

This study describes a ferroptosis/apoptosis-based treatment strategy via the cooperation between Fe2+ and doxorubicin.
Abstract
The rapid development of treatment resistance in tumors poses a technological bottleneck in clinical oncology. Ferroptosis is a form of regulated cell death with clinical translational potential, but the efficacy of ferroptosis-inducing agents is susceptible to many endogenous factors when administered alone, for which some cooperating mechanisms are urgently required. Here, we report an amorphous calcium carbonate (ACC)–based nanoassembly for tumor-targeted ferroptosis therapy, in which the totally degradable ACC substrate could synergize with the therapeutic interaction between doxorubicin (DOX) and Fe2+. The nanoplatform was simultaneously modified by dendrimers with metalloproteinase-2 (MMP-2)–sheddable PEG or targeting ligands, which offers the functional balance between circulation longevity and tumor-specific uptake. The therapeutic cargo could be released intracellularly in a self-regulated manner through acidity-triggered degradation of ACC, where DOX could amplify the ferroptosis effects of Fe2+ by producing H2O2. This nanoformulation has demonstrated potent ferroptosis efficacy and may offer clinical promise.
INTRODUCTION
The convergence of nanotechnology with modern biology and medicine has offered numerous opportunities for tumor therapy with great clinical significance, which has already revolutionized many aspects of the current tumor management principles (1–3). Among the emerging nanomaterials for biomedical applications, there is an ongoing interest in the development and implementation of inorganic-based nanoplatforms due to the cheap mass production, easy quality control, and multipurpose functionality (4–6). Nevertheless, the potential clinical translation of inorganic nanomaterials is usually held in check by the associated health and safety considerations, especially the problematic degradability of typical inorganic nanostructures in biologically relevant environment (7–10). Consequently, innovative inorganic nanomaterials are still urgently needed, which may retain those pharmacologically favorable properties while also fulfilling the required safety criteria.
Calcium carbonate (CaCO3) is an inorganic material commonly found in nature and attracts wide attention of researchers from biomedical science and other related disciplines due to the associated advantages, including low production cost, bioresorption, and intrinsic biocompatibility (11, 12). Nevertheless, its application as drug delivery system is impeded by the microscale size and low degradation rate in biological environment due to high crystallinity, which would severely undermine the drug delivery efficiency (13). These intrinsic limitations are eventually overcome by the development and implementation of amorphous calcium carbonate (ACC) nanostructures. Comparing to the crystalized form of CaCO3, ACC nanoparticles have higher energy levels due to the structural frustration and are extremely prone to hydrolysis in intracellular environment. The potential benefits of using ACC for drug delivery have already been preliminarily investigated in a series of studies (14–16). Typically, in one of our previous reports we co-condensed calcium precursors with doxorubicin (DOX) and obtained an ACC-based drug delivery system with intricate pH sensitivity, which could selectively release the therapeutic cargo within tumor cells (17). These studies are immediate evidence that ACC could be easily loaded with various bioactive molecules without the need of tedious synthesis procedures, which also suggests its great application potential when combined with diverse organic ligands and modifications such as tumor targeting moieties, tumor microenvironment–responsive linkages and PEGylation agents (18–20). It is anticipated that the combination of ACC-based nanosubstrate and tailored organic modification could further improve the biofunctionality and biocompatibility of the composite nanoformulations while retaining those favorable clinically relevant benefits of ACC.
It should be mentioned that the enhancement of tumor therapy relies not only on the development of biocompatible nanomaterials but also on the discovery and implementation of clinically capable therapeutic mechanisms. Currently, the therapeutic targets of most clinical tumor therapies are those apoptosis-based pathways (21, 22). For instance, DOX, an antitumor antibiotic that has been widely used in clinical practice, could induce tumor cell apoptosis via two primary mechanisms: (i) intercalating into the DNA of tumor cells and inhibiting the topoisomerase II–mediated DNA replication and (ii) entering mitochondria to form a semiquinone radical and activate the mitochondrial apoptosis pathways. By exploiting these cell damaging mechanisms, DOX has demonstrated clinically attractive efficacy against a plethora of tumor indications and also revealed emerging opportunities for the development of more advanced antitumor therapies by interacting with other treatment modalities (23, 24). One such candidate in this field is ferroptosis. Ferroptosis is a recently discovered mode of nonapoptotic cell death that is activated by the iron-dependent accumulation of cytotoxic lipid hydroperoxides, and increasing the intracellular iron level is an effective approach to induce ferroptosis in tumor cells (25, 26). It has been confirmed that iron plays a multifaceted role in the initiation and execution of ferroptosis. On one hand, iron is an intrinsic redox-active species and the intracellular labile iron pool (mostly in the form of Fe2+) could effectively catalyze the reactive oxygen species (ROS) generation via Fenton reaction to propagate lipid peroxidation. On the other hand, the intracellular iron levels may also affect the activity of other ROS-generating enzymes such as nicotinamide adenine dinucleotide phosphate oxidases (NOXs), lipoxygenases, and mitochondrial electron transport complexes and, thus, promote ferroptosis. Consequently, increasing the labile iron pool in tumor cells via Fe2+ delivery has become a promising strategy to initiate ferroptotic cell death. Tumor-targeted iron delivery is already widely used for Fenton reaction–mediated biocatalytic tumor treatment based on Fe2+/Fe3+ redox pair (27–29), but Fe3+ ions are kinetically unfavorable for catalyzing the generation of lipid ROS as they must first be reduced to Fe2+ to activate the ferroptotic cascade. Moreover, Fe2+ ions could also contribute to the ferroptosis via multiple pathways other than Fenton reactions including iron-catalyzed oxygenation of polyunsaturated fatty acids or the autoxidation of lipids (30, 31). Nevertheless, reports on its application in ferroptosis-based tumor therapy are still rare due to the relative high oxidation susceptibility of Fe2+ ions and the regulation of the ferroptosis process. In comparison with the rapid initiation and high acute cytotoxicity demonstrated by most of the Fenton catalyst–mediated tumor therapies, Fe2+-mediated ferroptosis therapy relies more on the regulation of those intrinsic signaling pathways and could last for a prolonged period of time for continuous treatment due to its self-sustainability and propagation capability, as the ferroptosis-inducing Fe2+ species could be recycled in physiological environment via the Haber-Weiss reaction chain and that the emerging phospholipid peroxyl radicals may further attack adjacent polyunsaturated fatty acids in the lipid bilayer in the presence of Fe2+ ions to initiate another lipid radical chain reaction and propagate the ferroptotic damage (32). Therefore, it would be of clinical significance to develop ferroptotic pathway–targeted nanotherapeutics with improved tumor specificity, controllable pharmacokinetic profiles, enhanced anticancer efficacy, and reduced health risks.
On the basis of the insights from the preliminary studies, we design a tumor-targeted ACC-based nanoformulation with complementary ferroptosis/apoptosis-inducing capability (Fig. 1A and fig. S1B). Here, DOX first chelates with ferrous ions and then co-condense with calcium-containing precursors to produce ACC-encapsulated Fe2+-DOX cores in an easily reproducible one-step approach (fig. S1B). The complexation of DOX and Fe2+ ions not only allows for the efficient loading of therapeutic agents into the ACC-based nanoassembly but also minimizes the susceptibility of ferrous ions to intrahost oxidative stress before their intracellular release. A thin layer of silica-CaCO3 (CaSi) hybrid was then deposited onto the surface of the core compartment, which was subsequently conjugated simultaneously with folate-modified and PEGylated polyamidoamine (PAMAM) dendrimers to confer targeting specificity to tumor cells. To achieve the balance between circulation stability and targeting capability, the PEG segment was conjugated to the PAMAM dendrimers via matrix metalloproteinase–2 (MMP-2)–cleavable peptides (GPLGVRGDGG) (33). Elevated expression level of MMP-2 in the extracellular environment is a defining feature of many invasive tumors, which has frequently been exploited as triggering signals for therapies that target the tumor microenvironment or those membrane components. As the molecular therapeutic targets of Fe2+ ions and DOX are both in the tumor intracellular environment, here, the MMP-2–cleavable peptide was used as the first-stage activating mechanism of the tumor targeting capabilities in the tumor microenvironment. The resultant dendrimeric conjugates could temporarily deactivate the bioactivity of the folate moieties while simultaneously preventing protein opsonization and phagocytosis after entering the biological environment. However, upon reaching the MMP-2–rich tumor microenvironment, these PEG moieties could be removed and restore the tumor-targeting ability of the folate ligands to facilitate the specific tumor uptake of the composite nanostructures. After the successful internalization by the tumor cells, the PAMAM dendrimers could facilitate the lysosomal escape of the Fe2+-DOX nanoformulation via the proton sponge effect (34). Meanwhile, the ACC cores would be sacrificed via hydrolysis and release ferrous ions and DOX into the cytosol. Compared with other ferroptosis-inducing strategies by solely increasing the intracellular iron accumulation, the concurrently delivered DOX not only serves as an adjuvant treatment modality but is also capable of increasing the ROS generation in the tumor cells (35). As ROS could contribute to the generation of lipoperoxides (36), the pH-triggered release of ferrous ions and DOX is anticipated to act synergistically to escalate the oxidative stress, leading to enhanced ferroptotic tumor cell death in an on-demand manner.
Fig. 1. Synthesis scheme and structural/compositional characterizations of the ACC-based nanoformulation.
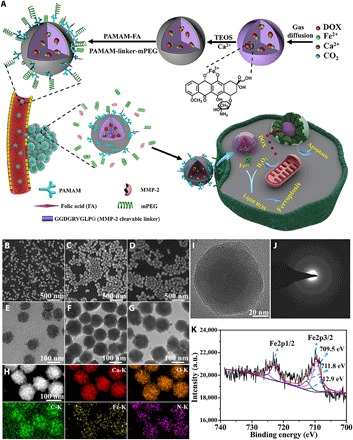
(A) Synthesis scheme of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG and its complementary ferroptosis/apoptosis-based therapeutic action. After intravenous injection, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG would accumulate in tumor tissues via the enhanced permeability and retention (EPR) effect and activated by the abnormally high MMP-2 levels at the tumor site, by which the exposed folic acid ligands could facilitate the tumor-specific uptake of the nanoformulation. (B to G) Scanning electron microscopy and TEM images of ACC@DOX.Fe2+ (B and E), ACC@DOX.Fe2+-CaSi (C and F), and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (D and G). (H) Scanning transmission electron microscopy image of ACC-DOX.Fe2+ and the corresponding energy-dispersive spectroscopy (EDS) analysis results. (I) High-resolution transmission electron microscopy images of ACC@DOX.Fe2+. (J) Selected area electron diffraction pattern of ACC@DOX.Fe2+, indicating the amorphous nature of ACC@DOX.Fe2+ nanoparticles. (K) Fe2p x-ray photoelectron spectroscopy (XPS) spectra of ACC@DOX.Fe2+. a.u., arbitrary units.
RESULTS
Construction and characterization of the tumor-targeting ACC-based Fe2+-DOX nanoformulation
The synthesis scheme of the tumor-targeting ACC-encapsulated Fe2+-DOX nanoformulation is illustrated in fig. S1. Mono-alkynylated dendrimers were first synthesized, which were functionalized individually with folic acid (FA) or MMP-2–sheddable PEG (fig. S1A). Their chemical structures were confirmed by the 1H nuclear magnetic resonance spectra, which revealed that FA was conjugated to the PAMAM dendrimers at a roughly 1:1 ratio (fig. S2). Meanwhile, the complexation between DOX and Fe2+ was characterized using fluorescence spectroscopy (Fig. 2A). It was found that fluorescence emission of DOX at 590 nm has decreased markedly after mixing with Fe2+ precursors in ethanol and dropped to the minimum when the molar ratio between DOX and Fe2+ was 1:3, and the ratio was used as a standard to prepare the DOX-Fe2+ complex for the subsequent experiments. The DOX-Fe2+ complex was then allowed to co-condense with calcium precursors to synthesize ACC-based DOX-Fe2+ cores (ACC@DOX.Fe2+) via a well-established gas diffusion method, followed by the coating of a degradable CaSi layer (ACC@DOX.Fe2+-CaSi) (fig. S1B). It was also observed that loading of the DOX-Fe2+ complex into the ACC substrate was very similar to that of free DOX in terms of both the operating procedures and loading capacity (5.4%) (17). Moreover, the loading efficiency of the DOX-Fe2+ complex was found to be around 65.9% according to the ultraviolet analysis (fig. S3C), which could be explained by the high affinity between DOX and ACC species. As shown by high-resolution transmission electron microscopy, selected area electron diffraction characterizations and x-ray diffraction (Fig. 1, I and J, and fig. S3B), the CaCO3 contents contained within the thin CaSi shells is in an amorphous state, potentiating rapid degradation when exposed to the tumor intracellular environment. In addition, the scanning electron microscopy and transmission electron microscopy (TEM) images revealed that the nanocores have a spherical shape with an average size of around 110 nm, and the thickness of the thin CaSi shell was revealed to be around 8 nm, which is suffice to prevent the premature hydrolysis of the ACC contents and their spontaneous crystallization before tumor uptake (Fig. 1, B, C, E, and F). The multifunctional dendrimer was conjugated onto the CaSi shell via MMP-2–cleavable peptide linkages to afford the finalized product of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, which remained uniformly spherical and monodispersive (Fig. 1, D and G). According to the dynamic light scattering results (fig. S3A), the average hydrodynamic diameter of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was around 122 nm. The changes in the chemical composition after each step of modification have also been investigated and confirmed using Fourier transform infrared spectroscopy (FTIR) (fig. S3D). Specifically, the peak at 878 cm−1 in the spectra confirmed the successful construction of the CaCO3 nanostructure. After the coating of silica shells, a new peak has appeared at 1086 cm−1, which was due to the stretching vibration of the Si─O bond. The silica-coated ACC further reacted with 3-aminopropyltriethoxysilane and resulted in the emergence of a new peak at 1656 cm−1, which confirmed the successful grafting of amine groups. Subsequently, the amine-modified nanoparticles were grafted with PEG-N3 and two new peaks appeared at 2110 and 2875 cm−1, which correspond to the azide and methylene groups on the PEG-N3, respectively. Eventually, after the conjugation of PAMAM-FA via click reactions, the azide peaks have disappeared as they were consumed during the reaction. Instead, two new peaks emerged near 1659 cm−1, which was assigned to the amine groups on the folate moiety. In addition, another two peaks have appeared at 1499 and 1431 cm−1, which also correspond to the aromatic rings of attached folate moieties. The modification-induced changes in the surface properties of the nanosamples were also consistently confirmed by fluorescamine spectra (fig. S3E) and zeta potential analysis (fig. S3F).
Fig. 2. DOX loading and the pH-triggered drug release features.
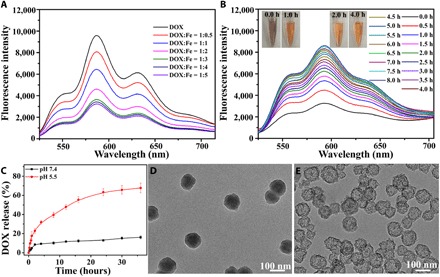
(A) Changes in DOX fluorescence after mixing DOX with Fe2+ at varied mix ratios in ethanol. It was observed that the intensity of DOX fluorescence reached the minimum when the DOX/Fe2+ ratio was 1:3. (B) Fluorescence spectroscopic analysis shows the dissociation of the DOX-Fe2+ complex in the buffer solution at pH 5.5, the color changes of the samples from 0 to 8 hours of incubation are indicated by the inserted images. Photo credit: Chen-Cheng Xue, Chongqing University. (C) The DOX release profiles from ACC@DOX- CaSi-PAMAM-FA/mPEG under pH 7.4 and pH 5.5. (D and E) Impact of pH on the release rate of DOX-Fe2+ complex from the ACC substrate, in which ACC@DOX.Fe2+-PAMAM-FA/mPEG was incubated in buffer solutions at different pH values for 24 hours. D, pH 7.4; E, pH 5.5.
In addition, the sample series have also been characterized with energy-dispersive spectroscopy (EDS) and x-ray photoelectron spectroscopy (XPS) to investigate their elemental composition. The EDS results visualized the distribution of calcium, carbon, oxygen, iron, and nitrogen within a single nanostructure (Fig. 1H), which confirm the formation of ACC@DOX.Fe2+ nanoassembly. Furthermore, the oxidation state of iron was analyzed quantitatively by comparing the corresponding integrated peak areas in XPS spectra, which provided a more accurate depiction of the Fe2+-stabilizing efficiency of the ACC-based nanocarrier against the potential oxidative stress in vivo. XPS is a widely used surface analysis technique with a quantitative accuracy of 90 to 95%, which enables the accurate assessment of the surface composition within experimental uncertainty (37). The spectra were first analyzed via peak fitting, and the fitting results of the Fe 2p3/2 spectra of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG showed three characteristic peaks at 709.5, 711.8, and 712.9 eV, respectively (Fig. 1K). As revealed by previous insights, the peak at 709.5 eV was attributed to Fe2+, while the other two peaks were caused by Fe3+. The relative ratio of Fe2+ to Fe3+ in the samples was further obtained by comparing the integrated peak areas, and it was observed that only around 9% of the encapsulated Fe2+ has been oxidized to Fe3+ during the co-condensation and modification process, and it was further confirmed that more than 80% of the encapsulated iron species were stabilized at low oxidation state even after being incubated in physiological buffer or serum for 24 hours (fig. S4, A to C). The high oxidation resistance of the Fe2+ species in the composite nanosystem could be explained by the stabilizing effect of DOX complexation and shell-isolation effect of the CaSi coating, which is highly favorable for the induction of ferroptosis in vivo.
Profiling release behaviors of DOX from ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG nanoformulation in buffer solutions
The realization of ferroptosis-dependent tumor therapy with the as-prepared composite nanostructure essentially relies on the efficient release of DOX and Fe2+ from ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG. Therefore, we first studied the release kinetics of DOX-Fe2+ complex in biomimetic environment by incubating in buffer solutions (Fig. 2, C to E, and fig. S4, G and H). Previous insights have revealed that ACC nanoassembly is highly prone to hydrolysis even in neutral environment, and our findings indicate that the coating of the CaSi shell has substantially enhanced its acid resistance. Specifically, under the physiological pH of 7.4, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG showed excellent chemical stability where the total DOX leakage after 35 hours of incubation was still low as 15% (Fig. 2C). In comparison, the ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG samples showed a distinct burst drug release pattern with an accumulative release percentage of around 70% when incubating at pH 5.5 for 36 hours, evidently suggesting the sensitivity of the nanoformulation to acidic pH (Fig. 2C). It was also observed that the DOX release rate became even faster after MMP-2 treatment, which could be explained by the decreased diffusion hindrance due to MMP-2–mediated removal of the PEG chains (fig. S4H). The release profile of Fe2+ ions was also measured via inductively coupled plasma mass spectrometry (ICP), and the trends were consistently identical. Specifically, only negligible Fe2+ ion leakage (~10%) was detected after incubating at pH 7.4 for 36 hours, while under pH 5.5, the release ratio increased to above 73% (fig. S4G). XPS analysis further revealed that most of the released iron ions were still at the valence state of +2 after incubating in phosphate-buffered saline (PBS) (around 91%), cell medium (around 90%), and serum (around 81%) (fig. S4, A to C), indicating the stabilizing effect of the nanoformulation against environmental oxidative stress. Moreover, the nanoformulation after incubation has also been collected for TEM observation, and it was found that the severity of structural degradation for ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG nanoformulation was positively correlated to the amount of accumulative drug release (Fig. 2, D and E). The TEM images indicated that the ACC cores have been completely dissolved after incubating in the buffer solution for 24 hours where only the CaSi shells remained. Moreover, even those CaSi shells have splintered into small pieces when the incubation period was extended to 72 hours, again validating the degradability of the whole composite nanostructure in physiological conditions (fig. S4, D to F). Furthermore, we also found that at the pH level of 5.5, the color of DOX-Fe2+ complex released from the nanoformulation changed gradually from dark purple to orange, accompanied by an increase in the characteristic DOX fluorescence at 595 nm (Fig. 2B). As the complexation between DOX and Fe2+ could quench the fluorescence emission of DOX, the color change and fluorescence recovery evidently show that the DOX-Fe2+ complex has been disassociated, and both components have reverted to the free form. Previous reports also suggest that the dissociation of DOX-Fe2+ complex in aqueous environment was due to the phenol protonation of DOX molecules, which may potentially reduce the cardiotoxicity of DOX chemotherapy (38, 39).
MMP-2 triggered PEG deshielding from ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG
In this study, we developed a cascade-targeting strategy through the rational combination of MMP-2–responsive linkers and active targeting ligands to avoid their accidental activation in off-target organs/tissues and drug leakage in the tumor extracellular environment. Specifically, during the PEGylation of those PAMAM dendrimers, we used an MMP-2–cleavable GPLGVRGDGG peptide linkers to conjugate the mPEG onto the dendrimer substrate. It is anticipated that the mPEG conjugates could temporarily shield the folate-based targeting ligands and the positively charged dendrimers to reduce the nonspecific absorption of the nanoformulation by healthy organs/tissues, as well as being effectively shed after entering the MMP-2–rich tumor microenvironment. As a result of the MMP-2–triggered PEG removal, the folate moieties and the positively charged substrate would be exposed to facilitate the preferential tumor cellular uptake via ligand-enabled targeting. The proposed mechanism was first studied in solution by monitoring the zeta potential changes of the nanocarriers after incubation with buffers containing varied concentrations of MMP-2 (fig. S3G). It could be observed that the zeta potential of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG after incubation in MMP-2–free buffer for 12 hours was as low as −7.29 eV, which became +37.93 eV when the ambient MMP-2 concentration increased to 400 ng/ml. The positive correlation between zeta potential values and MMP-2 concentration in the incubation medium evidently supports our hypothesis that the protective PEG conjugates could be removed efficiently in response to the increasing environmental MMP-2 concentration. In addition to the zeta potential changes, the shielding effect conveyed by the MMP-2 sheddable PEG was also consistently confirmed by the fluorescamine assay (fig. S3E), in which the nanoparticle fluorescence has dropped dramatically as a result of PEGylation but recovered after MMP-2 treatment.
Evaluation on the uptake efficiency of ACC@FITC.Fe2+-CaSi-PAMAM-FA/mPEG by tumor cells
The effectiveness of the sheddable PEG design in improving the tumor-targeted uptake of the nanoformulation was further investigated on 4T1 breast cancer cells and A375 melanoma cells, both of which show elevated levels of folate receptors (40). The sample series were all labeled with fluorescein isothiocyanate (FITC) and incubated with 4T1 or A375 cells for 12 and 24 hours and later analyzed using confocal laser scanning microscopy (CLSM) (Fig. 3A and fig. S6A, respectively). As shown by the CLSM results, 4T1 cells incubated with MMP-2–pretreated ACC@FITC-CaSi-PAMAM-FA/mPEG showed evidently stronger FITC fluorescence than original ACC@FITC-CaSi-PAMAM-FA/mPEG and ACC@FITC-CaSi-PEG after both incubation periods. According to the quantitative flow cytometric analysis (fig. S8, A and B), the FITC fluorescence intensity caused by the MMP-2–pretreated ACC@FITC-CaSi-PAMAM-FA/mPEG has increased by 93% in comparison with pristine ACC@FITC-CaSi-PAMAM-FA/mPEG after incubating for 12 hours. When the incubation increased to 24 hours, the discrepancy was slightly reduced due to the leveling effect of prolonged incubation and the relative increase dropped to 50%. The same trend has also been observed in A375 cells that have undergone similar treatment. The drastic fluorescence increases in the enzyme-preconditioned samples supported our hypothesis that MMP-2 could remove the anti-biofouling PEG conjugates and improve the uptake efficiency of the nanocarrier by tumor cells that are overexpressed with folate receptors.
Fig. 3. In vitro characterizations on the uptake and lysosomal release capabilities of the ACC-based nanoformulation.
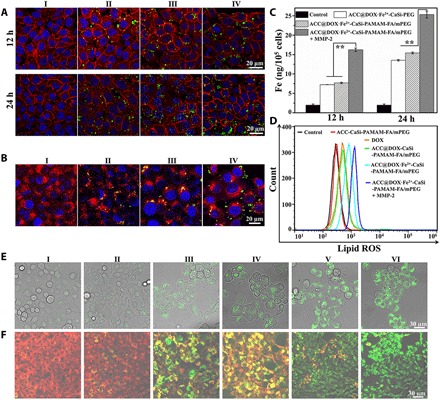
(A) CLSM images of 4T1 cells incubated with PBS (I), ACC@FITC-CaSi-PEG (II), ACC@FITC-CaSi-PAMAM-FA/mPEG (III) and MMP-2–treated ACC@FITC-CaSi-PAMAM-FA/mPEG (IV) for 12 and 24 hours. The blue, red, and green colors indicate cell nucleus, cell membrane, and the FITC-labeled nanoparticles, respectively. (B) CLSM evaluation on the lysosomal escape of nanoparticles where the 4T1 cells were incubated with PBS (I), ACC@FITC-CaSi-PEG (II), ACC@FITC-CaSi-PAMAM-FA/mPEG (III) and MMP-2–treated ACC@FITC-CaSi-PAMAM-FA/mPEG (IV) for 12 hours. The blue, red, and green colors indicate cell nucleus, lysosome, and the FITC-labeled nanoparticles, respectively. (C) ICP results on the intracellular iron levels of 4T1 cells after incubation with PBS, ACC@DOX.Fe2+-CaSi-PEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG for 12 or 24 hours. The double-asterisk symbol indicates significance at P < 0.01. (D) Flow cytometric analysis on the intracellular lipoperoxide levels in 4T1 cells incubated with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI) for 24 hours. The lipid ROS indicator was BODIPY-C11. (E) CLSM observation on the intracellular distribution of lipoperoxides in 4T1 cells after incubation with PBS, ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG for 24 hours. The green fluorescence is the lipid ROS after the staining with BODIPY-C11. (F) CLSM observation on the changes in the mitochondrial membrane potential of 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) for 24 hours.
In addition, the nanoformulation-induced intracellular iron accumulation was also investigated directly with ICP mass spectrometry (Fig. 3C). It could be observed that the trends in the intracellular iron levels were highly consistent with that of the FITC fluorescence, where the iron concentration in the ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG + MMP-2 group has increased by 124% compared to ACC@DOX.Fe2+-PEG and 111% to untreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG. The relative increase in the intracellular iron levels between ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG + MMP-2 and ACC@DOX.Fe2+-PEG or untreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG has dropped to 87.4 and 64.3% under the prolonged incubation period of 24 hours. The results regarding FITC fluorescence distribution and iron accumulation collectively demonstrate that ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG could switch to a high-affinity form against tumor cells after the mPEG contents were removed by MMP-2 enzyme, which offers the combined benefit of improved blood circulation stability and tumor specificity.
It is well established that the unique three-dimensional nanoarchitecture and strong positive charge of PAMAM dendrimers could result in the so-called proton sponge effect, which could be used to facilitate the escape of the nanoformulation from lysosomes after cellular uptake. Here, the lysosome escaping capability of the FITC-labeled samples was investigated using CLSM (Fig. 3B). The comparative analysis of the images demonstrates that for the two sample groups of ACC@FITC-CaSi-PEG and ACC@FITC-CaSi-PAMAM-FA/mPEG, most of the green areas (FITC fluorescence) still overlapped with the red ones (lysosome), indicating that most of the nanoformulation were still contained in the lysosomes. In comparison, for cells incubated with MMP-2–treated ACC@FITC-CaSi-PAMAM-FA/mPEG, strong fluorescence could be found outside the red areas, which immediately suggests that the nanoformulation has successfully entered the cytosol.
Investigation on the generation of lipoperoxide induced by ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG
Although the regulation mechanism of ferroptosis is still not fully understood, preliminary research has already identified several key components in the biological processes involved (Fig. 4D) (41–43). Typically, ferroptotic cell death is the direct result of the accumulation of lipid-based ROS, and its execution is intrinsically dependent on cellular iron overload. Several recent discoveries have found evidence that excessive iron could contribute to ferroptosis by participating the intracellular ROS metabolism and intensifying the lipid peroxidation (44). Therefore, simultaneously increasing the Fe2+ and H2O2 levels in tumor cells can be a promising strategy for the efficient initiation and execution of ferroptosis. In this study, the DOX-Fe2+ complex was chosen as the therapeutic cargo to fulfill this complementary task. It has already been demonstrated in previous sections that the DOX-Fe2+ complex could be readily dissociated in the tumor intracellular environment to afford bioactive DOX and Fe2+ species, and a variety of reports consistently revealed that DOX could not only directly damage DNA in tumor cell but also generate high level of ROS by activating the intracellular NADPH oxidase 4 (NOX4). NOX4 is a member of the NOX family of proteins, which is responsible for generating H2O2 by catalyzing the electron transfer from NADPH to oxygen. As the activity of NOX4 is primarily regulated by its abundance, it is anticipated that the DOX-induced up-regulation of NOX4 could enhance the generation of H2O2 in the intracellular environment and thus support the initiation and propagation of lipid peroxidation.
Fig. 4. Evaluations on the antitumor effect of the nanoformulation in vitro.
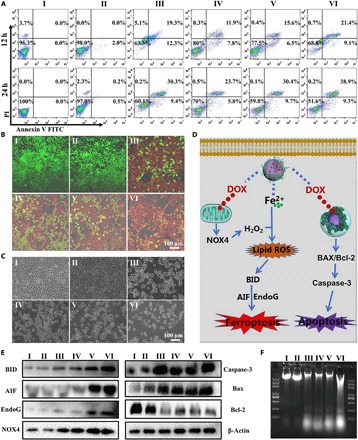
(A) Flow cytometric analysis on the apoptosis levels of 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI) for 12 and 24 hours. (B) CLSM observation apoptosis levels of 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI) for 24 hours. (C) Bright-field microscopy images of 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI) for 24 hours. (D) The proposed molecular mechanism for the nanoformulation-induced synergistic ferroptotic/apoptotic cell death. (E) Western blot analysis on the expression of key ferroptosis makers including BID, AIF, and EndoG, as well as apoptosis markers including NOX4, Caspase-3, BAX, and Bcl-2 in 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI). (F) DNA laddering assay on the DNA damage in 4T1 cells after incubation with PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV), ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V) and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (VI).
Here, we first investigated the effectiveness of DOX-induced ROS generation by incubating 4T1 and A375 cells with varying concentrations of DOX and subsequently monitoring the intracellular levels of different types of ROS. The results in figs. S7 (A to D) and S8 (C and D) collectively demonstrated that the intracellular levels of H2O2 in 4T1 and A375 cells have increased by 2.3- and 2.5-fold when the amount of administered DOX reached 1 μg/ml, while no apparent change was observed in the intracellular concentration of lipoperoxides at the same time. In comparison, when the 4T1 and A375 cells were incubated with the Fe2+-containing sample series, the lipoperoxide level has increased in a similar manner to the intracellular H2O2 concentration by 4.5- and 4.6-fold, respectively, which is apparently due to the proferroptotic effect of ferrous ions (Fig. 3D and figs. S7E and 8E). The MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG group showed the highest level of total lipid ROS in both tumor cell lines, which has increased by 3.6-fold compared to the free DOX group in 4T1 cells and 4.1-fold in A375 cells. Moreover, the strongest BODIPY-C11 fluorescence was found at the plasma membranes (Fig. 3E), which evidently supported the occurrence of lipid peroxidation. These observations are immediate evidence of the nanoformulation-induced ferroptotic damage and also support the enhanced uptake capability of the FA-modified nanoformulations by tumor cells.
Evaluation on the efficacy of the ferroptosis-based tumor therapy
The cytotoxic activity of the composite nanostructure on tumor cells was then quantitatively investigated using 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumro-mide (MTT) assay, in which the DOX concentrations were maintained at the same level. The results in fig. S5 reveal that the free DOX showed greater cytotoxicity to 4T1 cells than the nanoparticulate samples when the incubation period was only 12 hours, of which the relative cell viability of free DOX group was 67.3% compared to the 70.2% for cells treated with nanoformulation. However, as the incubation time was extended to 24 or 48 hours, the tumor cell inhibition effect by the nanoparticles has become more pronounced. Specifically, the survival rate of 4T1 cells incubated with MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was as low as 35.4% at the DOX concentration of 1 μg/ml, while the cell survival rate of the DOX group was still as high as 46.7% under the same concentration at 48 hours. The shifting in therapeutic potency could be explained by the difference between conventional chemotherapy and complementary ferroptosis/chemotherapy. DOX is a widely used anticancer therapeutic drug with acute cytotoxicity. In comparison, the onset of ferroptosis seems to require more time due to the complex biological activities involved in its initialization and execution. The discussion above was also supported by the MTT results on A375 cells, of which the survival patterns in different treatment groups were highly consistent.
The details revealed by the MTT assays were further interrogated using flow cytometry, bright-field microscopy, and CLSM (Fig. 4 and figs. S6 and S9).Typically, the flow cytometric analysis revealed that that the total ratio of dead/apoptotic cells in 4T1 cells after incubation with MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG for 24 hours was high as 48.2% in 4T1 and 50.7% for A375, while that in the DOX group was only 39.7% for 4T1 cells (Fig. 4A) and 39.8% for A375 cells (fig. S9). The apoptosis ratio revealed by the flow cytometry was also supported by the cell counting results based on bright-field microscopy images (Fig. 4C and fig. S6C). Similarly, the CLSM imaging after live cell staining also revealed that the number of live cells (green color) in the MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG group was visibly lower than the others for both tumor cell lines, suggesting that the more efficient tumor cell death was due to the intracellular release of the DOX-Fe2+ complex rather than DOX alone (Fig. 4B). All these results evidently demonstrated the superior therapeutic efficacy by combining ferroptosis therapy and DOX comparing to conventional chemotherapeutic strategies.
Investigation on the treatment-induced changes in the mitochondrial membrane potential
Mitochondrion is the primary site of energy production in most human cells and also the place where many programmed cell death pathways converge. For instance, mitochondrial dysfunction is a distinctive feature of apoptosis, which includes loss of mitochondrial membrane potential, enhanced membrane permeability, and leakage of cytochrome c. Recent studies revealed that ferroptosis is also accompanied by mitochondrial hyperpolarization due to the accumulation of ROS in the intracellular compartment (32). Consequently, we measured the changes in the mitochondrial membrane potential of tumor cells using a JC-1 assay kit, which shows red color when the membrane potential is positive but switches to green when the membrane potential decreases. Therefore, the color changes of the JC-1–stained mitochondria could be indicative of the severity of cellular damage caused by different treatments. According to the CLSM results in Fig. 3F and fig. S6B, the treatment with free DOX has caused moderated green fluorescence after 12 hours of incubation, resulting in a yellow-green color. In comparison, cells treated by MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG demonstrated strong green fluorescence, while red fluorescence could no longer be detected, which evidently suggests the highly negative mitochondrial membrane potential thereof due to the introduction of ferroptosis-inducing Fe2+ species. These observations are consistent with the lipoperoxide measurement and MTT results and support the successful activation of dual ferroptosis/apoptosis pathways.
Investigation on the therapeutic mechanism of the nanoformulation
We then sought to identify the cell death mechanisms involved in the therapeutic action of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG via multiple complementary techniques. From a mechanistic perspective, the treatment of DOX would up-regulate the expression level of NOX4 and increase the generation of H2O2 (45, 46), which could supply the Fe2+-mediated peroxidation of membrane phospholipids and further contribute to ferroptosis. Lipid peroxidation is an important event during the ferroptosis process, which may up-regulate the expression of BH3 interacting domain death agonist (BID) and downstream apoptosis-inducing factor/endonuclease G (AIF/EndoG) and eventually lead to DNA damage (Fig. 4D) (47, 48). The proposed molecular mechanism for the nanoformulation-induced synergistic ferroptotic/apoptotic cell death is well supported by the Western blot results. Specifically, the expression level of NOX4, BID, AIF, and EndoG levels in 4T1 cells have all increased after the treatment with MMP-2–processed ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG. It was found that the expression level of NOX4 has increased by 1.6-fold, which is consistent with the elevated H2O2 generation observed in fig. S7. Meanwhile, the amount of BID, AIF, and EndoG in these cells has increased by 4.1-, 5.4-, and 2.9-fold, respectively (Fig. 4Eand fig. S10, A and B), which immediately suggests that the Fe2+-coupled nanoformulation has substantially induced ferroptotic damage to the 4T1 cells. Meanwhile, it was observed that the expression level of caspase-3 has also increased by 2.1-fold, which is apparently due to the activation of the caspase-mediated apoptosis pathway by the concurrently delivered DOX. The foregoing results confirmed that the death of 4T1 cells after incubation with MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was caused by ferroptosis and DOX-mediated apoptosis combined while also supported the dual role of DOX as apoptosis inductor and ROS enhancer in this nanosystem. The western blot (WB) results also offered a possible explanation of the more pronounced tumor cell apoptosis by MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG revealed above. From a molecular viewpoint, BID and EndoG are not only involved in the onset and execution of ferroptosis process but also have facilitative effect on the DOX-mediated apoptosis. For instance, BID up-regulation could increase the permeability of the mitochondrial membrane and facilitate the release of AIF from the mitochondrial compartment, both of which would enhance the tumor cell apoptosis (49, 50). Alternatively, the mitochondrial EndoG would be released into the cytoplasm upon apoptosis-inducing stimulus and subsequently enter the cell nucleus to promote chromosomal DNA fragmentation (51), so the ferroptosis-induced EndoG up-regulation could also provide combinatorial benefit for enhancing the antitumor potency of DOX. It is thus anticipated that the complementary ferroptosis/apoptosis strategy in this study could further enhance the proapoptotic efficacy of DOX molecules.
In addition to the WB assay results shown above, we also carried out the DNA laddering assay to evaluate the severity of DNA damage. It is well established that DOX could interact with the DNA in tumor cells while inhibiting the activity of type II DNA topoisomerase, eventually breaking the DNA chains and preventing their replication. Consequently, a defining feature of DOX-induced DNA damage is the high rate of apoptotic DNA fragmentation, which is investigated in this study via DNA gel electrophoresis. As shown in Fig. 4F, DOX-treated 4T1 cells produced a visually discernible DNA ladder, whereas cells treated by ACC-CaSi-PAMAM-FA/mPEG did not produce any DNA ladder, which immediately suggested that the carrier itself has no proapoptotic activity. It was observed that the amount of DNA fragments in 4T1 cells treated by ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was greater than the group with DOX only, and the stimulated DNA fragmentation was attributed to the inclusion of Fe2+ species. The underlying mechanism is that the iron contents could activate lipid peroxidation to up-regulate the expression of AIF and EndoG, both of which could contribute to the DNA fragmentation according to some previous reports (51–53).
Pharmacokinetic analysis of the nanoformulation and the in vivo distribution patterns
Before studying the therapeutic performance of the nanoformulation on tumor-bearing mouse models, we first quantitatively investigated its pharmacokinetic profiles on mouse models (fig. S10C), and the results demonstrated that ACC@DOX-CaSi-PAMAM-FA/mPEG showed prolonged plasma half-life times than ACC@DOX-CaSi or pristine DOX, which has a half-life value of 13.1 ± 2 hours compared to the 5.6 ± 1 hours of ACC@DOX-CaSi or the 1.8 ± 0.7 hours of pristine DOX. The enhanced circulation stability of ACC@DOX-CaSi-PAMAM-FA/mPEG was apparently due to the modification of PEG segments, which is comparable to those nanoformulations currently under preclinical investigation (54) and facilitative for the passive targeting of tumors via enhanced permeability and retention (EPR) effect. The in vivo distribution patterns of the nanoformulation after intravenous injection were also investigated to determine their tumor specificity and delivery efficiency (Fig. 5). For the in vivo tracking of these nanoparticles, the samples were fluorescently stained with an near infrared–absorbing dye named Cy5. The imaging results demonstrated that ACC@Cy5-CaSi-PAMAM-FA/mPEG could escape renal elimination and mononuclear phagocyte system and be more effectively deposited into the tumor tissues compared to pristine Cy5 and ACC@Cy5-CaSi, and its superior tumor accumulation effect has been confirmed on both 4T1 and A375 tumor models. Quantitative analysis of the fluorescence patterns in 4T1 tumor–bearing mice revealed that the total Cy5 fluorescence in mice treated with ACC@Cy5-CaSi-PAMAM-FA/mPEG showed a 3.2-fold increase than the pristine Cy5 group and 1.6-fold increase than ACC@Cy5-CaSi. As for the A375 tumor models, the tumor accumulation of Cy5 fluorescence in ACC@Cy5-CaSi-PAMAM-FA/mPEG–treated mice increased by 3.4-fold than Cy5 and 1.7-fold than ACC@Cy5-CaSi. The improved tumor specificity and delivery efficacy of ACC@Cy5-CaSi-PAMAM-FA/mPEG could be explained by the optimized particle size and surface modification of PEG and FA ligands.
Fig. 5. Fluorescence investigation on the distribution patterns of the nanoformulation in 4T1 and A375 tumor–bearing nude mice.
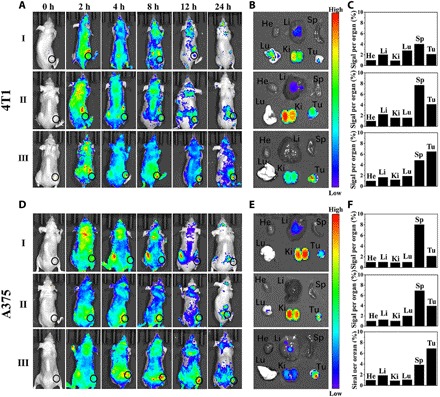
(A and D) Fluorescence images of the 4T1 and A375 tumor–bearing nude mice after the intravenous injection of Cy5 (I), ACC@Cy5-CaSi (II), and ACC@Cy5-CaSi-PAMAM-FA/mPEG (III) at different time points. (B and E) Ex vivo fluorescence images of organs and tumors harvested at 24 hours for mice bearing 4T1 or A375 tumors, respectively. (C and F) Quantitative analysis on the MFI (mean fluorescence intensity) of major organs and tumors in mice bearing 4T1 or A375 tumors, respectively. Data were collected 24 hours after intravenous injection.
In vivo evaluation on the antitumor efficacy of the complementary ferroptosis chemotherapy
After validating the ferroptosis/apoptosis-inducing effect of the Fe2+-coupled nanoformulation on tumor cells, we further studied its antitumor efficacy separately on 4T1 and A375 tumor mouse models. Briefly, nude mice with 4T1/A375 xenograft were divided into five groups (six mice each) and treated with PBS, ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, respectively, and it is ensured that equivalent amount of DOX or carrier substrates was used in each group. By comparing the volume of tumors shown in Fig. 6 (A and B), it was found that the tumor inhibition was the most pronounced in the ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG group. The observed trends in tumor volume were further statistically examined and shown in Fig. 6C, of which the results were highly consistent. Specifically, the average volume of 4T1 tumors after treatment with ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG for 21 days was only 158.7 mm3 with an average weight of 0.11 g, while the average volume and weight of the tumors in the free DOX group were 520.2 mm3 and 0.48 g, respectively. The superior antitumor efficacy of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG than free DOX was also confirmed in A375 tumor–bearing mouse models, in which the tumors treated by ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG have an average volume of 82.5 mm3 and an average weight of 0.09 g, while that of the DOX-only group was still as high as 702.3 mm3 and 0.56 g, respectively (Fig. 6, C and D). Note that even without the Fe2+ doping, the tumor inhibitory caused by ACC@DOX-CaSi-PAMAM-FA/mPEG was still better than free DOX in both tumor models, which could be explained by the tumor-targeting capability of the nanoformulation leading to higher DOX accumulation in the tumor cells. The superior treatment efficacy of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was also supported by the survival analysis (Fig. 6, E and F). Specifically, 4T1 tumor–bearing mice treated with ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG had a median survival time of 50 days, while those A375 tumor–bearing mice treated with ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG had a median survival time of 48 days, both of which were much longer than the control groups. Furthermore, the tumors were extracted and sliced for hematoxylin and eosin (H&E) and terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining (fig. S11C), from which the results showed that the tumor cell death/apoptosis was most severe in the ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG group. The data above consistently support our hypothesis that the ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG nanoformulation could effectively inhibit tumor growth on live animal models via complementary ferroptosis and chemotherapy.
Fig. 6. Therapeutic efficacy of the nanoformulation in vivo.
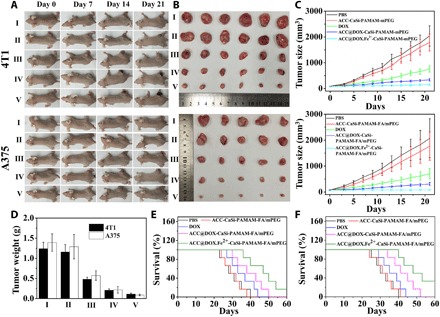
(A) Photographs of 4T1 and A375 tumor–bearing mice through the 21-day treatment period, PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV) and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V). (B) Comparison of tumor tissues extracted from 4T1 and A375 tumor–bearing mice after the 21-day treatment period, PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV) and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V). (C) Changes in the tumor volumes of the 4T1 tumor–bearing mice (six mice in each group) plotted against time, the tumor volume was measured every 2 days. (D) Final weight of tumor tissues extracted from 4T1 and A375 tumor–bearing mice after the 21-day treatment period. PBS (I), ACC-CaSi-PAMAM-FA/mPEG (II), DOX (III), ACC@DOX-CaSi-PAMAM-FA/mPEG (IV) and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (V). (E) Survival rate of 4T1 tumor–bearing mice in 60 days (six mice in each group). (F) Survival rate of A375 tumor–bearing mice in 60 days (six mice in each group). Photo credit: Chen-Cheng Xue, Chongqing University.
In addition to the evaluations on the antitumor efficacy, we also comparatively investigated the potential side effects of various samples after in vivo administration. As revealed by the changes of mouse weight through the experimental period (fig. S11, A and B), the DOX group showed apparent weight loss after around 3 days of incubation, and the final average weight was only 23.7 g, which was due to the severe toxicity caused by DOX. Contrastingly, the body weight for the ACC@DOX-CaSi-PAMAM-FA/mPEG and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG groups has remained at 24.5 and 24.4 g, respectively, which was at the same level to the PBS group (24.4 g). Similarly, it was also observed that the final average body weight of A375 tumor–bearing mice in the ACC@DOX-CaSi-PAMAM-FA/mPEG and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG groups has been maintained at 24.5 and 24.6 g, respectively, in comparison to the 24.6 g of the PBS group. The minimal weight loss in the ACC@DOX-PAMAM-FA/mPEG and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG groups immediately suggests that the iron complexation and ACC encapsulation could markedly ameliorate the detrimental effects induced by DOX. Consistently, histological analysis of the major organs harvested from the mice in each group via H&E staining demonstrates that the free DOX has caused severe damage to the heart muscle, while no apparent histological changes were registered for mice in the ACC@DOX-CaSi-PAMAM-FA/mPEG and ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG groups (fig. S11D). The body weight records and histological observations are direct evidence that the ACC-based DOX-Fe2+ nanoformulation has good biocompatibility, which may greatly facilitate their translation into potential clinical applications.
DISCUSSION
In summary, we report a tumor-targeting degradable therapeutic nanoformulation by encapsulating DOX-Fe2+ complex into ACC nanostructures and functionalized the surface using PAMAM dendrimers separately modified with folate or MMP-2–sheddable PEG. The resultant nanoformulation could be reactivated by tumor microenvironment and induce tumor cells death via complementary ferroptosis/apoptosis mechanism. The DOX-Fe2+ complexation within ACC could protect the Fe2+ from the oxidative stress in biological environment and minimize the premature drug leakage to optimize the therapeutic activity. Upon reaching the MMP-2–rich tumor microenvironment, the PEG segments could be shed at the GPLGVRGDGG peptide linker and consequently expose the folate moieties, thus enhancing the endocytosis of the nanoformulation by the tumor cells. The proton sponge effect of the PAMAM contents would subsequently facilitate the lysosomal escape of the nanoformulation and enable the cytosolic release of the therapeutic agents. Mechanistic studies demonstrated that ROS produced by DOX-induced NOX activation could act in synergy with the simultaneously released Fe2+ to amplify the ferroptotic damage, and the dual ferroptosis/apoptosis treatment eventually leads to potent tumor growth inhibition. The complementary treatment strategy introduced in this study could overcome the limitations of conventional antitumor modalities and offer promise for the development of effective tumor therapies.
MATERIALS AND METHODS
Material
Calcium chloride anhydrous (CaCl2), iron (II) chloride (FeCl2), FA, mPEG4000-N3, and FITC were purchased from Sigma-Aldrich. Ammonium bicarbonate (NH4HCO3), N-(3-dimethylaminopropyl)-N′-ethylcarbodi imide hydrochloride (EDC), n-hydroxysuccinimide (NHS), ammonium hydroxide (25%, v/v), copper sulfate pentahydrate (CuSO4·5H2O), ascorbic acid, and doxorubicin hydrochloride (DOX·HCl) were purchased from Aladdin China. Tetraethyl orthosilicate (TEOS) and (3-aminopropyl) trimethoxysilane were purchased from J&K Scientific Ltd., China. The MMP-2–cleavable peptide of alkynyl-GPLGVRGDGG was obtained from Biochem (Shanghai) Ltd. MTT was obtained from Solarbio Science & Technology Co. Ltd., China.
Synthesis of PAMAM dendrimers
The PAMAM dendrimer was synthesized using a multistep approach. To start with, methyl acrylate (2.60 ml, 28.80 mmol) was added dropwise into the methanolic solution (20.00 ml) of propargylamine (0.44 g, 8.00 mmol). The mixture solution was stirred continuously under argon protection for 48 hours, and the temperature was set at 30°C. The solvent was removed via evaporation when the reaction was complete, and the remaining substance was then subjected to silica gel column chromatography using the mixture of petroleum ether and ethyl acetate (EtOAc) (2/1), and the purified product was denoted as precursor 1. The precursor 1 (0.46 mg, 2.02 mmol) was subsequently solubilized in methanol (1.00 ml), into which ethylenediamine (2.50 ml, 37.50 mmol) was added for reaction. The solution was protected using argon atmosphere and stirred for 48 hours at 30°C. The solvent was also evaporated to obtain the solid residue, which were then precipitated three times with the mixture solution of CH3OH and Et2O for further purification. The oil-like product was the amine-terminated precursor 2 (0.55 g, 1.99 mmol), which was further dissolved in methanol (20.00 ml) and mixed with methyl acrylate dropwise (1.25 ml, 13.80 mmol). Argon atmosphere was used to protect the mixture solution, which was stirred at 30°C for 72 hours. The desired product was recovered through evaporation, and impurities were removed using silica gel column chromatography with the mixture solution of EtOAc and CH3OH (10/1), and precursor 3 was thus obtained. Precursor 3 (0.67 g, 1.08 mmol) was subsequently dissolved in methanol (2.50 ml) and again reacted with ethylenediamine (21.40 ml, 21.00 mmol) that was added dropwise under continuous stirring and argon protection. The reaction lasted for 48 hours while the temperature was maintained at 30°C, and the reaction progression was monitored by infrared spectroscopy. When the reaction was finished, the solvent was removed via evaporation, and the residual solids were allowed to precipitate in the mixture of CH3OH and Et2O for three times for complete purification. The PAMAM dendrimers were thus prepared.
Synthesis of PAMAM-FA
Synthesis of PAMAM-FA: To start with, FA (79.4 mg, 0.18 mmol) was dissolved in 10 ml of N,N′-dimethylformamide (DMF). Subsequently, EDC (0.4 mmol, 77.8 mg) and NHS (0.4 mmol, 46 mg) were added into the solution and stirred for 30 min to activate the terminal carboxyl groups. After the activation was complete, PAMAM (100 mg, 0.14 mmol) was added and the reaction lasted for 24 hours under stirring. The intermediate product was extracted via dialysis (molecular weight cut-off (MWCO): 1000 Da Millipore) and lyophilization and denoted as PAMAM-FA.
Synthesis of mPEG4000-GPLGVRGDGG-PAMAM
For the synthesis of PAMAM-GGPLGVRGDG-mPEG4000, the peptide would be first PEG via click reaction and then conjugate to the folate-modified PAMAM dendrimers. Typically, mPEG4000-N3 (0.025 mmol, 100 mg) and alkynyl-GPLGVRGDGG (0.03 mmol, 25.8 g) were dissolved in 10 ml of DMF, followed by the addition of CuSO4.5H2O (0.04 mmol, 10 mg) and ascorbic acid (0.08 mmol, 15 mg). The reaction was carried out in a single neck flask under nitrogen protection and would continue for 24 hours. The PEGylated peptide was obtained via dialysis (1000 Da, MWCO, Millipore) and denoted as mPEG4000-GPLGVRGDGG. Subsequently, the as-prepared mPEG4000-GPLGVRGDGG (0.02 mmol, 100 mg) was dispersed into 10 ml of DMF, where its carboxylic groups were activated after incubating with EDC (0.06 mmol, 11.5 mg) and NHS (0.06 mmol, 6.9 mg) for 30 min. PAMAM (0.02 mmol, 14.78 mg) was then added to the reaction mixture, and the incubation would continue for another 24 hours, of which the product was extracted via dialysis (2000 Da, MWCO, Millipore) and denoted as mPEG-GPLGVRGDGG-PAMAM.
Synthesis of ACC-DOX.Fe2+
The preparation of the ACC-encapsulated DOX-Fe2+ complex was achieved through a simple one-step gas diffusion method. Specifically, CaCl2 (150 mg), DOX (0.4 ml, 10 mg/ml), and anhydrous ferrous chloride (0.63 mg) were dissolved in 100 ml of absolute ethanol and extensively mixed. Subsequently, the mixture solution was transferred to a glass bottle and then wrapped with parafilm containing several small holes. The vial was placed in a desiccator together with another two bottles of ammonium bicarbonate and incubated at 30°C for 36 hours. The product was recovered via centrifugation at 8000 rpm, and the precipitate was washed twice with ethanol to remove residual reagents.
Preparation of ACC@DOX.Fe2+-CaSi
To coat the thin silica shell onto the ACC@DOX.Fe2+, 2 mg of ACC-DOX.Fe2+ was first dispersed into 20 ml of ethanol and then added with 25 μl of EDTA (12 mg/ml) and 400 μl of ammonia (25%, v/v). After stirring the mixture at 400 rpm for 15 min, 30 μl of TEOS and CaCl2 was added, followed by 400 μl of deionized water. The reaction would continue for another 24 hours, and the product was denoted as ACC@DOX.Fe2+-CaSi.
Preparation of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG and FITC labeling of nanosamples
To conjugate the multifunctional dendrimer onto the surface of the ACC-based cores, 20 mg of ACC@DOX.Fe2+-CaSi was first dispersed into anhydrous toluene and reacted with 10 μl of (3-aminopropyl) trimethoxysilane at 60°C for 24 hours under reflux. The reaction lasted for 24 hours, and the nanoparticle product was washed twice with ethanol. Subsequently, 20 mg of ACC@DOX.Fe2+-CaSi-NH2 was dispersed in 10 ml of ethanol and reacted with 5 mg of TOS-PEG4000-N3 at 45°C overnight under stirring to obtain ACC@DOX.Fe2+-CaSi-N3. The multifunctional dendrimers were eventually immobilized onto the nanoparticle surface via click reaction. Specifically, 20 mg of ACC@DOX.Fe2+-CaSi-PEG-N3, 20 mg of mPEG-GPLGVRGDGG-PAMAM, 2.1 mg of PAMAM-FA, 2 mg of pentahydrate copper sulfate, and 4 mg of ascorbic acid were mixed in 5 ml of DMF and stirred at room temperature for 24 hours under a nitrogen atmosphere. The nanoparticles were collected via centrifugation and washed twice with ethanol. The purified product was also stored in ethanol to prevent degradation.
Some of the nanopreparations were also labeled with FITC for supportive characterizations. The synthesis protocol of these samples was almost identical to that of ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, the only difference is that 4 mg of FITC was added for the co-condensation with ACC instead of the DOX·HCl and anhydrous ferrous chloride.
Structural and physical characterizations of the nanomaterial
The TEM images of the nanosamples were obtained by JEM-1400 (JEOL); high-resolution TEM and scanning transmission electron microscopy were performed on a Talos F200S (Thermo Fisher Scientific) system. The surface charges of the samples and size distribution were measured using by the multiangle particle size and highly sensitive zeta potential analyzer (Omni). XPS analysis was performed on an ESCALAB 250Xi x-ray photoelectron spectrometer.
Evaluation on the dissociation of DOX-Fe2+ complex and their release profiles
To study the dissociation of ACC@DOX-CaSi-PAMAM-FA/mPEG complex in response to pH changes and their release profiles in solution, ACC@DOX-CaSi-PAMAM-FA/mPEG was dispersed into buffer solutions of different pH values (pH 7.4 and pH 5.5) at a concentration of 0.4 mg/ml. After predetermined periods of time, 0.2 ml of the incubation solution was extracted and centrifuged at 8000 rpm for 5 min to remove undissolved nanoparticles. The concentration of DOX in the supernatant was detected by a fluorescence spectrophotometer. The total amount of loaded DOX was determined similarly by dissolving 4 mg of ACC@DOX-CaSi-PAMAM-FA/mPEG in 10 ml of acidic buffer pH 5.5 and sonicated for 1 min. At this condition the ACC@DOX-CaSi-PAMAM-FA/mPEG will degrade completely and release all the encapsulated contents. The DOX fluorescence was measured by extracting 0.5 ml of the incubation.
ICP analysis on the pH-triggered release of Fe2+ ions
ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG was dispersed into buffer solutions (15 ml) of different pH values (pH 7.4 and pH 5.5) at a concentration of 0.4 mg/ml. After a certain time period, 0.2 ml of the incubation solution was extracted and centrifuged at 8000 rpm for 5 min to remove undissolved nanoparticles, and the concentration of Fe in the supernatant was detected by ICP emission spectroscopy on an iCAP 6300 Duo analyzer (Thermo Fisher Scientific).
The complexation between DOX and Fe2+ was also evaluated using fluorescence spectroscopy. Specifically, FeCl2 was first solubilized in ethanol and then added into a series of ethanoic solutions of DOX (DOX concentration, 0.125 μg/ml) at varying amounts to study the effects of Fe2+ doses on DOX-Fe2+ complexation. The time-dependent dissociation of the DOX-Fe2+ complex under pH 5.5 was also studied using the same protocol as the release tests.
Fluorescamine assay on the MMP-2 dependent PEG shedding
Fluorescamine dye is a fluorescent stain that can rapidly react with primary amines and, therefore, has been widely used for the quantification of anime-containing agents. In this study, the fluorescamine assay was used to investigate the impact of the MMP-2–sheddable PEG on the availability of the multifunctional PAMAM dendrimers. Fluorescamine was first dissolved in high-performance liquid chromatography grade acetone (3 mg/ml) to obtain 1 mM solution. Subsequently, lysine external standards (0 to 500 μM) and nanoparticle solutions (100 to 500 μg/ml) were prepared using PBS (pH 7.4). Fluorescamine solution was then added to these as-prepared samples and mixed thoroughly. All samples were measured with an excitation wavelength of 390 nm and an emission wavelength of 460 nm after reacting for 2 hours.
XPS analysis on the valence state of the Fe2+ ions in ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG under different incubation conditions
ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (40 mg) was dispersed into 5 ml of PBS, cell culture medium, and serum and incubated for 24 hours. The nanoparticles were recovered via centrifugation under 8000 rpm for 10 min, and the potential valence changes of the encapsulated Fe2+ ions were analyzed on a x-ray photoelectron spectrometer (ESCALAB250Xi) according to the user manual.
MTT assay on the cytotoxicity of various samples
Two tumor cell lines were used in this study, which are the 4T1 breast cancer cells and A375 melanoma cells. They were both cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (Gibco), penicillin (100 U/ml), and streptomycin (100 μg/ml). The temperature was set at 37°C, and the incubation atmosphere contained 5% CO2. The culture media were refreshed every 2 days. The 4T1 cells were seeded onto a 96-well plate at density of 104 U per well and subsequently incubated overnight at 37°C under an atmospheric CO2 level of 5%. The incubation media were then replaced with fresh ones containing ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@ DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@DOX.Fe2+-CaSi -PAMAM-FA/mPEG (the concentration of MMP-2 was 400 ng/ml), while the tissue culture polystyrene (TCPS) group was used as the blank control. The concentration of nanosamples was 34 μg/ml, and the equivalent DOX concentration was maintained at 1 μg/ml. Each sample group contained six wells, and the incubation periods were set to 12, 24, and 48 hours. Fresh media containing MTT agents (0.5 mg/ml) were added into each well when the incubation was complete and incubated for 4 more hours. Dimethyl sulfoxide (100 μl) was then added to dissolve the emerging formazan crystals. The optical density of the samples at the excitation wavelength of 490 nm was measured on a SpectraMax i3x microplate reader.
Evaluation on the cellular uptake of the nanoagents
For the cellular uptake evaluations, the 4T1 or A375 cells were seeded into a six-well plate at a density of 105 U per well. When the cell confluence reached around 70%, fresh media containing ACC@FITC-CaSi-PEG, ACC@FITC-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@FITC-CaSi-PAMAM-FA/mPEG (MMP-2 concentration, 400 ng/ml) were used to replace the exhausted culture media, where TCPS was used as control. The nanoparticle concentration was maintained at 34 μg/ml, and the incubation periods were set to 12 and 24 hours. When the incubation was complete, the media were drained and the cells were washed three times with PBS. Non–EDTA-Na–containing trypsin was then added to detach the cells, which were further washed twice with PBS and concentrated by centrifugation. The intracellular FITC intensity of the purified cells was monitored by a CytoFLEX system (Beckman Coulter).
For the CLSM study, 4T1 or A375 cells were first seeded into the confocal dish at a density of 1 × 105 and incubated in 1 ml of culture media overnight at 37°C under an atmospheric CO2 level of 5%. The exhausted culture media were replaced with fresh ones containing TCPS (blank control), ACC@FITC-CaSi-PEG, ACC@FITC-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated nanoparticle concentration was maintained at 34 μg/ml and the incubation would continue for 12 or 24 hours. Fresh culture media containing trypan blue (200 μg/ml) was subsequently added to replace the old ones to quench the cytoplasmic fluorescence. The cell samples were then washed with nonserum-containing culture media, and the cell membrane was stained with rhodamine-labeled wheat germ agglutinin solubilized in nonserum-containing culture media (10 μg/ml). When the cell membrane staining was complete, the cells were washed three times with PBS and fixed by paraformaldehyde at 4°C for 30 min. The fixed cells were again washed three times with PBS, and the cell nuclei were stained using H33258 (10 μg/ml) for 5 min. The double-stained cells were washed three more times with PBS and mounted using glycerin. The mounted cells were observed on a Leica TCS SP8 confocal laser microscope.
Investigation on the PAMAM dendrimer induced lysosomal escape of nanocarriers
4T1 cells (105 U) were seeded into a confocal dish and incubated in 1 ml of culture media at 37°C under an atmospheric CO2 level of 5%. When the cell confluence reached 70%, the culture media were changed to fresh ones containing TCPS (blank control), ACC@FITC-CaSi-PEG, ACC@FITC-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@FITC-CaSi-PAMAM-FA/mPEG (MMP-2 concentration, 400 ng/ml) while maintaining the nanoparticle concentration at 34 μg/ml. The incubation would continue for 12 hours. The cytosolic fluorescence was then quenched by incubation with trypan blue (200 μg/ml), and then, the cell samples were washed with nonserum-containing culture media. The lysosomes were stained red by incubating with lyso-tracker red solubilized in nonserum-containing culture media (60 nM) at 37°C for 60 min. The supernatant was removed when the incubation was complete, and the cell samples were washed thrice with PBS. The cells were fixed with paraformaldehyde at 4°C for 30 min and washed again with PBS for three times. The cell nuclei were then stained using H33258 (10 μg/ml) for 5 min. The processed cell samples were washed three more times with PBS and mounted using glycerin. The mounted cells were observed on a Leica TCS SP8 confocal laser microscope.
Quantification of the intracellular iron level
4T1 cells were first seeded onto six-well plates at an initial cell density of 105 U per well, and the incubation conditions were kept the same with the in vitro experiments above. When the cell confluence reached around 70%, the previously added culture media were replaced by new ones containing TCPS (blank control), ACC@DOX.Fe2+-CaSi-PEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, or MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (MMP-2 concentration, 400 ng/ml). The concentration of nanosamples was 34 μg/ml, and the equivalent DOX concentration was maintained at 1 μg/ml. The incubation would continue for 12 or 24 hours. Subsequently, the culture medium in all wells was removed, and the cells were washed three times with PBS. The cells were then detached by trypsin without EDTA-Na and purified twice by repetitive centrifugation/PBS washing. Cell lysate (containing 1% SDS, 1% Triton X-100, and 40 mM tris acetate) was eventually added to lyse the cells, and the resultant solution was sonicated to ensure compete cell disintegration. The iron level was detected by ICP emission spectroscopy on an iCAP 6300 Duo (Thermo Fisher Scientific).
Monitoring the intracellular level of H2O2
The 4T1 cells were seeded into six-well plates using precisely the same protocol as above. When the cell confluence reached around 70%, the culture media in each well were replaced with fresh ones containing different concentrations of DOX (0.0625, 0.125, 0.25, 0.5,, and 2 μg/ml). The incubation would last for 24 hours, and then, intracellular hydrogen peroxide level was examined using the standard Fluorimetric Hydrogen Peroxide Assay Kit (Sigma-Aldrich), for which the fluorescent probe was first added to each well and the cells were further incubated for 20 min at 37°C. The red fluorescent product has an excitation wavelength of 540 nm and an emission wavelength of 590 nm, which were used for the observation of the intracellular H2O2 levels on CytoFLEX flow cytometry system (Beckman Coulter).
Evaluation of the intracellular lipoperoxide accumulation after various treatments
4T1 or A375 cells were seeded into six-well plates as described above and treated with various samples when the cell confluence reached 70%. The samples included TCPS (blank control), ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@ DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG (MMP-2 concentration, 400 ng/ml). The incubation lasted for 24 hours, and the concentration of nanosamples was 34 μg/ml, and the DOX concentration was at 1 μg/ml, which is to maintain both the nanoparticle and drug dosage at an equivalent level. When the incubation was complete, the cells were washed twice with PBS and incubated with DOPIBY C11 (Lipoperoxide indicator; concentration, 5 μM) for 30 min. The intracellular level of lipoperoxides was monitored using a CytoFLEX flow cytometry system (Beckman Coulter). A same experimental setup was also used for the CLSM observations.
Monitoring the changes in mitochondrial membrane potential
The 4T1 or A375 cells were seeded into confocal dishes and treated with various samples when the cell confluence reached 70%, which include TCPS, ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG. After incubation for 12 hours, the tumor mitochondria were stained with JC-1 dye following the procedures provided in the user manual and then observed by CLSM.
Flow cytometric analysis of the cell apoptosis
4T1 or A375 cells were seeded into a six-well plate at the density of 105 U per well. When the cell confluence reached 70%, the culture media were replaced with fresh ones containing TCPS (blank control), ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–pretreated ACC@ DOX.Fe2+-CaSi-PAMAM-FA-/mPEG (MMP-2 concentration, 400 ng/ml), and the incubation would continue for 12 or 24 hours. The concentration for all nanoparticles was 34 μg/ml, and the DOX concentration in each group was maintained at an equivalent level of 1 μg/ml. The media were drained when the incubation was complete, and the cells were washed three times with PBS and subsequently detached using non–EDTA-Na–containing trypsin. The detached cells were purified twice by repetitive washing and centrifugation. The cell apoptosis was investigated by flow cytometry using the Annexin V/PI cell assay kit (Invitrogen) via the protocol provided by the manufacturer.
Alternatively, 4T1 cells were also incubated into confocal dishes using the same procedures for the CLSM analysis of the cell apoptosis. The difference is that when the incubation was complete, the cell samples were incubated with fresh media containing fluorescein diacetate (5 μg/ml) for 30 min and then observed by a Leica TCS SP8 confocal laser microscope.
Western blot assays
To determine the expression levels of the apoptotic protein of Caspase-3 and the ferroptosis-related proteins of AIF and EndoG, 4T1 cells were seeded into six-well plates at a density of 105 U/cm2 and incubated overnight at 37°C under an atmospheric CO2 level of 5% until the cell confluence reached around 70%. Subsequently, the culture media were replaced by fresh ones containing TCPS (blank control), ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@DOX. Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG and further incubated for 24 hours. The concentration for all nanoparticles was 34 μg/ml, and the DOX concentration was 1 μg/ml, which is to ensure the comparability of the results. The cells were then lysed with Laemmli Sample Buffer (Bio-Rad), and the total protein was quantified by electrophoresis using a BCA protein kit (Beyotime) and 12% SDS–polyacrylamide gel electrophoresis. The proteins were then transferred from the gel onto polyvinylidene difluoride membrane (Immobilon P, Millipore) and blocked by primary and secondary antibodies. The images were captured on a molecular imager Versa Doc MP 4000 system (Bio-Rad).
Determination of treatment-induced DNA fragmentation via DNA laddering assay
DNA laddering assay was used to evaluate the DNA damage in 4T1 cells after treatment with TCPS (blank control), ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG, and MMP-2–treated ACC@DOX.Fe2+-CaSi-PAMAM-FA/mPEG. The incubation would last for 24 hours, and the DNA was extracted according to the user manual provided by the manufacturer of the Apoptotic DNA Ladder Isolation Kit (Invitrogen Co. Ltd.), and images were captured after gel electrophoresis with 1% agarose gel.
Fluorescence imaging for the in vivo tracking of the nanosamples
Mice with 4T1 or A375 tumor xenografts were intravenously injected with various sample solutions including Cy5, ACC@Cy5-CaSi, and ACC@Cy5-CaSi-PAMAM-FA/mPEG and imaged on a PerkinElmer in vivo imaging system (IVIS Lumina III) at certain time points. The equivalent Cy5 concentration was maintained at 5 mg/kg.
Analysis of the pharmacokinetic profiles
To determine the blood stability of the nanoformulation in vivo, different samples including DOX, ACC@DOX-CaSi, and ACC@DOX-CaSi-PAMAM-FA/mPEG were intravenously injected into mice (n = 3) with no tumor burden, and the equivalent DOX concentration was maintained at 5 mg/kg. Blood (20 μl) was extracted from each mouse at given time points, and the red blood cells were removed via low-speed centrifugation. The DOX fluorescence intensity in the blood samples was then measured on a fluorescence spectrometer.
Establishment of rodent tumor models and tumor therapy
The 4T1 or A375 cells were established by injecting 100 μl of PBS containing 107 U of 4T1 cells into the subcutaneous tissue of the mice. Various samples were then administrated when the tumor size reached 60 mm3, and the initial weight of all mice was maintained at 18.2 ± 0.2 g. Briefly, 30 4T1 tumor-bearing mice were randomly divided into five groups (each with six mice). The sample groups are PBS, ACC-CaSi-PAMAM-FA/mPEG, DOX, ACC@DOX-CaSi-PAMAM-FA/mPEG, and ACC@ DOX.Fe2+-CaSi-PAMAM-FA/mPEG. All samples were injected through the tail vein at an equivalent DOX concentration of 5 mg/kg. The injection was repeated every other day, and the body weight and tumor volume of nude mice were both recorded. The tumor volume was calculated as Vtumor = LW2/2 (L, maximum diameter of the tumor; W, minimum diameter of the tumor, both were measured using a digital vernier caliper). After 21 days of treatment, all mice were euthanized, and the tumors and major organs were harvested for the subsequent analysis. Typically, the organs and tissues were sectioned and embedded into paraffin after being fixed with 10% formalin at 4°C for 24 hours, and then, paraffin-embedded sections were stained with H&E to monitor the cytotoxicity induced by various samples. In addition, the tumor sections were also stained by the colorimetric TUNEL Apoptosis Assay Kit to determine the therapeutic effect. Both the H&E- and TUNEL-stained tissue sections were then observed with a microscope.
Survival improvement after various treatment
For the survival analysis, 30 4T1 tumor– or A375 tumor–bearing mice were treated using the above procedures. No more injection was given after 21 days, and the number of live mice in each group was recorded until day 60.
Supplementary Material
Acknowledgments
Funding: This work was financially supported by National Key R&D Program of China (2017YFB0702603), National Natural Science Foundation of China (grant nos. 51602034, 51603024, 51732011, 21431006, and 21761132008), Fundamental Research Funds for the Central Universities (2018CDQYSM0036 and 10611CDJXZ238826), and People’s Livelihood Special Innovation Projects of Chongqing CSTC (cstc2017shmsA1089). Ethics statement: The mice were purchased from Army Medical University and housed in Xinqiao Hospital of Chongqing. The study was approved by the Laboratory Animal Welfare and Ethics Committee of Army Medical University (SCXK-2017-0002) and strictly followed the animal experimentation guidelines (GB/T 35892-2018). Author contributions: S.H.Y. and Z.L. conceptualized this study and supervised the experiments. Yang Zhao and M.H.L. designed the experiments. C.-C.X., Yang Zhao, and M.H.L. performed experiments. All authors analyzed the data, discussed the results, and cowrote the manuscript. Yanli Zhao helped with the revision of the experiments and proofreading. All authors approved the final version of the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaax1346/DC1
REFERENCES AND NOTES
- 1.Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan W. C. W., Khademhosseini A., Parak W., Weiss P. S., Cancer: Nanoscience and nanotechnology approaches. ACS Nano 11, 4375–4376 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Rosenblum D., Joshi N., Tao W., Karp J. M., Peer D., Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 9, 1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darr J. A., Zhang J., Makwana N. M., Weng X., Continuous hydrothermal synthesis of inorganic nanoparticles: Applications and future directions. Chem. Rev. 117, 11125–11238 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Ni D., Bu W., Ehlerding E. B., Cai W., Shi J., Engineering of inorganic nanoparticles as magnetic resonance imaging contrast agents. Chem. Soc. Rev. 46, 7438–7468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwon H. J., Shin K., Soh M., Chang H., Kim J., Lee J., Ko G., Kim B. H., Kim D., Hyeon T., Large-scale synthesis and medical applications of uniform-sized metal oxide nanoparticles. Adv. Mater. 30, e1704290 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Ehlerding E. B., Chen F., Cai W., Biodegradable and renal clearable inorganic nanoparticles. Adv. Sci. 3, 1500223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L., Chueng S.-T. D., Li Y., Patel M., Rathnam C., Dey G., Wang L., Cai L., Lee K.-B., A biodegradable hybrid inorganic nanoscaffold for advanced stem cell therapy. Nat. Commun. 9, 3147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du B., Yu M., Zheng J., Transport and interactions of nanoparticles in the kidneys. Nat. Rev. Mater. 3, 358–374 (2018). [Google Scholar]
- 10.Soenen S. J., Parak W. J., Rejman J., Manshian B., (Intra)cellular stability of inorganic nanoparticles: Effects on cytotoxicity, particle functionality, and biomedical applications. Chem. Rev. 115, 2109–2135 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Mao L. B., Gao H. L., Yao H. B., Liu L., Cölfen H., Liu G., Chen S. M., Li S. K., Yan Y. X., Liu Y. Y., Yu S. H., Synthetic nacre by predesigned matrix-directed mineralization. Science 354, 107–110 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Qi C., Lin J., Fu L. H., Huang P., Calcium-based biomaterials for diagnosis, treatment, and theranostics. Chem. Soc. Rev. 47, 357–403 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Maleki Dizaj S., Barzegar-Jalali M., Zarrintan M. H., Adibkia K., Lotfipour F., Calcium carbonate nanoparticles as cancer drug delivery system. Expert Opin. Drug Deliv. 12, 1649–1660 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Min K. H., Min H. S., Lee H. J., Park D. J., Yhee J. Y., Kim K., Kwon I. C., Jeong S. Y., Silvestre O. F., Chen X., Hwang Y.-S., Kim E.-C., Lee S. C., pH-controlled gas-generating mineralized nanoparticles: A theranostic agent for ultrasound imaging and therapy of cancers. ACS Nano 9, 134–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong Z., Feng L., Hao Y., Chen M., Gao M., Chao Y., Zhao H., Zhu W., Liu J., Liang C., Zhang Q., Liu Z., Synthesis of hollow biomineralized CaCO3–Polydopamine nanoparticles for multimodal imaging-guided cancer photodynamic therapy with reduced skin photosensitivity. J. Am. Chem. Soc. 140, 2165–2178 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Wang C., Chen S. Q., Wang Y. X., Liu X. R., Hu F. Q., Sun J. H., Yuan H., Lipase-triggered water-responsive “Pandora’s Box” for cancer therapy: Toward induced neighboring effect and enhanced drug penetration. Adv. Mater. 30, e1706407 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Zhao Y., Luo Z., Li M., Qu Q., Ma X., Yu S.-H., Zhao Y., A preloaded amorphous calcium carbonate/doxorubicin@silica nanoreactor for pH-responsive delivery of an anticancer drug. Angew. Chem. Int. Ed. Engl. 54, 919–922 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Björnmalm M., Thurecht K. J., Michael M., Scott A. M., Caruso F., Bridging bio-nano science and cancer nanomedicine. ACS Nano 11, 9594–9613 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Shi Y. F., Du L. M., Lin L. Y., Wang Y., Tumour-associated mesenchymal stem/stromal cells: Emerging therapeutic targets. Nat. Rev. Drug Discov. 16, 35–52 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Dai Y. L., Xu C., Sun X. L., Chen X. Y., Nanoparticle design strategies for enhanced anticancer therapy by exploiting the tumour microenvironment. Chem. Soc. Rev. 46, 3830–3852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbst R. S., Morgensztern D., Boshoff C., The biology and management of non-small cell lung cancer. Nature 553, 446–454 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Cassetta L., Pollard J. W., Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 17, 887–904 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Vasan N., Baselga J., Hyman D. M., A view on drug resistance in cancer. Nature 575, 299–309 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tannock I. F., Cancer: Resistance through repopulation. Nature 517, 152–153 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., Fulda S., Gascón S., Hatzios S. K., Kagan V. E., Noel K., Jiang X., Linkermann A., Murphy M. E., Overholtzer M., Oyagi A., Pagnussat G. C., Park J., Ran Q., Rosenfeld C. S., Salnikow K., Tang D., Torti F. M., Torti S. V., Toyokuni S., Woerpel K. A., Zhang D. D., Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crielaard B. J., Lammers T., Rivella S., Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov. 16, 400–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang C., Bu W., Ni D., Zhang S., Li Q., Yao Z., Zhang J., Yao H., Wang Z., Shi J., Synthesis of iron nanometallic glasses and their application in cancer therapy by a localized fenton reaction. Angew. Chem. Int. Ed. Engl. 55, 2101–2106 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Ma P., Xiao H., Yu C., Liu J., Cheng Z., Song H., Zhang X., Li C., Wang J., Gu Z., Lin J., Enhanced cisplatin chemotherapy by iron oxide nanocarrier-mediated generation of highly toxic reactive oxygen species. Nano Lett. 17, 928–937 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Dai Y., Yang Z., Cheng S., Wang Z., Zhang R., Zhu G., Wang Z., Yung B. C., Tian R., Jacobson O., Xu C., Ni Q., Song J., Sun X., Niu G., Chen X., Toxic reactive oxygen species enhanced synergistic combination therapy by self-assembled metal-phenolic network nanoparticles. Adv. Mater. 30, 1704877 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Kagan V. E., Mao G., Qu F., Angeli J. P. F., Doll S., Croix C. S., Dar H. H., Liu B., Tyurin V. A., Ritov V. B., Kapralov A. A., Amoscato A. A., Jiang J., Anthonymuthu T., Mohammadyani D., Yang Q., Proneth B., Klein-Seetharaman J., Watkins S., Bahar I., Greenberger J., Mallampalli R. K., Stockwell B. R., Tyurina Y. Y., Conrad M., Bayır H., Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 13, 81–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R., Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. U.S.A. 113, E4966–E4975 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hassannia B., Vandenabeele P., Vanden Berghe T., Targeting ferroptosis to iron out cancer. Cancer Cell 35, 830–849 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Yao Q., Kou L., Tu Y., Zhu L., MMP-responsive ‘Smart’ drug delivery and tumor targeting. Trends Pharmacol. Sci. 39, 766–781 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Li X., Takashima M., Yuba E., Harada A., Kono K., PEGylated PAMAM dendrimer–Doxorubicin conjugate-hybridized gold nanorod for combined photothermal-chemotherapy. Biomaterials 35, 6576–6584 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Tacar O., Sriamornsak P., Dass C. R., Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 65, 157–170 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Song M., Sandoval T. A., Chae C.-S., Chopra S., Tan C., Rutkowski M. R., Raundhal M., Chaurio R. A., Payne K. K., Konrad C., Bettigole S. E., Shin H. R., Crowley M. J. P., Cerliani J. P., Kossenkov A. V., Motorykin I., Zhang S., Manfredi G., Zamarin D., Holcomb K., Rodriguez P. C., Rabinovich G. A., Conejo-Garcia J. R., Glimcher L. H., Cubillos-Ruiz J. R., IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 562, 423–428 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greczynski G., Hultman L., X-ray photoelectron spectroscopy: Towards reliable binding energy referencing. Prog. Mater. Sci. 107, 100591 (2020). [Google Scholar]
- 38.Munnier E., Cohen-Jonathan S., Linassier C., Douziech-Eyrolles L., Marchais H., Soucé M., Hervé K., Dubois P., Chourpa I., Novel method of doxorubicin–SPION reversible association for magnetic drug targeting. Int. J. Pharm. 363, 170–176 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Gautier J., Munnier E., Douziech-Eyrolles L., Paillard A., Dubois P., Chourpa I., SERS spectroscopic approach to study doxorubicin complexes with Fe2+ ions and drug release from SPION-based nanocarriers. Analyst 138, 7354–7361 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Alibolandi M., Abnous K., Hadizadeh F., Taghdisi S. M., Alabdollah F., Mohammadi M., Nassirli H., Ramezani M., Dextran-poly lactide-co-glycolide polymersomes decorated with folate-antennae for targeted delivery of docetaxel to breast adenocarcinima in vitro and in vivo. J. Control. Release 241, 45–56 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Angeli J. P. F., Shah R., Pratt D. A., Conrad M., Ferroptosis inhibition: Mechanisms and opportunities. Trends Pharmacol. Sci. 38, 489–498 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Jiang L., Kon N., Li T., Wang S.-J., Su T., Hibshoosh H., Baer R., Gu W., Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimada K., Skouta R., Kaplan A., Yang W. S., Hayano M., Dixon S. J., Brown L. M., Valenzuela C. A., Wolpaw A. J., Stockwell B. R., Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Z., Wang J., Xie R., Liu R., Lu Y., Mitochondria-derived reactive oxygen species play an important role in doxorubicin-induced platelet apoptosis. Int. J. Mol. Sci. 16, 11087–11100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elsherbiny N. M., El-Sherbiny M., Thymoquinone attenuates doxorubicin-induced nephrotoxicity in rats: Role of Nrf2 and NOX4. Chem. Biol. Interact. 223, 102–108 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Yang W. S., Stockwell B. R., Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 26, 165–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bogdan A. R., Miyazawa M., Hashimoto K., Tsuji Y., Regulators of iron homeostasis: New players in metabolism, cell death, and disease. Trends Biochem. Sci. 41, 274–286 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naghdi S., Slovinsky W. S., Madesh M., Rubin E., Hajnóczky G., Mitochondrial fusion and Bid-mediated mitochondrial apoptosis are perturbed by alcohol with distinct dependence on its metabolism. Cell Death Dis. 9, 1028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gahl R. F., Dwivedi P., Tjandra N., Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis. Cell Death Dis. 7, e2424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin J. L. J., Nakagawa A., Skeen-Gaar R., Yang W. Z., Zhao P., Zhang Z., Ge X., Mitani S., Xue D., Yuan H. S., Oxidative stress impairs cell death by repressing the nuclease activity of mitochondrial endonuclease G. Cell Rep. 16, 279–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kang Y., Tiziani S., Park G., Kaul M., Paternostro G., Cellular protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Commun. 5, 3672 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seiler A., Schneider M., Förster H., Roth S., Wirth E. K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G. W., Schweizer U., Conrad M., Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 8, 237–248 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Dogra P., Adolphi N. L., Wang Z., Lin Y.-S., Butler K. S., Durfee P. N., Croissant J. G., Noureddine A., Coker E. N., Bearer E. L., Cristini V., Brinker C. J., Establishing the effects of mesoporous silica nanoparticle properties on in vivo disposition using imaging-based pharmacokinetics. Nat. Commun. 9, 4551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaax1346/DC1