

Bacterial horizontal gene transfer provides functions that help explain the evolutionary success of aquatic primary producers.
Abstract
The extent and role of horizontal gene transfer (HGT) in phytoplankton and, more broadly, eukaryotic evolution remain controversial topics. Recent studies substantiate the importance of HGT in modifying or expanding functions such as metal or reactive species detoxification and buttressing halotolerance. Yet, the potential of HGT to significantly alter the fate of species in a major eukaryotic assemblage remains to be established. We provide such an example for the ecologically important lineages encompassed by cryptophytes, rhizarians, alveolates, stramenopiles, and haptophytes (“CRASH” taxa). We describe robust evidence of prokaryotic HGTs in these taxa affecting functions such as polysaccharide biosynthesis. Numbers of HGTs range from 0.16 to 1.44% of CRASH species gene inventories, comparable to the ca. 1% prokaryote-derived HGTs found in the genomes of extremophilic red algae. Our results substantially expand the impact of HGT in eukaryotes and define a set of general principles for prokaryotic gene fixation in phytoplankton genomes.
INTRODUCTION
The study of horizontal gene transfer (HGT) in eukaryotes is fundamentally more challenging than in prokaryotes due to the generally much larger eukaryote genome size, substantially greater complexity, and the sporadic and low frequency of foreign gene acquisition (1, 2). Many of these issues can be overcome with dense sampling of eukaryotic clades to accurately reconstruct the patterns of gene gain and loss in closely related species. In addition, the use of long-read sequencing to generate large individual reads [e.g., 10 to 50 kilo–base pair (kbp) in size] that encompass both foreign and native genes largely mitigates potential genome assembly issues resulting from incorporation of contaminant (prokaryotic or other) DNA (3). The recent availability of high-quality genome data from algae and related species begins to address these fundamental limitations (4–6). Here, we take on the challenge of quantifying prokaryote-derived HGT by focusing on the most taxonomically diverse eukaryote lineage known, the “CRASH” taxa that include cryptophytes, rhizarians, alveolates, stramenopiles, and haptophytes. Because of the taxonomic bias inherent in currently available high-quality genome data (i.e., favoring relatively small genomes of cultured, photosynthetic organisms), we included at least one lineage from each CRASH lineage in the analysis and focused our work on the stramenopiles for which the largest genome collection exists.
Among eukaryotic plankton none are more important than the CRASH, whose members include photosynthetic diatoms and dinoflagellates, as well as heterotrophs, saprobes, and the malaria parasite, many of which dominate marine environments and form the base of food webs or are important animal and plant pathogens. The diatoms alone contribute ca. 20% of global primary production (7). CRASH genomes are, however, challenging to study because they contain genes derived from one (due to the red algal plastid in many taxa) and most likely two algal endosymbioses (i.e., the other is an anciently derived, cryptic green plastid that was lost) that have left hundreds of algal genes in their genomes resulting from endosymbiotic gene transfers (EGTs), as well as independent HGTs (6, 8–10). For this reason, we limited our study of HGT to prokaryotic genes that have arisen via independent HGTs, not EGTs of putative bacterial origin arising from algal endosymbiosis. We recognize that the phylogeny of CRASH is unsettled and although the SAR forms a well-supported group, the other two phyla (cryptophytes and haptophytes) are of uncertain affiliation. Therefore, even though we used a reference tree to analyze HGT distribution, the results we describe are robust in the face of basal topological rearrangements in the CRASH phylogeny. Our work shows that 0.16 to 1.44% of the gene inventories in individual CRASH species constitute prokaryote-derived genes. We discuss these results in context to HGT in other eukaryote lineage and the potential role these foreign sequences play in functional diversification.
RESULTS AND DISCUSSION
Phylogenomic results
We gathered >524 K protein sequences from 23 species representing the five different CRASH lineages (Fig. 1 and data file S1). Using a combination of phylogenomics and a phylogeny-independent approach [Alien Index (AI)] (11, 12), we identified putative prokaryote-derived HGTs that originated after the divergence of each CRASH lineage (Fig. 1). Phylogenomics identifies genes in queried genomes that form monophyletic groups with prokaryotic homologs in phylogenetic trees (see Materials and Methods), whereas the AI approach identifies query genes that are more similar in sequence to prokaryotes than to eukaryotes (excluding the phyla that the query taxa belongs to; Materials and Methods). Genes supported by both methods have a high likelihood of being genuine prokaryote-derived HGTs. To validate these results, we mapped each HGT candidate to the relevant CRASH genome contig (i.e., assuming correct assembly) to study the phylogenetic history of flanking genes. We considered two or more HGT-derived genes that are physically linked to each other as being a single transfer event. Up to five flanking genes were used as queries to search the RefSeq database using BLASTp (e value cutoff = 1 × 10−3). Flanking genes were categorized as either prokaryotic or eukaryotic based on the BLASTp top hit (excluding self-hits). Species-specific genes were marked as “no hit.” Four types of HGT validation resulted from this analysis: Type 1 HGTs are flanked only by eukaryotic genes on both sides; type 2 are flanked by eukaryotic genes on one side and species-specific genes on the other; types 1 and 2 HGTs are therefore unlikely to be explained by contamination and represent 92% of all HGTs we report (fig. S1 and data file S2). Type 3 HGTs are located in contigs that lack flanking genes, and type 4 are flanked by a mixture of eukaryotic, species-specific, and prokaryotic genes. HGTs of types 3 and 4 are shared by more than one species of the same phylum (i.e., are not singletons in trees) and contain an average of 4.4 spliceosomal introns per gene (77 genes are, however, intron free), providing additional support for a eukaryotic (noncontaminant) origin (data file S3). Given the conservative nature of the approaches we have used, combined with unavoidable issues such as sporadic gene loss in queried genomes, changes in gene structure (e.g., domain fusions), and large variation in sequence divergence, our results most likely exclude many true HGTs and provide a reduced, high-confidence estimate. An example HGT we found is pantothenate kinase genes in stramenopiles that diverge deeply within the prokaryotic phylogeny and likely derive from an anciently split cyanobacterial lineage (Fig. 2A, left). Given the reference tree of life, invoking vertical transmission followed by “differential loss” as explanation for this tree topology would require the unlikely scenario of independent gene losses in four CRASH taxa, and two (or three) more losses in other eukaryotic lineages (Fig. 2A, right).
Fig. 1. Summary of prokaryotic-derived HGTs in 23 CRASH taxa.
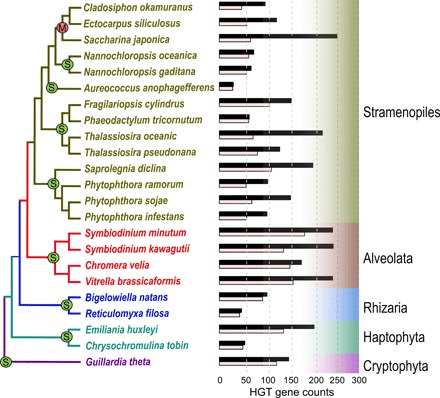
The bar plot indicates the amount of HGT detected in each species, with the black bar representing gene counts, and the pink bar, gene families. The red “M” and green “S” indicate multicellular and unicellular taxa, respectively.
Fig. 2. Putative origins of HGTs in CRASH taxa and phenotypes of HGT donors.
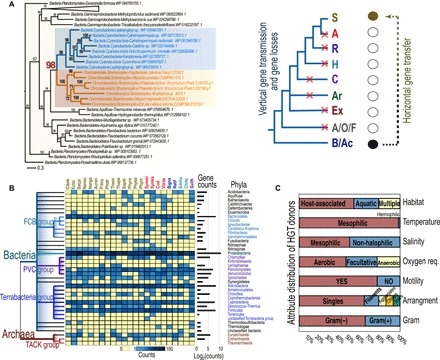
(A) Maximum likelihood tree of pantothenate kinase genes (left) and the two scenarios to explain the phylogeny (right). The simplified tree of life includes Cryptophyta (C), Rhizaria (R), Alveolata (A), stramenopiles (S), Haptophyta (H), Archaeplastida (Ar), Excavata (Ex), Amoebozoa/Opisthokonta/Fungi (A/O/F), and Bacteria/Archaea (B/Ac). Red crosses on the branches indicate putative gene losses in the tree. The dashed arrow marks the direction of gene transfer. (B) MMSH-based inference of HGT donors. Prokaryote phylogeny (left) was retrieved from the NCBI taxonomy database. The total HGT counts for each donor phylum (row) are shown as a bar plot (right edge). The TACK group includes Thaumarchaeota, Aigarchaeota, Chrenarchaeota and Korarcheaota. (C) MMSH-based phenotype of HGT donors. The phenotype data were retrieved from the NCBI microbial attributes database. The dominant types are indicated.
Our analysis identified broadly differing numbers of HGTs (29 to 254) in each CRASH lineage (3248 genes in total) (data files S1 and S3) with the frequency varying ~10-fold, accounting for 0.12 to 1.36% of the 23 studied nuclear gene inventories. These numbers differ from previous estimates made for red algae (~5%) (13), dinoflagellates Alexandrium tamarense (1 to 2%) (14), amoebozoa (<1%), and fungi (0.12%) (1), presumably due to differences in the databases used and the more narrow focus of these studies. Our results are, however, consistent with a recent analysis of 10 Galdieria (extremophilic red algae) genomes that showed ca. 1% prokaryotic HGTs in their gene inventories under the condition that the HGT was shared by at least two species (3). This consistency across two widely different groups of photosynthetic eukaryotes hints at a broader principle for prokaryotic HGT in phytoplankton genomes. Between 28 and 184 independent HGT events (each represented by a single HGT gene or a family of HGT paralogs) were found in the 23 species with 1.08 to 3.79 genes per family (data file S1). The largest families resulted from expansions of genes encoding prokaryote-derived mannuronan C-5-epimerase (76 copies) (fig. S2A) and polysaccharide lyase (23 copies) (fig. S2B) in brown algae and 120 self-repeat family proteins in the diatom (fig. S2C). The growth in number of mannuronan C-5-epimerase genes is potentially implicated in polysaccharide (alginate) biosynthesis in brown algae (5). In addition, the gene family expansion of prokaryote-derived C-5 cytosine-specific DNA methylase-encoding genes (55 copies in Symbiodinium kawagutii and 9 copies in Symbiodinium minutum) (data file S3) coincides with strong epigenetic activity in Symbiodinium species (15). Other interesting HGT cases include two genes in the polyamine metabolic pathway (S-adenosylmethionine decarboxylase and spermidine synthase) in Fragilariopsis cylindrus (fig. S2D). Polyamine is involved in stress response and cell division control, thereby promoting growth (16). The transfer of these two related gene functions was likely associated with the adaptation of F. cylindrus to cold polar waters. Similarly, two HGTs encoding a zinc-binding dehydrogenase and a CHC2 zinc finger in F. cylindrus may reflect adaptations to the relatively high zinc concentration in the Southern Ocean (17). To simplify the ensuing discussion, we will henceforth use HGT gene counts rather than gene family counts.
HGT origins in CRASH taxa
Consistent with the highly reticulate nature of bacterial genome evolution (and partially due to unresolved phylogenetic relationships), a majority of the HGTs (~59%; fig. S3) have unclear origins at the phylum level and are sister to monophyletic groups comprising two or more prokaryotic phyla (fig. S4A). Other HGTs (~20%) are positioned within groups from the same prokaryotic phylum, indicating potential donor sources (e.g., Cyanobacteria in Fig. 2A). These HGTs were derived from 24 different prokaryotic phyla (fig. S4B and data file S3). The remaining HGTs (~21%) formed monophyletic groups with a single prokaryotic taxon suggesting potential HGT sources at the species or strain level (fig. S4C). These latter HGTs were contributed by ca. 40 different prokaryotic phyla (unclassified phyla not included) (fig. S4C and data file S3). Although our database represents only a small fraction of prokaryotic sequences in nature and extensive HGT among these lineages very likely misleads the inference of HGT origins, our results suggest that CRASH taxa have recruited genes from a large diversity of prokaryotic sources. Of particular interest is 48 genes that were putatively derived from cyanobacteria corresponding to 29 separate HGT events (data file S4). In addition to the example shown in Fig. 2A, we identified two HGTs involving α-cyanobacteria, including an ABC transporter adenosine 5′-triphosphate (ATP)–binding protein encoding gene transferred from Prochlorococcus-like lineages to the dinoflagellate Symbiodinium (fig. S5A), and a DUF1254 domain–containing protein encoding gene transferred from Synechococcus-like lineages to several haptophytes (fig. S5B). The abundant marine picocyanobacterium Synechococcus is closely related to the donor of plastids in the thecate amoeba Paulinella spp. that resulted from a relatively recent endosymbiosis ~124 million years ago (18–20). It should be noted that the lack of a large number of HGTs from single donor sources contrasts with the obvious single-donor gene transfers often associated with endosymbiosis and EGT (8, 21).
Putative phenotypes of HGT donors
Given the unclear origins for the majority of the HGTs, we used a monophyletic most similar homolog (MMSH) approach to infer the putative phenotype and lifestyle of donor lineages. For a given HGT query, its MMSH is defined as the prokaryotic gene with the highest sequence identity to the query among all those within the smallest prokaryote-query monophyletic group (dark blue tick marks in fig. S4). The MMSH annotation for the complete HGT data revealed some overrepresented groups such as the FCB group, Proteobacteria, Cyanobacteria, and Firmicutes (Fig. 2B). Given these results, predominant features of donor prokaryotes included presence in aquatic habitats and a mesophilic (with respect to temperature and salinity) and aerobic lifestyle, consistent with the ecological distribution of most extant CRASH taxa (Fig. 2C). The predominant MMSH phenotype being unicellular, rod shaped, and motile (Fig. 2C) suggests that a shared lifestyle and morphology between the HGT donor and recipient species may increase the likelihood of gene transfer (22, 23). Many HGTs may have resulted from engulfment of prokaryotic cells as food sources (24). Whereas unicellular eukaryotes are expected to undergo HGT events more often than multicellular lineages due to the absence in the former of a sequestered germ line and frequent asexual reproduction, it is interesting that no significant difference is observed in HGT counts between unicellular and multicellular species (Phaeophyceae) in our study (Fig. 1). This may be explained by the single cell stages (e.g., spores, zygotes, and embryos) of multicellular organisms that are amenable to foreign DNA transfer (25, 26).
Recent HGTs in CRASH species
We identified 6 to 148 prokaryote-derived genes that were limited to single CRASH species or genera (e.g., Nannochloropsis and Phytophthora) (1002 in total), accounting for ~30% of the total number of HGTs identified in this study (data files S3 and S5). Under the differential loss hypothesis, these results would require a massive number of independent events to explain the data and is therefore considered highly unlikely. For example, the prokaryote-derived tetracycline efflux MFS (major facilitator superfamily) transporter Tet(C) encoding gene in Nannochloropsis oceanica (Fig. 3A) requires two additional losses in the well-sampled stramenopiles (nine gene losses in total; Fig. 3A). Another example represents the chlorophyll synthesis pathway protein BchC encoding a gene present in two Phytophthora species (Fig. 3B). Vertical transmission requires gene losses in oomycetes and photosynthetic stramenopiles (nine gene losses in total; Fig. 3B). Given their narrow taxonomic distribution in eukaryotes, these 1002 cases likely represent recent HGT events (data file S3).
Fig. 3. Examples of HGTs that occurred in limited CRASH species.
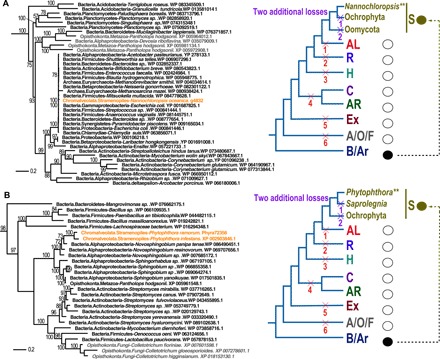
(A) Maximum likelihood tree of efflux MFS transporter Tet(C) gene. (B) Maximum likelihood tree of BchC. The inference of gene loss under the HGT and differential loss scenarios are as in Fig. 2. Eukaryotic sequences from CRASH are marked in orange, whereas the others are marked in gray.
There has been considerable discussion and understandable skepticism about the validity of prokaryote-derived gene transfers in single or a few eukaryotic genomes because these may have arisen from contamination (27). This view, however, ignores many bona fide HGT cases that have been experimentally validated [e.g., (28, 29)]. To address this issue, we identified 461 HGTs that are restricted to a single genus but shared by two or more different species (data file S3). These genes represent 152 separate HGT events (data file S6) after removing redundancy due to lineage splits. The BchC phylogeny shown in Fig. 3A is one such example where α-proteobacterial genes were transferred to the common ancestor of two Phytophthora species (Phyra72356 and Phytophthora infestans XP_002903846). These types of shared HGTs are primarily found in the broadly studied genera targeted in this study, including Phytophthora (Phytophthora sojae, Phytophthora ramorum, and Phytophthora parasitica) (Fig. 3B), Nannochloropsis (Nannochloropsis oceanica and Nannochloropsis gaditana) (fig. S5C), Symbiodinium (S. kawagutii and S. minutum) (fig. S5D). Given the low likelihood of contamination from the same prokaryotic genes in genome assemblies from different species (mostly produced at different locations), the more likely explanation for these results is HGT.
HGT-derived gene features
Following integration into host genomes, the fate of foreign genes largely depends on their features such as gene length, gene structure, and the surrounding genomic environment (30). We examined these features of HGT-derived genes using host “core” genes as the benchmark. The latter represents the most conserved CRASH gene inventory (see Supplementary Materials). The coding sequence (CDS) length of HGTs and their MMSHs are significantly shorter than that of the homologous host core genes (P < 0.01; Fig. 4A). No significant length difference between HGT-derived CDSs and their MMSHs was however detected (P > 0.1), suggesting the preservation of shorter prokaryotic CDS length following gene transfer into eukaryotic hosts. It has been widely reported that long fragments of foreign DNA undergo more rapid deactivation and are less likely to be fixed in recipient prokaryote genomes than shorter insertions (27, 30, 31). This is consistent with the significantly shorter CDS length of HGTs than host core genes (Fig. 4A), suggesting that size of the transferred prokaryote DNA is negatively correlated with its likelihood of fixation. HGTs tend to be located in gene-poor regions when compared with host core genes (Fig. 4B), consistent with findings in other studies (10). HGTs are more likely to be single-exon genes and contain fewer spliceosomal introns than core genes (Fig. 4, C and D, and data file S3), whereas intron lengths in HGT-derived genes are not significantly different than in core genes (fig. S6A). The acquisition of introns, most of which contain the canonical splice site motifs (GT-AG) (fig. S6B), suggests adaptation of HGTs to the eukaryotic splicing machinery (21, 32–34). In addition, intron gains are common in functional HGTs and likely drive their retention via increased gene expression (30).
Fig. 4. Gene structure and functional annotation of HGTs.
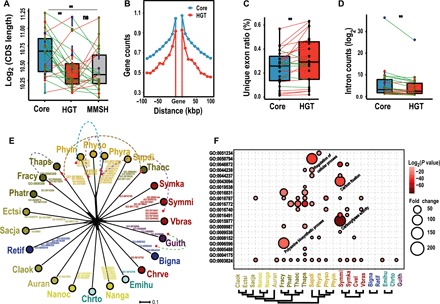
(A) The smaller CDS length in HGTs than in core genes is shown. The bar plots in blue, red, and gray indicate CRASH host core genes, HGTs, and their corresponding MMSH in prokaryotes, respectively. Each dot in the boxplot charts indicates the mean value for a CRASH species. Lines connect the same species across bar plots, with green color indicating decreases in value from left to right and red color indicating increases in value. The dots along the central vertical lines of bar plots are colored differentially for stramenopiles (brown), Alveolata (red), Rhizaria (blue), Haptophyta (green), and Cryptophyta (purple). (B) The lower gene density in genomic regions flanking HGTs than in CRASH core genes is shown. The 100 kbp upstream and downstream were surveyed in all 23 CRASH species; red line represents gene density of HGTs, whereas the blue line is for core genes. Every point stands for the average count of genes in the 10k length bin, using all data from 23 species. (C) The higher frequency of single-exon genes in HGTs than in core genes is shown. (D) Here, the lower intron number per gene in intron-bearing HGTs than in core genes is shown. The combined data from 23 CRASH species are shown as the blue dots. (E) The functional divergence of HGTs. Splits tree of 23 CRASH lineages based on the novel functions acquired in each lineage via HGT. The dashed lines connect gene functions present in two or more lineages. (F) Functional enrichment of HGTs. Only significantly enriched GO terms (P < 0.05 after multitest correction) are included. The probability value and fold change are indicated by the colors and circle sizes. **P < 0.001.
HGTs also differ significantly from core genes with respect to coding region guanine-cytosine (GC) content (fig. S6, C to M), codon usage, and gene expression level. Individual species or clades are typically characterized by lineage-specific GC content (35). Overall, CRASH core genes and HGTs differ significantly in GC content, whereas they both show much broader GC distributions (0.4 to 0.7) than the corresponding MMSHs (4.8 to 5.7) (fig. S6C). In each species, GC content in HGT-derived genes largely reflects those in the core genes (fig. S6C, lines in green and red), consistent with their ongoing genomic domestication. GC content at third codon positions is significantly higher in HGT-derived than in core genes (fig. S6I). The same is true for C content (C3s; fig. S6E) but not for G content (G3s; fig. S6G). Codon usage is an important factor that determines the fate of HGTs due to the need for compatibility with the transcription machinery and temporal RNA (tRNA) pool in the host (30, 36). HGTs differ from CRASH core genes with respect to having significantly lower CAI (codon adaptation index), CBI (codon bias index), and Fop (frequency of optimal codon usage) values (fig. S6, N to P; for the full dataset of CRASH data). The CAI quantifies similarity in synonymous codon usage between test genes and those representing a set of highly expressed genes and therefore approximates the likelihood of heterologous gene expression in a given species (37). Consistent with observations in other species (38), a significant positive correlation between CAI and gene expression level is found when analyzing ~600 CRASH transcriptomes (fig. S6Q). HGTs generally have lower CBI and Fop values than core genes, reflecting their suboptimal codon usage in terms of gene expression (fig. S6, O and P). These codon biases suggest incompatibility between HGT-derived genes and the tRNA pool in their CRASH hosts and therefore likely negatively affect foreign gene expression by limiting transcription speed.
Given this prediction, we explored the functions of HGTs by analyzing ~600 CRASH transcriptome datasets (data file S11). We compared gene expression profiles between host core genes and HGTs that have shared Gene Ontology (GO)/Kyoto Encyclopedia of Genes and Genomes (KEGG)/Pfam terms (fig. S7). For each species, the mean difference in gene expression between the two gene categories was assessed within each annotated GO/KEGG/Pfam term using the Student’s t test. GO/KEGG/Pfam terms with less than five genes in either gene category were ignored in this analysis. The differences in expression dispersal (coefficient of variation: SD across genes or samples/mean value) and expression specificity {frequencies of a gene to be detected as unexpressed [defined as transcripts per million (TPM), <1] in any condition} were assessed in a similar way. Given the variable experimental conditions associated with different transcriptome data for each species, gene expression values for a gene were used regardless of the condition. Compared with CRASH core genes, HGTs generally show lower expression levels (fig. S7, A, D, and G), consistent with their compromised gene expression activity as predicted by lower CAI, CBI, and Fop values (fig. S6, N to Q). In contrast, gene expression (estimated using protein and mRNA levels) is a major date determinant of sequence evolution (36). The evolutionary rate of a protein is strongly negatively correlated with its mRNA abundance (39). Lower expression of HGT-derived genes suggests the likely rapid evolutionary rate, which may be beneficial in terms of accelerating adaptation of codon usage. Moreover, HGT genes display higher expression dispersal and higher expression specificity (see Materials and methods) compared with CRASH core genes (fig. S7, B, C, E, F, H, and I), supporting their likely roles in driving lineage-specific adaptation and species divergence within the CRASH assemblage. These results indicate that HGTs might be implicated in functions that are important in specific conditions such as response to environmental stressors. Given these results, the expectation is that over time, selection on CRASH lineages will lead to the adaptation of HGT-derived genes to their domestic tRNA pools and regulatory machineries via alterations in GC content, intron length, and codon usage.
In general, synonymous mutations are due to neutral selection without functional implications, whereas fixation of nonsynonymous mutations often results from positive selection. We calculated the number of nonsynonymous mutations (Ka), synonymous mutations (Ks), and the ratio of these values (Ka/Ks) when comparing the HGTs and their prokaryotic putative most closely related homologs (MMSHs). We found that most HGT-derived genes have undergone purifying selection (i.e., Ka/Ks < 1; fig. S8A). Synonymous mutations therefore likely contributed primarily to CDS divergence between HGT genes and the relevant putative closest homologs in prokaryotes. Another contributing factor is the divergence in CDSs between paralogs. However, we found a positive correlation between Ks and CDS identity between HGTs and their MMSHs. That is, candidate HGT genes in eukaryotes tend to be more similar to their putative closest homologs in bacteria when they had higher synonymous substitution rates. This is the opposite pattern from what we would expect, given that higher substitution rates lead to increased sequence divergence. Yet, the negative correlation (as one would expect) applies only to a limited number of genes with high CDS identities (green dots in fig. S8B). This is explained by the unreliability of Ks measurements due to substitution saturation when distantly related sequences are compared. In contrast, nonsynonymous substitutions are more likely to be preserved when favored by natural selection. Thus, divergent paralogs tend to maintain more measurable nonynonymous substitutions, which is supported by the significant negative correlation between the Ka and CDS local identity (fig. S8C). We recognize that the comparison of HGT-prokaryotic putative closest homolog does not distinguish between sequence evolution that occurred before and after gene transfer. Nevertheless, the observed strong sequence conservation suggests functional constraints on HGTs that ultimately may play a role in driving the divergence of CRASH species. In summary, the differences between HGT-derived and native core genes are consistent with the prokaryotic provenance of the former and their ongoing domestication in CRASH taxa.
Functional divergence among HGTs
Two explanations exist for HGT fixation, sporadic gains and gene replacements whose adaptive potential is hard to decipher, or the more obvious gain of function or modification of existing pathways that result in novel phenotypes (30). With these alternative explanations in mind, we categorized the HGTs with respect to putative function using standard annotation databases (see Materials and methods). In 18/23 taxa, HGTs encoded novel GO terms (1 to 9) that were not present in the corresponding CRASH host gene inventories. These could be explained by the transfer of previously absent functions into the host or replacement of equivalent functions (data file S7) (28). Several functions were independently transferred into two or more lineages, such as amine metabolic process and sulfuric ester hydrolase activity (data file S7). Neighbor-net–based analysis of these novel functions (i.e., GO terms) resulted in a star-like topology, suggesting independent acquisitions among lineages (Fig. 4E). The acquisition of these novel functions via HGT might be associated with lineage-specific adaptations (1). Regarding functions present in both HGTs and host gene inventories, we identified significant HGT enrichment in 15 taxa [false discover rate (FDR) < 0.05] (Fig. 4F and data file S8). The highest enrichments include carboxylyase activity (>325×), carbon fixation (122×), and cellular metabolic compound salvage (97×) in S. minutum, polyamine biosynthetic process (114×) in the diatom F. cylindrus, and regulation of cellular process (109×) in the oomycete Saprolegnia diclina (data file S8). Unexpectedly, catalytic activity is significantly enriched in all of these 15 taxa (two- to sixfold, P = 1.73 × 10–8 to 0.03, and FDR < 0.05). This is followed by hydrolase and transferase activity that are enriched in eight taxa (data file S8). As shown in Fig. 4E, the overall functions encoded by HGTs (including both specific to HGTs and shared with host genes) largely followed the trend of independent evolution, in contrast to CRASH native core genes (fig. S9). Whereas the significance of HGTs to specific aspects of lineage adaptation remains to be investigated using genetic tools, it is clear that this process has contributed greatly to functional diversification among CRASH lineages. This was apparently not the case for carbohydrate-active enzymes and metabolite transporters (fig. S10).
In summary, despite the long-standing controversy about the existence and extent of HGTs in eukaryotes (27, 40), our results show that prokaryote-derived HGT is prevalent in the CRASH assemblage. These findings argue against the alternative differential loss hypothesis for the presence of prokaryote-derived genes in eukaryotes. Many of the HGTs we found appear to drive functional divergence among lineages and, overall, show a different expression pattern than native genes. Our study highlights the power of broad taxonomic sampling and integration of structural and functional data in investigating HGTs in eukaryotes. We predict that additional prokaryote-derived HGTs will be identified as a larger number of high-quality phytoplankton and other eukaryote genomes based on long-read sequencing become available in the coming years.
MATERIALS AND METHODS
Experimental design
To include as much genome data as possible into our analysis of HGT, we first downloaded protein sequences from the National Center for Biotechnology Information (NCBI) RefSeq database (version 82) (ftp://ftp.ncbi.nlm.nih.gov/refseq/). Sequences associated with unknown species or derived from environmental studies were discarded. Given the underrepresentation of algal lineages in RefSeq, we then included the extensive algal protein data from the MMETSP (Marine Microbial Eukaryotic Transcriptome Sequencing Project) (41) and other public sources (data file S1). The collected protein sequences were combined into a central database (“refseq+algae”), followed by removal of highly similar sequences (sequence identity ≥90%) from each order (e.g., Brassicales or primates) using CD-HIT version 4.5.4 (42). This resulted in a protein database comprising 39.9 million sequences from >7786 taxa (data file S9) with reasonable coverage of CRASH lineages (data file S10). These data were the target of the statistical analyses described below.
Statistical analysis
Algal phylogenomics
To search for genes with potential prokaryotic origins, we used the CRASH protein sequences as queries to search against the refseq+algae database using USEARCH under the default settings (43). All sequences with detectable hits associated with prokaryotes (regardless of rank in the sorted output) were retained for downstream phylogenomic analyses as done in previous studies (13, 44). Briefly, to build single-gene phylogenetic trees, CRASH query proteins were searched against the refseq+algae database using BLASTp (e value cutoff = 1 × 10−5). For each query, the top 1000 significant hits, sorted by bit-score in a descending order (by default) were recorded. Up to 60 sequences corresponding to the BLASTp hits were then retrieved from the database with no more than 3 sequences for each genus and no more than 12 sequences for each phylum (scripts are available at https://github.com/hqiu17/HGTools). The significant hits (with query-hit alignment length ≥120 amino acids) were then re-sorted according to query-hit identity in descending order. A second set of homologous sequences (up to 60) was retrieved from the database following the aforementioned procedure. The two sets of homologous sequences plus the query were then combined and were aligned using MUSCLE version 3.8.31 under default settings (45). The resulting alignments were trimmed using TrimAI version 1.2 in an automated mode (-automated1) (46). The trimmed alignments (≥50 amino acids) were used for construction of phylogenetic trees using FastTree version 2.1.7 (47) under a “WAG + CAT” model with four rounds of minimum-evolution subtree pruning and regrafting (SPR) moves (-spr 4) and exhaustive maximum likelihood (ML) nearest-neighbor interchanges (-mlacc 2 -slownni). Branch support values were estimated using the Shimodaira-Hasegawa (SH) test (48) with FastTree version 2.1.7 (47).
Tree-based HGT inference
Phylogenetic trees were searched for topologies with CRASH query sequences being nested among prokaryotic sequences as in previous studies (13, 28). A nested position is defined as two or more monophyletic clades comprising queries and prokaryotic sequences supported by different nodes in a tree (13). Species belonging to the same phylum as query taxa were allowed in the monophyletic clades. Monophyletic clades were treated as one if they contained the same group of prokaryotic sequences but differed in sequences from optional taxa. Only nested positions that were supported with ≥0.85 SH test by at least one supporting node were retained. To validate the potential HGT candidates, phylogenetic trees were rebuilt using maximum likelihood method with IQ-Tree (49). The best evolutionary models were selected on the fly using the built-in ModelFinder (50). Branch support was estimated using 1200 ultrafast bootstrap (UFBoot) replicates (51). On the basis of the standard output of IQ-Tree analyses, we discarded queries that showed significantly different amino acid composition (P < 0.05) than the remaining sequences in the alignment. CRASH queries nested among prokaryotic sequences (supported by >85% UFBoot at one or more supporting nodes) were retained. A Java implementation of the phylogenetic tree-based scanning tool (NestedIn.jar) is available for download (https://github.com/hqiu17/NestedIn). We removed trees derived from short alignments (≤50 amino acids) or alignments with less than four prokaryotic sequences. We also examined the genomic locations of putative singleton HGTs (i.e., not shared with any other species from the same phylum) and removed those that are present in short contigs with three or less genes. Genomic contigs comprising ≥50% HGTs were discarded as contamination. A variable degree of contamination (from few to over a hundred genes) was found in a majority of studied genomes. Notably, dozens of contigs (comprising >5 K genes) were found in Cladosiphon okamuranus genome (data file S12).
AI analysis
We calculated the AI score for each query gene following previous methods (11). Using the BLASTp search results generated from phylogenomic pipeline as inputs, the AI score is calculated with the following formula
Using Saccharina japonica queries as an example, bbhG is the e value of best Basic Local Alignment Search Tool (BLAST) hit within group lineage (non-Stramenopiles eukaryotes), whereas bbhO is the e value of best BLAST hit to species outside of group (prokaryotes). When no significant BLAST hits were detected, the corresponding bbhG or bbhO was set to 1. AI score ranges from 461 (no eukaryotic hit and prokaryotic sequence identical to query) to −461 (no prokaryotic hit and identical eukaryotic hit to query). An AI score >0 indicates a better BLAST hit to the query species in prokaryotes. The higher the AI score is, the more similar is the queries to their prokaryotic homologs than to eukaryotic homologs. Since all BLASTp searches were queried against the same database (refseq+algae), it is reasonable to apply a single cutoff to all 23 CRASH query taxa. Because the AI score is combined with phylogenomics to infer HGTs, we arbitrarily used a less-stringent cutoff (AI > 10) than previous studies (11). HGTs supported by both the tree-based approach and the AI approach were considered for downstream analyses.
Orthologous gene family analysis, CRASH phylogeny construction
We did orthologous clustering of ~524 K proteins encoded in 23 sampled CRASH genomes (data file S1). All-against-all BLAST searches (version 2.2.28; e value cutoff = 1 × 10−10; local identity cutoff = 20%) were conducted for all combinations of any two genomes. Sequences were clustered using OrthoFinder (52) based on the BLAST table outputs with the default parameters and modifications [percentMatchCutoff = 20, evalueExponentCutoff = −10, and Markov Clustering (MCL) = 1.5]. This clustering resulted in 27,631 orthologous families with ≥2 members. A total of 1520 families containing genes from more than two-thirds of all included genomes (15 genomes) were considered as CRASH core gene families.
Gene function annotation
The CRASH protein sequences were used as queries to search against GO database using Blast2GO (53) and KEGG database using blastKOALA (http://www.kegg.jp/blastkoala/) with default parameters. For each annotated GO/KEGG term, the full gene sets of each species were set as the background to test for enrichment in HGT genes using Fisher’s exact test (P value cutoff = 0.01) with the Python module from SciPy software (https://docs.scipy.org/doc/scipy/reference/stats.html). In addition, we used HGT and core proteins to query the SEED (http://www.theseed.org/wiki/Home_of_the_SEED), IPR2GO (http://www.ebi.ac.uk/interpro/search/sequence-search), eggNOG (http://eggnogdb.embl.de/#/app/home), and Pfams, respectively. To visualize and explore the recombination networks of species based on the functions of core genes and HGT genes, unrooted neighbor-joining trees were built using SplitsTree (54). The data matrix of gene function term distribution in all species was used as the input data.
Functional prediction and enrichment analysis of CAZymes and transporters
Carbohydrate-active enzymes were annotated using the dbNCAN meta server (http://bcb.unl.edu/dbCAN2/blast.php) using the HMMER default option (55), and metabolite transporters were annotated using a BLASTp (e value cutoff = 1 × 10−10) search against a transporter database (56). The full gene set of each species (excluding HGTs) was used as background to test the enrichment in HGT genes of cazymes and particular transporters using the Student’s t test (P value cutoff = 0.05). The distribution of CAZymes and transporters was sporadic (fig. S10), and the enriched functions were all due to gene duplications following gene transfer.
Calculation of Ka/Ks and local CDS identity
In general, synonymous mutations are due to neutral selection without functional implications, whereas maintained nonsynonymous mutation tend to be caused by positive selection. We calculated Ka, Ks, and Ka/Ks values, comparing HGTs and their prokaryotic MMSHs. To construct back-translated nucleotide alignments, protein alignments between HGTs and the corresponding MMSHs were generated using MUSCLE version 3.8.31 under the default settings (45); thereafter, protein-coding DNA alignments were guided by their protein alignments and limited to ungapped regions using ParaAT (Parallel Alignment and back-Translation tool) (57). The Ka, Ks, and ω (Ka/Ks) between HGTs and the corresponding MMSHs were calculated using KaKs_Calculator 2.0 (https://sourceforge.net/projects/kakscalculator2) under the model-selected setting (58). Sequence identities with the coding DNA alignments (defined as local CDS identity) were calculated using BLAST with the default parameters (59). We found that most HGT genes have undergone purifying selection (i.e., Ka/Ks < 1) (fig. S8, for all species combined; fig. S26, for each species separately).
Codon usage
Indices of codon usage and GC content were calculated using CodonW 1.4.4 (http://codonw.sourceforge.net). Correlation tests between CAI and gene expression were carried out using the Spearman’s rank correlation analysis tool (P. Wessa, Free Statistics Software, Office for Research Development and Education, version 1.1.23-r7; https://www.wessa.net/).
Gene expression analysis
RNA sequencing data generated from ~600 samples corresponding to 19 CRASH species were downloaded from the NCBI Sequence Read Archive database (data file S11). We used FastQC (Babraham Institute; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to examine the overall sequencing quality of the raw reads. The FASTQ data were cleaned and trimmed using Trimmomatic (60) (SLIDINGWINDOW: 4:15, LEADING:3, TRAILING:3, ILLUMINACLIP: adapter.fa: 2: 30: 10). All short reads (< 36 base pairs) and broken pairs were discarded. The resulting high-quality reads were aligned to the corresponding genome assemblies using HISAT2 (61) for the Illumina data and SHRiMP (62) for the ABI (Applied Biosystems) solid data with the default parameters. The resulting mapping files (SAM format) were sorted and transformed to BAM format using SAMTools (63). TPM was calculated using the BAM files and the corresponding gene model annotation (GFF3 files) using StringTie (64) under the default settings.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Key Research and Development Program of China (2018YFD0900703), a Major Scientific and Technological Innovation Project of the Shandong Provincial Key Research and Development Program (2019JZZY020706); the Marine S&T Fund of Shandong Province for the Pilot National Laboratory for Marine Science and Technology (Qingdao) (2018SDKJ0406-3); the Central Public-interest Scientific Institution Basal Research Fund, CAFS(2020TD27); the China Agriculture Research System (CARS-50); the Financial Fund of the Ministry of Agriculture and Rural Affairs, P.R. China (NFZX2018); Taishan Scholars Funding and Talent Projects of Distinguished Scientific Scholars in Agriculture. We are grateful to the Center for High Performance Computing and System Simulation, the Pilot National Laboratory for Marine Science and Technology (Qingdao), and to the High Performance Scientific Computing and System Simulation Platform of the National Supercomputing Center (Jinan). D.B. was supported by a grant from the National Aeronautics and Space Administration (80NSSC19K0462) and a NIFA-USDA Hatch grant (NJ01170). Author contributions: N.Y. designed the project with contributions from X.F. H.Q., N.Y., and X.F. performed bioinformatics analysis. H.Q., X.F., and D.B. wrote the manuscript. All authors read and approved the manuscript before submission. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaba0111/DC1
REFERENCES AND NOTES
- 1.Schönknecht G., Weber A. P. M., Lercher M. J., Horizontal gene acquisitions by eukaryotes as drivers of adaptive evolution. Bioessays 36, 9–20 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Lacroix B. T., Citovsky V., Transfer of DNA from bacteria to Eukaryotes. mBio 7, e00863–16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rossoni A. W., Price D. C., Seger M., Lyska D., Lammers P., Bhattacharya D., Weber A. P. M., The genomes of polyextremophilic cyanidiales contain 1% horizontally transferred genes with diverse adaptive functions. eLife 8, e45017 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Price D. C., Goodenough U. W., Roth R., Lee J.-H., Kariyawasam T., Mutwil M., Ferrari C., Facchinelli F., Ball S. G., Cenci U., Chan C. X., Wagner N. E., Yoon H. S., Weber A. P. M., Bhattacharya D., Analysis of an improved Cyanophora paradoxa genome assembly. DNA Res. 26, 287–299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ye N., Zhang X., Miao M., Fan X., Zheng Y., Xu D., Wang J., Zhou L., Wang D., Gao Y., Wang Y., Shi W., Ji P., Li D., Guan Z., Shao C., Zhuang Z., Gao Z., Qi J., Zhao F., Saccharina genomes provide novel insight into kelp biology. Nat. Commun. 6, 6986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mock T., Otillar R. P., Strauss J., Mcmullan M., Paajanen P., Schmutz J., Salamov A., Sanges R., Toseland A., Ward B. J., Allen A. E., Dupont C. L., Frickenhaus S., Maumus F., Veluchamy A., Wu T., Barry K. W., Falciatore A., Ferrante M. I., Fortunato A. E., Glöckner G., Gruber A., Hipkin R., Janech M. G., Kroth P. G., Leese F., Lindquist E. A., Lyon B. R., Martin J., Mayer C., Parker M., Quesneville H., Raymond J. A., Uhlig C., Valas R. E., Valentin K. U., Worden A. Z., Armbrust E. V., Clark M. D., Bowler C., Green B. R., Moulton V., van Oosterhout C., Grigoriev I. V., Evolutionary genomics of the cold-adapted diatom Fragilariopsis cylindrus. Nature 541, 536–540 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Malviya S., Scalco E., Audic S., Vincent F., Veluchamy A., Poulain J., Wincker P., Iudicone D., de Vargas C., Bittner L., Zingone A., Bowler C., Insights into global diatom distribution and diversity in the world’s ocean. Proc. Natl. Acad. Sci. U. S. A. 113, E1516–E1525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalier-Smith T., Principles of protein and lipid targeting in secondary symbiogenesis: Euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J. Eukaryot. Microbiol. 46, 347–366 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Moustafa A., Beszteri B., Maier U. G., Bowler C., Valentin K., Bhattacharya D., Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 324, 1724–1726 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Archibald J. M., Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 25, R911–R921 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Gladyshev E. A., Meselson M., Arkhipova I. R., Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Alexander W. G., Wisecaver J. H., Rokas A., Hittinger C. T., Horizontally acquired genes in early-diverging pathogenic fungi enable the use of host nucleosides and nucleotides. Proc. Natl. Acad. Sci. U. S. A. 113, 4116–4121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiu H., Price D. C., Yang E. C., Yoon H. S., Bhattacharya D., Evidence of ancient genome reduction in red algae (Rhodophyta). J. Phycol. 51, 624–636 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Chan C. X., Soares M. B., Bonaldo M. F. F., Wisecaver J. H., Hackett J. D., Anderson D. M., Erdner D. L., Bhattacharya D., Analysis of Alexandrium tamarense (Dinophyceae) genes reveals the complex evolutionary history of a microbial eukaryote. J. Phycol. 48, 1130–1142 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H., Stephens T. G., González-Pech R. A., Beltran V. H., Lapeyre B., Bongaerts P., Cooke I., Aranda M., Bourne D. G., Forêt S., Miller D. J., van Oppen M. J. H., Voolstra C. R., Ragan M. A., Chan C. X., Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 1, 95 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin H. Y., Lin H. J., Polyamines in microalgae: Something borrowed something new. Mar. Drugs 17, E1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber T., John S., Tagliabue A., DeVries T., Biological uptake and reversible scavenging of zinc in the global ocean. Science 361, 72–76 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Vlasova A., Capella-Gutiérrez S., Rendón-Anaya M., Hernández-Oñate M., Minoche A. E., Erb I., Câmara F., Prieto-Barja P., Corvelo A., Sanseverino W., Westergaard G., Dohm J. C., Pappas G. J. Jr., Saburido-Alvarez S., Kedra D., Gonzalez I., Cozzuto L., Gómez-Garrido J., Aguilar-Morón M. A., Andreu N., Aguilar O. M., Garcia-Mas J., Zehnsdorf M., Vázquez M. P., Delgado-Salinas A., Delaye L., Lowy E., Mentaberry A., Vianello-Brondani R. P., García J. L., Alioto T., Sánchez F., Himmelbauer H., Santalla M., Notredame C., Gabaldón T., Herrera-Estrella A., Guigó R., Genome and transcriptome analysis of the Mesoamerican common bean and the role of gene duplications in establishing tissue and temporal specialization of genes. Genome Biol. 17, 32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delaye L., Valadez-Cano C., Pérez-Zamorano B., How really ancient is paulinella chromatophora? PLOS Curr. 8, ecurrents.tol.e68a099364bb1a1e129a17b4e06b0c6b (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lhee D., Ha J.-S., Kim S., Park M. G., Bhattacharya D., Yoon H. S., Evolutionary dynamics of the chromatophore genome in three photosynthetic Paulinella species. Sci. Rep. 9, 2560 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Husnik F., Nikoh N., Koga R., Ross L., Duncan R. P., Fujie M., Tanaka M., Satoh N., Bachtrog D., Wilson A. C., von Dohlen C. D., Fukatsu T., McCutcheon J. P., Horizontal gene transfer from diverse bacteria to an insect genome enables a tripartite nested mealybug symbiosis. Cell 153, 1567–1578 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Xu F., Jerlstöm-Hultqvist J., Kolisko M., Simpson A. G. B., Roger A. J., Svärd S. G., Andersson J. O., Erratum to: On the reversibility of parasitism: Adaptation to a free-living lifestyle via gene acquisitions in the diplomonad Trepomonas sp. PC1. BMC Biol. 14, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowack E. C. M., Price D. C., Bhattacharya D., Singer A., Melkonian M., Grossman A. R., Gene transfers from diverse bacteria compensate for reductive genome evolution in the chromatophore of Paulinella chromatophora. Proc. Natl. Acad. Sci. U.S.A. 113, 12214–12219 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gogarten J. P., Doolittle W. F., Lawrence J. G., Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Huang J., Horizontal gene transfer in eukaryotes: The weak-link model. Bioessays 35, 868–875 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doolittle W. F., You are what you eat: A gene transfer ratchet could account for bacterial genes in eukaryotic nucleaar genomes. Trends Genet 14, 307–311 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Ku C., Martin W. F., A natural barrier to lateral gene transfer from prokaryotes to eukaryotes revealed from genomes: The 70% rule. BMC Biol. 14, 89 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu H., Price D. C., Weber A. P. M., Reeb V., Yang E. C., Lee J. M., Kim S. Y., Yoon H. S., Bhattacharya D., Adaptation through horizontal gene transfer in the cryptoendolithic red alga Galdieria phlegrea. Curr. Biol. 23, R865–R866 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Shumaker A., Putnam H. M., Qiu H., Price D. C., Zelzion E., Harel A., Wagner N. E., Gates R. D., Yoon H. S., Bhattacharya D., Genome analysis of the rice coral Montipora capitata. Sci. Rep. 9, 2571 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Husnik F., McCutcheon J. P., Functional horizontal gene transfer from bacteria to eukaryotes. Nat. Rev. Microbiol. 16, 67–79 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Ku C., Nelson-Sathi S., Roettger M., Garg S., Hazkani-Covo E., Martin W. F., Endosymbiotic gene transfer from prokaryotic pangenomes: Inherited chimerism in eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 112, 10139–10146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moran Y., Fredman D., Szczesny P., Grynberg M., Technau U., Recurrent horizontal transfer of bacterial toxin genes to eukaryotes. Mol. Biol. Evol. 29, 2223–2230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu B., Novelli J., Jiang D., Dailey H. A., Landmann F., Ford L., Taylor M. J., Carlow C. K. S., Kumar S., Foster J. M., Slatko B. E., Interdomain lateral gene transfer of an essential ferrochelatase gene in human parasitic nematodes. Proc. Natl. Acad. Sci. U.S.A. 110, 7748–7753 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcet-Houben M., Gabaldón T., Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 26, 5–8 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Ravenhall M., Škunca N., Lassalle F., Dessimoz C., Inferring horizontal gene transfer. PLOS Comput. Biol. 11, e1004095 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J., Yang J. R., Determinants of the rate of protein sequence evolution. Nat. Rev. Genet. 16, 409–420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Puigbò P., Bravo I. G., Garcia-Vallvé S., E-CAI: A novel server to estimate an expected value of codon adaptation index (eCAI). BMC bioinforamtics 9, 65 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou W. J., Yang J. K., Mao L., Miao L. H., Codon optimization, promoter and expression system selection that achieved high-level production of Yarrowia lipolytica lipase in Pichia pastoris. Enzyme Microb. Technol. 71, 66–72 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Liao B. Y., Scott N. M., Zhang J., Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol. Biol. Evol. 23, 2072–2080 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Ku C., Nelson-Sathi S., Roettger M., Sousa F. L., Lockhart P. J., Bryant D., Hazkani-Covo E., McInerney J. O., Landan G., Martin W. F., Endosymbiotic origin and differential loss of eukaryotic genes. Nature 524, 427–432 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Keeling P. J., Burki F., Wilcox H. M., Allam B., Allen E. E., Amaralzettler L. A., Armbrust E. V., Archibald J. M., Bharti A. K., Bell C. J., Beszteri B., Bidle K. D., Cameron C. T., Campbell L., Caron D. A., Cattolico R. A., Collier J. L., Coyne K., Davy S. K., Deschamps P., Dyhrman S. T., Edvardsen B., Gates R. D., Gobler C. J., Greenwood S. J., Guida S. M., Jacobi J. L., Jakobsen K. S., James E. R., Jenkins B., John U., Johnson M. D., Juhl A. R., Kamp A., Katz L. A., Kiene R., Kudryavtsev A., Leander B. S., Lin S., Lovejoy C., Lynn D., Marchetti A., McManus G., Nedelcu A. M., Menden-Deuer S., Miceli C., Mock T., Montresor M., Moran M. A., Murray S., Nadathur G., Nagai S., Ngam P. B., Palenik B., Pawlowski J., Petroni G., Piganeau G., Posewitz M. C., Rengefors K., Romano G., Rumpho M. E., Rynearson T., Schilling K. B., Schroeder D. C., Simpson A. G., Slamovits C. H., Smith D. R., Smith G. J., Smith S. R., Sosik H. M., Stief P., Theriot E., Twary S. N., Umale P. E., Vaulot D., Wawrik B., Wheeler G. L., Wilson W. H., Xu Y., Zingone A., Worden A. Z., The marine microbial eukaryote transcriptome sequencing project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLOS Biol. 12, e1001889 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.W. Li, Fast program for clustering and comparing large sets of protein or nucleotide sequences, Encyclopedia of Metagenomics: Genes, Genomes and Metagenomes: Basics, Methods, Databases and Tools, K. E. Nelson, Ed. (Springer, Boston, MA, 2015), pp. 173–177.
- 43.Edgar R. C., Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Qiu H., Cai G., Luo J., Bhattacharya D., Zhang N., Extensive horizontal gene transfers between plant pathogenic fungi. BMC Biol. 14, 41 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar R. C., MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Capella-Gutiérrez S., Silla-Martinez J. M., Gabaldón T., trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price M. N., Dehal P. S., Arkin A. P., FastTree 2–Approximately maximum-likelihood trees for large alignments. PLOS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimodaira H., Hasegawa M., Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999). [Google Scholar]
- 49.Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q., IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalyaanamoorthy S., Minh B. Q., Wong T. Q. F., von Haeseler A., Jermiin L. S., ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minh B. Q., Nguyen M. A., von Haeseler A., Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emms D. M., Kelly S., OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M., Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Huson D. H., Bryant D., Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Saier M. H. Jr., Reddy V. S., Tamang D. G., Västermark Å., The transporter classification database. Nucleic Acids Res. 42, D251–D258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang H., Yohe T., Huang L., Entwistle S., Wu P., Yang Z., Busk P. K., Xu Y., Yin Y., dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 46, W95–W101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pertea M., Kim D., Pertea G. M., Leek J. T., Salzberg S. L., Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z., Li J., Zhao X. Q., Wang J., Wong G. K., Yu J., KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4, 259–263 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rice P., Longden I., Bleasby A., EMBOSS: The european molecular biology open software suite. Trends Genet. 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 60.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rumble S. M., Lacroute P., Dalca A. V., Fiume M., Sidow A., Brudno M., SHRiMP: Accurate mapping of short color-space reads. PLOS Comput. Biol. 5, e1000386 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pertea M., Pertea G. M., Antonescu C. M., Chang T. C., Mendell J. T., Salzberg S. L., StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaba0111/DC1