

Abstract
Viral glycoproteins fold and oligomerize in the endoplasmic reticulum of the host cell. They employ the cellular machinery and receive assistance from cellular folding factors. During the folding process, they are retained in the compartment and their structural quality is checked by the quality control system of the endoplasmic reticulum. A special characteristic that distinguishes viral fusion proteins from most cellular proteins is the extensive conformational change they undergo during fusion of the viral and cellular membrane. Many viral proteins fold in conjunction with and dependent on a viral partner protein, sometimes even synthesized from the same mRNA. Relevant for folding is that viral glycoproteins from the same or related virus families may consist of overlapping sets of domain modules. The consequences of these features for viral protein folding are at the heart of this review.
Keywords: Endoplasmic reticulum, protein folding, viral glycoprotein
Enveloped viruses acquire their envelope, the protein‐packed membrane surrounding the genetic material of the virus, by budding through a membrane of the infected cell. Bunyaviruses, for instance, bud from the Golgi complex [1] and coronaviruses and orthopox viruses bud through membranes from the intermediate compartment between endoplasmic reticulum (ER) and Golgi [1, 2]. Most enveloped viruses, however, assemble their envelope on the plasma membrane. These include the alphaviruses, such as Semliki Forest Virus (SFV), the influenza viruses, and the lentiviruses, such as the Human Immunodeficiency Virus (HIV).
To position the envelope glycoproteins in the proper membrane, they are synthesized by polysomes bound to the ER. Co‐translational translocation places them into the ER lumen, where they fold and assemble into oligomers, before being transported to the budding compartment. In the ER, they fold as the endogenous proteins do: they undergo the same covalent modifications, they use the same folding machinery, and they are subjected to the same quality control process that determines whether a newly synthesized host cell protein is correctly folded and assembled before its release from the ER.
The functions of viral glycoproteins are diverse. For infectivity of enveloped viruses, the primary requirements of viral attachment, fusion and entry need to be fulfilled by at least one of the envelope proteins. For some viruses, all these functions are combined in a single glycoprotein [3, 4, 5]. Examples are the hemagglutinin (HA) of Influenza A Virus [4], Vesicular Stomatitis Virus (VSV) G protein [1] and the hemagglutinin‐esterase‐fusion (HEF) protein of influenza C [3]. Other functions of viral glycoproteins are to destroy the cellular receptor after binding and to mediate immune evasion. In particular, the poxviruses and herpes viruses are known for their numerous strategies to escape the host's immune system [6]. Because RNA viruses have relatively small genomes, they can only afford the essential functions, often merged into even fewer proteins consisting of domains with individual activities.
Folding of Glycoproteins in the ER
Newly synthesized proteins start to fold during synthesis. Secondary structure elements such as alpha helices and beta strands will start to form as soon as amino acids are strung into a chain. It is not known whether the ribosome channel and the Sec61 translocon allow any conformational changes, but the folding of growing nascent chains in the ER lumen has been well‐documented [7, 8]. N‐linked glycan chains are added to asparagine residues in consensus glycosylation sequences some 12–14 residues from the ER membrane [9], and disulfide bond formation starts early as well. During the folding process, native as well as non‐native disulfide bonds may form, a process that is catalyzed by disulfide isomerases in the ER. The unscrambling of wrong cysteine bridges into native ones is assisted by the same enzymes. Competition between disulfide bond formation and glycosylation [10] suggests that folding may start in the translocon. A viral example is the Newcastle disease virus HN protein, in which deletion of cysteines number 13 or 14 allows the use of one additional neighboring glycosylation site [11].
Folding and disulfide bond formation continue after termination of the nascent chain and release from the ribosome. The process may take from minutes to days to complete, and during this time the newly synthesized protein is retained in the ER. Purified proteins can fold into the correct conformation in isolation, since the three‐dimensional structure is solely determined by the primary amino acid sequence. Correct in vitro folding usually requires low protein concentrations and low temperatures. The crowdedness in the ER at physiological temperatures makes folding assistance invaluable. Folding enzymes such as disulfide isomerases and prolyl‐peptidyl cis‐trans isomerases catalyze covalent changes that may be rate‐limiting to folding. In addition, a large number of molecules act by preventing undesirable interactions between the newly synthesized immature proteins. They are suitably called molecular chaperones [12].
Two major classes of molecular chaperones have been identified up to now. The first class consists of chaperones that have affinity for hydrophobic stretches or patches of amino acids, which are especially prevalent in unfolded and misfolded proteins. These include members of the heat shock protein 70 class, in the ER predominantly BiP. The second class consists of two members so far: the lectin chaperones calnexin and calreticulin. These chaperones associate with monoglucosylated N‐linked glycans.
Chaperones and folding enzymes bind to immature and misfolded proteins and thereby prevent their exit from the ER [13]. The ER folding factors may thus constitute the quality control system of the organelle as well. Up to now, the lectin chaperones are the only ones for which this connection is firmly established [13]. Calnexin and calreticulin themselves serve as retention factors; they do not control the quality of the product they bind. Quality is checked by the enzyme UDP‐glucose:glycoprotein glucosyltransferase, which adds a single glucose to the mannose chain, provided some hydrophobic residues are exposed on the protein in addition to the innermost GlcNAc residue of the glycan [14]. Reglucosylation results in renewed binding to calnexin and calreticulin, which is preceded and followed by glucosidase II‐mediated deglucosylation. Alternating glucosidase II and glucosyltransferase activities establish a cycle of binding and release to the lectin chaperones, during which the substrate folds and is checked for structural quality [15]. All proteins with glycan chains that have been examined so far interact with at least one of the lectin chaperones. This suggests that in the choice of a newly synthesized protein for a particular chaperone, the affinity of the lectins for the glycan chain is dominant.
Are Viral Glycoproteins Different?
All viral glycoproteins studied so far associate with calnexin and/or calreticulin [e.g. [16]] and do not display any difference from endogenous proteins ( Fig. 1). However, the comparison is difficult to make because literature on the maturation of viral glycoproteins is much more abundant than reports on the folding of glycoproteins. One difference that emerges is the more extensive glycosylation most viral glycoproteins undergo. This could serve to protect the virus and cover potentially immunogenic epitopes. Glycosylation sites are easily added or deleted during viral evolution and the diversity of glycan modifications, which depends on the location of the site in the protein and on the cell type, adds complexity to these structures [17].
Figure 1.
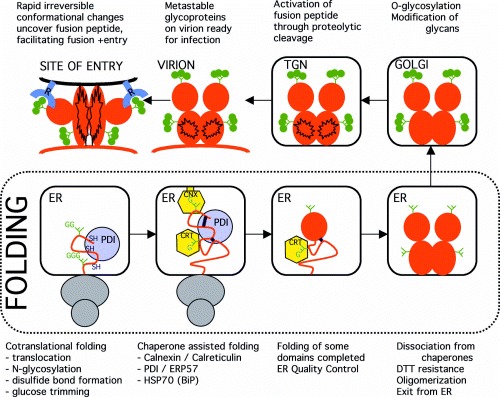
Schematic representation of the foolding and the post folding conformational changes of a typical viral fusion protein.
As soon as a folding protein has an N‐linked glycan, calnexin or calreticulin will bind. VSV G protein, which contains two glycans, binds in vivo to calnexin but not to calreticulin [18]. In an in vitro binding assay, however, calreticulin can associate with VSV G [15], suggesting that the membrane attachment of calnexin determine the preference of VSV G in the intact ER. Deletion of one of the two carbohydrate chains on VSV G protein still allows an interaction with calnexin, albeit inefficient [19]. The single glycan on SFV E1 protein is different: it allows an interaction with either calnexin or calreticulin [20]. Proteins that are more heavily glycosylated, such as influenza A virus HA [21] and HIV‐1 envelope glycoprotein [22], associate with both chaperones. The lectins can bind simultaneously to one folding HA molecule, showing specificity for particular glycans [21]. Of the two glycans in the hepatitis B virus M protein, only one was shown to bind calnexin [23]. Calnexin and calreticulin apparently can bind the same glycan, but whether they associate in vivo depends on the location of the glycan in the folding protein, probably in relation to the ER membrane.
The chaperone–ligand complex is large because of the additional presence of the protein disulfide isomerase‐like protein Erp57, which forms a heterodimer with calnexin and calreticulin [24]. Erp57 and PDI, the most prominent disulfide isomerase in the ER, form a transient covalent complex with the E1 and p62 glycoproteins of SFV [20], in addition to the noncovalent complex to calnexin and calreticulin. This implies that Erp57 and PDI assist native disulfide bond formation during folding of E1 and p62, seconded by the lectin chaperones. Aside from the lectins, newly synthesized viral glycoproteins may bind BiP and other folding factors. The sequence and timing with which they associate and the particular chaperone that is used first, depend on the substrate molecule that is folding. Whereas HA binds to both lectin chaperones, and only associates with BiP if this first‐choice binding is prevented [25], VSV G protein associates with calnexin but not calreticulin and requires transfer to BiP to complete folding [18]. Glucosidase inhibitors (e.g. casta‐nospermin) prevent binding to calnexin and calreticulin, but this rarely abolishes correct folding completely [20, 26, 27, 28], indicative of redundancy of folding factor activity in the ER.
Because of the redundancy of ER chaperone activities, and because viral proteins use the cellular protein folding machinery in the ER, antiviral strategies targeting the ER might fail. Surprisingly perhaps, glucosidase inhibitors do show an effect on virus replication. Hepatitis B virus [29] and bovine viral diarrhea virus, a pestivirus model for hepatitis C virus [30], both display altered glycosylation and dramatically reduced viral yields upon incubation of infected cells with glucosidase inhibitors. In the same cells, general protein maturation in the ER is not affected. Rudd and Dwek [17] suggest that the specificity for viral proteins is due to the icosahedral structure of the affected viruses. A small conformational change in one molecule may not be tolerated in the stringent lattice of the virus particle. In favor of this argument is the lack of effect of glucosidase inhibitors on the Newcastle disease virus, which does not have a strict symmetric particle structure [1]. The folding rate of its HN protein, which normally binds calnexin, is decreased by a factor two in the presence of glucosidase inhibitors, but the protein still passes the ER quality control, reaches the cell surface, and has biological activity [28].
Viral Glycoproteins are Different from Cellular Proteins
The suggestion of a strict structural requirement for various viral glycoproteins tells us that, indeed, viral proteins may be different from cellular proteins. The crystalline shell surrounding many viral nucleocapsids necessitates a wealth of regular interactions between the structural proteins in the envelope. It is insufficient for the individual viral proteins to display biological activity per se. In addition, the conformation needs to be such that not only oligomeric interactions between polypeptide chains arise, but also that larger complexes can be formed. Altogether, the folding of many viral glycoproteins may allow fewer deviations from the native path than that of cellular proteins. Mammalian proteins ‘only’ need to be biologically active, and they need to pass the quality control in the ER.
A second feature that distinguishes viral from cellular proteins is the complex evolutionary history of viral proteins. Viruses evolve much more rapidly than their hosts do, and recombination events are frequent. As a consequence, protein modules from different viruses may combine into one multifunctional protein. This is a survival strategy of many RNA viruses, because of their small genome. One or two glycoproteins on the viral envelope then need to perform all the essential functions of the virus. A beautiful example is the influenza C virus spike protein HEF [3]. It consists of three domains, each having its own biological activity. It combines a receptor‐binding domain R, an esterase domain E, and a fusion domain F. Domain R is placed in a loop of domain E and domain E is placed in a loop of domain F. Domains R and F have close structural similarity to the HA protein of influenza A virus. The only difference is the lack of E in influenza A; the receptor‐binding domain is inserted directly into a loop of the fusion domain ( Fig. 2). In each domain as well as in the complete proteins, the N‐ and C‐termini are adjacent. The domains basically form loops, sometimes stabilized by a disulfide bond between the N‐ and C‐terminus [31]. Examination of crystal structures of other viral glycoproteins, e.g. of retroviruses, raises the suggestion that this may be a more general phenomenon. The core of the soluble receptor‐binding subunit gp120 of HIV envelope has adjacent N‐ and C‐termini; the envelope protein of a mouse retrovirus was shown to tolerate large insertions and deletions in its hypervariable domain [32]; the same was seen in a variant of a pseudorabies virus glycoprotein [33].
Figure 2.

Schematic representation of the folding process for multidomain viral glycoproteins. Different domains are inserted into each other, most likely during virus evolution [3]. The interrupted domain must fold first, which brings the separate polypeptide chains of the next domain close enough to allow its folding. Etcetera.
The modular construction of viral glycoproteins implies that each domain must fold independently of the others. Two of the three domains in influenza C virus HEF are composed of interrupted portions of the polypeptide chain. To fold properly, these chains need to combine during the folding process. Influenza A HA starts to fold co‐translationally and the first domain to fold is the top domain [8, 34]. This is the receptor‐binding domain, similar to R in influenza C, and the only domain consisting of one continuous polypeptide chain. The stem domain, comparable with fusion domain F in influenza C, folds next, after the folding of the top domain is complete [8, 34]. The polypeptide chain of influenza A HA forms a loop, and folding starts at the tip of the loop. We predict that this is the same for the influenza C HEF protein ( Fig. 2), and probably also for the other viral proteins that consist of domain modules inserted into each other.
Loop formation needs to precede tertiary structure formation in the modular proteins. This could imply that the N‐terminal part of the protein needs to be kept unfolded during synthesis until its partner chain has been synthesized. Chaperone binding might be more important for this type of protein than for proteins with sequential domains formed from N‐ to C‐terminus. Disulfide bond formation in the loop structures may involve transient non‐native disulfide cross‐links that need to be unscrambled by PDI and its family members. Up to now, however, there is no evidence for a different requirement for folding factors or for a difference in the number of non‐native disulfide bridges during folding. This could be due to a shortage of available information, but it is more likely that there is no difference at all, because the folding process of virtually all proteins studied so far occurs mostly post‐translationally. When folding continues for minutes to hours after synthesis, it becomes irrelevant whether contacts in the native protein are local or distant in the polypeptide chain.
On the other hand, keeping the N‐terminus immobilized during synthesis may facilitate formation of the loop structure. The timing of cleavage of the signal sequence that directs proteins to the ER may play a role. Tampering with the cleavage site is known to affect proper folding of several proteins, for instance for the VSV G protein [35]. For HIV envelope, signal sequence cleavage was shown to occur late [36], i.e. long after synthesis was completed, instead of co‐translationally, as seen for all other proteins examined so far. Based on the structure of influenza A HA, Wilson et al. [5] proposed in 1981 that signal sequence removal would be late in HA. We still do not know when it occurs, except that it is a co‐translational process. Data on more proteins are needed to determine the exact role of the signal peptide and its removal during glycoprotein folding.
Special: Viral Fusion Proteins
A third characteristic of viral glycoproteins concerns the fusion proteins, which mediate fusion of the viral membrane with the membrane of the cell during infection. The best studied fusion protein is the influenza A virus HA [31, 37], which shows many similarities with other viral fusion proteins [38, 39, 40, 41]. A common theme is the immense irreversible conformational change fusion proteins undergo during the fusion process, sometimes even involving disulfide bond isomerization [53]. The fusion protein is synthesized as an inactive precursor protein. It folds in the ER into a stable conformation and oligomerizes; influenza virus and retrovirus spikes assemble into a homotrimer. At some point before or after incorporation of the protein into the virion, the fusion protein is activated by a proteolytic cleavage which results in a homotrimer of subunits consisting of a disulfide‐linked (influenza A HA) or noncovalently associated (HIV Env) dimer. The conformation of the cleaved protein is metastable: the protein has fusogenic activity. Consequently, receptor‐binding (such as for HIV) or a change in pH after entry into endosomes (such as for influenza virus), triggers a conformational change that brings the viral and cellular membranes close enough to fuse. This post‐fusion structure cannot be reversed anymore, and was shown to be more stable than the pre‐fusion structure [ Fig. 1, see [31]].
The membrane‐bound subunit of HA after fusion (pH change) is a rod‐shaped coiled coil, also found in other viral fusion proteins and in SNAREs, proteins involved in fusion of transport vesicles with target membranes in eukaryotic cells [41]. In several viruses, such as tick‐borne encephalitis virus and SFV, the conformational change of the spike glycoproteins is not limited to the oligomer, but induces a rearrangement between subunits, resulting in different oligomeric contacts than the ones formed during folding [42, 43]. The question is whether this immense conformational change means that these proteins fold in a different manner than proteins that remain stable after folding. The cleaved precursor protein is metastable, and the conformational change is irreversible, because the new conformation has a lower free energy. Irrespective, the protein that folds in the ER is the uncleaved precursor, essentially a different protein. Ingenious as the fusion machines that have evolved may be, it does not exclude their precursors from the ordinary treatment they receive in the ER ( Fig. 1).
Many Viral Glycoproteins may Depend on Each Other for Folding
General ER folding factors assist proteins that fold in the ER. In addition, some specific proteins are known that play a role in the folding of one particular protein or family of proteins [13]. To pass the ER quality control system, oligomers usually need to be assembled, although often one of the subunits can escape without association [13, 44]. Rare for cellular proteins, but quite common among viral glycoproteins, is the need for co‐translational assembly to reach a proper structure. Relatively well‐characterized examples are the envelope proteins from hepatitis C virus and the alphaviruses Sindbis and SFV. The alphaviruses present the genes encoding for the structural proteins on a 26S subgenomic mRNA [43]. This is translated as a precursor polyprotein in which the proteins are separated by co‐translational cleavage events. The cytosolic capsid protein folds, cleaves itself off, and the following set of proteins is translocated into the ER lumen and forms the envelope spike [see [20]]. Essentially, two structural proteins are synthesized next, in addition to one small 6K protein, which has not yet been very well characterized. The p62 and E1 protein of SFV are similar to the pE2 and E1 protein of Sindbis virus. E1 and p62 form a heterodimer. Cleavage of p62 into E2 and E3 in the TGN renders the spike protein fusogenic. Upon fusion, the interactions change completely and a trimer of E1 subunits is the result.
The glycoproteins in the precursor protein are synthesized in the order E3–E2–6K–E1. ER signal peptidases release the individual proteins from each other during synthesis [43]. SFV p62 and E1, when expressed from separate mRNAs, display different behavior: p62 folds normally but E1 misfolds and accumulates in aggregates [45]. This implies that E1 needs to associate co‐translationally with p62 for productive folding. For Sindbis E1 and pE2, the group of Brown [46, 47, 48] carried out extensive folding studies. The E1 protein proceeds through three differentially disulfide‐linked folding intermediates E1α, E1β and E1Γ before it reaches the native state E1ɛ. Two other species are detected, but these seem to be unrelated to the correct folding pathway. Folding of E1 beyond E1β depends on dimerization with pE2 [46]. E1 associates transiently with BiP, until pE2 takes over. The assembly of E1 with pE2 and its release from BiP coincide with the acquisition of a conformation that is compact enough to be resistant to reduction by DTT, indicative of an important maturation step. pE2, on the other hand, is only found in a complex with BiP when it is misfolded [48]. Neither calnexin nor grp94 have been found to co‐immunoprecipitate with the Sindbis glycoproteins. This may be a technical issue, because SFV p62 and E1 both associate with calnexin and calreticulin as well as the disulfide isomerases PDI and Erp57 [20]. Although p62 can fold quite well when expressed from a different mRNA than E1, its ‘homologue’ in Sindbis virus, pE2, does aggregate when a misfolded E1 is co‐expressed [47]. This suggests that in both SFV and Sindbis virus the two envelope proteins associate before folding of p62 or pE2 is complete.
Although the hepatitis C virus originates from a different family of viruses, the flaviviridae, its co‐cistronically expressed glycoproteins show similar dependence on each other as the alphavirus proteins. The hepatitis C virus E2 protein folds rapidly: its folding is virtually complete at the moment of precursor cleavage and is hence independent of E1 [49]. The E1 protein, with which it dimerizes, folds slower; it needs the association with E2 to fold correctly. Both proteins associate transiently with calnexin until some time after dimerization [50], but calreticulin and BiP are found only in complex with misfolded dimers [51].
The feline herpesvirus proteins gD and gI are co‐cistronic but gI is independent of gD for folding. Instead, folding of gI is dependent on its partner gE, although gE and gI are synthesized from different mRNAs. Proper folding and subsequent exit from the ER is impossible for either gI or gE without the other. This is seen as well for various other herpesvirus protein combinations [52]. Apparently, early co‐translational association is not needed for the mutual dependence phenotype.
A picture emerges that viral proteins, as with cellular proteins, have their own specific requirements during the folding process. Preference for particular chaperones cannot easily be predicted. In most cases, the absence of a detectable association is likely to have a technical rather than a physiological reason. What makes viral envelope proteins special is the intricate network of functions and associations of which they are part of: the activation of precursor proteins into metastable structures, the changing interactions during cell attachment and cell entry, the tight regulation of subunit synthesis, making proteins dependent on each other for folding. Moreover, in contrast to cellular proteins, a higher yield of folded viral protein may not always be favorable for the virus, because viral replication and virus production require a relatively healthy host cell for prolonged periods of time. It is clear that more studies on viral envelope glycoproteins are needed to determine whether folding in the ER can be a target for antiviral strategies.
References
- 1.Fields B, Knipe D, Howley P (Eds.) Fields Virology. Philadelphia, PA: Lippincott‐Raven Publishers, 1996.
- 2. Garoff H, Hewson R, Opstelten D. Virus maturation by budding. Microbiol Mol Biol Rev 1998;62: 1171 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenthal P, Zhang X, Formanowski F, Fitz W, Wong C‐H, Meier‐Ewert H, Skehel J, Wiley D. Structure of the haemagglutinin‐esterase‐fusion glycoprotein of influenza C virus. Nature 1998;396: 92 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrison S, Skehel J, Wiley D. Virus structure In: Fields B, Knipe D, Howley P. (Eds.), Fields Virology. Philadelphia, PA: Lippincott‐Raven Publishers, 1996: 59 99. [Google Scholar]
- 5. Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981;289: 366 373. [DOI] [PubMed] [Google Scholar]
- 6. Ploegh H. Viral strategies of immune evasion. Science 1998;280: 248 253. [DOI] [PubMed] [Google Scholar]
- 7. Bergman LW & Kuehl WM. Formation of an intrachain disulfide bond on nascent immunoglobulin light chains. J Biol Chem 1979;254: 8869 8876. [PubMed] [Google Scholar]
- 8. Chen W, Helenius J, Braakman I, Helenius A. Cotranslational folding and calnexin binding during glycoprotein synthesis. Proc Natl Acad Sci USA 1995;92: 6229 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilsson IM & Von Heijne G. Determination of the distance between the oligosaccharyltransferase active site and the endoplasmic reticulum membrane. J Biol Chem 1993;268: 5798 5801. [PubMed] [Google Scholar]
- 10. Allen S, Naim HY, Bulleid NJ. Intracellular folding of tissue‐type plasminogen activator. Effects of disulfide bond formation on N‐linked glycosylation and secretion. J Biol Chem 1995;270: 4797 4804. [DOI] [PubMed] [Google Scholar]
- 11. McGinnes L & Morrison T. Disulfide bond formation is a determinant of glycosylation site usage in the hemagglutinin‐neuraminidase glycoprotein of Newcastle disease virus. J Virol 1997;71: 3083 3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gething M‐J & Sambrook J. Protein folding in the cell. Nature 1992;355: 33 45. [DOI] [PubMed] [Google Scholar]
- 13. Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science 2000;286: 1882 1888. [DOI] [PubMed] [Google Scholar]
- 14. Sousa M & Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP‐Glc:glycoprotein glucosyltransferase. EMBO J 1995;14: 4196 4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peterson R & Helenius A. In vitro reconstitution of calreticulin‐substrate interactions. J Cell Sci 1999;112: 2775 2784. [DOI] [PubMed] [Google Scholar]
- 16. Yamashita Y, Shimokata K, Mizuno S, Daikoku T, Tsurumi T, Nishiyama Y. Calnexin acts as a molecular chaperone during the folding of glycoprotein B of human cytomegalovirus. J Virol 1996;70: 2237 2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rudd P & Dwek R. Glycosylation: heterogeneity and the 3D structure of proteins. Crit Rev Biochem Molec Biol 1997;32: 1 100. [DOI] [PubMed] [Google Scholar]
- 18. Hammond C & Helenius A. Folding of VSV G protein: sequential interaction with BiP and calnexin. Science 1994;266: 456 458. [DOI] [PubMed] [Google Scholar]
- 19. Cannon K, Hebert D, Helenius A. Glycan‐dependent and ‐independent association of vesicular stomatitis virus G protein with calnexin. J Biol Chem 1996;271: 14280 14284. [DOI] [PubMed] [Google Scholar]
- 20. Molinari M & Helenius A. Glycoproteins form mixed disulphides with oxidoreductases during folding in living cells. Nature 1999;402: 90 93. [DOI] [PubMed] [Google Scholar]
- 21. Hebert DN, Zhang J‐X, Chen W, Foellmer B, Helenius A. The number and location of glycans on influenza hemagglutinin determine folding and association with calnexin and calreticulin. J Cell Biol 1997;139: 613 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Otteken A & Moss B. Calreticulin interacts with newly synthesized human immunodeficiency virus type 1 envelope glycoprotein, suggesting a chaperone function similar to that of calnexin. J Biol Chem 1996;271: 97 103. [DOI] [PubMed] [Google Scholar]
- 23. Werr M & Prange R. Role for calnexin and N‐linked glycosylation in the assembly and secretion of hepatitis B virus middle envelope protein particles. J Virol 1998;72: 778 782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oliver J, Roderick H, Llewellyn D, High S. ERp57 functions as a subunit of specific complexes formed with the ER lectins calreticulin and calnexin. Mol Biol Cell 1999;10: 2573 2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang J‐X, Braakman I, Matlack KES, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C‐terminal truncations. Mol Biol Cell 1997;8: 1943 1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hammond C, Braakman I, Helenius A. Role of N‐linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc Natl Acad Sci USA 1994;91: 913 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gaudin Y. Folding of rabies virus glycoprotein: epitope acquisition and interaction with endoplasmic reticulum chaperones. J Virol 1997;71: 3742 3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McGinnes L & Morrison T. Role of carbohydrate processing and calnexin binding in the folding and activity of the HN protein of Newcastle disease virus. Virus Res 1998;53: 175 185.DOI: 10.1016/s0168-1702(97)00144-5 [DOI] [PubMed] [Google Scholar]
- 29. Mehta A, Zitzmann N, Rudd P, Block T, Dwek R. Alpha‐glucosidase inhibitors as potential broad based anti‐viral agents. FEBS Lett 1998;430: 17 22.DOI: 10.1016/s0014-5793(98)00525-0 [DOI] [PubMed] [Google Scholar]
- 30. Zitzmann N, Mehta A, Carrouee S, Butters T, Platt F, McCauley J, Blumberg B, Dwek R, Block T. Imino sugars inhibit the formation and secretion of bovine viral diarrhea virus, a pestivirus model of hepatitis C virus: implications for the development of broad spectrum anti‐hepatitis virus agents. Proc Natl Acad Sci USA 1999;96: 11878 11882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen J, Lee K, Steinhauer D, Stevens D, Skehel J, Wiley D. Structure of the haemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998;95: 409 417. [DOI] [PubMed] [Google Scholar]
- 32. Kayman S, Park H, Saxon M, Pinter A. The hypervariable domain of the murine leukemia virus surface protein tolerates large insertions and deletions, enabling development of a retroviral particle display system. J Virol 1999;73: 1802 1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Klupp B & Mettenleiter T. Glycoprotein gL‐independent infectivity of pseudorabies virus is mediated by a gD‐gH fusion protein. J Virol 1999;73: 3014 3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braakman I, Helenius J, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J 1992;11: 1717 1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shaw AS, Rottier PJM, Rose JK. Evidence for the loop model of signal‐sequence insertion into the endoplasmic reticulum. Proc Natl Acad Sci USA 1988;85: 7592 7596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Luo L, Thomas DY, Kang CY. Control of expression, glycosylation, and secretion of HIV‐1 gp120 by homologous and heterologous signal sequences. Virol 1994;204: 266 278. [DOI] [PubMed] [Google Scholar]
- 37. Bullough P, Hughson F, Skehel J, Wiley D. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994;371: 37 43. [DOI] [PubMed] [Google Scholar]
- 38. Weissenhorn W, Wharton S, Calder L, Earl P, Moss B, Aliprandis E, Skehel J, Wiley D. The ectodomain of HIV‐1 env subunit gp41 forms a soluble, alpha‐helical, rod‐like oligomer in the absence of gp120 and the N‐terminal fusion peptide. EMBO J 1996;15: 1507 1514. [PMC free article] [PubMed] [Google Scholar]
- 39. Lu M, Ji H, Shen S. Subdomain folding and biological activity of the core structure from human immunodeficiency virus type 1 gp41: implications for viral membrane fusion. J Virol 1999;73: 4433 4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Malashkevich V, Schneider B, McNally M, Milhollen M, Pang J, Kim P. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9‐Å resolution. Proc Natl Acad Sci USA 1999;96: 2662 2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Skehel J & Wiley D. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell 1998;95: 871 874. [DOI] [PubMed] [Google Scholar]
- 42. Allison S, Schalich J, Stiasny K, Mandl C, Kunz C, Heinz F. Oligomeric rearrangement of tick‐borne encephalitis virus envelope proteins induced by an acid pH. J Virol 1995;69: 695 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strauss J & Strauss E. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 1994;58: 491 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hurtley SM & Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 1989;5: 277 307. [DOI] [PubMed] [Google Scholar]
- 45. Andersson H, Barth B, Ekstrom M, Garoff H. Oligomerization‐dependent folding of the membrane fusion protein of Semliki Forest virus. J Virol 1997;71: 9654 9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carleton M & Brown D. Disulfide bridge‐mediated folding of Sindbis virus glycoproteins. J Virol 1996;70: 5541 5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carleton M & Brown D. The formation of intramolecular disulfide bridges is required for induction of the Sindbis virus mutant ts23 phenotype. J Virol 1997;71: 7696 7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mulvey M & Brown D. Involvement of the molecular chaperone BiP in maturation of Sindbis virus envelope glycoproteins. J Virol 1995;69: 1621 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Michalak J, Wychowski C, Choukhi A, Meunier J, Ung S, Rice C, Dubuisson J. Characterization of truncated forms of hepatitis C virus glycoproteins. J Gen Virol 1997;78: 2299 2306. [DOI] [PubMed] [Google Scholar]
- 50. Dubuisson J & Rice C. Hepatitis C virus glycoprotein folding: disulfide bond formation and association with calnexin. J Virol 1996;70: 778 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choukhi A, Ung S, Wychowski C, Dubuisson J. Involvement of endoplasmic reticulum chaperones in the folding of hepatitis C virus glycoproteins. J Virol 1998;72: 3851 3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mijnes J, Van Der Horst L, Van Anken E, Horzinek M, Rottier P, De Groot R. Biosynthesis of glycoproteins E and I of feline herpesvirus: gE‐gI interaction is required for intracellular transport. J Virol 1996;70: 5466 5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stegman T. Membrane fusion mechanisms: The influence of influenza hemagglutinin paradigm and its implications for intracellular fusion. Traffic 2000 (in press);1. [DOI] [PubMed] [Google Scholar]