl (PFB) ring to biological nucleophiles, a 10 mM solution of AES-135 in dimethyl sulfoxide (DMSO) was mixed with a 100-fold excess of reduced l-glutathione in (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer, pH 7.4, and monitored by analytical high-performance liquid chromatography (HPLC) at regular intervals. No discernible reaction was observed even after 25 h (Figures S7 and S8, Supporting Information). This was corroborated by 19F NMR studies, in which no displacement of fluoride was observed after 16 h of immersion in 100-fold excess glutathione (Figure S9, Supporting Information).
Given the structural origin of AES-135 from 1, we confirmed that the cytotoxicity observed with the former was not due to STAT3/5 inhibition. Western blot studies were performed in MDA-MB-231 breast cancer cells (STAT3-overexpressing) and MV4–11 AML cells (STAT5-overexpressing). The results showed that AES-135 did not inhibit activation of STAT3 or STAT5 signaling via immunoblotting for Y694 phosphorylation on STAT5b and Y705 phosphorylation on STAT3 (Figure S10, Supporting Information). In MDA-MB-231 cells, AES-135 returned an IC50 of 2.72 ± 0.60 μM (n = 4), yet even at 10-fold of this concentration, neither total STAT3 nor pY705 STAT3 was significantly reduced. Similar results were observed in MV4–11 cells, where AES-135 had an IC50 of 1.88 ± 0.89 μM (n = 4), yet failed to suppress total STAT5b or pY694 STAT5b, even at 10 μM. Evidence from Western blots in pancreatic Pa03C cancer cells and multiple BTSC glioblastoma multiforme (GBM) lines was subsequently obtained, further supporting the conclusion that AES-135 was not a STAT3/5 inhibitor (Figures S11–S14, Supporting Information).
To confirm that the N-hydroxamic acid group in AES-135 was responsible for HDAC targeting, the compound was screened against seven metal-dependent HDACs, representing classes I, II, and IV, at a fixed concentration (Table 2). As a negative control, an N-methylhydroxamic acid, 60, was prepared and assessed in parallel. The synthetic route is described in Scheme 2. Briefly, starting from O-benzylhydroxylamine (55), Boc protection of the amino group was followed by methylation and acid-mediated removal of the Boc group to furnish O-benzyl-N-methylhydroxylamine (58). Coupling of this compound with 37, followed by hydrogenation, as previously described, yielded 60.
Table 2.
Percentage Inhibition of HDACs 1, 3, 4, 6, 8, 10, and 11 by AES-135 and 60 at 10 μM (EMSA, n = 2)
| % inhibition | ||
|---|---|---|
| HDAC | AES-13S | 60 |
| 1 | 72 | 0 |
| 3 | 98 | 0 |
| 4 | 15 | 0 |
| 6 | 98 | 12 |
| 8 | 94 | 0 |
| 10 | 70 | 0 |
| 11 | 97 | 64 |
Scheme 2.
Synthesis of N-Methylhydroxamic Acid 60a
a(a) Boc2O, THF, 24 h, RT; (b) MeI, NaH (60%), DMF, 24 h, RT; (c) (i) CF3CO2H/CHCl3 (1:3), 22 h, RT; (ii) 1 M NaOH; (d) (i) (COCl)2, THF, DMF, 2 h, 0 °C, N2; (ii) 58, iPr2NEt, THF, 16 h, RT, N2; (e) H2, 10% Pd/C, THF/MeOH (2:1), 16 h, RT.
AES-135 and control compound 60 were screened against full recombinant human HDACs 1, 3, 4, 6, 8, 10, and 11 using an electrophoretic mobility shift assay (EMSA) at 10 μM. AES-135 inhibited HDACs 3, 6, 8, and 11 (>90%) and showed moderate inhibition of HDACs 1 and 10 (≥70%), with HDAC4 not being affected (<20%). HDAC inhibition was highly sensitive to modification of the hydroxamic acid motif, with compound 60 demonstrating only modest inhibition of HDAC11 (64%) but negligible activity against the remaining HDACs (Table 2).
To evaluate in vitro stability, AES-135 and 8 were incubated with mouse hepatocytes for 2 h to assess the rate of intrinsic clearance. The calcium channel blocker Verapamil was used as a positive control. AES-135 reported an intrinsic clearance rate of 36.0 μL/min per 106 cells, almost half the rate of Verapamil (63.3 μL/min per 106 cells), and a half-life of 38.5 min, which was almost twice that of Verapamil (21.9 min) (Table S4 and Figures S15–S20, Supporting Information). Derivative 8, possessing a CF in the R1 3 position, performed similarly to AES-135 (R1 = t-Bu), returning a clearance rate of 37.4 μL/min per 106 cells and a half-life of 37.1 min, suggesting that the t-Bu group of AES-135 was not being targeted for oxidation in mouse hepatocytes. The observed stability of AES-135 supported the in vitro findings that the PFB ring was a relatively stable substituent. A protein binding study using AES-135 in mouse plasma found that the compound was 99.6% bound after 6 h of incubation. The low recovery (16–20%) of AES-135 after this time indicated the compound to be susceptible to metabolism in plasma (Table S5, Supporting Information). In a separate study, the experimental Log D7.4 for AES-135 was calculated using a 1-octanol/phosphate-buffered saline (PBS) system, returning a value of 4.15 (Table S6, Supporting Information).
To investigate the permeability profiles of AES-135 and 8 through the blood–brain barrier, the compounds were tested using a parallel artificial membrane permeability assay (PAMPA), which assesses the ability of a compound to cross a lipid-infused artificial membrane and has been shown to correlate well with performance in crossing in vivo barriers. Testosterone and the antimetabolite Methotrexate were used as positive and negative controls, respectively. In this assay, a permeability coefficient (−Log Pe) <6 defined the compound as having high permeability through a lipid membrane, whereas a −Log Pe > 6 meant the compound had low permeability. Results from this assay showed that AES-135 and 8 were poorly permeable compounds, returning −Log Pe values of 7.73 and 7.02, respectively. By comparison, testosterone gave a value of 4.61 and Methotrexate >8.5, where the degree of membrane permeation was below the limit of detection (Tables S7–S11, Supporting Information). The results indicated that AES-135 would be poorly efficacious against cancers surrounded by undamaged membranes, for example, GBM, despite impressive in vitro potency.
AES-135 was also analyzed in a Caco-2 assay to gauge its permeability through a monolayer of tightly packed epithelial cells, an in vitro model of the human small intestinal membrane. The Caco-2 cells also express several transporter proteins, for example, P-glycoprotein, and can thus provide information on the efflux rate of compounds from a cell. Propranolol, Digoxin, and Prazosin were used as controls with low, high, and medium efflux rates, respectively. Results from this assay supported those from the PAMPA, showing AES-135 to have poor permeation through the monolayer, with an apparent permeability coefficient, Papp (A-B), of 0.27 × 10−6 cm/s. Compared to Propranolol and Prazosin, with respective Papp (A-B) values of 15.41 and 19.94 × 10−6 cm/s, AES-135 was significantly less permeable. In addition, the Papp (B-A) for AES-135 was 1.02 × 10−6 cm/s, calculating an efflux ratio of 3.83 (Tables S12 and S13, Supporting Information). This suggested that AES-135 was transported out of the cell approximately 4-fold faster than it was being absorbed, meaning that it would struggle to achieve suitable intestinal absorption in vivo if administered orally. The data also indicated that despite potent in vitro activity, AES-135 would not be efficacious if used to treat cancers requiring penetration of bone marrow, for example, AML.
To assess the pharmacokinetic (PK) properties of AES-135 in vivo, NSG mice were dosed with a single 20 mg/kg intraperitoneal (IP) injection, and blood was taken at 0.5, 1, 2, 4, 8, and 24 h. AES-135 achieved μM concentrations in the blood, reaching Cmax 7452 ng/mL (10.74 μM) within 30 min, which was sustained for 8 h, with significant clearance observed only after 24 h (Table 3). In these mice, AES-135 had a calculated half-life of 5.0 h, with a clearance rate of 0.004 L/h, assuming bioavailability to be 100% (Tables S14 and S15, Supporting Information).
Table 3.
Serum Concentrations of AES-135 Following One Dose at 20 mg/kg, IP (n = 2)
 | |
|---|---|
| time (h) | [AES-135] (ng/mL) |
| 0.5 | 7452 ± 1354 |
| 1 | 5397 ± 1079 |
| 2 | 5391 ± 2418 |
| 4 | 3655 ± 400 |
| 8 | 6392 ± 222 |
| 24 | 265 ± 29 |
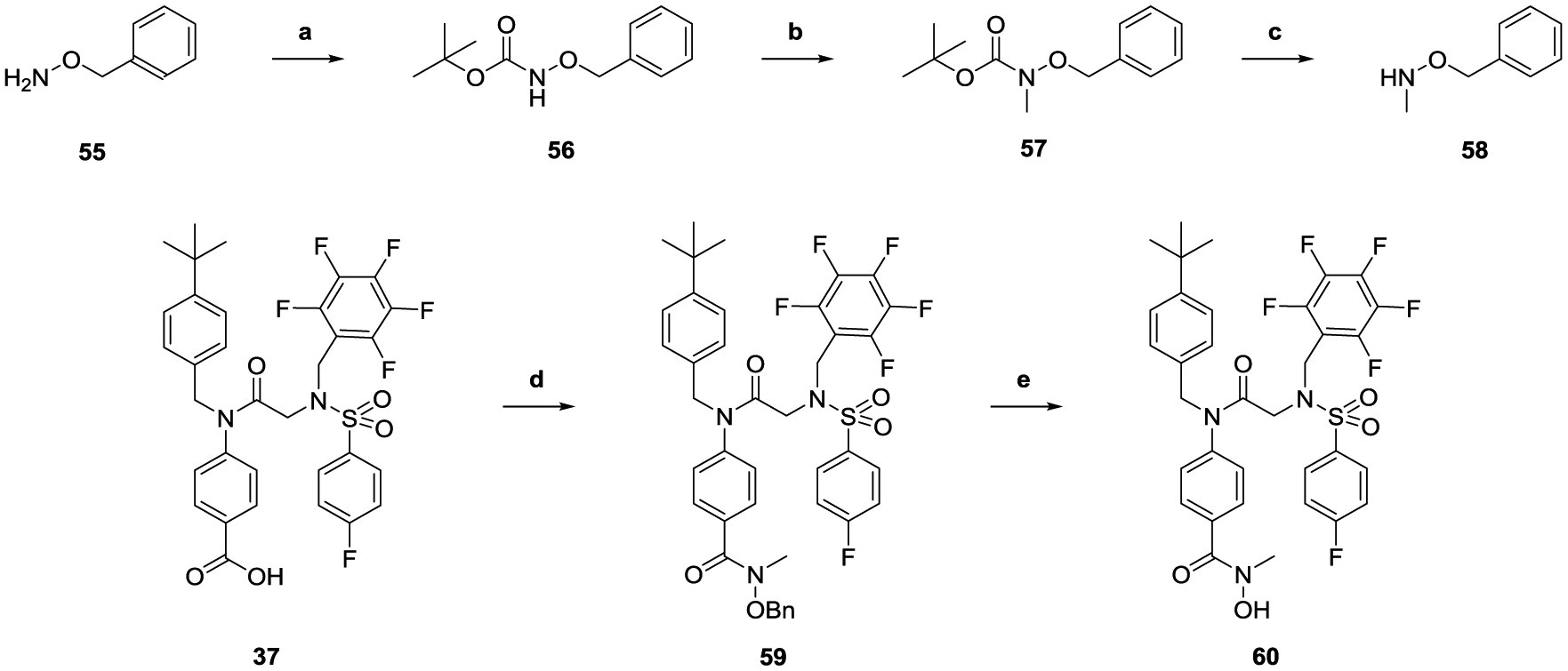
In a follow-up study, AES-135 was administered at 10 and 40 mg/kg once a day for 5 days. Blood was collected from each mouse 5 h following the final injection (Figure 2A). Encouragingly, the results showed that the blood concentration of AES-135 was dose dependent, achieving an average of 323 ng/mL (0.47 μM) with 10 mg/kg dosing and 1829 ng/mL (2.64 μM) with 40 mg/kg. No visible toxicity associated with either dose, based on percentage weight loss compared to vehicle control, was observed. This represents an approximate 5.7-fold increase in blood concentration from quadrupling the dose.
Figure 2.
(A) Serum concentrations of AES-135 in NSG mice following 10 and 40 mg/kg injections daily for 5 days, IP (n = 6, ±standard deviation (SD)). (B) Toxicity study with AES-135 in NSG mice administered over 5 days, IP (n = 6, ±SD).
To evaluate AES-135 toxicity in vivo, NSG mice were dosed by IP daily with a range of concentrations for 4–5 days (n = 6). Mice were weighed prior to, and following, administration of the compound and toxicity assessed via weight loss (Figure 2B). At 60 mg/kg, the mice showed no significant weight loss, indicating AES-135 to be nontoxic at the highest concentration.
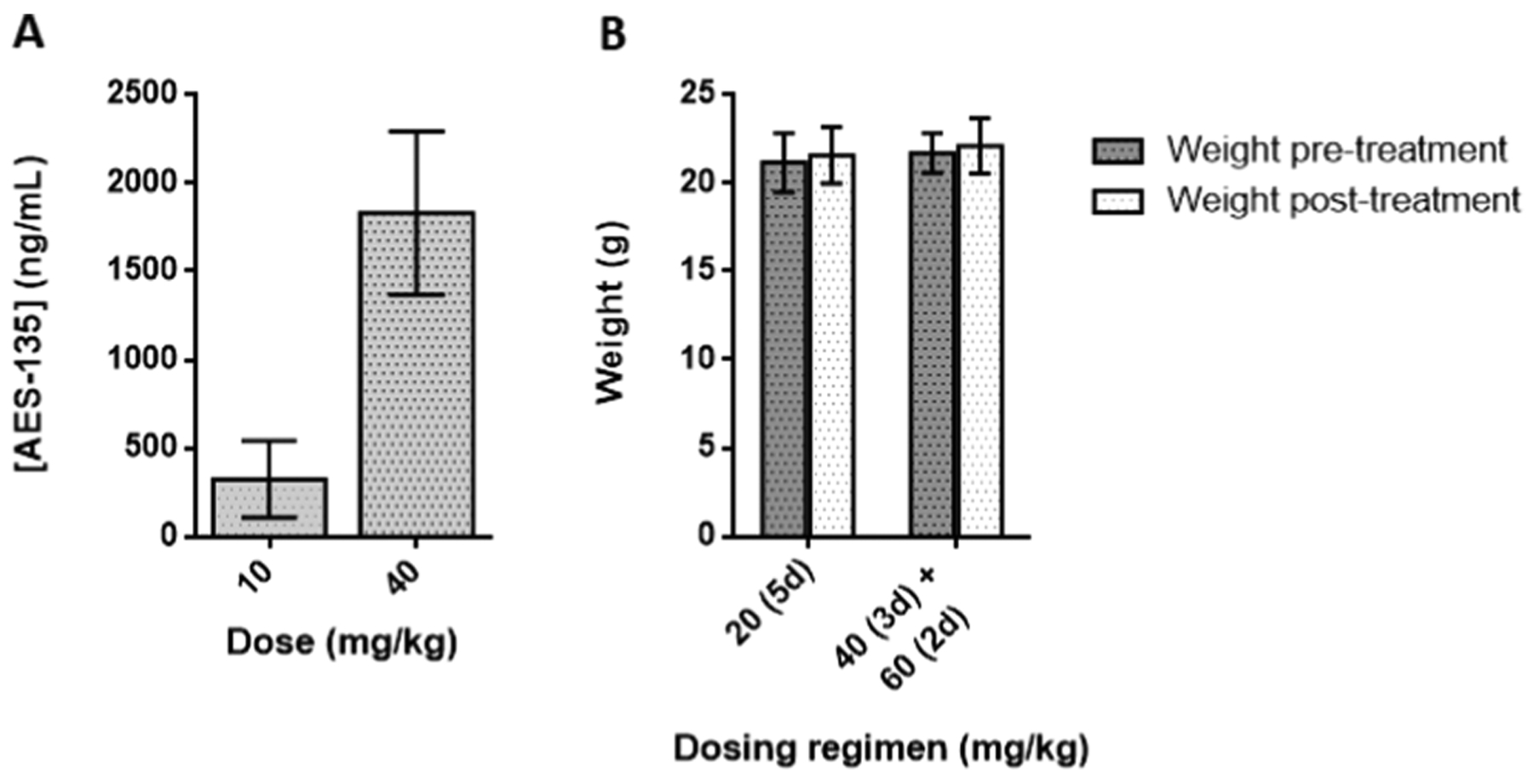
AES-135 was consistently cytotoxic in multiple low-passage patient-derived pancreatic cancer cell lines, namely, Pa03C, Pa02C, and Panc10.05 cells (the latter hereto referred to as 10.05). IC50 values were in the low μM range (1–4 μM) in monolayer proliferation-based assays of these tumor lines. The efficacy of AES-135 was also assessed in KPC tumor cells, which are derived from the “gold standard” genetically engineered mouse model of PDAC (KrasLSL.G12D/+; Trp53R172H/+; Elas-CreER).41 Interestingly, the tumor cells generated from the KPC42 genetically engineered PDAC mouse model were extremely sensitive to AES-135 and had an IC50 of 1.3 μM in the monolayer, compared to 8.5 μM for the pan-HDAC inhibitor Panobinostat, which was used as a positive control (Figure 3). KPC mice develop premalignant lesions called pancreatic intraepithelial neoplasia (PanIN), which progress to visible carcinomas with 100% penetrance and display a morphology similar to that observed in human PDAC. Metastases arise in 80% of KPC mice, primarily in the liver and lungs, the most common metastatic sites in humans. The KPC tumors possess intricate genomic rearrangements, a sign of genomic instability, making this one of the most aggressive PDAC models used in preclinical research. They are notoriously resistant to standard-of-care therapy; only 12% of tumors demonstrate sensitivity toward gemcitabine.43
Figure 3.
Dose-dependent reduction by (A) AES-135 and (B) Panobinostat in cell proliferation in pancreatic cancer cells in the monolayer. Average of at least three experiments ± standard error (SE).
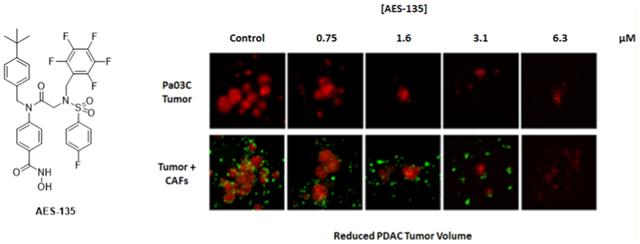
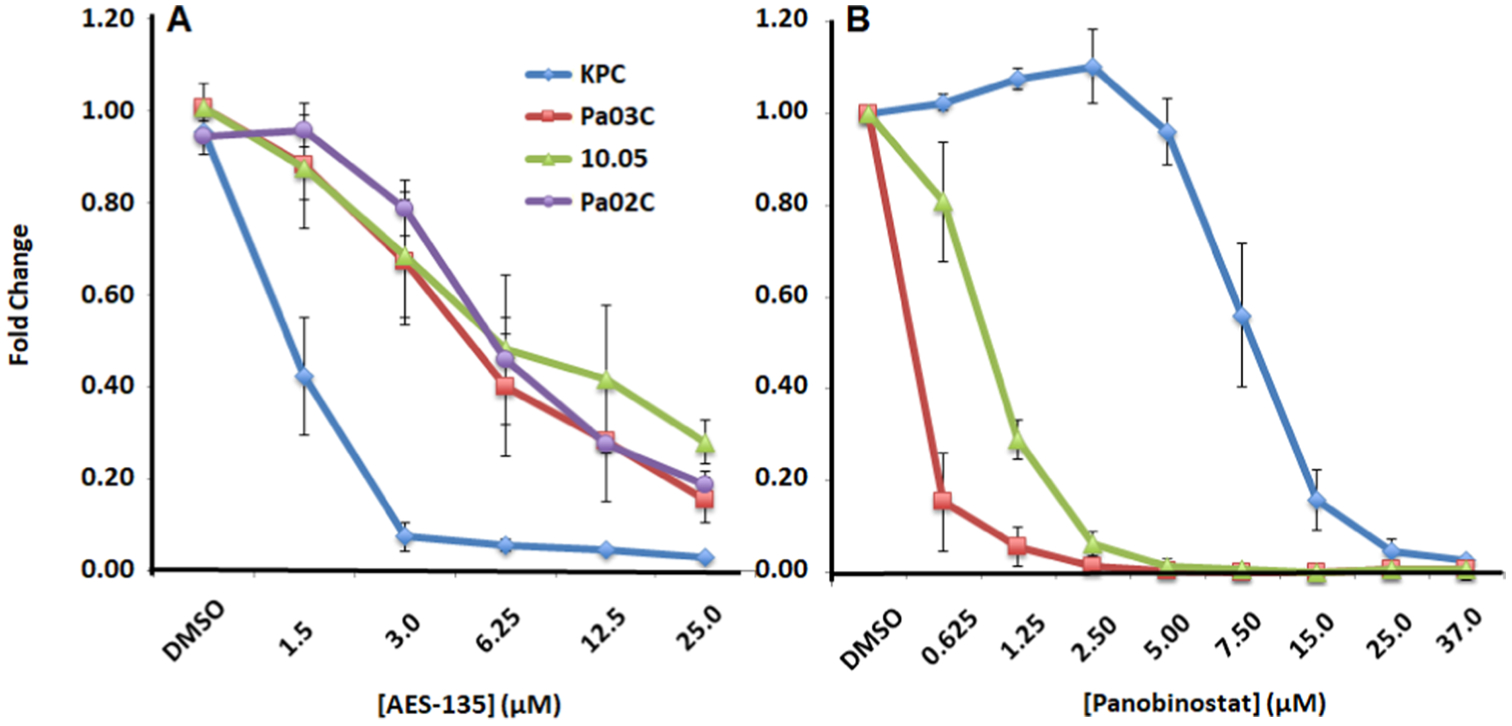
Next, we evaluated the efficacy of AES-135 in preclinical predictive three-dimensional (3D) tumor models of pancreatic cancer using patient-derived tumor cells as well as CAFs. Pancreatic cancers are difficult to treat effectively in part because of the CAFs that surround the tumor and impede access by chemotherapeutics. Additionally, CAFs facilitate tumor growth through the secretion of growth factors, for example, vascular endothelial growth factor, interleukin 6, and transforming growth factor β, promoting invasion and metastasis.47 AES-135 reduced pancreatic tumor spheroids, even with a protective CAF microenvironment, and showed 5-to 6-fold greater selectivity for the tumor cells over the CAFs (Figure 4). Single-digit μM to high nM potencies were demonstrated in the analogous 3D tumor models, in reducing both tumor area and intensity (Table 4).
Figure 4.
Dose-dependent reduction in tumor spheroid intensity in patient-derived pancreatic cancer cells Pa03C (red, n = 3, ±SE) and 10.05 (blue, n = 3, ±SE); fold change compares the treated tumor only spheroids to media control.
Table 4.
IC50 Values for AES-135 in Several Monolayer and 3D Human-Derived PDAC Cell Lines (n = 3–5)
| monolayer PDAC cells | ||
|---|---|---|
| cell line | IC50 (μM) | |
| Pa02C | 4.6 | |
| Pa03C | 3.4 | |
| 10.05 | 3.9 | |
| KPC | 1.3 | |
| 3D PDAC cells | ||
| cell line | scan type | IC50 (μM) |
| Pa03C | areaa | 1.22 |
| Pa03C + CAFs | area | 1.41 |
| CAF coculture | area | 7.80 |
| Pa03C | intensityb | 1.33 |
| Pa03C + CAFs | intensity | 1.56 |
| CAF coculture | intensity | 4.50 |
| 10.05 | area | 0.97 |
| 10.05 + CAFs | area | 0.94 |
| CAF coculture | area | 4.70 |
| 10.05 | intensity | 0.60 |
| 10.05 + CAFs | intensity | 0.50 |
| CAF coculture | intensity | 3.40 |
Area: the μ2 of objects that exceed a minimum tomato red intensity threshold in the well.
The sum of all intensity values for pixels marked as χ2 objects (i.e., total red OR green fluorescence in the well, after background removal).
Due to the high sensitivity of the KPC cells to AES-135 treatment, we tested the in vivo potency of AES-135 in a syngeneic orthotopic model. KPC2 cells were orthotopically implanted in the pancreas of C57Bl/6 mice, which were subsequently treated with either 50 mg/kg AES-135 or vehicle control. A murine mouse model was utilized in the in vivo studies due to the role of HDACs in the modulation of immune cell function.44,45 Furthermore, in lung and renal cell carcinoma mouse models, the HDAC inhibitor Entinostat (SNDX-275; MS-275) potentiated the effects of PD-1 inhibition, and this effect was partially mediated by functional inhibition of MDSCs.45 HDAC inhibitors would be effective in blockading tumor cell proliferation and cell-cycle progression and also have immunomodulatory effects.46,47 Mice treated with AES-135 showed significantly increased survival, with a median survival rate of 36.5 days compared to 29.5 days for the vehicle mice (Figure 5, p = 0.0146). The ability to provide a survival advantage in this aggressive PDAC model illustrates the potential of AES-135 as a hit-to-lead compound. This effect was only observed in immunocompetent mice; the equivalent immunodeficient mice showed no obvious survival advantage (Figure S21, Supporting Information).
Figure 5.
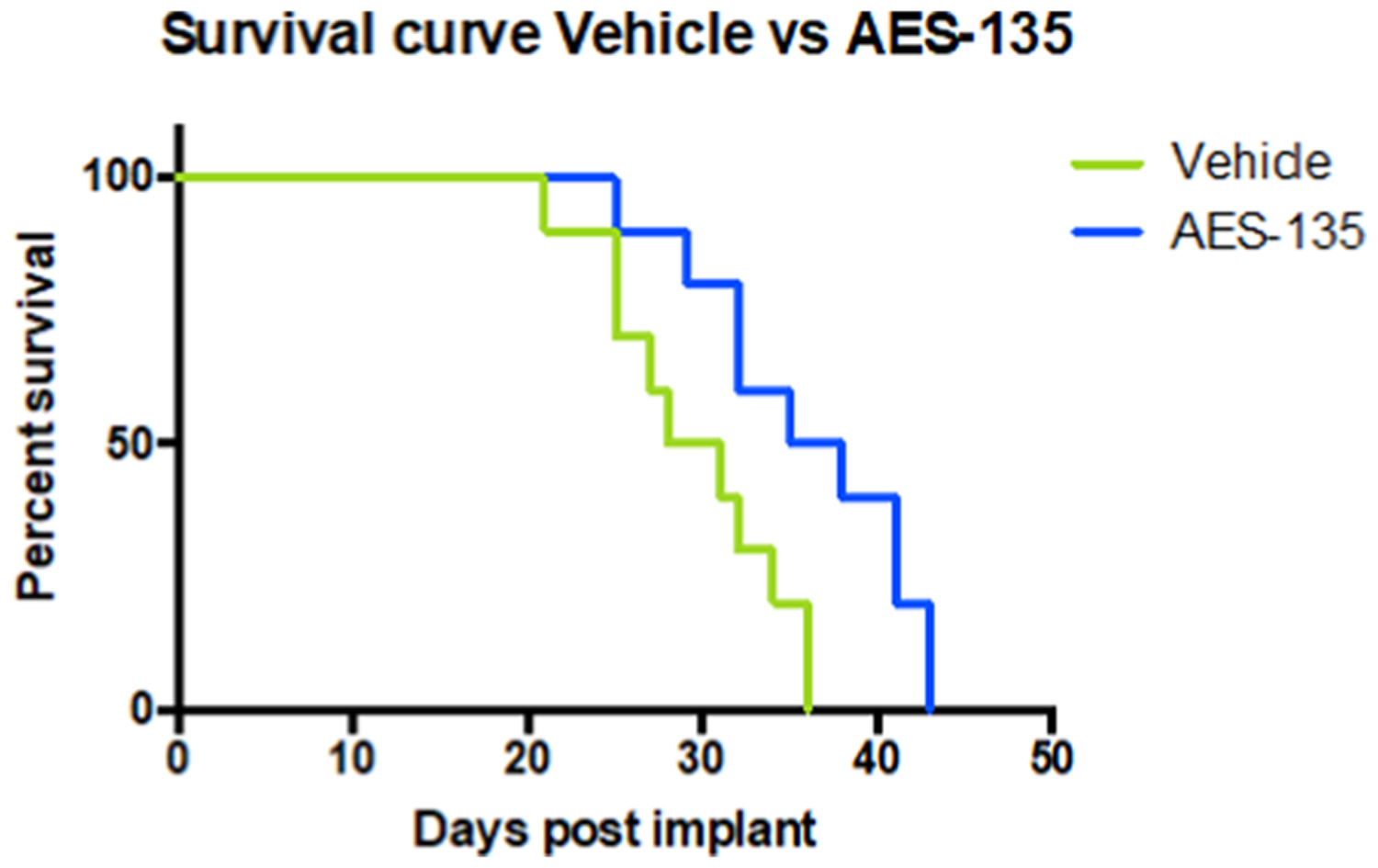
Increased survival of C57Bl/6 mice implanted with KPC tumor cells following AES-135 treatment. Mice treated with 50 mg/ kg AES-135, IP daily (blue, n = 10) exhibited a statistically significant survival advantage compared to mice treated with vehicle (green, n = 10), p = 0.0146 (log-rank test). Treatment started on day 7 with a cycle of 5 days on, 2 days off and continued until day 36.
CONCLUSIONS
Several reviews have described the potential of HDAC inhibitors to effectively treat PDAC,48–51 but to date, no compound has been published demonstrating suitable potency and druglike properties against this aggressive disease. We have presented a set of structurally novel hydroxamic acid-containing molecules displaying nM inhibition of HDACs in a target-based assay. The lead compound, AES-135, demonstrated potent inhibition of HDACs 3, 6, 8, and 11 and high cytotoxicity in a variety of cancer cell lines, most notably in pancreatic tumor lines. AES-135 was consistently more active in PDAC tumor models, both monolayer and 3D, than STAT inhibitor 1, even showing single-digit μM IC50 values in the highly aggressive KPC model, which was superior to the FDA-approved HDAC inhibitor Panobinostat. AES-135 showed an impressive PK profile in mice, with an in vivo half-life of 5.0 h, prolonged blood concentration above its IC50 value, and unremarkable toxicity, as assessed by a brief study. Subsequent in vivo evaluation in immunocompetent mice found that AES-135 extended the life of the mice significantly. To effectively treat pancreatic cancer, single agent therapy has not been effective; therefore, individualized combinations of targeted therapies will be necessary for making therapeutic advances in this devastating disease. Combination studies including AES-135 in animal models are crucial in order to determine whether the addition of HDAC inhibitors to standard-of-care agents or a new combination, such as immune checkpoint inhibitors, will dramatically extend survival. NMR and X-ray crystallographic studies are ongoing to determine the exact binding mechanism of AES-135 with HDACs to identify more potent and selective binding agents for preclinical evaluation.
EXPERIMENTAL SECTION
Materials and Methods.
A 400 MHz Bruker NMR was utilized to obtain 1H, 13C, and 19F NMR spectra in CDCl3 (99.8 atom % D), CD3CN (99.8 atom % D), or MeOH-d4 (99.8 atom % D), as indicated (1H at 400 MHz, 13C at 100 MHz, and 19F at 54 MHz). Chemical shifts (δ) are reported in parts per million (ppm), after calibration to the residual isotopic solvent, and coupling constants (J) are reported in hertz (Hz). Low-resolution mass spectrometry (LRMS) was carried out using Waters LC–MS in electrospray ionization (ESI) mode, fitted with Micromass ZQ MS and Alliance 2690 LC. High-resolution mass spectrometry (HRMS) was carried out using an Agilent 6538 UHD quadrupole-time-of-flight mass spectrometry in ESI mode with a mass accuracy of ±1 mDa. Thin-layer chromatography was conducted on Merck silica gel 60F254 on aluminum sheets. All sheets were dried after use and visualized using short-wave (254 nm) and long-wave (365 nm) UV light and/or staining with KMnO4. Column chromatography was carried out at room temperature using Biotage Isolera One and Isolera Prime purification systems, with industry-standard SNAP cartridges loaded with 40–60 μm silica gel (average pore size 60 Å). Semipreparative HPLC was conducted using a Waters 2487 Dual λ absorbance detector, equipped with a Symmetry C18 4.6 mm × 150 mm cartridge. Inhibitor purity was evaluated at room temperature by a Hewlett Packard Series 1100 analytical HPLC system fitted with a Phenomenex Luna 5.0 μm C18 4.6 mm × 150 mm cartridge using gradient mixtures of (A) Milli-Q water with 0.1% (v/v) TFA and (B) HPLC-grade acetonitrile. Biologically evaluated compounds are ≥95% chemically pure, as measured by HPLC. Chemicals and solvents were purchased from Sigma-Aldrich (MilliporeSigma), VWR International, Alfa Aesar, Combi-Blocks, Caledon Laboratory Chemicals, and Promega and were used as supplied.
General Procedure for the Synthesis of Compounds AES-135, 2–8, 36–43, and 60.
The benzyl or hydroxamate ester (1.0 equiv) was dissolved in THF/methanol (2:1) (0.1 M) and purged with nitrogen. Then, 10% Pd/C (0.04 equiv) was added after 15 min, and the flask was purged with hydrogen for 10 min. The reaction was allowed to stir at RT under hydrogen. After 16 h, the reaction was filtered through celite, washed with EtOAc, and concentrated in vacuo. Semipreparative HPLC, followed by lyophilization at −50 °C, isolated the target compound.
4-(N-(4-(tert-Butyl)benzyl)-2-((4-fluoro-N-((perfluorophenyl)-methyl)phenyl)sulfonamido)acetamido)-N-hydroxybenzamide (AES-135).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 42.8–44.0 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give AES-135 as a white solid (52.2 mg, 56%); 1H δ/ppm (400 MHz, CDCl3) 1.29 (s, 9H, t-Bu), 3.86 (s, 2H, CH2), 4.61 (s, 2H, CH2), 4.73 (s, 2H, CH2), 6.97 (d, J = 7.8 Hz, 2H, 2 CH), 7.08 (d, J = 7.3 Hz, 2H, 2 CH), 7.14 (t, J = 8.5 Hz, 2H, 2 CH), 7.27 (d, J = 7.9 Hz, 2H, 2 CH), 7.75 (d, J = 7.1 Hz, 2H, 2 CH), 7.83–7.87 (m, 2H, 2 CH), hydroxamic acid NH and OH protons were not observed; 13C δ/ppm (100 MHz, CDCl3) 31.1, 34.4, 39.6, 49.2, 53.0, 101.4, 115.9, 116.1, 124.4, 125.4, 128.3, 128.7, 130.3, 130.4, 132.8, 134.87, 134.91, 143.8, 144.2, 146.8, 150.9, 163.9, 164.8, 166.2, 166.5; 19F δ/ppm (54 MHz, CDCl3) −162.3 (td, J = 6.1 and 20.9 Hz, 2F), −153.9 (t, J = 20.5 Hz, 1F), −142.2 (dd, J = 7.0 and 22.5 Hz, 2F), −105.4 to −105.3 (m, 1F); LRMS (ESI+) m/z calcd for [C33H30F6N3O5S]+ : 694.67, found: 694.36; calcd for [C33H29F6N3O5SNa]+: 716.65, found: 716.34; HRMS (ESI+) m/z calcd for [C33H30 F6N3O5S]+: 694.1798, found: 694.1805; HPLC (I) tR = 23.55 min (97.9%); HPLC (II) tR = 38.44 min (99.9%).
4-(N-(4-(tert-Butyl)benzyl)-2-((3, 4-difluoro-N-(2-(trifluoromethyl)benzyl)phenyl)sulfonamido)acetamido)-N-hydroxybenzamide (2).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 42.1–43.6 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 2 as a white solid (83.9 mg, 68%); 1H δ/ppm (400 MHz, CDCl3) 1.29 (s, 9H, t-Bu), 3.68 (s, br, 2H, CH2), 4.70 (s, 2H, CH2), 4.74 (s, 2H, CH2), 6.90 (d, J = 7.3 Hz, 2H, 2 CH), 6.94 (d, J = 8.1 Hz, 2H, 2 CH), 7.26 (d, J = 8.2 Hz, 2H, 2 CH), 7.30 (d, J = 8.3 Hz, 1H, CH), 7.36 (t, J = 8.3 Hz, 1H, CH), 7.46 (t, J = 7.4 Hz, 1H, CH), 7.61–7.77 (m, 6H, 6 CH), hydroxamic acid NH and OH protons were not observed; 13C δ/ppm (100 MHz, CDCl3) 31.3, 34.5, 47.4, 48.0, 53.1, 117.7, 117.87, 117.90, 118.0, 122.7, 124.87, 124.91, 124.94, 125.0, 125.4, 125.5, 125.75, 125.81, 125.86, 125.92, 128.0, 128.4, 128.5, 128.65, 128.74, 130.1, 132.5, 132.9, 134.1, 136.5, 143.9, 148.8, 151.0, 151.9, 165.8, 166.0; 19F δ/ppm (54 MHz, CDCl3) −134.0 (dt, J = 8.5 and 20.7 Hz, 1F), −129.3 to −129.1 (m, 1F), −59.1 (s, 3F); LRMS (ESI+) m/z calcd for [C34H33F5N3O5S]+: 690.71, found: 690.45; calcd for [C34H32F5N3O5SNa]+: 712.69, found: 712.43; HRMS (ESI+) m/z calcd for [C34H33F5N3O5S]+: 690.2064, found: 690.2056; HPLC (I) tR = 24.65 min (99.9%); HPLC (II) tR = 40.23 min (99.9%).
4-(2-((3,4-Difluoro-N-(2-(trifluoromethyl)benzyl)phenyl)-sulfonamido)-N-(4-(trifluoromethyl)benzyl)acetamido)-N-hydroxybenzamide (3).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 37.9–39.4 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 3 as a white solid (82.2 mg, 71%); 1H δ/ppm (400 MHz, CDCl3) 3.70 (s, 2H, CH2), 4.71 (s, 2H, CH2), 4.79 (s, 2H, CH2), 6.91 (d, J = 7.6 Hz, 2H, 2 CH), 7.17 (d, J = 7.9 Hz, 2H, 2 CH), 7.29–7.33 (m, 1H, CH), 7.37 (t, J = 7.7 Hz, 1H, CH), 7.47 (t, J = 7.7 Hz, 1H, CH), 7.51 (d, J = 8.1 Hz, 2H, 2 CH), 7.61–7.76 (m, 6H, 6 CH), 9.19 (s, br, 1H, NH), hydroxamic acid OH proton was not observed; 13C δ/ppm (100 MHz, CDCl3) 47.4, 48.0, 52.9, 117.6, 117.8, 117.9, 118.1, 122.6, 122.7, 124.8, 124.87, 124.91, 125.0, 125.3, 125.4, 125.57, 125.59, 125.63, 125.7, 125.8, 125.9, 128.2, 128.3, 128.5, 128.6, 128.9, 129.0, 130.2, 130.4, 131.2, 132.6, 133.9, 136.5, 140.0, 143.5, 151.2, 165.2, 166.4; 19F δ/ppm (54 MHz, CDCl3) −133.9 (dt, J = 8.7 and 20.6 Hz, 1F), −129.0 to −128.9 (m, 1F), −62.6 (s, 3F), −59.0 (s, 3F); LRMS (ESI+) m/z calcd for [C31H23F8N3O5S]+: 702.60, found: 702.36; calcd for [C31H23F8N3O5SNa]+: 724.58, found: 724.34; HRMS (ESI+) m/z calcd for [C31H23F8N3O5S]+: 702.1308, found: 702.1303; HPLC (I) tR = 23.11 min (99.9%); HPLC (II) tR = 37.67 min (99.9%).
4-(N-(4-(tert-Butyl)benzyl)-2-((2, 4-difluoro-N-(2-(trifluoromethyl)benzyl)phenyl)sulfonamido)acetamido)-N-hydroxybenzamide (4).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 42.0–43.7 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 4 as a white solid (57.4 mg, 69%); 1H δ/ppm (400 MHz, CDCl3) 1.29 (s, 9H, t-Bu), 3.72 (s, 2H, CH2), 4.68 (s, 2H, CH2), 4.88 (s, 2H, CH2), 6.91–6.98 (m, 6H, 6 CH), 7.24 (d, J = 8.2 Hz, 2H, 2 CH), 7.34 (t, J = 7.6 Hz, 1H, CH), 7.46 (t, J = 7.5 Hz, 1H, CH), 7.60–7.66 (m, 4H, 4 CH), 7.85–7.91 (m, 1H, CH), 9.14 (s, br, 1H, NH), hydroxamic acid OH proton was not observed; 13C δ/ppm (100 MHz, CDCl3) 31.3, 34.5, 47.7, 48.0, 53.0, 105.5, 105.7, 106.0, 111.4, 111.6, 124.7, 125.4, 125.5, 125.7, 125.75, 125.81, 125.9, 127.9, 128.4, 128.5, 128.6, 128.7, 128.75, 128.77, 128.84, 130.1, 131.8, 131.9, 132.5, 133.1, 134.7, 144.0, 150.9, 164.1, 164.6, 165.4; 19F δ/ppm (54 MHz, CDCl3) −101.3 to −101.2 (m, 1F), −100.7 to −100.6 (m, 1F), −59.1 (s, 3F); LRMS (ESI+) m/z calcd for [C34H32F5N3O5SNa]+: 712.69, found: 712.56; HRMS (ESI+) m/z calcd for [C34H33F5N3O5S]+: 690.2063, found: 690.2056; HPLC (I) tR = 24.11 min (95.3%); HPLC (II) tR = 39.18 min (98.8%).
4-(2-((2,4-Difluoro-N-(2-(trifluoromethyl)benzyl)phenyl)-sulfonamido)-N-(4-(trifluoromethyl)benzyl)acetamido)-N-hydroxybenzamide (5).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 38.3–40.1 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 5 as a white solid (95.2 mg, 80%); 1H δ/ppm (400 MHz, CDCl3) 3.74 (s, 2H, CH2), 4.77 (s, 2H, CH2), 4.85 (s, 2H, CH2), 6.92–6.99 (m, 4H, 4 CH), 7.14 (d, J = 8.0 Hz, 2H, 2 CH), 7.35 (t, J = 7.6 Hz, 1H, CH), 7.46 (t, J = 7.6 Hz, 1H, CH), 7.49 (d, J = 8.2 Hz, 2H, 2 CH), 7.59–7.66 (m, 4H, 4 CH), 7.86–7.91 (m, 1H, CH), hydroxamic acid NH and OH protons were not observed; 13C δ/ppm (100 MHz, CDCl3) 47.6, 48.0, 52.9, 105.4, 105.7, 105.9, 111.45, 111.49, 111.68, 111.71, 122.8, 124.58, 124.62, 124.7, 124.8, 125.3, 125.5, 125.7, 125.78, 125.83, 125.9, 127.3, 128.0, 128.6, 128.7, 128.97, 129.00, 130.2, 131.8, 131.9, 132.0, 132.5, 134.4, 140.1, 143.6, 161.6, 164.5, 166.3, 167.1; 19F δ/ppm (54 MHz, CDCl3) −101.6 to −101.5 (m, 1F), −100.3 to −100.2 (m, 1F), −62.6 (s, 3F), −59.0 (s, 3F); LRMS (ESI+) m/z calcd for [C31H24F8N3O5S]+: 702.60, found: 702.48; calcd for [C31H24F8N3O5SNa]+: 724.58, found: 724.40; HRMS (ESI+) m/z calcd for [C31H24F8N3O5S]+: 702.1307, found: 702.1303; HPLC (I) tR = 22.62 min (99.9%); HPLC (II) tR = 36.62 min (97.2%).
4-(N-(4-(tert-Butyl)benzyl)-2-((4-fluoro-N-(2-(trifluoromethyl)-benzyl)phenyl)sulfonamido)acetamido)-N-hydroxybenzamide (6).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 41.0–42.5 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 6 as a white solid (90.4 mg, 68%); 1H δ/ppm (400 MHz, CDCl3) 1.29 (s, 9H, t-Bu), 3.69 (s, 2H, CH2), 4.69 (s, 2H, CH2), 4.75 (s, 2H, CH2), 6.91 (d, J = 8.0 Hz, 2H, 2 CH), 6.94 (d, J = 8.2 Hz, 2H, 2 CH), 7.18 (t, J = 8.5 Hz, 2H, 2 CH), 7.25 (d, J = 8.2 Hz, 2H, 2 CH), 7.34 (t, J = 7.6 Hz, 1H, CH), 7.45 (t, J = 7.6 Hz, 1H, CH), 7.62 (t, J = 7.8 Hz, 2H, 2 CH), 7.66 (d, J = 8.0 Hz, 2H, 2 CH), 7.90–7.94 (m, 2H, 2 CH), 9.14 (s, br, 1H, NH), hydroxamic acid OH proton was not observed; 13C δ/ppm (100 MHz, CDCl3) 31.3, 34.5, 47.5, 47.9, 53.0, 116.0, 116.2, 125.5, 125.7, 125.76, 125.81, 125.9, 127.9, 128.0, 128.3, 128.5, 128.6, 128.7, 130.0, 130.5, 130.6, 130.7, 132.5, 133.0, 134.45, 134.47, 135.7, 135.8, 150.9, 164.0, 166.13, 166.16, 166.5; 19F δ/ppm (54 MHz, CDCl3) −104.84 to −104.77 (m, 1F), −59.2 (s, 3F); LRMS (ESI+) m/z calcd for [C34H34F4N3O5S]+: 672.72, found: 672.50; calcd for [C34H34F4N3O5SNa]+: 694.70, found: 694.42; HRMS (ESI+) m/z calcd for [C34H34F4N3O5S]+: 672.2157, found: 672.2150; HPLC (I) tR = 23.66 min (99.9%); HPLC (II) tR = 38.58 min (99.9%).
4-(2-((4-Fluoro-N-(2-(trifluoromethyl)benzyl)phenyl)-sulfonamido)-N-(4-(trifluoromethyl)benzyl)acetamido)-N-hydroxybenzamide (7).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 37.1–38.6 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 7 as a white solid (87.9 mg, 77%); 1H δ/ppm (400 MHz, CDCl3) 3.70 (s, 2H, CH2), 4.73 (s, 2H, CH2), 4.79 (s, 2H, CH2), 6.91 (d, J = 7.6 Hz, 2H, 2 CH), 7.15 (d, J = 8.5 Hz, 2H, 2 CH), 7.19 (t, J = 8.5 Hz, 2H, 2 CH), 7.35 (t, J = 7.6 Hz, 1H, CH), 7.45 (t, J = 7.6 Hz, 1H, CH), 7.49 (d, J = 8.1 Hz, 2H, 2 CH), 7.61 (d, J = 8.5 Hz, 2H, 2 CH), 7.65 (d, J = 8.2 Hz, 2H, 2 CH), 7.90–7.94 (m, 2H, 2 CH), hydroxamic acid NH and OH protons were not observed; 13C δ/ppm (100 MHz, CDCl3) 47.4, 47.9, 52.9, 116.1, 116.3, 122.7, 125.3, 125.47, 125.53, 125.57, 125.64, 125.8, 125.85, 125.88, 128.0, 128.3, 128.5, 128.6, 128.9, 128.97, 129.00, 130.1, 130.4, 130.5, 132.5, 134.2, 135.66, 135.69, 140.1, 143.7, 164.0, 166.55, 166.64; 19F δ/ppm (54 MHz, CDCl3) −104.6 to −104.5 (m, 1F), −62.6 (s, 3F), −59.0 (s, 3F); LRMS (ESI+) m/z calcd for [C31H24F7N3O5SNa]+: 706.59, found: 706.33; HRMS (ESI+) m/z calcd for [C31H25F7N3O5S]+: 684.1399, found: 684.1398; HPLC (I) tR = 22.20 min (99.9%); HPLC (II) tR = 36.13 min (97.1%).
4-(2-((4-Fluoro-N-((perfluorophenyl)methyl)phenyl)-sulfonamido)-N-(4-(trifluoromethyl)benzyl)acetamido)-N-hydroxybenzamide (8).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 37.3–37.9 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 4 mL) and lyophilized overnight at −50 °C to give 8 as a white solid (14.0 mg, 46%); 1H δ/ppm (400 MHz, CDCl3) 3.85 (s, 2H, CH2), 4.60 (s, 2H, CH2), 4.84 (s, 2H, CH2), 7.10 (d, J = 7.8 Hz, 2H, 2 CH), 7.16 (t, J = 8.5 Hz, 2H, 2 CH), 7.21 (d, J = 7.8 Hz, 2H, 2 CH), 7.52 (d, J = 8.0 Hz, 2H, 2 CH), 7.77 (d, J = 7.7 Hz, 2H, 2 CH), 7.84 (dd, J = 5.1 and 8.6 Hz, 2H, 2 CH), hydroxamic acid NH and OH protons were not observed; 13C δ/ppm (100 MHz, CDCl3) 39.7, 49.3, 53.0, 109.7, 116.1, 116.3, 125.6, 125.65, 125.71, 125.74, 128.6, 128.7, 129.00, 129.04, 129.07, 129.09, 130.1, 130.4, 130.5, 131.1, 133.6, 135.0, 135.1, 140.0, 143.6, 164.1, 165.1, 166.7, 166.8; 19F δ/ppm (54 MHz, CDCl3) −161.1 (td, J = 7.4 and 21.5 Hz, 2F), −152.5 (t, J = 20.9 Hz, 1F), −141.3 (dd, J = 8.0 and 22.2 Hz, 2F), −104.1 to −104.0 (m, 1F), −62.6 (s, 3F); LRMS (ESI−) m/z calcd for [C30H19F9N3O5S]−: 704.54, found: 704.35; HRMS (ESI+) m/z calcd for [C30H21F9N3O5S]+: 706.1053, found: 706.1044; HPLC (I) tR = 21.96 min (99.9%); HPLC (II) tR = 35.74 min (99.9%).
4-(N-(4-(tert-Butyl)benzyl)-2-((4-fluoro-N-((perfluorophenyl)-methyl)phenyl)sulfonamido)acetamido)-N-hydroxy-N-methylbenzamide (60).
Semipreparative HPLC using acetonitrile/0.1% (v/v) TFA in Milli-Q water (0:1 → 1:0, 50 min → 10 min) eluted the target compound at 47.2–48.4 min. The product was suspended in acetonitrile/Milli-Q water (1:3, 2 mL) and lyophilized overnight at −50 °C to give 60 as a white solid (19.1 mg, 77%); 1H δ/ppm (400 MHz, CDCl3) 1.31 (s, 9H, t-Bu), 3.42 (s, 3H, CH3), 3.87 (s, 2H, CH2), 4.61 (s, 2H, CH2), 4.75 (s, 2H, CH2), 6.99 (d, J = 8.0 Hz, 2H, 2 CH), 7.09 (d, J = 7.8 Hz, 2H, 2 CH), 7.17 (t, J = 8.6 Hz, 2H, 2 CH), 7.29 (d, J = 8.0 Hz, 2H, 2 CH), 7.57 (d, J = 8.2 Hz, 2H, 2 CH), 7.85–7.89 (m, 2H, 2 CH), hydroxamic acid OH proton was not observed; 13C δ/ppm (100 MHz, CDCl3) 31.3, 34.6, 38.0, 39.7, 49.5, 53.2, 116.0, 116.2, 125.5, 127.9, 128.5, 128.56, 128.61, 129.9, 130.5, 130.6, 132.5, 133.0, 135.2, 151.0, 161.1, 164.1, 166.4, 167.2; 19F δ/ppm (54 MHz, CDCl3) −161.3 (td, J = 7.4 and 21.7 Hz, 2F), −151.9 (t, J = 21.0 Hz, 1F), −141.1 (dd, J = 7.7 and 22.4 Hz, 2F), −104.5 to −104.4 (m, 1F); LRMS (ESI+) m/z calcd for [C34H32F6N3O5S]+: 708.70, found: 708.25; calcd for [C34H31F6N3O5SNa]+: 730.68, found: 730.25; (ESI−) m/z calcd for [C34H30F6N3O5S]−: 706.68, found: 706.36; HRMS (ESI+) m/z calcd for [C34H32F6N3O5S]+: 708.1969, found: 708.1961; HPLC (I) tR = 24.49 min (99.9%); HPLC (II) tR = 40.09 min (99.9%).
General Procedure for the Synthesis of Compound 10.
The appropriate benzoic acid (1.0 equiv) and cesium carbonate (1.2 equiv) were suspended in DMF (0.7 M) and stirred at RT for 20 min in air, before addition of benzyl bromide (1.0 equiv) in one go, and the reaction was stirred at RT. After 24 h, the solvent was removed in vacuo at 80 °C, and the resulting solid was partitioned using EtOAc with saturated aqueous NaHCO3 and distilled water (1:1). The layers were separated, and the organic layer was washed with saturated aqueous NaHCO3 and distilled water (1:1) before drying (MgSO4), filtering, and concentrating in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compounds 11 and 12.
The appropriate aniline (1.0 equiv) and anhydrous MgSO4 (excess) were suspended in THF (0.5 M) in air at RT and charged with the appropriate benzaldehyde in one go. After 16 h, the mixture was filtered in vacuo, washed with EtOAc, and concentrated in vacuo, before suspending in TFE or methanol (0.2 M) and mixing with sodium borohydride (4.0 equiv) portionwise at RT in air. After 16 h, the reaction was concentrated to a low volume in vacuo and partitioned using EtOAc with saturated aqueous NaHCO3 and distilled water (1:1). The layers were separated, and the aqueous layer was extracted with EtOAc. The combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compounds 14–17.
The amine salt (1.0 equiv) and the sulfonyl chloride (1.1 equiv) were dissolved in dichloromethane (DCM) (0.2 M) under nitrogen and cooled to 0 °C, before adding anhydrous diisopropylethylamine (3.0 equiv) dropwise. The solution was stirred at 0 °C for 10 min before being allowed to reach RT. After 16 h, the reaction was quenched with 1 M HCl, and the layers were partitioned and separated. The aqueous layer was extracted with DCM, and the combined organic layers were dried (MgSO4), filtered, and concentrated in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compounds 18–22.
The sulfonamide (1.0 equiv) was charged with cesium carbonate (2.0 equiv) and dissolved in acetonitrile (0.2 M) in air before stirring at RT to 50 °C for 20 min. The benzyl or alkyl bromide (1.1–1.5 equiv) was added in one go, and the reaction was stirred at RT. After 16 h, the reaction was concentrated in vacuo and partitioned between EtOAc and distilled water. The layers were separated, and the organic layer was washed with distilled water. The combined aqueous layer was extracted with EtOAc, and the combined organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compounds 23–27.
The tert-butyl ester (1.0 equiv) was dissolved in DCM or chloroform in air at RT before mixing with trifluoroacetic acid (3:1, 2:1, or 1:1) (final concentration, 0.2 M). After 3 h, the reaction was concentrated in vacuo, azeotroping with DCM, to isolate the target compound without further purification.
General Procedure for the Synthesis of Compounds 28–35.
The carboxylic acid (1.2 equiv) and dichlorotriphenylphosphorane (2.4 equiv) were dissolved in chloroform (0.15–0.2 M) under nitrogen and stirred vigorously at RT for 15 min, prior to addition of the aniline (1.0 equiv), neat or as a solution in chloroform (0.3 M), and the vial was irradiated at 100 °C for 1 h (high absorbance). The solution was concentrated in vacuo, and column chromatography isolated the target compound.
General Procedure for the Synthesis of Compounds 44–51 and 59.
The benzoic acid (1.0 equiv) was dissolved in THF (0.05 M) under nitrogen and cooled to 0 °C before mixing with oxalyl chloride (5.0 equiv) and DMF (one to two drops). After 2 h, the reaction was concentrated in vacuo, repurged with nitrogen, and dissolved in THF (0.05 M). Diisopropylethylamine (4.0 equiv) and O-benzylhydroxylamine (2.0 equiv) were added, and the reaction was stirred at RT. After 16 h, the reaction was quenched with 1 M HCl and partitioned with EtOAc. The layers were separated, and the organic layer was washed with 1 M HCl. The combined aqueous layer was extracted with EtOAc, and the combined organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compound 56.
Di-tert-butyl dicarbonate (1.0 equiv) was added, as a solution in THF (7.0 M), to the amine (2.0 equiv) in THF (0.5 M) in air at RT. After 24 h, the reaction was partitioned between EtOAc and 0.5 M HCl. The layers were separated, and the organic layer was washed with 0.5 M HCl. The combined aqueous layer was extracted with EtOAc, and the organic layer was dried (MgSO4), filtered, and concentrated in vacuo to give the target compound without further purification.
General Procedure for the Synthesis of Compound 57.
Sodium hydride (60% in mineral oil) (3.0 equiv) was added, in one go, to the carbamate (1.0 equiv) in DMF (0.40 M) at RT, followed by iodomethane (1.5 equiv) after 15 min. After 24 h, the reaction was quenched with distilled water and extracted with diethyl ether. The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Column chromatography isolated the target compound.
General Procedure for the Synthesis of Compound 58.
The carbamate (1.0 equiv) was dissolved in chloroform in air at RT and mixed with trifluoroacetic acid (3:1) (final concentration, 0.2 M). After 22 h, the reaction was concentrated in vacuo and partitioned between EtOAc and 1 M NaOH. The layers were separated, and the aqueous layer was extracted with EtOAc. The organic layer was dried (MgSO4), filtered, and concentrated in vacuo to give the target compound without further purification.
Cytotoxicity Assays in MDA-MB-231 and MDA-MB-468 Breast Cancer Cells, MV4–11 and MOLM-13 Acute Myeloid Leukemia (AML) Cells, K562 Chronic Myeloid Leukemia (CML) Cells, and MRC-9 Human Lung Fibroblasts.
MDA-MB-231 and MDA-MB-468 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich). MV4–11, MOLM-13, K562, and MRC-9 cells were maintained in Iscove’s modified Dulbecco’s media and Roswell Park Memorial Institute (RPMI)-1640 media, separately, and supplemented with 10% FBS (Sigma-Aldrich). Next, 10 000 cells were plated per well in 96-well flat-bottom sterile culture plates with low-evaporation lids (Costar #3997). After 24 h, inhibitors and a vehicle control (0.5% DMSO) were added (final concentration 100 μM), and the cells were incubated for 72 h at 37 °C in 5% CO2. Inhibitors were examined in triplicate at a maximal concentration of 50.0 μM, followed by 50% dilutions in subsequent wells (25.0, 12.5, 6.25, 3.125, 1.5625, 0.78125, 0.390625, 0.195313, and 0.097656 μM). After 72 h, wells were treated with CellTiter-Blue (Promega #G808A) (20 μL/well), and the plates were incubated using standard cell culture conditions for 1–4 h. Plates were shaken for 10 s, and fluorescence was recorded at 560/590 nm using a Cytation 3 spectrophotometer. IC50 values were determined using nonlinear regression analysis with GraphPad Prism 6.0 (GraphPad Software Inc.).
Cytotoxicity Assays in AR230 and AR230R CML Cells.
Cells were maintained in RPMI-1640 culture media with l-glutamine (Gibco #11875) supplemented with 10% FBS. In addition, AR230R cells were cultured in the presence of 5.0 μM imatinib. Inhibitors were diluted 1000-fold in 100% DMSO into 96-well flat-bottom polystyrene tissue culture (TC)-treated culture plates (Falcon #353072). AR230 cells were plated at 5000 cells/well, and AR230R cells were plated at 15 000 cells/well in culture media (100 μL) and incubated with inhibitors at 37 °C and 5% CO2 for 48 h. Inhibitors were tested in duplicate at a maximum concentration of 31.6 μM, followed by half-logarithmic dilutions between wells (10.0, 3.16, 1.00, 0.316, 0.100, 0.032, 0.010, 0.0032, and 0.001 μM). A vehicle lane (0.1% DMSO) was also included. Following incubation, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium reagent (CellTiter96, Promega) (20 μL) was added to each well, followed by incubation at 37 °C and 5% CO2 for 3–4 h. Absorbance for each well was measured using an Epoch spectrophotometer (Biotek) at 490 nm, and IC50 values were determined using nonlinear regression analysis with GraphPad Prism 6.0 (GraphPad Software Inc.).
Cytotoxicity Assays in Glioblastoma Brain Tumor Stem Cells (GBM BTSCs).
Cells were cultured from tumor surgical specimens obtained following consent from adult GBM patients during operative procedures and approved by the University of Calgary Ethics Review Board. BTSC cultures were initiated in serum-free culture media (SFM), containing tissue culture water (150 mL), 10× DMEM (Gibco #12100–046) and F12 (Gibco #21700–075) (20 mL), Hormone Mix (20 mL), 30% glucose (Sigma-Aldrich #G7528) (4 mL), 7.5% NaHCO3 (Sigma-Aldrich #S5761) (3 mL), and 1 M HEPES (Sigma-Aldrich #H4034) (1 mL). Nonadherent spheres were formed after 7–21 days in culture. BTSC cultures were passaged until they stabilized (5–10 passages) before being cryopreserved in 10% DMSO (Sigma-Aldrich) in SFM until required. All cultures were used within 15 passages after thawing.
BTSC spheres were dissociated to single cells by incubating with Accumax (Innovative Technologies) (1 mL per T25 flask of cells, 5 min, 37 °C), seeded at 2500 cells/well in TC-grade low-adherence 96-well culture plates (Nalgene), and treated with either vehicle (DMSO) or inhibitor (stock concentration 10 mM in DMSO) 1 day after plating. Inhibitors were administered in logarithmic or half-logarithmic serial dilutions, with eight concentrations measured between 100 nM and 20 μM, and cell viability was assessed after 48 h using the alamarBlue assay according to the manufacturer’s instructions. Experiments were performed in triplicate with a minimum of three wells per condition. IC50 values were determined using nonlinear regression analysis with GraphPad Prism 6.0 (GraphPad Software Inc.).
Cytotoxicity Assays in D425 (Primary) and D458 (Recurrent) Medulloblastoma Cells.
Cells were cultured in DMEM high glucose (Life Technologies #11965–118) supplemented with 1% penicillin– streptomycin and 20% FBS. To evaluate the IC50 concentration of each inhibitor, 1000 cells were plated into each well of a tissue-culture-treated 96-well flat-bottom plate (Falcon) with 150 μL of DMEM high glucose with 1% FBS and 50 μL of serially diluted inhibitor. The inhibitor was plated at a concentration of 20 μM, following twofold dilutions, resulting in a final tested concentration of 39 nM. The cells were allowed to proliferate for 3 days at 37 °C in the presence of the inhibitor or DMSO before 20 μL of Presto Blue (Life Technologies), a fluorescent cell metabolism indicator, was added to each well approximately 4 h prior to the readout time point. Fluorescence was measured using a FLUOstar Omega Fluorescence 556 Microplate reader (BMG LABTECH) at excitation and emission wavelengths of 540–570 nm, respectively. Readings were analyzed by Omega software by plotting percent cell viability versus log dilutions of the inhibitors to determine the IC50 value.
Cytotoxicity Assays in Pancreatic Cancer Pa03C, Pa02C, 10.05, and KPC Cells.
Patient-derived tumor cells and CAF19 cells were a kind gift from Dr. Anirban Maitra (The Johns Hopkins University), and KPC cells (TB32908 male) were a kind gift from Drs. David Tuveson and Christopher Frese. All cell lines were authenticated via short tandem repeat analysis (IDEXX BioResearch) and checked routinely for mycoplasma contamination. The proliferative capacities of Pa03C, 10.05, Pa02C, and KPC cells in monolayer were assessed using alamarBlue. For alamarBlue assays, PDAC cells were plated at 2000 cells/well in 96-well plates and treated with AES-135 for 72 h. Assays were performed in at least triplicate.
Glutathione Stability Assay Using HPLC.
Assays were run using a Hewlett Packard Series 1100 analytical HPLC system fitted with an Agilent ZORBAX 3.5 μm Eclipse XDB-C18 4.6 mm × 75 mm column at room temperature. Eluent flow was set to 1.200 mL/min using gradient mixtures of (A) Milli-Q water with 0.1% (v/v) TFA and (B) HPLC-grade acetonitrile. Glutathione conjugation was measured by performing a linear elution gradient—A/B (1:0 → 0:1, 8.0 min → 2.0 min)—with UV detection set to 254 nm. Changes in the absorbance profile of the inhibitor were measured across time, with reductions in the HPLC peak area corresponding to a decay in the concentration of the parent compound.
Glutathione Stability Assay Using 19F NMR.
One-dimensional 19F NMR experiments were recorded at 37 °C on a 600 MHz spectrometer with an H(F)CN room-temperature probe (number of transients, 800) (scan width, 150 ppm). Compounds were prepared at a final concentration of 100 μM in a solution comprising 100 mM HEPES, pH 7.4, 100 μM 5-fluorotryptophan, 1 mM reduced l-glutathione (in blank samples, an equivalent volume of HEPES solution was added), 40% DMSO, and 10% D2O. All samples were incubated at 37 °C for 16 h, and the data was processed and analyzed using MestreNova 10.0.
Western Blotting in MDA-MB-231 Breast Cancer Cells and MV4–11 AML Cells.
All cells were lysed with radioimmunoprecipitation assay (RIPA) buffer: 20 mM Tris, pH 7.4, 150 mM NaCl, 0.5% deoxycholate, 1% Triton X-100, and 0.1% sodium dodecyl sulfate (SDS). Total protein was measured using a bicinchoninic acid assay (Sigma-Aldrich). In each assay, clarified protein was resolved on a 4–15% polyacrylamide–SDS gel and transferred to a poly(vinylidene difluoride) (PVDF) membrane (Bio-Rad). The membranes were blocked with a 5% solution of skimmed milk powder in TBST and incubated for ≥1 h followed by an overnight incubation at 4 °C in primary antibody 1:1000 dilution. Blots were probed with antibodies against pSTAT5, total STAT5, c-myc, Bcl2, and cleaved PARP. β actin (Santa Cruz Biotechnology, #SC-835) was used as a loading control. The PVDF membrane was washed with TBST (three times, 5 min each).
A horseradish peroxidase (HRP)-conjugated goat antimouse immunoglobulin G (IgG) secondary antibody (Cell Signaling, #7076S), a fluorescently labeled secondary antibody antimouse IgG (H+L), F(ab′)2 fragment (Alexa Fluor 488 Conjugate, #4408), or an antirabbit IgG (H+L), F(ab′)2 fragment (Alexa Fluor 647 Conjugate, #4414) was applied to the membrane at a 1:5000 dilution and incubated for 1 h at room temperature. The blots were then rinsed again three times in TBST for 10 min each. Bands were visualized using clarity western enhanced chemilumescent (ECL) substrate luminol/enhancer solution and peroxide solution (1:1) for HRP secondary antibody according to the manufacturer’s instructions (Bio-Rad) and analyzed using Image Lab software (Bio-Rad).
Western Blotting in Glioblastoma Brain Tumor Stem Cells (GBM BTSCs).
For protein analysis following drug treatment, BTSCs were dissociated to single cells and treated with drug or vehicle (DMSO) for 24 h. Cell pellets were lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Na deoxycholate, and 1% NP40) and cOmplete Protease Inhibitor Cocktail Tablets (Roche); 20 μg of protein lysate was separated by SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane according to standard protocols. Membranes were blocked in Tris-buffered saline with 5% nonfat dry milk and incubated overnight with a primary antibody at 4 °C followed by a 1 h incubation with the appropriate horseradish peroxidase-conjugated secondary antibody. Images were acquired on an Amersham imager 600 using Amersham ECL Select Western Blotting Detection Reagent. Primary antibodies: pSTAT3 (anti-phosphotyrosine 705 STAT3 antibody, Cell Signaling #9145S); STAT3 (1:1000, Santa Cruz Biotechnology, SC-8019); actin (1:2000; Santa Cruz Biotechnology, SC-1615). Secondary antibodies: donkey antimouse (1:5000, Millipore); donkey antirabbit (1:5000, Millipore); donkey antigoat (1:5000, Millipore).
Determination of Half-Life and Intrinsic Clearance in Mouse Hepatocytes.
Bioanalytical evaluation of in vitro half-lives and rates of intrinsic clearance in mouse hepatocytes was performed at Pharmaron using a liquid chromatography system (Shimadzu) and an API 5500 mass spectrometer (AB Inc., Canada) with an electrospray ionization (ESI) interface. The LC system was equipped with a Phenomenex Synergi 4 μm Hydro-PR 80A (2.0 × 30 mm2) column, through which 5 μL injections were made, eluting at 0.65 mL/min at 25 °C, with a mobile phase consisting of (A) Milli-Q water with 0.1% (v/v) formic acid and (B) acetonitrile with 0.1% (v/v) formic acid. Gradients were run over 2.0 min as follows: (A/B, 95:5, 0.0–0.3 min → 0:100, 0.3–0.8 min → 95:5, 1.2–1.5 min → 2.0 min). The MS was equipped with a turbo spray ion source, detecting samples with ionspray voltages of +5500 V (positive MRM) and −4500 V (negative MRM), and using the additional instrument parameters of temperature 500 °C, collision gas 6.0 L/min, curtain gas 30 L/min, nebulize gas 50 L/min, and auxiliary gas 50 L/min. Hepatocytes were sourced from male ICR/CD-1 mice (BioreclamationIVT #M00505, Lot no. XNN) and cryopreserved until used. Calculations were carried out using Excel (Microsoft), and peak areas were determined using the extracted ion chromatograms. The in vitro half-lives of each compound were calculated using regression analysis of the %parent disappearance versus time curve and the following equation: t1/2 = 0.693/k, where t1/2 is the half-life (min) and k is the rate constant (min−1). Conversion of the half-life to the in vitro intrinsic clearance (CLint, μL/min per 106 cells) was done using the following equation: CLint = kV/N, where V is the incubation volume (200 μL) and N is the number of hepatocytes per well (0.5 × 106).
Mouse Plasma Protein Binding Assay.
Determination of protein binding in mouse plasma was performed at Pharmaron using a liquid chromatography system (Shimadzu) and an API 4000 mass spectrometer (AB Inc., Canada) with an electrospray ionization (ESI) interface. The LC system was equipped with a Phenomenex Synergi 4.0 μm Hydro-RP 80A (2.0 × 30 mm2) new column, through which 10 μL injections were made, eluting at 0.65 mL/min at room temperature. The mobile phase consisted of (A) Milli-Q water with 0.1% (v/v) formic acid and (B) acetonitrile with 0.1% (v/v) formic acid. Gradients were run over 1.4 min and proceeded as follows: (A/B, 95:5 → 0:100, 0.0–0.8 min, 0:100, 0.8–1.1 min, 0:100 → 95:5, 1.1–1.2 min, 95:5, 1.2–1.4 min). The MS was equipped with a turbo spray ion source, detecting samples with an ionspray voltage of −4500 V (negative MRM), and using the additional instrument parameters of temperature 500 °C, collision gas 6.0 L/min, curtain gas 30 L/min, nebulize gas 50 L/min, and auxiliary gas 50 L/min. Plasma from male and female CD-1 mice (BioreclamationIVT) was stored at −80 °C until required. Ketoconazole was used as a control. Experiments were run in duplicate, and calculations were carried out using Microsoft Excel. Concentrations of the test compound in the buffer and plasma chambers were determined from peak area ratios.
Determination of Experimental Log D7.4.
Determination of experimental Log D7.4 was performed at Pharmaron using a liquid chromatography system (Shimadzu) and an API 4000 mass spectrometer (AB Inc., Canada) with an electrospray ionization (ESI) interface. The LC system was equipped with a Phenomenex Synergi 4.0 μm Hydro-RP 80A (2.0 × 30 mm2) new column coupled with a preguard, through which 10 μL injections were made, eluting at 0.65 mL/min at room temperature. The mobile phase consisted of (A) Milli-Q water with 0.1% (v/v) formic acid and (B) acetonitrile with 0.1% (v/v) formic acid. Gradients were run over 1.4 min and proceeded as follows: (A/B, 95:5 → 0:100, 0.0–0.8 min, 0:100, 0.8–1.1 min, 0:100 → 95:5, 1.1–1.2 min, 95:5, 1.2–1.4 min). The MS was equipped with a turbo spray ion source, detecting samples with an ionspray voltage of +5500 V (positive MRM), and using the additional instrument parameters of temperature 500 °C, collision gas 10 L/min, curtain gas 30 L/min, nebulize gas 55 L/min, and auxiliary gas 55 L/min. Progesterone was used as a control, and experiments were performed in duplicate.
Permeability Determination Using a Lipid-PAMPA.
Determination of compound cell permeability using a parallel artificial membrane permeability assay (PAMPA) was performed at Pharmaron using a liquid chromatography system (Shimadzu) and an API 4000 mass spectrometer (AB Inc., Canada) with an electrospray ionization (ESI) interface. The LC system was equipped with a Phenomenex Synergi 4 μm Hydro-PR 80A (2.0 × 30 mm2) column, through which 10 μL injections were made, eluting at 0.65 mL/min at 25 °C, with a mobile phase consisting of (A) Milli-Q water with 0.1% (v/v) formic acid and (B) acetonitrile with 0.1% (v/v) formic acid. Two gradients were run over 1.4 min (run 1) and 2.0 min (run 2). Run 1 proceeded as follows: (A/B, 95:5 → 0:100, 0.0–0.3 min, 0:100 → 95:5, 0.8–1.1 min, 95:5, 1.1–1.4 min). Run 2 proceeded as follows: (A/B, 95:5, 0.0–0.3 min, 95:5 → 0:100, 0.3–0.8 min, 0:100 → 95:5, 1.2–1.5 min, 95:5, 1.5–2.0 min). The MS was equipped with a turbo spray ion source, detecting samples with ionspray voltages of +5500 V (positive MRM) and −4500 V (negative MRM), and using the additional instrument parameters of temperature 500 °C, collision gas 6.0 L/ min, curtain gas 30 L/min, nebulize gas 50 L/min, and auxiliary gas 50 L/min. Experiments were conducted in triplicate, and methotrexate and testosterone were used as positive controls.
Permeability Determination Using a Caco-2 Assay.
Determination of compound cell permeability using a Caco-2 cell line was performed at Pharmaron using a liquid chromatography system (Shimadzu) and API 5500 and API 4000 mass spectrometers (AB Inc., Canada) with an electrospray ionization (ESI) interface. The LC systems were equipped with a Phenomenex Kinetex 1.7 μm C8 100A (2.1 × 30 mm2) column and a Phenomenex Kinetex 1.7 μm C18 100A (2.1 × 30 mm2) column, through which 10 and 3.0 μL injections were made, eluting at 0.65 mL/min at 40 and 25 °C. The mobile phase consisted of (A) Milli-Q water with 0.1% (v/v) formic acid and (B) acetonitrile with 0.1% (v/v) formic acid. Two gradients were run over 2.0 min (run 1) and 1.4 min (run 2). Run 1 (10 μL injection) proceeded as follows: (A/B, 95:5, 0.0–0.3 min, 95:5 → 0:100, 0.3–0.8 min, 0:100 → 95:5, 1.2–1.5 min, 95:5, 1.5–2.0 min). Run 2 (3.0 μL injection) proceeded as follows: (A/B, 95:5 → 0:100, 0.0–0.8 min, 0:100 → 95:5, 1.1–1.2 min, 95:5, 1.2–1.4 min). The MS was equipped with a turbo spray ion source, detecting samples with ionspray voltages of +5500 V (positive MRM) and −4500 V (negative MRM), and using the additional instrument parameters of temperature 500 °C, collision gas 6.0 L/min, curtain gas 30 L/min, nebulize gas 50 L/min, and auxiliary gas 50 L/min. Transepithelial electrical resistance (TEER) was measured across the monolayer using a Millicell Epithelial Volt-Ohm measuring system (Millipore), and the plate was returned to the incubator. TEER values were calculated using the following equation: TEER (Ω cm2) = TEER measurement (Ω) × membrane area (cm2). Studies were run in duplicate, and Digoxin, Prazosin, and Propranolol were used as control compounds. Internal standards consisted of 100 nM alprazolam with 200 nM labetalol (positive mode) and 2.0 μM ketoprofen with 200 nM labetalol (negative mode). Lucifer Yellow fluorescence to monitor monolayer integrity was measured in a fluorescence plate reader at 485 nm excitation and 530 nm emission.
Inhibition of Histone Deacetylases (HDACs).
Biochemical HDAC assays were performed at Nanosyn using microfluidic detection technology (electrophoretic mobility shift assay). Full-length recombinant human HDACs 3, 6, 8, and 11 were produced in an SF9 baculoviral system. Reactions were assembled in 384-well plates (total volume 20 μL), and the test compounds were serially prediluted in DMSO and added by an acoustic dispenser (Labcyte550) directly to the reaction buffer comprising 100 mM HEPES (pH 7.5), 25 mM KCl, 0.1% bovine serum albumin, 0.01% Triton X-100, and enzyme. Final concentrations of HDACs 3, 6, 8, and 11 were 0.5, 60, 5.0, and 10 nM, respectively. Concentration of DMSO was equalized at 1% in all samples. Reactions were initiated by addition of the fluorescently FAM-labeled acetylated peptide substrate to a final concentration of 1 μM with HDACs 3, 6, and 8 and 100 μM with HDAC11. Change in the relative fluorescence intensity of the substrate and product peaks is the parameter measured, reflecting the enzyme activity. Activity in each test sample was determined as the product sum ratio (PSR): P/ (S + P), where P is the peak height of the product and S is the peak height of the substrate. For each compound, enzyme activity was measured at 12 concentrations spaced by 3× dilution intervals, ranging from 30.0 to 0.0001694 μM. Reference compound JNJ-26481585 (Quisinostat) was tested in an identical manner. Negative control samples (0% inhibition in the absence of inhibitor, DMSO only) and positive control samples (100% inhibition in the absence of enzyme) were assembled in replicates of four and were used to calculate % inhibition values in the presence of compounds. Percent inhibition (Pinh) was determined using the following equation: Pinh = (PSR0% – PSRinh)/(PSR0% – PSR100%) × 100, where PSRinh is the product sum ratio in the presence of inhibitor, PSR0% is the product sum ratio in the absence of inhibitor, and PSR100% is the product sum ratio in 100% inhibition control samples. To determine IC50 values, the inhibition curves (Pinh vs inhibitor concentration) were fitted by a four-parameter sigmoid dose-response model using XLfit software (IDBS).
Molecular Modeling.
Receptor and ligand preparation protocols utilized molecular visualization software PyMOL v.1.7.4.5, advanced cross-platform molecular editing software Avagadro v.1.2.0, as well as graphical user interface software AutoDockTools (ADT) v.4.2.6. The docking simulations were performed by AutoDock Vina v.1.1.2. Analyses of the docking results were also visualized by PyMOL v.1.7.4.5. Three-dimensional crystal structures of the human HDAC isoforms were retrieved from the RCSB protein data bank (www.rcsb.org): hHDAC3 (PDB 4A69), hHDAC6 (PDB 5EDU), and hHDAC8 (PDB 1T64). ADT was used to remove water molecules, assign polar hydrogens, unite atom Kollman charges, and assign Gasteiger charges and solvation parameters. ADT does not naturally recognize charged inorganic heteroatoms; hence, the charges on zinc in all three enzymes were manually modeled to +2. These studies utilized three distinct ligands: AES-135, 6, and SAHA (Vorinostat). Energy minimization calculations utilizing molecular mechanics and the steepest descent algorithm were used to produce low-energy conformers of each ligand. The grid size was set to 40 × 40 × 40 xyz points, with a grid spacing of 0.497 Å. Binding poses with the most favorable free energy of binding values were visualized using PyMOL.
Pharmacokinetic (PK) Studies.
All animal studies were conducted under the guidelines of the National Institute of Health and were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine. Animals were maintained under pathogen-free conditions and a 12 h light–dark cycle. NOD SCID γ (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) or NSG mice were administered 20 mg/kg AES-135 IP in Cremophor EL/EtOH (1:1, 4% final volume)/sterile saline, and blood was collected via the tail vein at multiple timepoints between 0.5 and 24 h following administration. AES-135 was quantified in plasma using an internal standard (sorafenib), liquid–liquid extraction with ethyl acetate, and HPLC–MS/MS (Agilent HPLC, Applied Biosystems API 4000). The HPLC was run in isocratic mode using acetonitrile/5 mM ammonium acetate (20:80, v/v). The API 4000 was run in negative mode for AES-135 (Q1/Q3: 692/192) and positive mode for sorafenib (Q1/ Q3, 465/270). The lower limit of quantification was 1 ng/mL using 20 μL of blood or plasma.
Pharmacokinetic parameters for AES-135, including area under the curve (AUC), area under the moment curve (AUMC), and t1/2, were estimated using noncompartmental methods with Excel. The maximum plasma concentration (Cmax) and time of Cmax (tmax) were obtained from the data. The AUC from zero to infinity (AUC0-∞) was estimated from the AUC0-t (time zero to the last quantifiable concentration Clast) and the AUC from Clast to infinity, Clast/kel, where kel is the rate constant of elimination. The AUMC0–∞ was estimated in an analogous manner. The systemic clearance (Cl/F, where F = bioavailability) of AES-135 was calculated from the dose and AUC0–∞. The apparent volume of distribution (Vdss) was estimated by the following equation: (dosage/AUC0–∞) × (AUMC0–∞/AUC0–∞).
Tumor and Cancer-Associated Fibroblast (CAF) 3D Cocultures.
Patient-derived tumor cells and CAF19 cells were a kind gift from Dr. Anirban Maitra (The Johns Hopkins University), and KPC cells (TB32908 male) were a kind gift from Drs. David Tuveson and Christopher Frese. TdTomato-labeled PDAC cells and EGFP-labeled CAFs were resuspended in DMEM media containing 3% Reduced Growth Factor Matrigel (BD Biosciences) and 5% FBS at a cell ratio of 1:4 (tumor/CAF) and fed or treated with AES-135 on days 4 and 8 following plating. Both cell populations were quantitated for intensity and area via Thermo ArrayScan at day 12 of coculture.
Orthotopic Tumor Treatment.
All animal studies were conducted under the guidelines of the National Institute of Health and were approved by the Institutional Animal Care and Use Committee of Indiana University School of Medicine. Animals were maintained under pathogen-free conditions and a 12 h light–dark cycle. C57Bl/6 mice (Jackson Laboratory) were orthotopically implanted with 5 × 104 KPC2 cells. The mice were randomized into two groups of 10 mice each just before commencing treatment (7 days post implantation). The treatment regime consisted of 50 mg/kg AES-135 IP prepared in Cremophor EL/EtOH (1:1, 8% final volume) in sterile PBS. The vehicle mice received Cremophor EL/EtOH (1:1, 8% final volume) in sterile PBS. Both groups were treated 5 days a week for 1 month. Mice were euthanized when they exhibited signs of deterioration such as lack of grooming and appetite, loss of weight and activity, etc. Data was analyzed using Kaplan–Meier curves (GraphPad Prism 6), statistical significance was determined using the log-rank test, and p values <0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Leukemia and Lymphoma Society of Canada (LLSC) and a Mitacs Accelerate grant from the University of Toronto (A.E.S and P.T.G.), as well as support from the Indiana Clinical and Translational Sciences Institute funded, in part, by Award Number UL1TR001108 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award (M.L.F.), by the National Cancer Institute CA122298 (M.L.F.), CA167291, to M.L.F., and Jeff Gordon Children’s Foundation (M.L.F.). H.A.L., S.W., and P.T.G. shared grants from the Stem Cell Network and Alberta Innovates Health Solutions. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors also would like to thank Lubna Abu-Jazar from the University of Toronto for technical assistance, Nanosyn (Santa Clara, CA) for EMSA data, and Pharmaron (Beijing, China) for various PK analyses.
ABBREVIATIONS
- HAT
histone acetyltransferase
- STAT
signal transducer and activator of transcription
- BTSC
brain tumor stem cell
- PFB
pentafluorobenzyl/perfluorobenzyl
- GBM
glioblastoma multiforme
- MDB
medulloblastoma
- EMSA
electrophoretic mobility shift assay
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid)
- CAF
cancer-associated fibroblast
- KPC
KrasLSL.G12D/+, p53R172H/+, PdxCretg/+
- PDAC
pancreatic ductal adenocarcinoma
- PanIN
pancreatic intraepithelial neoplasia
- LRMS
low-resolution mass spectrometry
- HRMS
high-resolution mass spectrometry
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01957.
Chemicals and solvents, analytical techniques and chromatography methods; cytotoxicity results of top compounds in breast, AML, CML, MDB, and pancreatic cancer cell lines, GBM BTSCs, and noncancerous human lung fibroblasts; glutathione stability data by HPLC and 19F NMR; Western blots in breast cancer cells, AML cells, and GBM BTSCs; in vitro half-life and intrinsic clearance rates of top compounds in mouse hepatocytes; mouse plasma protein binding assay data; determination of experimental Log D7.4; PAMPA and Caco-2 permeability data; in vivo PK data and orthotopic tumor treatment data; HDAC inhibition assay data; in silico modeling/docking data; chemical synthesis procedures for all compounds with NMR, LRMS, HRMS, and HPLC data; 1H NMR spectra for all final compounds (PDF)
Molecular formula strings for final compounds (CSV)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Manji GA; Olive KP; Saenger YM; Oberstein P Current and Emerging Therapies in Metastatic Pancreatic Cancer. Clin. Cancer Res 2017, 23, 1670–1678. [DOI] [PubMed] [Google Scholar]
- (2).Rahib L; Smith BD; Aizenberg R; Rosenzweig AB; Fleshman JM; Matrisian LM Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- (3).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2016. CA—Cancer J. Clin 2016, 66, 7–30. [DOI] [PubMed] [Google Scholar]
- (4).Jones S; Zhang X; Parsons DW; Lin JC; Leary RJ; Angenendt P; Mankoo P; Carter H; Kamiyama H; Jimeno A; Hong SM; Fu B; Lin MT; Calhoun ES; Kamiyama M; Walter K; Nikolskaya T; Nikolsky Y; Hartigan J; Smith DR; Hidalgo M; Leach SD; Klein AP; Jaffee EM; Goggins M; Maitra A; Iacobuzio-Donahue C; Eshleman JR; Kern SE; Hruban RH; Karchin R; Papadopoulos N; Parmigiani G; Vogelstein B; Velculescu VE; Kinzler KW Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Biankin AV; Waddell N; Kassahn KS; Gingras M-C; Muthuswamy LB; Johns AL; Miller DK; Wilson PJ; Patch A-M; Wu J; Chang DK; Cowley MJ; Gardiner BB; Song S; Harliwong I; Idrisoglu S; Nourse C; Nourbakhsh E; Manning S; Wani S; Gongora M; Pajic M; Scarlett CJ; Gill AJ; Pinho AV; Rooman I; Anderson M; Holmes O; Leonard C; Taylor D; Wood S; Xu Q; Nones K; Fink JL; Christ A; Bruxner T; Cloonan N; Kolle G; Newell F; Pinese M; Mead RS; Humphris JL; Kaplan W; Jones MD; Colvin EK; Nagrial AM; Humphrey ES; Chou A; Chin VT; Chantrill LA; Mawson A; Samra JS; Kench JG; Lovell JA; Daly RJ; Merrett ND; Toon C; Epari K; Nguyen NQ; Barbour A; Zeps N; Australian Pancreatic Cancer Genome Initiative;Kakkar N; Zhao F; Wu YQ; Wang M; Muzny DM; Fisher WE; Brunicardi FC; Hodges SE; Reid JG; Drummond J; Chang K; Han Y; Lewis LR; Dinh H; Buhay CJ; Beck T; Timms L; Sam M; Begley K; Brown A; Pai D; Panchal A; Buchner N; De Borja R; Denroche RE; Yung CK; Serra S; Onetto N; Mukhopadhyay D; Tsao M-S; Shaw PA; Petersen GM; Gallinger S; Hruban RH; Maitra A; Iacobuzio-Donahue CA; Schulick RD; Wolfgang CL; Morgan RA; Lawlor RT; Capelli P; Corbo V; Scardoni M; Tortora G; Tempero MA; Mann KM; Jenkins NA; Perez-Mancera PA; Adams DJ; Largaespada DA; Wessels LF; Rust AG; Stein LD; Tuveson DA; Copeland NG; Musgrove EA; Scarpa A; Eshleman JR; Hudson TJ; Sutherland RL; Wheeler DA; Pearson JV; McPherson JD; Gibbs RA; Grimmond SM Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rucki AA; Foley K; Zhang P; Xiao Q; Kleponis J; Wu AA; Sharma R; Mo G; Liu A; Van Eyk J; Jaffee EM; Zheng L Heterogeneous Stromal Signaling within the Tumor Microenvironment Controls the Metastasis of Pancreatic Cancer. Cancer Res. 2017, 77, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Chang Q; Jurisica I; Do T; Hedley DW Hypoxia Predicts Aggressive Growth and Spontaneous Metastasis Formation from Orthotopically Grown Primary Xenografts of Human Pancreatic Cancer. Cancer Res. 2011, 71, 3110–3120. [DOI] [PubMed] [Google Scholar]
- (8).Ceccacci E; Minucci S Inhibition of Histone Deacetylases in Cancer Therapy: Lessons from Leukaemia. Br. J. Cancer 2016, 114, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Barneda-Zahonero B; Parra M Histone Deacetylases and Cancer. Mol. Oncol 2012, 6, 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lombardi PM; Cole KE; Dowling DP; Christianson DW Structure, Mechanism, and Inhibition of Histone Deacetylases and Related Metalloenzymes. Curr. Opin. Struct. Biol 2011, 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Verdin E; Ott M 50 Years of Protein Acetylation: from Gene Regulation to Epigenetics, Metabolism and Beyond. Nat. Rev. Mol. Cell Biol 2015, 16, 258–264. [DOI] [PubMed] [Google Scholar]
- (12).de Ruijter AJM; van Gennip AH; Caron HN; Kemp S; van Kuilenburg BP Histone Deacetylases (HDACs): Characterization of the Classical HDAC Family. Biochem. J 2003, 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Falkenberg KJ; Johnstone RW Histone Deacetylases and their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat. Rev. Drug Discovery 2014, 13, 673–691. [DOI] [PubMed] [Google Scholar]
- (14).Furdas SD; Kannan S; Sippl W; Jung M Small Molecule Inhibitors of Histone Acetyltransferases as Epigenetic Tools and Drug Candidates. Arch. Pharm 2012, 345, 7–21. [DOI] [PubMed] [Google Scholar]
- (15).Mariño-Ramírez L; Kann MG; Shoemaker BA; Landsman D Histone Structure and Nucleosome Stability. Expert Rev. Proteomics 2005, 2, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kumagai T; Wakimoto N; Yin D; Gery S; Kawamata N; Takai N; Komatsu N; Chumakov A; Imai Y; Koeffler HP Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid (Vorinostat, SAHA) Profoundly Inhibits the Growth of Human Pancreatic Cancer Cells. Int. J. Cancer 2007, 121, 656–665. [DOI] [PubMed] [Google Scholar]
- (17).Piekarz RL; Frye R; Turner M; Wright JJ; Allen SL; Kirschbaum MH; Zain J; Prince HM; Leonard JP; Geskin LJ; Reeder C; Joske D; Figg WD; Gardner ER; Steinberg SM; Jaffe ES; Stetler-Stevenson M; Lade S; Fojo AT; Bates SE Phase II Multi-Institutional Trial of the Histone Deacetylase Inhibitor Romidepsin As Monotherapy for Patients With Cutaneous T-Cell Lymphoma. J. Clin. Oncol 2009, 27, 5410–5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Qian X; Ara G; Mills E; LaRochelle WJ; Lichenstein HS; Jeffers M Activity of the Histone Deacetylase Inhibitor Belinostat (PXD101) in Preclinical Models of Prostate Cancer. Int. J. Cancer 2008, 122, 1400–1410. [DOI] [PubMed] [Google Scholar]
- (19).Ellis L; Pan Y; Smyth GK; George DJ; McCormack C; Williams-Truax R; Mita M; Beck J; Burris H; Ryan G; Atadja P; Butterfoss D; Dugan M; Culver K; Johnstone RW; Prince HM Histone Deacetylase Inhibitor Panobinostat Induces Clinical Responses with Associated Alterations in Gene Expression Profiles in Cutaneous T-Cell Lymphoma. Clin. Cancer Res 2008, 14, 4500–4510. [DOI] [PubMed] [Google Scholar]
- (20).NPI-0052 and Vorinostat in Patients with Non-small Cell Lung Cancer, Pancreatic Cancer, Melanoma or Lymphoma. ClinicalTrials.gov; Identifier NCT00667082, 2016. (accessed Aug 16, 2017). [Google Scholar]
- (21).Vorinostat Plus Radiation Therapy in Pancreatic Cancer. ClinicalTrials.gov; Identifier NCT00831493, 2012. (accessed Aug 16, 2017). [Google Scholar]
- (22).Vorinostat With XRT and 5-FU for Locally Advanced Adenocarcinoma of the Pancreas. ClinicalTrials.gov; Identifier NCT00948688, 2017. (accessed Aug 16, 2017). [Google Scholar]
- (23).Capecitabine, Vorinostat, and Radiation Therapy in Treating Patients With Nonmetastatic Pancreatic Cancer. ClinicalTrials.gov; Identifier NCT00983268, 2015. (accessed Aug 16, 2017). [Google Scholar]
- (24).Neoadjuvant Chemotherapy Followed by Radiation Therapy and Gemcitabine/Sorafenib/Vorinostat in Pancreatic Cancer. ClinicalTrials.gov; Identifier NCT02349867, 2017. (accessed Aug 16, 2017). [Google Scholar]
- (25).MS-275 in Treating Patients With Advanced Solid Tumors or Lymphoma. ClinicalTrials.gov; Identifier NCT00020579, 2012. (accessed Aug 16, 2017). [Google Scholar]
- (26).Panobinostat & Bortezomib in Pancreatic Cancer Progressing on Gemcitabine Therapy. ClinicalTrials.gov; Identifier NCT01056601, 2012. (accessed Aug 16, 2017). [Google Scholar]
- (27).Abdelfatah E; Kerner Z; Nanda N; Ahuja N Epigenetic Therapy in Gastrointestinal Cancer: the Right Combination. Ther. Adv. Gastroenterol 2016, 9, 560–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Novotny-Diermayr V; Hart S; Goh KC; Cheong A; Ong L-C; Hentze H; Pasha MK; Jayaraman R; Ethirajulu K; Wood JM The Oral HDAC Inhibitor Pracinostat (SB939) is Efficacious and Synergistic with the JAK2 Inhibitor Pacritinib (SB1518) in Preclinical Models of AML. Blood Cancer J. 2012, 2, No. e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Venugopal B; Baird R; Kristeleit RS; Plummer R; Cowan R; Stewart A; Fourneau N; Hellemans P; Elsayed Y; Mcclue S; Smit JW; Forslund A; Phelps C; Camm J; Evans TRJ; de Bono JS; Banerji U A Phase I Study of Quisinostat (JNJ-26481585), an Oral Hydroxamate Histone Deacetylase Inhibitor with Evidence of Target Modulation and Antitumor Activity, in Patients with Advanced Solid Tumors. Clin. Cancer Res 2013, 19, 4262–4272. [DOI] [PubMed] [Google Scholar]
- (30).Knipstein J; Gore L Entinostat for Treatment of Solid Tumors and Hematologic Malignancies. Expert Opin. Invest. Drugs 2011, 20, 1455–1467. [DOI] [PubMed] [Google Scholar]
- (31).Krämer OH; Mahboobi S; Sellmer A Drugging the HDAC6–HSP90 Interplay in Malignant Cells. Trends Pharmacol. Sci 2014, 35, 501–509. [DOI] [PubMed] [Google Scholar]
- (32).Kroesen M; Gielen PR; Brok IC; Armandari I; Hoogerbrugge PM; Adema GJ HDAC Inhibitors and Immunotherapy; a Double Edged Sword? Oncotarget 2014, 5, 6558–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Haftchenary S; Luchman HA; Jouk AO; Veloso AJ; Page BDG; Cheng XR; Dawson SS; Grinshtein N; Shahani VM; Kerman K; Kaplan DR; Griffin C; Aman AM; Al-awar R; Weiss S; Gunning PT Potent Targeting of the STAT3 Protein in Brain Cancer Stem Cells: A Promising Route for Treating Glioblastoma. ACS Med. Chem. Lett 2013, 4, 1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Li ZH; Haftchenary S; Croucher D; Gunning PT; Trudel S Abstract C246: SH-4–54, a Novel Small-Molecule Inhibitor of STAT3, Demonstrates Significant Anti-Tumor Activity Against Multiple Myeloma. Mol. Cancer. Ther 2013, 12, C246. [Google Scholar]
- (35).Ali AM; Gomez-Biagi RF; Rosa DA; Lai P-S; Heaton WL; Park JS; Eiring AM; Vellore NA; de Araujo ED; Ball DP; Shouksmith AE; Patel AB; Deininger MW; O’Hare T; Gunning PT Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities. ChemMedChem 2016, 11, 850–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cumaraswamy AA; Lewis AM; Geletu M; Todic A; Diaz DB; Cheng XR; Brown CE; Laister RC; Muench D; Kerman K; Grimes HL; Minden MD; Gunning PT Nanomolar-Potency Small Molecule Inhibitor of STAT5 Protein. ACS Med. Chem. Lett 2014, 5, 1202–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Page BDG; Croucher DC; Li ZH; Haftchenary S; Jimenez-Zepeda VH; Atkinson J; Spagnuolo PA; Wong YL; Colaguori R; Lewis AM; Schimmer AD; Trudel S; Gunning PT Inhibiting Aberrant Signal Transducer and Activator of Transcription Protein Activation with Tetrapodal, Small Molecule Src Homology 2 Domain Binders: Promising Agents Against Multiple Myeloma. J. Med. Chem 2013, 56, 7190–7200. [DOI] [PubMed] [Google Scholar]
- (38).Wang D-F; Helquist P; Wiech NL; Wiest O Toward Selective Histone Deacetylase Inhibitor Design: Homology Modeling, Docking Studies, and Molecular Dynamics Simulations of Human Class I Histone Deacetylases. J. Med. Chem 2005, 48, 6936–6947. [DOI] [PubMed] [Google Scholar]
- (39).Giannini G; Marzi M; Marzo MD; Battistuzzi G; Pezzi R; Brunetti T; Cabri W; Vesci L; Pisano C Exploring Bis-(Indolyl)Methane Moiety as an Alternative and Innovative CAP Group in the Design of Histone Deacetylase (HDAC) Inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 2840–2843. [DOI] [PubMed] [Google Scholar]
- (40).Salmi-Smail C; Fabre A; Dequiedt F; Restouin A; Castellano R; Garbit S; Roche P; Morelli X; Brunel JM; Collette Y Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J. Med. Chem 2010, 53, 3038–3047. [DOI] [PubMed] [Google Scholar]
- (41).Liu X; Pitarresi JR; Cuitino MC; Kladney RD; Woelke SA; Sizemore GM; Nayak SG; Egriboz O; Schweickert PG; Yu L; Trela S; Schilling DJ; Halloran SK; Li M; Dutta S; Fernandez SA; Rosol TJ; Lesinski GB; Shakya R; Ludwig T; Konieczny SF; Leone G; Wu J; Ostrowski MC Genetic Ablation of Smoothened in Pancreatic Fibroblasts Increases Acinar-Ductal Metaplasia. Genes Dev. 2016, 30, 1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hingorani SR; Wang L; Multani AS; Combs C; Deramaudt TB; Hruban RH; Rustgi AK; Chang S; Tuveson DA Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [DOI] [PubMed] [Google Scholar]
- (43).Olive KP; Jacobetz MA; Davidson CJ; Gopinathan A; McIntyre D; Honess D; Madhu B; Goldgraben MA; Caldwell ME; Allard D; Frese KK; DeNicola G; Feig C; Combs C; Winter SP; Ireland-Zecchini H; Reichelt S; Howat WJ; Chang A; Dhara M; Wang LF; Ruckert F; Grutzmann R; Pilarsky C; Izeradjene K; Hingorani SR; Huang P; Davies SE; Plunkett W; Egorin M; Hruban RH; Whitebread N; McGovern K; Adams J; Iacobuzio-Donahue C; Griffiths J; Tuveson DA Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Licciardi PV; Ververis K; Tang ML; El-Osta A; Karagiannis TC Immunomodulatory Effects of Histone Deacetylase Inhibitors. Curr. Mol. Med 2013, 13, 640–647. [DOI] [PubMed] [Google Scholar]
- (45).Orillion A; Hashimoto A; Damayanti N; Shen L; Adelaiye-Ogala R; Arisa S; Chintala S; Ordentlich P; Kao C; Elzey B; Gabrilovich D; Pili R Entinostat Neutralizes Myeloid Derived Suppressor Cells and Enhances the Anti-Tumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res 2017, 23, 5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Markowitz J; Brooks TR; Duggan MC; Paul BK; Pan X; Wei L; Abrams Z; Luedke E; Lesinski GB; Mundy-Bosse B; Bekaii-Saab T; Carson WE Patients with Pancreatic Adenocarcinoma Exhibit Elevated Levels of Myeloid-Derived Suppressor Cells upon Progression of Disease. Cancer Immunol. Immunother 2015, 64, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mei L; Du W; Ma WW Targeting Stromal Microenvironment in Pancreatic Ductal Adenocarcinoma: Controversies and Promises. J. Gastrointest. Oncol 2016, 7, 487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Schneider G; Kramer OH; Fritsche P; Schuler S; Schmid RM; Saur D Targeting Histone Deacetylases in Pancreatic Ductal Adenocarcinoma. J. Cell. Mol. Med 2010, 14, 1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Koutsounas I; Giaginis C; Patsouris E; Theocharis S Current Evidence for Histone Deacetylase Inhibitors in Pancreatic Cancer. World J. Gastroenterol 2013, 19, 813–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Feng W; Zhang B; Cai D; Zou X Therapeutic Potential of Histone Deacetylase Inhibitors in Pancreatic Cancer. Cancer Lett. 2014, 347, 183–190. [DOI] [PubMed] [Google Scholar]
- (51).Zhang H; Shang Y-P; Chen H-Y; Li J Histone Deacetylases Function as Novel Potential Therapeutic Targets for Cancer. Hepatol. Res 2017, 47, 149–159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.