

Backpacks enable a new strategy to control cellular phenotypes in vivo by attaching to macrophages and guiding their phenotypes.
Abstract
Adoptive cell transfers have emerged as a disruptive approach to treat disease in a manner that is more specific than using small-molecule drugs; however, unlike traditional drugs, cells are living entities that can alter their function in response to environmental cues. In the present study, we report an engineered particle referred to as a “backpack” that can robustly adhere to macrophage surfaces and regulate cellular phenotypes in vivo. Backpacks evade phagocytosis for several days and release cytokines to continuously guide the polarization of macrophages toward antitumor phenotypes. We demonstrate that these antitumor phenotypes are durable, even in the strongly immunosuppressive environment of a murine breast cancer model. Conserved phenotypes led to reduced metastatic burdens and slowed tumor growths compared with those of mice treated with an equal dose of macrophages with free cytokine. Overall, these studies highlight a new pathway to control and maintain phenotypes of adoptive cellular immunotherapies.
INTRODUCTION
Adoptive cell therapy has revolutionized clinical approaches to treat cancer. The most prominent example to date is chimeric antigen receptor (CAR) T cell therapy, which consists of engineered T cells that express CARs. CAR T cell therapies are on the cusp of a clinical revolution (1), leading to a full recovery in over 90% of patients with some blood-borne cancers (2). However, the success of CAR T cell therapy generally depends on (i) a prior knowledge and presence of tumor-specific antigens, (ii) tumors that are not solid (i.e., liquid cancers), and (iii) several weeks to prepare and expand cell populations (3). In contrast, macrophages are able to kill tumor cells where tumor-specific antigens are spare or unknown in a more immediate fashion, giving them the potential to succeed where T cell therapies have fallen short (4). However, a major hurdle that has slowed the adoption of macrophages in cancer immunotherapy is their tendency to shift to protumoral phenotypes once injected into the body.
Macrophages are perhaps the most plastic cell type in the hematopoietic system. This plasticity allows them to assume many roles like defending against foreign pathogens, aiding in wound healing, and regulating tissue homeostasis (5). Furthermore, the phenotypic plasticity of macrophages makes them excellent candidates for addressing a range of diseases (4). Macrophages rely on soluble cues from the tissue microenvironment to guide their polarization into the appropriate phenotype. Polarization is best described as a multidimensional spectrum (6), which ostensibly can be simplified into classically activated (M1) and alternatively activated (M2) phenotypes. M1 macrophages produce nitric oxide (NO), reactive oxygen species (ROS), tumor necrosis factor–α (TNFα), interleukin-12 (IL-12), and other cytokines that generate an inflammatory response (7). M2 macrophages, on the other hand, are associated with a broad range of phenotypes typically associated with wound healing and tissue regeneration. However, when tissues become dysfunctional, macrophages can develop phenotypes that promote disease pathogenesis (8). In the case of cancer, tumor-associated macrophages (TAMs) typically adopt M2 (tumor-promoting) phenotypes due to the immunosuppresive microenvironment of solid tumors (9), which is associated with tumor growth, angiogenesis, chemotherapy resistance, and metastasis (10). To address these challenges, several clinical trials emerged in the 1990s to adoptively transfer macrophages polarized ex vivo with proinflammatory cytkines (4). These strategies ultimately failed, as macrophages eventually reverted to M2 phenotypes once embedded in the tumor microenvironment (Fig. 1A). Thus, for macrophage-based therapies to induce robust therapeutic effects in the clinic, strategies must be developed to control phenotypes of adoptively transferred macrophages in vivo.
Fig. 1. Schematic illustration of cellular backpacks for maintaining proinflammatory phenotypes of adoptive MΦ therapies.
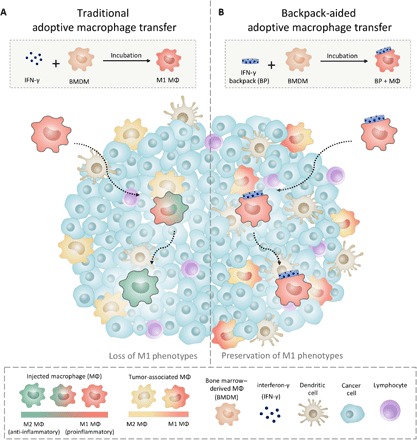
(A) MΦs polarized with IFN-γ ex vivo quickly shift from proinflammatory to anti-inflammatory phenotypes after penetrating a solid tumor. (B) MΦs carrying IFN-γ–loaded backpacks maintain their proinflammatory phenotypes deep within the tumor microenvironment, altering the phenotypes of endogenous TAMs.
We report a class of soft discoidal particles called “backpacks” capable of regulating the phenotype of macrophages in vivo (Fig. 1B). This work builds on the discovery that target geometry plays a deterministic role in the phagocytic fate of particles and that anisotropically shaped particles can evade phagocytosis for prolonged durations (11). Macrophages phagocytose particles through actin-mediated membrane motion. We have previously shown that particle shape determines coordination of polymerized actin network and that anisotropic shapes frustrate the formation of actin structures necessary to complete phagocytosis. Phagocytosis-resistant backpacks have formed the basis of several innovative demonstrations of drug delivery in the last several years (12–16). Previously reported particle-based cell therapies are mostly designed to transport payloads to target sites, whereby the payloads do not interact with the carrier cell (17). An interesting exception was demonstrated by Irvine and co-workers, which used IL-15 super-agonist to boost the activity of injected CAR T cells (18). Here, we report a class of backpacks that robustly bind to macrophages, which are cells of the innate immune system, and can provide an antigen-agnostic benefit. We demonstrate that backpack-loaded macrophages can maintain their phenotypes deep within the immunosuppressive neoplasm of solid tumors and potentiate a robust antitumor response.
RESULTS
Backpack fabrication, characterization, and monocyte interactions
Backpacks were prepared from biodegradable polymers using microcontact printing (see fig. S1) (16, 19). Each backpack contained a cell-adhesive layer, a poly(lactic-co-glycolic) acid (PLGA) layer, a polyvinyl alcohol (PVA) layer, and a second PLGA layer (Fig. 2A, i and ii). PVA was chosen as the interior layer due to its hydrophilicity, enabling facile incorporation of cytokine. We chose interferon-γ (IFN-γ) due to its potency in stimulating proinflammatory macrophages and its robust antitumor activity (20). PLGA was chosen to provide structural support to the PVA layer. The cell-adhesive layer was made by layer-by-layer (LBL) assembly. It comprised two sets of alternating layers of hyaluronic acid modified with aldehyde (HA-Ald) and poly(allylamine) hydrochloride (PAH). After 1.5 hours of incubation, we found that backpacks with a cell-adhesive layer bound to 86.9% of bone marrow–derived macrophages (BMDMs) from BALB/c mice, whereas backpacks without a cell-adhesive layer bound to only 63.4% of BMDMs (see fig. S2) (21). Backpacks displayed an average stiffness of 292 ± 67 MPa, an average thickness of 1.49 ± 0.14 μm, and an average width of 7.56 ± 0.37 μm, as determined by atomic force microscopy (AFM) (Fig. 2A, iii; see fig. S3).
Fig. 2. Backpack preparation, characterization, and monocyte interactions.
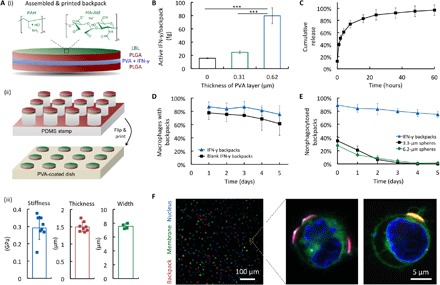
(A) Schematic illustrations of a backpack (i) and its method of printing (ii); graphs of average backpack stiffness, thickness, and width (n ≥ 4) (iii). (B) Amount of active IFN-γ per backpack, determined by ELISA (n = 5). ***P < 0.001. (C) Cumulative release of IFN-γ from backpacks over 60 hours (n = 3). (D) Association of backpacks with primary murine macrophages over time in vitro (n = 3). (E) Proportion of backpacks that evaded phagocytosis over time compared with spheres of similar volume (n = 5). (F) Confocal micrographs of leukocytes (nucleus, blue; membrane, green) displaying PLGA discs (red).
We investigated the role of PVA in the interior of the backpacks on stabilizing IFN-γ. We found that increased thicknesses of PVA improved the activity of IFN-γ, despite the same loading of IFN-γ per backpack (Fig. 2B). This is likely because the PVA stabilized the IFN-γ when the second layer of PLGA dissolved in acetone was deposited. While thicker PVA layers improved the activity of IFN-γ, we fixed the thickness to 0.62 μm for the remainder of the study, as higher PVA content reduced printing efficiency (see fig. S4A). Next, we investigated the release of IFN-γ from the backpacks into serum media at 37°C over time (Fig. 2C). We found that backpacks released IFN-γ for at least 60 hours. We also found that backpacks maintained activity of IFN-γ after printing and storage for 3 months at −80°C (see fig. S5).
Next, we evaluated the interaction of backpacks with primary BMDMs using two techniques. First, we examined the association of fluorescent backpacks with cells using flow cytometry, which included both surface-bound and phagocytosed backpacks (Fig. 2D). We found that backpacks encapsulating IFN-γ displayed a higher affinity to BMDMs than those without, which is likely due to the enhanced activity of macrophages when stimulated by IFN-γ. Over 5 days, the association of IFN-γ backpacks reduced from 83.6 to 75.4%, whereas the association of blank backpacks reduced from 77.5 to 61.2%. Second, we examined the resistance of IFN-γ backpacks to phagocytosis compared with spheres of similar volumes using fluorescence microscopy (Fig. 2E). We compared the number of surface-bound backpacks (VBP = 49.8 μm3) to the number of surface-bound 3.3-μm spheres (V3.3 = 18.8 μm3) and 6.2-μm spheres (V6.2 = 124.8 μm3). Over 5 days, the proportion of backpacks that remained surface bound reduced from 89.1 to 77.3%, whereas spheres of both sizes were nearly completely internalized after 3 days (<5% remained surface-bound). Together, both sets of data suggest that the majority of cell-associated backpacks evaded phagocytosis for at least 5 days. We also imaged cells (labeled with NucBlue, blue; coumarin 6, green) displaying backpacks made from rhodamine B PLGA discs (red) using confocal microscopy (Fig. 2F).
IFN-γ backpacks stimulate polarization of macrophages toward M1 phenotypes in vitro
To assess the potency of IFN-γ backpacks to potentiate a durable shift in polarization, we evaluated the expression of several markers associated with M1 and M2 phenotypes (Fig. 3). BMDMs were cultured from murine bone marrow progenitor cells, and IFN-γ backpacks were added to cells in a ratio of 3:2, respectively. After 1.5 hours, unbound backpacks were removed, and cells were cultured for 24 to 120 hours. In addition to BMDMs with IFN-γ backpacks in standard culture conditions, we also cultured BMDMs (i) without backpacks (no IFN-γ), (ii) with blank backpacks (no IFN-γ), and (iii) without backpacks yet with an equivalent dose of free IFN-γ (16 ng/ml). We also cultured BMDMs with IFN-γ backpacks in tumor-mimicking conditions [i.e., in hypoxia (1% O2) and 10 volume % 4T1-conditioned media]. Each day for 5 days, cells were harvested, stained, and analyzed for molecular expression by flow cytometry. Expression of each biomarker was normalized to unpolarized BMDMs.
Fig. 3. Phenotypic evaluation of macrophages (MΦs) carrying IFN-γ backpacks in vitro.
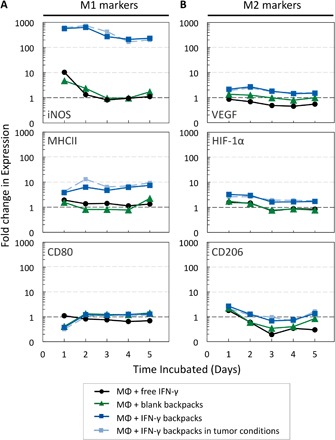
BMDMs were cultured for 5 days with free IFN-γ (16 ng/ml; black lines), blank backpacks (0 ng/ml IFN-γ; green lines), and IFN-γ backpacks (16 ng/ml equivalent) in normoxia (dark blue lines) and tumor-mimicking conditions (1% O2 and 10 volume % tumor-conditioned media; light blue lines). Cellular expression of representative (A) M1 markers (iNOS, MHCII, and CD80) and (B) M2 markers [vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1α (HIF-1α), and CD206], relative to that of unpolarized macrophages (without IFN-γ or backpacks). Graphs are logarithmic (n = 10,000 events per data point).
Macrophages carrying IFN-γ backpacks strongly exhibited traits of M1 phenotypes. We investigated the relative expression of M1 biomarkers, including inducible NO synthase (iNOS), major histocompatibility complex class II (MHCII), and CD80, because of their important role in innate immunity. iNOS is involved in the production of NO, which serves as a potent tumoricidal and antimicrobial agent (22). MHCII proteins are involved in antigen presentation to T cells to facilitate adaptive immunity (23). MHCII is expressed on macrophages with M1 and M2 phenotypes, but it is overexpressed on cells with M1 polarizations. CD80 is a costimulatory molecule used to trigger an adaptive immune response in the presence of an antigen-presenting cell (24).
Macrophages displaying IFN-γ backpacks showed marked increases in iNOS, MHCII, and CD80 expression relative to unpolarized cells (Fig. 3A). Here, we make several important observations. First, expression of both iNOS and MHCII in cells displaying IFN-γ backpacks was synergistic. Specifically, iNOS expression was 629.3-fold higher in the IFN-γ backpack group compared with only 2.4- and 1.3-fold higher in groups treated with blank backpacks and IFN-γ alone, respectively, after 48 hours. Similarly, MHCII expression was 6.3-folder higher in the IFN-γ backpack group compared with only 0.8- and 1.4-fold higher in groups treated with blank backpacks and IFN-γ alone, respectively, after 48 hours. While the origins of this apparent synergy needs further investigation, the differential protein expressions may arise, at least in part, from local and sustained concentration gradients of IFN-γ formed near the cells to which the backpacks are bound, thus enhancing the activity of IFN-γ. Second, the data suggest that the presence of backpacks without IFN-γ (blank backpacks) induces modest, but non-negligible phenotypic shifts toward M1 phenotypes, as evidenced by increased expressions of iNOS, MHCII, and CD80. This effect could be due to frustrated phagocytosis, whereby macrophages enhance their inflammatory phenotypes upon encountering large foreign objects (25, 26). Third, the expression of M1-related markers in BMDMs carrying IFN-γ backpacks was more durable than that of BMDMs cultured with free IFN-γ. Specifically, the relative expression of iNOS decreased by 89.1% after 5 days in cells treated with free IFN-γ, but only by 59.1% in cells with IFN-γ backpacks. Further, the relative expression of MHCII and CD80 decreased by 30.1 and 37.6%, respectively, after 5 days for cells treated with free IFN-γ; however, the relative expression of MHCII and CD80 in cells treated with IFN-γ backpacks actually increased by 95.7 and 248.4%, respectively, after 5 days. Last, no major differences in marker expression were observed between BMDMs with IFN-γ backpacks in standard culture conditions versus tumor-mimicking conditions, which we hypothesize will be critical to allow BMDMs to maintain M1 phenotypes in vivo. Overall, these data suggest that IFN-γ backpacks potentiate a shift in macrophage polarization toward M1 phenotypes that is more potent and durable than free IFN-γ.
We also investigated the expression of markers associated with M2 phenotypes: vascular endothelial growth factor (VEGF), hypoxia-inducible factor 1α (HIF-1α), and CD206. VEGF is often overexpressed in TAMs, which serves as a source of angiogenic cytokines and proteases to promote tumor vascularization (27). HIF-1α is also overexpressed by TAMs, which suppresses T cell function and promotes tumor progression (28). CD206 is the mannose receptor, which has been linked to immunosuppression, angiogenesis, and metastasis (29). Cells displaying IFN-γ backpacks showed elevated levels of all three M2 markers relative to untreated controls; however, the magnitude of this increase was modest. The highest fold changes observed were 2.7, 3.3, and 2.6 for VEGF, HIF-1α, and CD206, respectively. These changes were less substantial than those observed for M1 markers, and the relative expression of all three M2 markers returned to values near the expression of untreated controls after 5 days.
IFN-γ backpacks enable macrophages to maintain M1 phenotypes in vivo
Next, we sought to test our central hypothesis that macrophages carrying IFN-γ backpacks can maintain their M1 phenotypes in vivo. We chose orthotopic 4T1 breast tumors as a model immunosuppressive environment due to its association with chemotherapy resistance, tumor metastasis, and lack of tumor-specific antigens, making them challenging targets for CAR T cell therapy (10, 30, 31). To distinguish injected macrophages from TAMs, we stained BMDMs with VivoTrack 680 (see the Supplementary Materials). Macrophages were injected intratumorally. Distributions of injected cells were monitored each day for 5 days using an in vivo imaging system (IVIS) (see fig. S6). After 5 days, a second injection was administered as before. Mouse body weight, tumor growth, tumor radiance, and necrosis were monitored to the end of the study (see fig. S7). Two days after the second injection, mice were euthanized, and their tumors were extracted, digested, and tumor-associated immune cells (CD45+) were isolated. Dendritic cells and macrophages were stained and identified by hierarchical gating (see fig. S8). Macrophages were phenotyped as before (see table S1), except markers were used for arginase 1 (arg-1) instead of VEGF. Arg-1 affects NO synthase and down-regulates NO production (32). Expression of each marker was normalized to that of endogenous TAMs in mice treated with saline.
Macrophages carrying IFN-γ backpacks retained M1 polarizations in solid tumors for at least 48 hours (Fig. 4A). Relative to the TAMs of control mice (i.e., mice injected with saline), the expression of iNOS, MHCII, and CD80 in injected macrophages displaying IFN-γ backpacks was significantly higher than that of injected cells displaying blank backpacks or injected cells with free IFN-γ. The relative increase in MHCII and CD80 expression of cells carrying IFN-γ backpacks surpassed that of cells carrying IFN-γ backpacks in vitro (12.3- and 3.0-fold in vivo versus 6.3- and 1.3-fold in vitro for MHCII and CD80, respectively; Figs. 3A and 4A). However, the relative increase in iNOS was less substantial in vivo (7.2-fold in vivo versus 629.3-fold in vitro). The reduction was due to an elevated basal expression of iNOS in the TAMs of control mice compared with untreated control cells in vitro. This observation is consistent with findings by others that iNOS expression can increase in M2-polarized TAMs (33). We also found no statistically significant differences in the relative expression of HIF-1α and CD206 between macrophages carrying IFN-γ backpacks and those carrying free backpacks or injected with free IFN-γ. Although cells carrying IFN-γ backpacks displayed significantly higher levels of Arg-1 relative to those carrying blank backpacks or injected with free IFN-γ, they did not show a significant increase in the expression of Arg-1 relative to untreated TAMs.
Fig. 4. IFN-γ backpacks promote proinflammatory phenotypes in solid tumors.
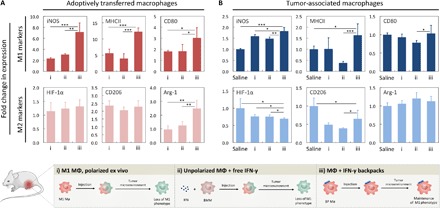
(A) Polarization of adoptively transferred macrophages (MΦs) 48 hours after injection. BMDMs were polarized ex vivo for 24 hours with IFN-γ (16 ng/ml) (i), left unpolarized and injected with 50 ng of free IFN-γ (ii) or left unpolarized, bound to IFN-γ backpacks at a dose of 50 ng equivalent IFN-γ and injected (iii). Bar graphs indicate the fold change in the median expression of representative M1 biomarkers (iNOS, MHCII, and CD80; top row) and M2 biomarkers (HIF-1α, CD206, and Arg-1; bottom row), relative to their native expression in endogenous TAMs. (B) Polarization of endogenous TAMs 48 hours after injection of groups described in (A). Bar graphs indicate the fold change in the median expression of representative M1 biomarkers (top row) and M2 biomarkers (bottom row) relative to the native expression of endogenous TAMs [leftmost bars in (B)]. For all bar graphs, n = 5. *P < 0.05; **P < 0.01; ***P < 0.001.
IFN-γ backpacks shift the polarization of TAMs toward M1 phenotypes
After demonstrating that the IFN-γ backpacks allowed macrophages to maintain their phenotypes in vivo, we sought to evaluate the phenotype of TAMs in response to adoptive transfer of macrophages carrying IFN-γ backpacks (see fig. S8 for hierarchical gating). An emergent therapeutic strategy to attack tumorous tissues is via repolarizing TAMs toward M1 phenotypes (34–39). TAMs affect cancer progression in a manner that is dependent on their polarization (40–42). Macrophages possessing M1 phenotypes have been shown to improve outcomes in cancer therapy due to their antigen-dependent and antigen-independent facets. This gives macrophages the potential to be useful in tumors that lack the tumor-specific antigens typically required for adoptive T cell therapy (43–45). This has been demonstrated by others through the delivery of nanoparticles with payloads that inhibit colony stimulating factor 1 receptor (CSF1-R) and Src homology region 2 (SH2) domain-containing phosphatase 1 (SHP2) pathways on macrophages (46) as well as the delivery of nanoparticles encapsulating microRNA-125b (47). Still, supplying a sufficient concentration of immunomodulatory factors to repolarize TAMs while minimizing toxicity remains a major challenge.
We administered two intratumoral injections of macrophages with 50 ng of IFN-γ per mouse, which is 100-fold lower than the maximum total dose (MTD) administered in other studies (48). Our motivation for the comparatively low dose was to supply sufficient IFN-γ to maintain the M1 polarization of adoptively transferred macrophages while minimizing toxic side effects (49). Here, mice received two equivalent injections of saline (control) and (i) macrophages polarized ex vivo for 24 hours in IFN-γ (20 ng/ml; M1 polarized), (ii) unpolarized macrophages injected with 50 ng of free IFN-γ, and (iii) macrophages carrying IFN-γ backpacks that encapsulated 50 ng of IFN-γ (Fig. 4B). Administrations were separated by 5 days.
We found that TAMs of mice treated with the IFN-γ backpack therapy were polarized toward M1 phenotypes, as evidenced by significantly increased expressions of iNOS (1.8-fold) and MHCII (1.6-fold) compared with TAMs of mice treated with saline (Fig. 4B). Second, the relative increase in iNOS expression in TAMs of mice treated with the IFN-γ backpack therapy was significantly higher than in TAMs of mice treated with macrophages polarized ex vivo (1.8- versus 1.0-fold, respectively) (ii). Third, the relative increase in CD80 expression in TAMs of mice treated with the IFN-γ backpack therapy was significantly higher than in TAMs of mice treated with macrophages plus free IFN-γ (1.03- versus 0.78-fold, respectively) (iii).
We also investigated the relative expression of M2 markers in TAMs. We found that relative HIF-1α expression in TAMs of mice treated with the IFN-γ backpack therapy was significantly lower than in all other groups (Fig. 4B). This finding was particularly interesting, as relative HIF-1α expression in macrophages displaying IFN-γ backpacks was higher in vitro (Fig. 3B). TAM expression of CD206 was also significantly lower for mice treated with IFN-γ backpacks than saline. However, the group that displayed the lowest relative expression of CD206 in TAMs was in mice treated with macrophages plus free IFN-γ. No significant differences were observed in the relative expression of Arg-1. Overall, these data show that macrophages carrying IFN-γ backpacks can shift the polarization TAMs toward M1 phenotypes at a markedly reduced dose, 100-fold lower than the MTD (48). In addition, the same dose of free IFN-γ was not able to potentiate a shift in TAM polarization. Given this these findings, we sought to examine the therapeutic efficacy of macrophages with IFN-γ backpacks.
Antitumor efficacy of macrophages carrying IFN-γ backpacks
To evaluate the therapeutic efficacy of IFN-γ backpacks, we investigated the formation of metastases, tumor growth kinetics, and overall survival of immunocompetent BALB/c mice burdened with 4T1-Luc cells. 4T1-Luc cells were chosen because of their high luciferase expression, enabling bioluminescence imaging to visualize the formation of metastatic colonies in the chest cavities by radiance using an IVIS. We administered the same low dose of IFN-γ as before to understand the influence of the IFN-γ backpacks. After tumors became palpable (~50 mm3), mice received two equivalent injections (separated by 5 days) of (i) saline, (ii) unpolarized macrophages with 50 ng of free IFN-γ, and (iii) macrophages carrying IFN-γ backpacks encapsulating 50 ng of IFN-γ.
We found that mice treated with the IFN-γ backpack therapy had significantly fewer metastatic nodules than control mice (Fig. 5A). Chest cavities of mice given the IFN-γ backpack therapy showed 5.2-fold lower radiance compared with that of mice treated with saline and 4.9-fold lower radiance compared with that of mice treated with macrophages and free IFN-γ (Fig. 5B). This suggests that, even at a low dose, IFN-γ backpacks are able to significantly inhibit the formation of metastatic colonies. To assess toxicity, peripheral blood was isolated via cardiac puncture immediately after euthanasia, and serum was analyzed for cytokines (see Methods in the Supplementary Materials). Analysis revealed that all treatments were well tolerated, and IFN-γ levels were below the limit of detection in all groups. This result was expected given that the dose of IFN-γ was 100-fold lower than the MTD used previously (48). One exception was that significantly higher IL-6 was found in the serum of mice treated with macrophages plus free IFN-γ (see fig. S9). Increased expression of IL-6 has been correlated with tumor metastasis (50); however, IL-6 is also secreted by macrophages with anti-inflammatory phenotypes (51).
Fig. 5. Efficacy of IFN-γ backpacks for reducing metastasis and tumor burden of 4T1 mammary carcinomas.
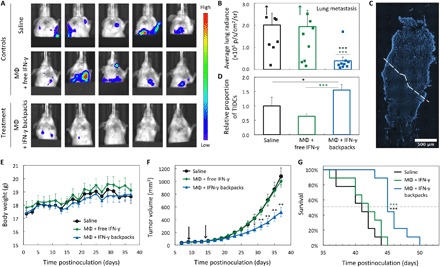
(A) In vivo bioluminescence imaging of metastatic colony formation in the chest cavities of mice burdened with 4T1-Luc cells 32 days after inoculation (primary tumor outside of view). Five representative images per treatment group are shown. (B) Average radiance from bioluminescence in the chest cavities of the mice in (A) (n = 9). (C) Representative histological section of a 4T1 tumor treated with macrophages carrying IFN-γ backpacks. Dotted line separates regions of cleared (top) and intact tumorous tissue (bottom). (D) Relative proportion of tumor-infiltrating dendritic cells (TIDCs) in solid 4T1 tumors revealed through tumor-associated immune cell phenotyping (determined by CD45+, SYTOX−, and CD11c+; n = 5). (E) Weight changes of mice burdened with 4T1-Luc tumors in different groups (n = 9). (F) Growth kinetics of tumors in the groups shown in (E). Black arrows indicate days of therapeutic injections. (G) Survival of mice in (E). Statistical significance was determined via a log-rank test. *P < 0.05; **P < 0.01; ***p < 0.001.
We also assessed tumor morphology and dendritic cell infiltration. For both analyses, tumors from BALB/c mice burdened with 4T1 breast cancer (from the previous study; Fig. 4B) were isolated and cut into four vertical portions. One portion was sectioned for histology, and the remaining three portions were digested and stained for phenotypic evaluation by flow cytometry. The top half of the tumor revealed large areas of digested tissue, whereas the bottom half remained largely intact (Fig. 5C). This finding suggests that the areas of highest tumor clearance occurred in regions where the injected cells resided, as all treatments were injected toward the top each tumor. We found that mice treated with the IFN-γ backpack therapy had significantly higher infiltration of CD11c+ dendritic cells (Fig. 5D), as determined by the gating schema shown in fig. S8A (see the Supplementary Materials). While not studied here, we believe this could be a promising future direction of study, as higher dendritic cell populations could be used to instruct adaptive immunity as a cancer vaccine (52, 53).
Last, we evaluated the progression of tumor growth and overall survival of mice treated with the IFN-γ backpack therapy (Fig. 5, E to G). Consistent with the metastasis data, mice injected with the IFN-γ backpack therapy showed significantly smaller tumors than the two controls 14 to 23 days after the second therapeutic injection. By 37 days after inoculation, tumors of mice receiving the IFN-γ backpack therapy were 51.9 and 48.3% smaller than those of mice receiving injections of saline and macrophages with free IFN-γ, respectively. Mice receiving the IFN-γ backpack therapy showed significantly improved survival, as determined by a log-rank test. The average time of survival for mice treated with saline, macrophages with free IFN-γ, and macrophages carrying IFN-γ backpacks was 30.7, 31.7, and 35.9 days after inoculation, respectively. Together, the slowed tumor growth, smaller tumor volumes, and decrease in serum IL-6 of mice treated with the IFN-γ backpack therapy likely potentiated the reduced metastatic burdens and improved overall survival compared with controls.
DISCUSSION
In summary, we have developed a particle-based strategy, referred to as backpacks, that can regulate the phenotype of adoptively transferred macrophages. We demonstrate that IFN-γ backpacks (i) securely attach to macrophage surfaces and evade phagocytosis for several days, (ii) show favorable release kinetics of encapsulated cytokines to induce potent and durable shifts in macrophage polarization, and (iii) allow adoptively transferred macrophages to maintain their phenotypes deep within the immunosuppressive milieu of solid tumors. Backpacks were prepared from biodegradable materials that enable facile preparation, long-term storage, and simple metabolic clearance, all of which are favorable for clinical translation. Furthermore, injected macrophages were allogeneic, which reduces the time scale of preparing cell transfers from weeks [i.e., for CAR T cell therapy (54)] to several hours.
In addition to validating our central hypothesis, we also show that low doses of IFN-γ can induce a shift in the polarization of TAMs and potentiate an antitumor response against 4T1 triple negative breast tumors. While the doses reported here are not optimized, we show that the slowed tumor growth suppresses formation of metastases and improves overall survival. Future studies will investigate the optimal loading of IFN-γ into backpacks and their release kinetics to enhance this therapeutic efficacy against solid tumors. In addition, future work can combine backpacks with adjuvant therapies to enhance therapeutic effects.
Overall, this work offers a strategy to regulate the phenotype of adoptively transferred macrophages, which can be used to address a broad range of inflammatory diseases, including cancer, autoimmune disorders, and infectious disease. While the backpacks described here bind to macrophage surfaces, other designs can be conceived to attach to other circulatory cells with higher chemotactic sensitivity (17, 18, 55). Also, a range of immunomodulatory payloads can be considered, including those that facilitate adaptive immune responses toward a backpack-based vaccine (52) or promote anti-inflammatory phenotypes to aid in tissue regeneration or repair for autoimmune diseases (56, 57).
MATERIALS AND METHODS
Materials
4T1 mammary carcinoma cells and 4T1-Fluc-Neo/eGFP-Puro cells expressing firefly luciferase were obtained from the American Type Culture Collection and Imanis Life Sciences, respectively. RPMI 1640 media, Dulbecco’s modified Eagle’s medium (DMEM) F12 media, fetal bovine serum (FBS), penicillin and streptomycin (Pen Strep), mouse IFN-γ recombinant protein, Gibco Type 1 Collagenase, SYTOX blue dead cell stain, NucBlue stain, coumarin 6 membrane dye, heparin-coated plasma preparation tubes, and UltraComp eBeads compensation beads were obtained from Thermo Fisher Scientific. Materials for backpack fabrication, including polydimethylsiloxane (PDMS), PAH, HA, PVA, PLGA, and trichloro(1H,1H,2H,2H-perfluorooctyl)silane (FDTS) were obtained from Millipore Sigma. Red blood cell lysing buffer Hybri-Max, Trypan blue, DNAse I, trypsin, and all solvents used were obtained from Millipore Sigma. Cell culture flasks, plates, and conical tubes were obtained from Corning. Mouse T helper cell type 1 (TH1)/TH2/TH17 cytokine quantification kits, cell fixation/permeabilization kits, and cell strainers (40 and 70 μm) were obtained from BD Biosciences. A QuadroMACS separator, a CD45+ leukocyte isolation kit, and a mouse tumor dissociation kit were obtained from Miltenyi Biotec. Bambanker cell freezing media and OCT (optimum cutting temperature) compound were obtained from VWR International. Recombinant murine macrophage CSF (M-CSF) was obtained from PeproTech. Murine IFN-γ enzyme-linked immunosorbent assay (ELISA) kits were obtained from R&D Systems. Female BALB/c mice (6 to 8 weeks old) were obtained from Charles River. Information about the antibodies and their related clones and fluorophores are detailed in the Supplementary Materials (table S1).
PDMS template preparation
PDMS templates were prepared by soft lithography using methods similar to those described previously (16). Before use, silicon molds were fabricated by monolithic photolithography and passivated with a thin film of FDTS by vapor deposition (see Methods in the Supplementary Materials). A 10:1 weight ratio of PDMS base to cross-linker from a Sylgard 184 kit was thoroughly mixed and poured on top of the silicon molds in separate petri dishes (~20 g per mold). PDMS was degassed in a desiccator at 25°C until no visible bubbles remained. Dishes were then placed into an oven at 65°C overnight to cure the PDMS. After curing, PDMS templates were removed from the molds by cutting the petri dishes and peeling away the PDMS.
Cell-adhesive coating
HA (2500 kDa) was modified with aldehyde (HA-Ald) (see Methods in the Supplementary Materials). An aqueous solution of HA-Ald (2 mg/ml) was prepared in 150 mM NaCl (pH 6.8), and a aqueous solution of PAH (2 mg/ml; 17.5 kDa) in 150 mM NaCl (pH 6.8) was prepared. HA-Ald, PAH, and 150 mM NaCl (pH 6.8) solutions were separately poured into weigh boats. PDMS templates were rinsed with isopropyl alcohol and dried by a steady stream of air. Templates were then placed patterned side down in the HA-Ald solution for 15 min. Care was taken to ensure the templates were floating to maximize contact of the patterned PDMS with the solution. Templates were transferred to the NaCl solution for 2 min and were then rinsed with deionized (DI) water to remove free HA-Ald. Templates were transferred to the PAH solution for 15 min in the same fashion and then transferred to new weigh boats with the NaCl solution for 2 min. Templates were rinsed with DI water, and the entire process was repeated once more to form an LBL coating of HA-Ald/PAH/HA-Ald/PAH. Coated templates were rinsed with DI water for 30 s and dried by a stream of air. Templates were stored in petri dishes, patterned side up, at 4°C.
Backpack fabrication
An 8% w/v solution of PLGA in acetone was prepared from a 100:1 weight ratio of nonfluorescent PLGA (7 to 17 kDa; Resomer 502 H) and fluorescent PLGA (10 to 30 kDa; LG 50:50 rhodamine B; PolySciTech). PDMS templates with LBL coatings were cut into quadrants and spin coated with 225 μl of PLGA solution per quadrant at 2000 rpm for 20 s (at a 200 rpm/s ramp). Quadrants were then plasma ashed with O2 for 60 s. A 0.5 weight % solution of PVA (146 to 186 kDa, 99 + % hydrolyzed) in phosphate-buffered saline (PBS) was prepared with IFN-γ (25 μg/ml). Immediately after plasma treatment, 100 μl of the PVA solution was evenly spread onto each quadrant by pipette. Quadrants were then placed in a desiccator under vacuum with Drierite desiccant (W.A. Hammond Drierite Co.) until dry, making a 0.6-μm-thick PVA film. A second PLGA layer was deposited using the same procedure as the first.
Microcontact printing
PVA-coated dishes were prepared by making a 3% w/v solution of PVA (13 to 23 kDa, 87% hydrolyzed) in DI water. The solution was stirred at 80°C for several hours, and excess crystals were filtered using a 0.22-μm filter. Sterile petri dishes were coated with 2.5 ml of solution, and placed in an oven at 60° to 75°C until dry. Backpacks were printed using techniques similar to those described previously (14). Briefly, a beaker was filled with DI H2O and heated to 65°C. The coated side of a PVA-coated dish was held ~2 cm over the beaker for 6 to 12 s. A PDMS quadrant containing backpacks was immediately pressed onto the warmed PVA dish, and consistent pressure was applied for 15 to 20 s. The quadrant was then peeled away, leaving a coating of backpacks on the dish. This was repeated until the material had fully transferred. Backpacks were then stored at −80°C until needed. To harvest backpacks, dishes were covered with 2.5 ml of PBS and were gently washed. This was repeated until the surface appeared mostly clear (typically twice). The solution was collected, passed through a 40-μm cell strainer, and centrifuged at 2500g for 5 min. Backpacks were resuspended in 5 ml of BMM− (i.e., 500 ml of DMEM F12, 50 ml of FBS, 5 ml of Pen Strep, and 25 ml of 200 mM GlutaMAX) or serum-free BMM− (i.e., BMM− sans FBS), depending on the application.
Bone marrow isolation
Progenitor cells were isolated from murine bone marrow following methods described previously (58). Briefly, 6- to 8-week-old BALB/c mice were euthanized via CO2 inhalation. Sterile surgical scissors were used to extract the tibias, femurs, and humeri. Isolated bones were submerged in 70% ethanol, rinsed with PBS, and then transferred to a separate PBS solution. In a sterile environment, epiphyses of each bone were cut, and the bones were flushed with PBS via a syringe with a 31-gauge needle into a 50-ml collection tube. The solution was mixed thoroughly, passed through a 40-μm cell strainer, and centrifuged at 400g for 10 min at 4°C. Cells were resuspended in Bambanker (2 ml per mouse equivalent; Lymphotec Inc.) and stored in cryovials at −80°C until needed.
BMDM culture
BMDMs were cultured from murine bone marrow progenitor cells following methods described previously (9). Briefly, frozen bone marrow was thawed and mixed with 4°C BMM− at 1:5 ratio by volume. The solution was centrifuged, the liquid was aspirated, cells were resuspended in BMM+ [i.e., BMM− with M-CSF (20 ng/ml)], and cells were counted with a hemocytometer. Approximately 4 × 106 bone marrow cells were added to non–tissue culture (TC)–treated T175 flasks containing 25 ml of BMM+. Cells were incubated under standard culture conditions. Additional BMM+ (25 ml) was added to the flasks on days 3 and 7. On day 8, media were aspirated from the flasks, and cells were washed once with 10 ml of PBS. To dislodge the cells, PBS was aspirated and replaced with 10 ml of Accumax (Innovative Cell Technologies) at 4°C. Cells were incubated with the Accumax at 37°C for 10 min. The flask was then removed from the incubator and vigorously thumped several times. More Accumax (10 ml) was added to the flask, and cells were incubated for an additional 10 min and thumped again. The suspension of BMDMs was added to a 50-ml conical tube with an equal volume of BMM− and centrifuged. The supernatant was aspirated and replaced with BMM+. BMDMs were counted and plated on non-TC-treated 12-well plates at a concentration of 2.5 × 105 cells per well in a volume of 1 ml per well and incubated under standard conditions for 24 hours. All centrifugation steps were performed at 400g for 10 min at 4°C.
Binding backpacks to BMDMs
Backpacks were harvested and centrifuged at 2500g for 5 min at 4°C and resuspended in serum-free BMM−. Meanwhile, BMDMs cultured in 12-well plates for 24 hours were removed from the incubator, and BMM+ was exchanged with serum-free BMM− using a serial dilution technique [see Methods in the Supplementary Materials (9)]. Backpacks were counted using a hemocytometer, and 0.375 × 105 backpacks were added to each well of each 12-well plate (yielding a 3:2 ratio of backpacks:cells). Plates were then centrifuged at 300g for 7.5 min to allow backpacks to gather along the bottom of the plate. Plates were then placed in a cell culture incubator for 1.5 hours to allow BMDMs to bind to backpacks. After 1.5 hours, serum-free BMM− was exchanged with BMM− via serial dilution. Plates were then incubated in either standard culturing conditions (normoxia; 74% N2, 5% CO2, and 21% O2) or hypoxic conditions (94% N2, 5% CO2, and 1% O2), depending on the study (see Methods in the Supplementary Materials). In cases where plates were stored in a hypoxia chamber, 100 μl of BMM− from each well was replaced with 100 μl of tumor-conditioned media, obtained from culture with 4T1 cells. In lieu of backpacks, free IFN-γ was sometimes added at a concentration of 16 ng/ml to the appropriate wells.
Phenotyping in vitro cultures of BMDMs
Serial dilution was performed to replace media in each well of the 12-well plates with Hank’s balanced salt solution (HBSS). Then, 500 μl of HBSS was aspirated from each well and replaced with 2 ml of Accumax. Plates were incubated at 37°C and 5% CO2 for 10 to 15 min. Plates were then thumped to release BMDMs, and the respective groups were collected into separate 50-ml tubes with an equal volume BMM−. Cells were centrifuged and pellets were resuspended in 1 ml of stain buffer, comprising 1% FBS in PBS without Mg2+ or Ca2+ (pH 7.4 to 7.6). Cells were transferred into 1.5-ml Eppendorf tubes, where they were centrifuged again. Pellets were resuspended in 99 μl of stain buffer with 1 μl of Fc block and were incubated for 15 min at 4°C. After incubation, samples were diluted with 1 ml of stain buffer, centrifuged, and resuspended in 1 ml of stain buffer. Each sample was then split into two groups of 500 μl for surface marker staining and intracellular staining. For surface staining, samples were centrifuged and resuspended in an antibody mixture of anti-CD80, anti-MHCII, anti-VEGF, and stain buffer (at concentrations suggested by the manufacturer) in the dark at 4°C. After 30 min, cells were washed with 1 ml of stain buffer, centrifuged, resuspended in 300 μl, and stored in the dark at 4°C until use. For intracellular staining, samples were fixed and permeabilized following instructions from the manufacturer (BD Biosciences). Cells were centrifuged and resuspended in 100 μl of an antibody solution comprising anti-iNOS, anti-HIF-1α, anti-CD206, and Perm/Wash Buffer (at concentrations suggested by the manufacturer) in the dark at 25°C. After 30 min, cells were diluted with 1 ml of Perm/Wash Buffer, centrifuged, resuspended in 300 μl of stain buffer, and stored in the dark at 4°C until use. All centrifugation steps were performed at 350g for 5 min at 4°C. Compensation and voltage settings were determined 1 day prior using sets of compensation beads, each stained with one antibody. Up to 10,000 events were collected for each sample. Data were analyzed using FCS Express 6 Software (De Novo Software).
Tumor model establishment
Experiments involving animals were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Harvard University. Two orthotopic breast cancer models were used in mice, 4T1 and 4T1-Fluc-Neo/eGFP-Puro cells expressing firefly luciferase (4T1-Luc). 4T1 cells were cultured in DMEM supplemented 10% FBS and 1% Pen Strep. 4T1-Luc cells were cultured in RPMI 1640 media supplemented with 10% FBS, 1% Pen Strep, and G418 (0.1 mg/ml). Both lines were cultured in a humidified incubator maintained at 37°C and 5% CO2. Cells were passaged twice before inoculation. Cells were released via trypsin, centrifuged, and resuspended in physiological saline. Mice were inoculated with 1 × 105 4T1 cells or 1 × 106 4T1-Luc cells (>98% cell viability) in 50 μl by subcutaneous injection into the lower left inguinal mammary fat pad of BALB/c mice 42 to 56 days in age using a 25-gauge needle. Tumor-bearing mice were randomized before treatments and monitored for tumor growth and body weight changes throughout the study. Each mouse model received two treatments, which began 14 days after inoculation in the 4T1 model (tumor volume, ~100 mm3) and 9 days after inoculation in the 4T1-Luc model (tumor volume, ~ 50 mm3). Tumor volumes were calculated using the formula: V = ½ L × W2, where L and W were the longest and shortest dimensions of the tumor, respectively. Mice harboring 4T1 tumors were used for tumor-immune cell phenotyping. These mice were euthanized if L exceeded 15 mm or if body weight loss exceeded 15%. Mice in the 4T1-Luc model were enrolled in a survival study and were left alive until they succumbed to tumor burden or were euthanized with CO2 if they became moribund.
Intratumoral injections
Mice received two intratumoral treatment injections, occurring 14 and 18 days after inoculation in the 4T1 model and 9 and 14 days after inoculation in the 4T1-Luc model. IFN-γ was limited to 50 ng per administration (59). Mice requiring injected macrophages each received the same number of cells (i.e., 0.78 × 106 macrophages per mouse per injection). Numbers were determined based on the number of macrophages necessary to deliver 50 ng worth of IFN-γ backpacks. Determinations were based on ELISA data, which revealed ~85 fg IFN-γ/backpack, and flow cytometry, which revealed ≥75% of macrophages were labeled with ≥1 backpack after the 1.5 hours incubation period. In all groups (i.e., saline, free IFN-γ, or groups with macrophages), injection volumes were 10 μl.
In vivo tracking of adoptively transferred macrophages
Mice inoculated with 4T1 cells were treated with macrophages labeled with a near-infrared dye (VivoTrack 680, PerkinElmer). Seven days before tumor inoculations through to the end of the study, mice were fed alfalfa-free diets to reduce background fluorescence levels (Picco Rodent 5V5R 50IF irradiated pelleted, Scott Pharma Solutions). Before imaging, hair over top and near the tumor was removed using a topical formulation (Nair, Church & Dwight). Mice were imaged under anesthesia (from isoflurane) using IVIS each day after the first therapeutic administration for a total of 5 days.
In vivo monitoring of lung metastases
Lung metastases were evaluated 32 days after inoculation with 4T1-Luc. Mice were injected with 150 μl of XenoLight D-Luciferin potassium salt bioluminescence substrate (30 mg/ml) (PerkinElmer) in saline via intraperitoneal injection. Fifteen minutes after injection, mice were imaged under anesthesia (from isoflurane) using IVIS. Primary tumors were covered with strips of black paper to eliminate signal washout from the main tumors, which were brighter than the metastatic colonies in the chest cavities.
Phenotyping tumor-associated immune cells
Procedures for isolating and staining tumor-associated immune cells were similar to those described previously (30). Briefly, mice were euthanized via CO2 inhalation 2 days after administration of the second treatment. Primary tumors (from 4T1 cells) were harvested, cut into small pieces (<5 mm thick), and enzymatically degraded using a mouse tumor dissociation kit with a gentleMACS dissociator (Miltenyi Biotec). Cells were centrifuged and resuspended in ACK red cell lysis buffer supplemented with DNAse I (50 U/ml) for 5 min. Cells were again centrifuged and resuspended in PBS to quantify the remaining intact cells. Leucocytes were isolated from the general population using a CD45+ isolation kit, following instructions from the manufacturer (Miltenyi Biotec). For the remainder of the study, 1 × 106 cells per animal were used, and all steps were performed in 100 μl of fluorescence-activated cell sorting (FACS) buffer (PBS with 3% FBS) supplemented with additional reagents as necessary. Cells were blocked for 30 min in a solution consisting of 5% rat serum, 5% mouse serum, and 1% anti-mouse CD16/32 antibody. Cells were stained with test and control antibodies (see fig. S8 and table S1) for 30 min at 25°C and for 20 min on ice in a dark enclosed space. Cells were then washed twice with ice-cold FACS buffer and resuspended in 500 μl of PBS. Following instructions from the manufacturer, cells were stained with SYTOX blue to measure cell viability at the end of all treatment steps. Stained cells were then analyzed by flow cytometry (BD LSRII). Compensation and voltage settings were determined 1 day prior to using sets of compensation beads, each stained with one antibody. Up to 100,000 events were collected for each sample. Data were analyzed using FCS Express 6 Software. All centrifugation steps were performed at 350g for 5 min.
Statistical analysis
Unless otherwise indicated, the data were represented as means ± SE using GraphPad (Prism 8.0). For determination of statistical significance, multiple t tests or one-way analysis of variance (ANOVA) with Tukey’s multiple comparison tests were used, as applicable. Significance was determined at the following cutoff points (*P < 0.05, **P < 0.01, ***P < 0.001). Significance from the survival time was quantified using a log-rank test.
Supplementary Material
Acknowledgments
We thank Z. Niziolek and J. Nelson of the Bauer Core at Harvard University for technical assistance with flow cytometry, D. Pan of Harvard University for sharing biological samples, D. Vogus of Harvard University for advice on the animal model, and J. Alvarenga of the Wyss Institute for Biologically Inspired Engineering at Harvard University for technical assistance with AFM. Funding: We acknowledge support from the NIH through grant R01 HL143806 and the Wyss Institute for Biologically Inspired Engineering. Author contributions: C.W.S., M.A.E., L.L.-W.W., and S.M. designed the research. C.W.S., M.A.E., L.L.-W.W., N.B., S.I., D.W., Z.Z., A.U., and A.P. performed the research. C.W.S., M.A.E., L.L.-W.W., N.B., S.I., and S.M. analyzed the data. C.W.S. and S.M. wrote the paper with input from all authors. S.M. conceived the project. Competing interests: A patent application has been filed by Harvard University based on the technology described in this study (WO/2019/139892). S.M., M.A.E., and C.W.S. are inventors on this patent application. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz6579/DC1
REFERENCES AND NOTES
- 1.Khalil D. N., Smith E. L., Brentjens R. J., Wolchok J. D., The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 13, 273–290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miliotou A. N., Papadopoulou L. C., CAR T-cell therapy: A new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 19, 5–18 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Hucks G., Rheingold S. R., The journey to CAR T cell therapy: The pediatric and young adult experience with relapsed or refractory B-ALL. Blood Cancer J. 9, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee S., Kivimäe S., Dolor A., Szoka F. C., Macrophage-based cell therapies: The long and winding road. J. Control. Release 240, 527–540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wynn T. A., Chawla A., Pollard J. W., Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez F. O., Gordon S., The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 6, 13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown J. M., Recht L., Strober S., The Promise of Targeting Macrophages in Cancer Therapy. Clin. Cancer Res. 23, 3241–3250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultze J. L., Schmieder A., Goerdt S., Macrophage activation in human diseases. Semin. Immunol. 27, 249–256 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Evans M. A., Huang P. J., Iwamoto Y., Ibsen K. N., Chan E. M., Hitomi Y., Ford P. C., Mitragotri S., Macrophage-mediated delivery of light activated nitric oxide prodrugs with spatial, temporal and concentration control. Chem. Sci. 9, 3729–3741 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams C. B., Yeh E. S., Soloff A. C., Tumor-associated macrophages: Unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer 2, 15025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Champion J. A., Mitragotri S., Role of target geometry in phagocytosis. Proc. Natl. Acad. Sci. U.S.A. 103, 4930–4934 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doshi N., Swiston A. J., Gilbert J. B., Alcaraz M. L., Cohen R. E., Rubner M. F., Mitragotri S., Cell-based drug delivery devices using phagocytosis-resistant backpacks. Adv. Mater. 23, H105–H109 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Anselmo A. C., Gilbert J. B., Kumar S., Gupta V., Cohen R. E., Rubner M. F., Mitragotri S., Monocyte-mediated delivery of polymeric backpacks to inflamed tissues: A generalized strategy to deliver drugs to treat inflammation. J. Control. Release 199, 29–36 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Xia J., Wang Z., Huang D., Yan Y., Li Y., Guan J., Asymmetric biodegradable microdevices for cell-borne drug delivery. ACS Appl. Mater. Interfaces 7, 6293–6299 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Klyachko N. L., Polak R., Haney M. J., Zhao Y., Gomes Neto R. J., Hill M. C., Kabanov A. V., Cohen R. E., Rubner M. F., Batrakova E. V., Macrophages with cellular backpacks for targeted drug delivery to the brain. Biomaterials 140, 79–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang P., Guan J., Fabrication of multilayered microparticles by integrating layer-by-layer assembly and microcontact printing. Small 7, 2998–3004 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Huang B., Abraham W. D., Zheng Y., Bustamante López S. C., Luo S. S., Irvine D. J., Active targeting of chemotherapy to disseminated tumors using nanoparticle-carrying T cells. Sci. Transl. Med. 7, 291ra294 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang L., Zheng Y., Melo M. B., Mabardi L., Castaño A. P., Xie Y.-Q., Li N., Kudchodkar S. B., Wong H. C., Jeng E. K., Maus M. V., Irvine D. J., Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 36, 707–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shields C. W. IV, Wang L. L.-W., Evans M. A., Mitragotri S., Materials for Immunotherapy. Adv. Mater. 2019, e1901633 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Schroder K., Hertzog P. J., Ravasi T., Hume D. A., Interferon-γ: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Vasconcellos F. C., Swiston A. J., Beppu M. M., Cohen R. E., Rubner M. F., Bioactive polyelectrolyte multilayers: Hyaluronic acid mediated B lymphocyte adhesion. Biomacromolecules 11, 2407–2414 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Tripathi P., Tripathi P., Kashyap L., Singh V., The role of nitric oxide in inflammatory reactions. FEMS Immunol. Med. Microbiol. 51, 443–452 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Steimle V., Siegrist C., Mottet A., Lisowska-Grospierre B., Mach B., Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 265, 106–109 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Wu C., Xue Y., Wang P., Lin L., Liu Q., Li N., Xu J., Cao X., IFN-γ primes macrophage activation by increasing phosphatase and tensin homolog via downregulation of miR-3473b. J. Immunol. 193, 3036–3044 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Labrousse A. M., Meunier E., Record J., Labernadie A., Beduer A., Vieu C., Safta T. B., Maridonneau-Parini I., Frustrated phagocytosis on micro-patterned immune complexes to characterize lysosome movements in live macrophages. Front. Immunol. 2, 51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.A. Mularski, F. Marie-Anaïs, J. Mazzolini, F. Niedergang, Observing Frustrated Phagocytosis and Phagosome Formation and Closure Using Total Internal Reflection Fluorescence Microscopy (TIRFM), G. Rousselet, Ed. (Humana Press New York, NY, 2018). [DOI] [PubMed] [Google Scholar]
- 27.Mittal K., Ebos J., Rini B., Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin. Oncol. 41, 235–251 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Doedens A. L., Stockmann C., Rubinstein M. P., Liao D., Zhang N., DeNardo D. G., Coussens L. M., Karin M., Goldrath A. W., Johnson R. S., Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 70, 7465–7475 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scodeller P., Simón-Gracia L., Kopanchuk S., Tobi A., Kilk K., Säälik P., Kurm K., Squadrito M. L., Kotamraju V. R., Rinken A., de Palma M., Ruoslahti E., Teesalu T., Precision Targeting of Tumor Macrophages with a CD206 Binding Peptide. Sci. Rep. 7, 14655 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pusuluri A., Krishnan V., Wu D., Shields C. W. IV, Wang L. W., Mitragotri S., Role of synergy and immunostimulation in design of chemotherapy combinations: An analysis of doxorubicin and camptothecin. Bioeng. Transl. Med. 4, e10129 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng R., Wang S. M., Yin T., Ye T. H., Shen G. B., Li L., Zhao J. Y., Sang Y. X., Duan X. G., Wei Y. Q., Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol. Res. 20, 473–482 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Durante W., Johnson F. K., Johnson R. A., Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharmacol. Physiol. 34, 906–911 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrotta C., Cervia D., di Renzo I., Moscheni C., Bassi M. T., Campana L., Martelli C., Catalani E., Giovarelli M., Zecchini S., Coazzoli M., Capobianco A., Ottobrini L., Lucignani G., Rosa P., Rovere-Querini P., de Palma C., Clementi E., Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with syntaxin 4 and acid sphingomyelinase inhibition. Front. Immunol. 9, 1186 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choo Y. W., Kang M., Kim H. Y., Han J., Kang S., Lee J. R., Jeong G. J., Kwon S. P., Song S. Y., Go S., Jung M., Hong J., Kim B. S., M1 macrophage-derived nanovesicles potentiate the anticancer efficacy of immune checkpoint inhibitors. ACS Nano 12, 8977–8993 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Wang Y., Lin Y.-X., Qiao S.-L., An H.-W., Ma Y., Qiao Z.-Y., Rajapaksha R. P. Y. J., Wang H., Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials 112, 153–163 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Song M., Liu T., Shi C., Zhang X., Chen X., Bioconjugated manganese dioxide nanoparticles enhance chemotherapy response by priming tumor-associated macrophages toward m1-like phenotype and attenuating tumor hypoxia. ACS Nano 10, 633–647 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zanganeh S., Hutter G., Spitler R., Lenkov O., Mahmoudi M., Shaw A., Pajarinen J. S., Nejadnik H., Goodman S., Moseley M., Coussens L. M., Daldrup-Link H. E., Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat. Nanotechnol. 11, 986–994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guerriero J. L., Sotayo A., Ponichtera H. E., Castrillon J. A., Pourzia A. L., Schad S., Johnson S. F., Carrasco R. D., Lazo S., Bronson R. T., Davis S. P., Lobera M., Nolan M. A., Letai A., Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li C.-X., Zhang Y., Dong X., Zhang L., Liu M.-D., Li B., Zhang M.-K., Feng J., Zhang X.-Z., Artificially reprogrammed macrophages as tumor-tropic immunosuppression-resistant biologics to realize therapeutics production and immune activation. Adv. Mater. 31, e1807211 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Edin S., Wikberg M. L., Dahlin A. M., Rutegård J., Öberg Å., Oldenborg P. A., Palmqvist R., The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLOS ONE 7, e47045 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan A., Hsiao Y.-J., Chen H.-Y., Chen H.-W., Ho C.-C., Chen Y.-Y., Liu Y.-C., Hong T.-H., Yu S.-L., Chen J. J.-W., Yang P.-C., Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci. Rep. 5, 14273 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., Cheng S., Zhang M., Zhen L., Pang D., Zhang Q., Li Z., High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLOS ONE 8, e76147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mills C. D., Lenz L. L., Harris R. A., A Breakthrough: Macrophage-Directed Cancer Immunotherapy. Cancer Res. 76, 513–516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Sullivan T., Saddawi-Konefka R., Vermi W., Koebel C. M., Arthur C., White J. M., Uppaluri R., Andrews D. M., Ngiow S. F., Teng M. W., Smyth M. J., Schreiber R. D., Bui J. D., Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J. Exp. Med. 209, 1869–1882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tremble L. F., Forde P. F., Soden D. M., Clinical evaluation of macrophages in cancer: Role in treatment, modulation and challenges. Cancer Immunol. Immunother. 66, 1509–1527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramesh A., Kumar S., Nandi D., Kulkarni A., CSF1R- and SHP2-inhibitor-loaded nanoparticles enhance cytotoxic activity and phagocytosis in tumor-associated macrophages. Adv Mater 31, e1904364 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Parayath N. N., Parikh A., Amiji M. M., Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating MicroRNA-125b. Nano Lett. 18, 3571–3579 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Liu Y., Liang X., Yin X., Lv J., Tang K., Ma J., Ji T., Zhang H., Dong W., Jin X., Chen D., Li Y., Zhang S., Xie H. Q., Zhao B., Zhao T., Lu J., Hu Z. W., Cao X., Qin F. X. F., Huang B., Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 8, 15207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Miller C. H., Maher S. G., Young H. A., Clinical Use of Interferon-gamma. Ann. N. Y. Acad. Sci. 1182, 69–79 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Razidlo G. L., Burton K. M., McNiven M. A., Interleukin-6 promotes pancreatic cancer cell migration by rapidly activating the small GTPase CDC42. J. Biol. Chem. 293, 11143–11153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arango Duque G., Descoteaux A., Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 5, 491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carreno B. M., Magrini V., Becker-Hapak M., Kaabinejadian S., Hundal J., Petti A. A., Ly A., Lie W. R., Hildebrand W. H., Mardis E. R., Linette G. P., A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang T., Wang D., Yu H., Feng B., Zhou F., Zhang H., Zhou L., Jiao S., Li Y., A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 9, 1532 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheung A. S., Zhang D. K. Y., Koshy S. T., Mooney D. J., Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat. Biotechnol. 36, 160–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tong H. I., Kang W., Davy P. M. C., Shi Y., Sun S., Allsopp R. C., Lu Y., Monocyte trafficking, engraftment, and delivery of nanoparticles and an exogenous gene into the acutely inflamed brain tissue - evaluations on monocyte-based delivery system for the central nervous system. PLOS ONE 11, e0154022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raimondo T. M., Mooney D. J., Functional muscle recovery with nanoparticle-directed M2 macrophage polarization in mice. Proc. Natl. Acad. Sci. U.S.A. 115, 10648–10653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shahbazi M.-A., Sedighi M., Bauleth-Ramos T., Kant K., Correia A., Poursina N., Sarmento B., Hirvonen J., Santos H. A., Targeted reinforcement of macrophage reprogramming toward M2 polarization by IL-4-loaded hyaluronic acid particles. ACS Omega 3, 18444–18455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang X., Goncalves R., Mosser D. M., The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. Chapter 14, Unit 14.1 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andreesen R., Scheibenbogen C., Brugger W., Krause S., Meerpohl H. G., Leser H. G., Engler H., Löhr G. W., Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating blood monocytes: A new approach to cancer immunotherapy. Cancer Res. 50, 7450–7456 (1990). [PubMed] [Google Scholar]
- 60.Weis M., Shan J., Kuhlmann M., Jungst T., Tessmar J., Groll J., Evaluation of hydrogels based on oxidized hyaluronic acid for bioprinting. Gels 4, E82 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz6579/DC1