

MicroRNA-based screening identifies the novel regulators of EV secretion from cancer cells.
Abstract
Extracellular vesicles (EVs) are involved in intercellular communication during cancer progression; thus, elucidating the mechanism of EV secretion in cancer cells will contribute to the development of an EV-targeted cancer treatment. However, the biogenesis of EVs in cancer cells is not fully understood. MicroRNAs (miRNAs) regulate a variety of biological phenomena; thus, miRNAs could regulate EV secretion. Here, we performed high-throughput miRNA-based screening to identify the regulators of EV secretion using an ExoScreen assay. By using this method, we identified miR-26a involved in EV secretion from prostate cancer (PCa) cells. In addition, we found that SHC4, PFDN4, and CHORDC1 genes regulate EV secretion in PCa cells. Furthermore, the progression of the PCa cells suppressing these genes was inhibited in an in vivo study. Together, our findings suggest that miR-26a regulates EV secretion via targeting SHC4, PFDN4, and CHORDC1 in PCa cells, resulting in the suppression of PCa progression.
INTRODUCTION
Extracellular vesicles (EVs) include a wide variety of small membrane-bound vesicles that are actively released from almost all types of cells (1), and play important roles in intercellular communication. EVs transfer functional molecules, including microRNAs (miRNAs), mRNAs, proteins, and lipids, into the recipient cells. Through the transfer of these contents, EVs not only function in normal physiological processes (2) but are also associated with the pathogenesis of various diseases. Particularly in the field of cancer, a number of studies have shown that EVs play important roles in tumor progression (3). In prostate cancer (PCa), some reports have shown that EVs contribute to drug resistance or the progression of metastasis (4–6).
Recently, several reports have shown that the reduction in cancer-derived EVs may have therapeutic value by inhibiting cancer proliferation and dissemination (3). For instance, human epidermal growth factor receptor 2 (HER2) expressed on the surface of breast cancer–derived EVs has been shown to interfere with therapy and is associated with cancer progression (7). In addition, Marleau et al. (8) described a therapeutic strategy for the removal of circulating EVs by developing a hemofiltration system to capture HER2-positive EVs. Furthermore, we recently showed that the administration of antibodies against human-specific CD9 and CD63, which are enriched on the surface of EVs, significantly decreased metastasis in a human breast cancer xenograft mouse model (9). These reports provide promising evidence that the inhibition of circulating EVs could be a novel strategy for cancer treatment. EV secretion from cancer cells is higher than that from normal cells, suggesting that cancer cells have a gene regulation network to promote EV production and/or EV secretion (10). Therefore, understanding this regulatory network will have therapeutic value in cancer treatment. However, despite recent advances in understanding the role of EVs in cancer progression, investigations of the biogenesis of EVs in cancer cells remain obscure. Therefore, the identification of the mechanisms of EV biogenesis will have significant therapeutic potential for treating cancer.
miRNAs are small noncoding RNAs of 20 to 25 nucleotides in length that posttranscriptionally regulate the expression of thousands of genes, and a growing body of evidence has shown that miRNAs are the key regulators of several biological processes. miRNAs are closely associated with tumorigenesis and several stages of metastasis (11). In noncancer cells, miRNAs systematically regulate RNA molecular networks; however, in cancer cells, aberrantly expressed miRNAs disrupt the otherwise tightly regulated relationship between miRNAs and mRNAs, leading to progression and metastasis (12). As shown previously, EV is involved in cancer progression; thus, we hypothesized that miRNAs could regulate EV secretion in cancer cells.
In this study, we found a novel mechanism of EV secretion in PCa cells by investigating miRNAs that are involved in EV secretion. To perform the screening of nearly 2000 species of miRNAs, we used our established EV detection method, ExoScreen, which can directly detect EVs in the conditioned medium (CM) based on an amplified luminescent proximity homogeneous assay (13). We comprehensively screened miRNAs using an miRNA library and found that miR-26a, which was reported as a tumor-suppressive miRNA in PCa (14), negatively regulates EV secretion in PCa cells. In addition, we identified three target genes that were involved in EV secretion in PCa cells. Furthermore, reduced expression of miR-26a and up-regulation of target genes were shown in PCa tumors compared with normal tissues. An in vivo study demonstrated that reduced expression of these three genes inhibited PCa tumor growth, and this change was partially rescued by the injection of EVs from PCa cells. These results suggest novel insights into miRNA-mediated tumor suppression through inhibiting EV biogenesis, which may provide novel approaches for PCa treatment.
RESULTS
Establishment of a high-throughput compatible EV biogenesis assay
The PC3M cells were seeded in 96-well plates and transfected with each miRNA mimic. Twenty-four hours after transfection, the medium was changed to serum-free medium and incubated for another 48 hours. After that, the CM was collected to evaluate the EV level using an ExoScreen, which can directly detect EVs based on an amplified luminescent proximity homogeneous assay using photosensitizer beads and two specific antigens residing on EVs (Fig. 1A) (13). We performed immunoblotting of conventional EV markers (15) and confirmed that EVs derived from PC3M were CD9 and CD63 positive by immunoblotting (Fig. 1B); therefore, we used CD9 antibodies to detect the change in CD9/CD9 double-positive EV secretion in this first screen. We found a correlation between particle number detected via nanoparticle tracking analysis (NTA) and the ExoScreen (fig. S1). In addition, to exclude the effect of miRNAs on cellular proliferation, we performed a colorimetric 3-(4,5-dimethylthiazol-2-yl)-5(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay, as shown in Fig. 1A. To assess the quality of transfection in each plate, several controls were used. The effectiveness of the small interfering RNA (siRNA) controls on EV secretion was almost the same between the plates, which validated the quality of transfection (fig. S2, A and B). EV secretion was calculated through the ExoScreen and MTS assays, and the values were evaluated as the fold change relative to the negative control.
Fig. 1. Screening of miRNAs regulating EV secretion in PCa.
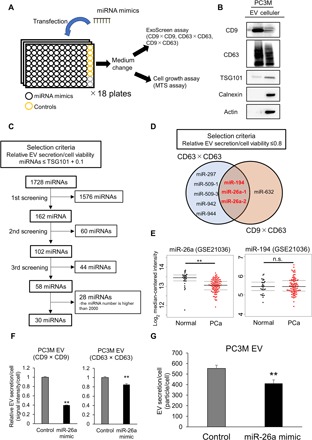
(A) Schematic illustration of a high-throughput compatible EV biogenesis assay to detect EV biogenesis–regulating miRNAs. (B) Immunoblot analysis of the conventional EV markers. A 1-μg sample of EVs and a 10-μg sample of cell lysate from PC3M cells were loaded into each lane. (C) Flow diagram of miRNAs used for selecting candidate miRNAs. (D) Venn diagram showing miRNAs that suppress EV secretion. The miRNAs whose relative EV secretion/cell viability was lower than 0.8 were selected in each assay. The secretion of EVs was evaluated via ExoScreen, and the cell viability was measured via MTS assay. (E) Expression levels of miR-26a and miR-194 in PCa clinical specimens (GSE21036). **P < 0.01; and n.s., not significant. (F) Effect of the miR-26a mimic on EV secretion per PC3M cell. The amount of secretion of EVs per cell was evaluated by the signal intensity of ExoScreen per cell. The values are depicted as the fold change relative to the nonspecific miRNA mimic (control). The values are the means ± SE (n = 3). **P < 0.01. (G) Effect of the miR-26a mimic on EV secretion per PC3M cell. The amount of EV secreted per cell was evaluated using a nanoparticle tracking system. The values are the means ± SE (n = 3). **P < 0.01.
Quantitative high-throughput analysis of candidate miRNAs in PCa cells
An miRNA mimic library was screened to investigate the modulatory effects of various types of miRNAs on EV biogenesis. The effectiveness of each miRNA on the secretion of CD9/CD9-positive EVs was evaluated by ExoScreen, and cell proliferation by colorimetric MTS assays. miRNAs were selected according to the criteria shown in Fig. 1C. We performed screenings three times and selected 58 miRNAs. After excluding miRNAs whose numbers were higher than 2000, 30 miRNAs were selected (Fig. 1C). Next, to further validate the initial screening, the secretion of CD63/CD63-positive EVs and CD9/CD63–double-positive EVs was assessed by ExoScreen in these 30 miRNAs (Fig. 1A). In this set, miRNAs were selected that showed the relative value of EV secretion/cell viability, evaluated by the ExoScreen and MTS assays, which was lower than 0.8. Since the relative value of EV secretion/cell viability by silencing TSG101, which is known to regulate the biogenesis of EVs (16), was 0.77, as evaluated by CD9/CD63–double-positive EVs (fig. S2C), it was expected that the miRNAs could suppress the secretion of EVs, similar to TSG101. Then, we selected miR-26a and miR-194 as candidate miRNAs to regulate EV secretion (Fig. 1D). To select miRNAs that can clinically regulate EV secretion, we investigated a public database (GSE21036). Principal component analysis (PCA) maps with 373 miRNAs suggested that the miRNA profiles differed between the PCa and normal adjacent benign prostate tissues (fig. S3A). In addition, as shown in the heat map displaying the 59 differentially expressed miRNAs repressed by more than 1.25-fold in PCa tissue, relative to normal adjacent benign prostate tissue, with P < 0.001, there were obvious differences in miRNA expression, including miR-26a. However, no difference in the expression of miR-194 in PCa tissue relative to normal adjacent benign prostate tissue was identified (Fig. 1E and fig. S3B). These results suggest that miR-26a is involved in the EV secretion of PCa. Furthermore, it was confirmed via ExoScreen and NTA that the particle number of EVs secreted by each PCa cell transfected with the miR-26a mimic also decreased (Fig. 1, F and G, and fig. S3, C to E). Therefore, miR-26a was selected for further detailed analysis, and whether miR-26a regulates EV secretion in PCa was investigated.
Selection of candidate genes regulating EV secretion in PCa cells
miRNAs are known to regulate hundreds of mRNA targets, providing global changes in the cellular phenotype of cells (17). To further elucidate the molecular mechanisms of miR-26a in EV secretion, the target genes of miR-26a were identified. mRNA microarray analysis in PC3M and PC3 was performed after the transfection of the miR-26a mimic or control. For the genes that could be targeted by miR-26a picked up by TargetScan, it was found that overexpression of miR-26a in PCa cells down-regulated 88 genes, compared with the control cells, by miRNA expression (Fig. 2A). Then, to select genes regulating EV secretion, high-throughput screening was performed using ExoScreen. The PC3M cells were seeded on a 96-well plate and transfected with each candidate siRNA of the 88 genes. Twenty-four hours after transfection, the medium was changed to serum-free medium for 48 hours of incubation. From the transfected PC3M cells, the CM was collected to evaluate the EV levels through ExoScreen and MTS assays (Fig. 2B). CD9- and CD63-positive EVs were evaluated via ExoScreen. The criteria of the selected genes are described in Fig. 2C. The results of each screening are shown in fig. S4. Last, four genes, SHC4, PFDN4, CHORDC1, and PRKCD, were identified as candidate genes regulating EV secretion (Fig. 2C).
Fig. 2. SHC4, PFDN4, and CHORDC1 are involved in miR-26a–mediated EV secretion.

(A) Venn diagram of predicted miR-26a targets (TargetScan) and transcripts that were experimentally repressed >2-fold by miR-26a overexpression in PCa cells (PC3M or PC3) relative to control conditions. (B) Schematic of the high-throughput compatible EV biogenesis assay to choose EV biogenesis–regulating genes. (C) Venn diagram showing genes that suppress EV secretion evaluated by ExoScreen. The genes whose relative EV secretion/cell viability was lower than that of miR-26a plus 0.3 were selected in each assay. The secretion of EV was evaluated by ExoScreen, and the cell viability was measured through the MTS assay. (D) Effect of siRNAs against candidate genes on EV secretion in PC3M cells. The EV secretion per cell was evaluated by the signal intensity of ExoScreen per cell. The values are depicted as the fold change relative to the negative control siRNA (control). The values are the means ± SE (n = 3). *P < 0.05; **P < 0.01; and n.s., not significant. (E) Effect of siRNAs against candidate genes on EV secretion per PC3M cell. The particle number of EVs was measured using a nanoparticle tracking system. The values are the means ± SE (n = 3). *P < 0.05; n.s., not significant. (F) Effect of SHC4, PFDN4, and CHORDC1 siRNA on the mRNA expression level of each gene. β-Actin was used as an internal control. Error bars represent the SE deduced by Student’s t test (*P < 0.05 and **P < 0.01). n.s., no significant difference. The data are representative of at least three independent experiments. The values are the means ± SE (n = 3). **P < 0.01.
SHC4, PFDN4, and CHORDC1 regulate EV secretion in PCa
Next, the effects of these genes on the secretion of EVs derived from PCa cells after treatment with siRNA were confirmed. The EV levels secreted by each PCa cell decreased after transfection with the siRNAs of the three genes, SHC4, PFDN4, and CHORDC1, indicating that these genes are regulators of EV secretion (Fig. 2, D and E, and fig. S5, A and B). The down-regulation of three genes in PCa cells transfected with the siRNA of each gene was confirmed using quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Fig. 2F). These results suggest that SHC4, PFDN4, and CHORDC1 contributed to the up-regulation of EV secretion in PCa.
miR-26a suppressed EV secretion in PCa cancer cells by targeting SHC4, PFDN4, and CHORDC1
First, it was confirmed that miR-26a suppressed the expression levels of SHC4, PFDN4, and CHORDC1 in PCa cells by qRT-PCR (fig. S6, A and B) and immunoblot analysis (Fig. 3A). Then, to address whether miR-26a directly regulated these genes, a luciferase reporter assay was performed (Fig. 3B). The ectopic expression of miR-26a significantly suppressed the luciferase activity of the wild-type SHC4, PFDN4, and CHORDC1 3′ untranslated regions (3′UTRs), but not their mutant 3′UTRs (Fig. 3C). These results provide experimental evidence that miR-26a can directly repress the translation initiation of SHC4, PFDN4, and CHORDC1 and the down-regulation of miR-26a–promoted EV secretion. In addition, using a public database (GSE6099), the expression levels of these genes in PCa tissue were investigated. It was confirmed that the expression levels of PFDN4 and CHORDC1 were significantly up-regulated in PCa tissue compared with normal tissue (Fig. 3D).
Fig. 3. miR-26a directly regulates the expression levels of SHC4, PFDN4, and CHORDC1.

(A) Immunoblot analysis of PC3M cells transfected with nonspecific miRNA mimic (negative control mimic) or miR-26a mimic. A 25-μg sample of cell lysate derived from transfected PCa cells was loaded for the detection of SHC4, CHORDC1, PFDN4, and actin. (B) Summary of miR-26a target sites and mutated sites (shown in red) in the 3′UTRs of SHC4, PFDN4, and CHORDC1. (C) Target validation of SHC4, PFDN4, and CHORDC1 was confirmed in the luciferase reporter assay. The values are depicted as the fold change relative to the negative control siRNA (control). wt, wild type; mut, mutant. The values are the means ± SE (n = 8, ×3 independent experiments). **P < 0.01. (D) Expression levels of SHC4, PFDN4, and CHORDC1 in PCa and normal prostate tissue clinical specimens (GSE6099). *P < 0.05; and n.s., not significant.
PFDN4, SHC4, and CHORDC1 regulate EV secretion and promote tumorigenesis in vivo
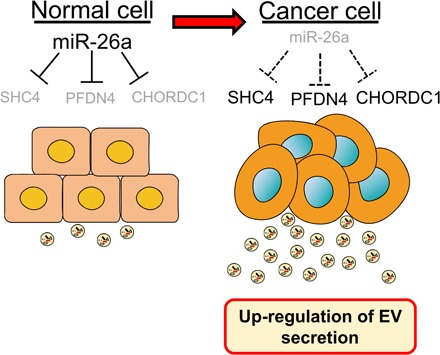
miR-26a was shown to suppress the tumor formation of PCa; thus, this antitumor activity might be due to the suppression of EV secretion from PCa cells by miR-26a (14, 18–20). To confirm the role of these genes targeted by miR-26a in PCa-derived EVs, in vivo experiments were performed. Initially, a PC3M cell line with stable SHC4, PFDN4, or CHORDC1 depletion was established by using each short hairpin RNA (shRNA), and the common aspects of tumorigenesis were evaluated. Depletion of these genes repressed EV secretion (Fig. 4A). The effect of SHC4, PFDN4, and CHORDC1 down-regulation in this model was assessed, and it was found that mice bearing PC3M xenografts with depletion of these genes had smaller tumors that weighed less than those of the control mice (Fig. 4, B and C). In addition, the tumor tissues of nude mice injected with PC3M-derived EVs showed partially rescued tumor sizes and weights (Fig. 4D and fig. S7). The above data suggest a signaling network that links miR-26a with its targets SHC4, PFDN4, and CHORDC1, and demonstrate the novel mechanism of miR-26a–regulated EV secretion in PCa (Fig. 5).
Fig. 4. Down-regulation of EV secretion inhibits cancer progression in vivo.
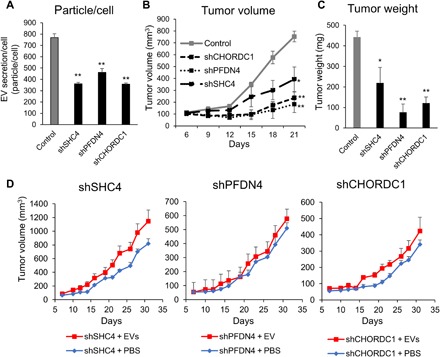
(A) Establishing the PC3M cell line with stable SHC4, PFDN4, and CHORDC1 depletion using shRNAs and evaluation of EV secretion. The values are the means ± SE (n = 3). **P < 0.01. (B) The tumor volumes were measured every 3 days after tumor inoculation. The values are the means ± SE (n = 3). *P < 0.05; **P < 0.01. (C) The tumor weights in nude mice at day 21 were determined. The values are the means ± SE (n = 3). *P < 0.05; **P < 0.01. (D) The tumor volumes were measured every other day before the injection of EVs. The values are the means ± SE (n = 6).
Fig. 5. Schematic model of the regulation of EV secretion in PCa.
DISCUSSION
Although EVs have been reported to modulate cancer progression for approximately 10 years (21), the biogenesis of EV in cancer cells remains unclear. The endosomal sorting complex required for transport (ESCRT) machinery and their associated proteins, including TSG101 (16), Alix (22), and VPS4 (23), have been implicated in EV secretion. In addition, we showed that neutral sphingomyelinase-2 (nSMase2), which is required for the synthesis of ceramide, regulates EV secretion in breast cancer (10). However, the down-regulation of nSMase2 did not inhibit EV secretion in PCa (24). These data suggest that the biogenesis of EV secretion is different between each cancer type. In addition, to establish a novel therapeutic strategy for PCa by inhibiting EV secretion, the mechanism of PCa-specific EV secretion should be revealed.
In the present study, on the basis of a comprehensive miRNA analysis, it was shown that miR-26a and miR-194 regulate the secretion of EVs in PCa. miR-26a was focused on because its expression level was reduced in PCa tissue compared with normal prostate tissue. Previous studies have demonstrated the suppressive role of miR-26a in PCa growth (14, 18–20), and this study reports the novel role of miR-26a. That is, that miR-26a not only inhibits tumorigenesis but also prevents the secretion of EVs. As mentioned above, the suppression of EV secretion may be a novel therapeutic strategy. It is already known that EV secretion in cells is regulated by a variety of genes not only involved in pathological status but also in physiological situations. For instance, EVs from immune cells are indispensable for a proper immune response (25). Therefore, targeted suppression of the genes that specifically regulate EV secretion in cancer cells but not of those from normal cells could be a suitable strategy for cancer treatment. In this study, the cancer-specific EV secretion pathway was identified through screening, and the findings suggest that miR-26a and their target genes represent potential therapeutic targets to inhibit the progression of PCa by inhibiting EV communication.
miRNAs regulate hundreds of mRNA targets, providing global changes in the cellular phenotypes of cells (17). To investigate the target genes of miR-26a that suppress EV secretion, high-throughput screening was performed, and four candidate genes, PRKCD, SHC4, PFDN4, and CHORDC1, were selected. Although the knockdown of PRKCD in PCa decreased the secretion of total EVs, it significantly decreased PCa cell proliferation. Therefore, the secretion of EVs by each cell was not decreased. PRKCD is implicated in the growth, migration, and invasion of cancer cells, including PCa (26); therefore, the down-regulation of PRKCD in PCa cells is critical for cell viability.
In this study, it is revealed that miR-26a directly targets SHC4, PFDN4, and CHORDC1, and the down-regulation of these genes suppressed the secretion of EVs in PCa. Although the precise mechanism of these genes in regulating EV secretion is not reported in the present study, several previous studies have suggested a relationship between EV biogenesis and these genes. SHC4 is a protein of the Src homology and collagen family: ShcA, ShcB, ShcC, and SHC4 (or ShcD), in chronological order of discovery. Shc proteins engage in the epidermal growth factor receptor (EGFR) internalization process (27), and ShcD alters steady-state EGFR trafficking dynamics, which reduces cellular ligand sensitivity by recruiting the EGFR into juxtanuclear vesicles identified as Rab-11–positive endocytic recycling compartments (28). Rab-11 contributes to the endocytic pathway through transportation of the endocytosed cargo to the endocytic recycling compartment (29). In addition, Rab-11 is essential for EV secretion (30). These data suggest that ShcD may also affect the endocytic pathway and contribute to EV secretion. Morgana, which is coded by the CHORDC1 gene, binds to the heat shock protein 90 (Hsp90) chaperone protein, behaving as a cochaperone (31). Recently, Lauwers et al. (32) reported that Hsp90 directly binds and deforms membranes, thereby promoting the fusion of multivesicular bodies with the plasma membrane and the release of EVs. Therefore, CHORDC1 may support the secretion of EVs via stabilizing Hsp90. Compared with SHC4 and CHORDC1, much less is known about the function of PFDN4. The PFDN4 gene is a transcriptional factor that regulates the cell cycle (33). In breast cancer, PFDN4 plays a role in cancer behavior; however, biological analysis of this remains to be performed (34). The precise mechanism of these genes in EV secretion should be investigated in a future study; however, our data suggest, for the first time, that SHC4, PFDN4, and CHORDC1 contribute to EV secretion in PCa and could be novel therapeutic targets.
In addition, in vivo experiments showed that knocking down these genes decreased tumor progression at the primary site. Although it was not possible to confirm whether the tumor-suppressive effect was caused exclusively by the inhibition of EV secretion, the tumor xenografts of nude mice injected with PC3M-derived EVs partially rescued the tumor size and weight of the gene-depleted PC3M xenografts. SHC4, PFDN4, and CHORDC1 are expected to be beneficial for the inhibition of cell proliferation and cell-to-cell communication via EVs in PCa.
In summary, this study extensively screened miRNAs that regulate EV secretion in PCa and found that miR-26a regulates EV secretion by targeting SHC4, PFDN4, and CHORDC1. This finding reveals a novel insight into miRNA-mediated tumor suppression by inhibiting EV biogenesis, which may provide novel approaches for PCa treatment.
MATERIALS AND METHODS
Reagents
The following antibodies were used as primary antibodies in immunoblotting: mouse monoclonal anti-human CD9 antibody (clone 12A12, dilution 1:1000) and CD63 antibody (clone 8A12, dilution 1:1000) from Cosmo Bio (Tokyo, Japan). Rabbit polyclonal anti-human calnexin (#2433, dilution 1:1000) was obtained from Cell Signaling Technology. Mouse monoclonal anti-human TSG101 (clone 51/TSG101, dilution 1:200) was obtained from BD Biosciences. Rabbit polyclonal anti-human PFDN4 (16045-1-AP, dilution 1:200) was obtained from the ProteinTech Group. Mouse monoclonal anti-human actin antibody (clone C4, dilution 1:1000) was obtained from Merck Millipore. Mouse monoclonal anti-human SHC4 (clone 2F5, dilution 1:300) was obtained from Sigma-Aldrich. Rabbit polyclonal anti-human CHORDC1 (NBP1-78304, dilution 1:2500) was purchased from Novus Biologicals (Littleton, CO). Secondary antibodies [horseradish peroxidase–linked anti-mouse immunoglobulin G (IgG), NA931, or horseradish peroxidase–linked anti-rabbit IgG, NA934, dilution 1:5000] were purchased from GE Healthcare.
The following antibodies were used for the ExoScreen analysis: mouse monoclonal anti-human CD9 (clone 12A12) and CD63 antibodies (clone 8A12). These antibodies were used to modify either the acceptor bead or biotin following the manufacturer’s protocol.
The following miRNA mimic or siRNA was used for the transient transfection assay: miR-26a mimic (MC10249), miR-194-5p mimic (MC10004), miRNA mimic negative control #1 (4464058), TSAP6 siRNA (4392422), Rab27a siRNA (S11693), Rab27b siRNA (S11697), and nSMase2 siRNA (S30925) were purchased from Ambion (Austin, TX, USA). ALL STAR negative control siRNA (SI03650318) and TSG101 siRNA (SI02655184) were purchased from Qiagen (Hilden, Germany). PFDN4 siRNA (siGENOME SMARTpool siRNA M-013012), SHC4 siRNA (siGENOME SMARTpool siRNA M-031768), CHORDC1 siRNA (siGENOME SMARTpool M-019998), and PRKCD siRNA (siGENOME SMARTpool M-003524) were purchased from Dharmacon (Lafayette, CO, USA).
Cell culture
The human PCa epithelial metastatic cell line PC3 (ATCC CRL-1435) was purchased from the American Type Culture Collection (ATCC). PC3 and PC-3M-luc-C6 (PC3M) (Xenogen, Alameda, CA) were cultured in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum and an antibiotic-antimycotic (Gibco) solution at 37°C. For routine maintenance, each cell line was grown as a monolayer at 37°C with 5% carbon dioxide and 95% relative humidity. In total, 100,000 cells were seeded in 18 ml of RPMI complete medium and incubated for 3 to 4 days. When the cells were about 80% confluent, they were divided. Cells at a passage number lower than 20 were used in this study.
Preparation of conditioned media and EVs
The cells were washed with phosphate-buffered saline (PBS), and the culture medium was replaced with advanced RPMI 1640 medium (Gibco) for PC3M and PC3, containing an antibiotic-antimycotic and 2 mM l-glutamine (Gibco). EVs from the CM were isolated by a differential ultracentrifugation protocol, as we previously reported (35, 36). Briefly, the CM was centrifuged at 2000g for 10 min to remove contaminating cells. The resulting supernatants were then transferred to fresh tubes and filtered through a 0.22-μm filter (Millipore). The filtered CM was centrifuged for 70 min at 110,000g to pellet the enriched EVs (Beckman Coulter, Brea, CA, USA). The pellets were washed with 11 ml of PBS and ultracentrifuged at 110,000g for another 70 min. To prevent adherence to the tube wall, the isolated EVs were stored in a PROKEEP low protein adsorption tube (15) in a refrigerator at 4°C until use. The collected EVs were used within 2 days for the in vitro study and within 1 week for the animal study.
Immunoblotting
The EV fraction or the transfected PCa cells was measured for protein content using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Wilmington, DE). Equal amounts of protein were loaded onto 4 to 20% Mini-PROTEAN TGX gels (Bio-Rad, Munich, Germany). Following electrophoresis (100 V, 30 mA), the proteins were transferred to a polyvinylidene difluoride membrane. The membranes were blocked with Blocking One solution (Nacalai Tesque, Kyoto, Japan) and then incubated with primary antibodies. After washing, the membrane was incubated with horseradish peroxidase–conjugated sheep anti-mouse IgG or donkey anti-rabbit IgG. Bound antibodies were visualized by chemiluminescence using ImmunoStar LD (Wako).
ExoScreen
AlphaLISA reagents (PerkinElmer Inc., Waltham, MA, USA) consisted of AlphaScreen streptavidin–coated donor beads (6760002), AlphaLISA unconjugated acceptor beads (6062011), and AlphaLISA universal buffer (AL001F). AlphaLISA assays were performed in 96-well half area white plates (6005560) and read in an EnSpire Alpha 2300 Mutilabel Plate Reader (PerkinElmer Inc.). A 96-well white plate was filled with 5 μl of EVs or 10 μl of CM, 10 μl of 5 nM biotinylated antibodies, and 10 μl of AlphaLISA acceptor bead–conjugated antibodies (50 μg/ml) in the universal buffer. To detect CD9/CD9-positive EVs, biotinylated anti-human CD9 antibodies and AlphaLISA acceptor bead–conjugated anti-human CD9 antibodies were used. For CD9/CD63–double-positive EVs, biotinylated anti-human CD9 antibodies and AlphaLISA acceptor bead–conjugated anti-human CD63 antibodies were used. In addition, biotinylated anti-human CD63 antibodies and AlphaLISA acceptor bead–conjugated anti-human CD63 antibodies were used for the detection of CD63/CD63-positive EVs. The plate was then incubated for 1 hour at 37°C. After incubation, 25 μl of AlphaScreen streptavidin–coated donor beads (80 μg/ml) was added. The reaction mixture was incubated in the dark for another 30 min at 37°C. Then, the plate was read on the EnSpire Alpha 2300 Multilabel Plate Reader using an excitation wavelength of 680 nm and emission detection set at 615 nm. Background signals obtained from filtrated advanced RPMI or PBS were subtracted from the measured signals.
Cell growth assay
The cellular viability was determined through a colorimetric assay using the Cell Counting Kit-8 (Dojindo Molecular Technologies Inc.). Plates were read with a spectrophotometer at a wavelength of 450 nm.
miRNA-based high-throughput screening using ExoScreen and cell growth assay
High-throughput miRNA screening (total of 1728 miRNAs) was performed using the AccuTarget Human miRNA mimic library constructed on the basis of miRBase version 21 (Cosmo Bio, Tokyo, Japan). A 100-μl PC3M cell suspension of 5000 cells per well (in RPMI containing 10% serum without antibiotics) was seeded into 96 wells and incubated for 24 hours. Then, transfections of 10 nM miRNA mimics or siRNAs were accomplished with the DharmaFECT Transfection Reagent 1 (Dharmacon) according to the manufacturer’s protocol. After 24 hours, the medium was changed to advanced RPMI 1640 medium containing 2 mM l-glutamine without an antibiotic-antimycotic. Next, 48 hours after medium change, ExoScreen and cell growth assays, which are described above, were performed. Briefly, after 10 μl of CM was collected for the ExoScreen assay, cellular viability was examined through a cell growth assay. The values of EV secretion/cell viability were calculated using the results of the ExoScreen and cell growth assays. Then, the relative values of each well were calculated by comparing to the negative control well.
miRNA and siRNA transient transfection assay
A 2-ml PC3M or PC3 cell suspension of 1.0 × 105 cells per well was seeded into six-well plates and incubated for 24 hours. Then, transfections of 10 nM miRNA mimics or siRNAs were accomplished with the DharmaFECT Transfection Reagent 1 according to the manufacturer’s protocol. The miR-26a mimic or negative control miRNA (miRNA mimic negative control #1, Ambion) was used at a final concentration of 10 nM to investigate the effect of miR-26a on EV secretion. PFDN4 siRNA, CHORDC1 siRNA, SHC4 siRNA, PRKCD siRNA, or ALL STAR negative control siRNA was used at a final concentration of 10 nM to investigate the effect of these genes on EV secretion. After 24 hours, the CM was changed to advanced RPMI medium containing an antibiotic-antimycotic and 2 mM l-glutamine. Then, 48 hours after the medium change, total RNA was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions, and PFDN4, CHORDC1, and SHC4 expression were then determined by qPCR. The CM was collected and purified for EVs by ultracentrifugation.
Analysis of EV particles by NTA
Isolated EV suspensions diluted in PBS were analyzed by NanoSight particle tracking analysis (LM10, with software version 2.03). For particle tracking, at least five 60-s videos were taken of each sample with a camera level 14. The analysis settings were optimized and kept constant between samples. EV concentrations were calculated as particle/cell of the culture to obtain net vesicle secretion rates.
Microarray and bioinformatics
To perform an mRNA microarray, 1.0 × 105 PC3M and PC3 cells were seeded into six-well plates, and 24 hours later, the transfection of miR-26a mimic or controls was accomplished with DharmaFECT Transfection Reagent 1 (Dharmacon) according to the manufacturer’s protocol. After 24 hours, the CM was changed to advanced RPMI medium containing 2 mM l-glutamine without an antibiotic-antimycotic. Total mRNAs were extracted from the PC3M and PC3 cells 48 hours after the medium change.
Total RNA was amplified and labeled with Cy3 using the Low Input Quick Amp Labeling Kit, one color (Agilent Technologies), following the manufacturer’s instructions. Briefly, 100 ng of total RNA was reverse transcribed to double-stranded complementary DNA (cDNA) using a poly dT-T7 promoter primer. The primer, template RNA, and quality control transcripts of a known concentration and quality were first denatured at 65°C for 10 min and incubated for 2 hours at 40°C with 5× first-strand buffer, 0.1 M dithiothreitol, 10 mM deoxynucleotide triphosphate mix, and AffinityScript RNase Block Mix. The AffinityScript enzyme was inactivated at 70°C for 15 min. cDNA products were then used as templates for in vitro transcription to generate fluorescent complementary RNA (cRNA). cDNA products were mixed with a transcription master mix in the presence of T7 RNA polymerase and Cy3-labeled CTP (cytidine 5′-triphosphate) and incubated at 40°C for 2 hours. Labeled cRNA was purified using RNeasy Mini Spin Columns (Qiagen) and eluted in 30 μl of nuclease-free water. After amplification and labeling, cRNA quantity and cyanine incorporation were determined using a NanoDrop ND-1000 spectrophotometer and an Agilent Bioanalyzer. For each hybridization, 0.60 μg of Cy3-labeled cRNA was fragmented and hybridized at 65°C for 17 hours to an Agilent SurePrint G3 Human GE v3 8x60K Microarray (design ID: 072363). After washing, the microarray chips were scanned using an Agilent DNA microarray scanner. The intensity values of each scanned feature were quantified using the Agilent Feature Extraction software version 11.5.1.1, which performs background subtractions. Only features flagged as no errors (detected flags) were used, and features that were not positive, not significant, not uniform, not above background, saturated, and population outliers (compromised and not detected flags) were excluded. Normalization was performed with Agilent GeneSpring version 14.9.1 (per chip: normalization to 75th percentile shift). There are a total of 58,201 probes on the Agilent SurePrint G3 Human GE v3 8x60K Microarray (design ID: 072363) without control probes. The altered transcripts were quantified using the comparative method. Raw and normalized microarray data are available in the Gene Expression Omnibus database (accession number GSE132161).
Unsupervised clustering and heat map generation using Pearson’s correlation in Ward’s method for linkage analysis and PCA were performed using Partek Genomics Suite 6.6. GSEA (www.broadinstitute.org/gsea) to compare the gene expression of PC3M and PC3 after transfection of miR-26a with those of the control (miRNA mimic negative control #1).
RNA extraction and qPCR analysis
Total RNA was extracted from cultured cells using QIAzol and the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. The purity and concentration of all RNA samples were quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific). The reverse transcription reaction was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) and a random hexamer primer. Real-time PCR analyses were performed using StepOne Plus and TaqMan Universal PCR Master Mix (Thermo Fisher Scientific). The expression of mRNA was normalized to β-actin. TaqMan probes for PFDN4, SHC4, CHORDC1, and β-actin were purchased from Applied Biosystems.
Construction of shRNA vector and establishment of stable cell lines
Knockdown vectors expressing shRNAs were constructed by subcloning an annealed oligonucleotide into the pBAsi-hU6-Pur vector (TaKaRa Bio). Oligonucleotide sequences encoding the shRNA hairpin are as follows: CHORDC1_sense, 5′-GATCCGAAGCAAATAGCACATTGTTAAATCTGTGAAGCCACAGATGGGATTTAACAATGTGCTATTTGCTTCTTTTTTA-3′; CHORDC1_antisense, 5′-AGCTTAAAAAAGAAGCAAATAGCACATTGTTAAATCCCATCTGTGGCTTCACAGATTTAACAATGTGCTATTTGCTTCG-3′; PFDN4_sense, 5′-GATCCGAAGAAATTGACGCCTTAGAATCCCTGTGAAGCCACAGATGGGGGATTCTAAGGCGTCAATTTCTTCTTTTTTA-3′; PFDN4_antisense, 5′-AGCTTAAAAAAGAAGAAATTGACGCCTTAGAATCCCCCATCTGTGGCTTCACAGGGATTCTAAGGCGTCAATTTCTTCG-3′; SHC4_sense, 5′-GATCCGCCCCAGAAACCAGTTTAAGTAGGCTGTGAAGCCACAGATGGGCCTACTTAAACTGGTTTCTGGGGCTTTTTTA-3′; SHC4_antisense, 5′-AGCTTAAAAAAGCCCCAGAAACCAGTTTAAGTAGGCCCATCTGTGGCTTCACAGCCTACTTAAACTGGTTTCTGGGGCG-3′.
Hind III (TOYOBO #HND-311) and Bam HI (TOYOBO #BAH-111) were used to digest the pBAsi-hU6-Pur vector and the double-stranded shRNA oligonucleotide cassette insert followed by ligation with T4 DNA ligase. The template oligonucleotides were purchased from Thermo Fisher Scientific. Inserted sequences were confirmed by Sanger sequencing.
Stable PC3M SHC4-, PFDN4-, or CHORDC1-modified cell lines that expressed shRNA against each human gene were generated by selection with puromycin (2 μg/ml). PC3M cells at 90% confluency were transfected with 0.5 μg of the vector in 24-well dishes using the Lipofectamine LTX Reagent in accordance with the manufacturer’s instructions (Life Technologies). Cells were replaced in a 15-cm dish 12 hours after transfection, followed by selection with puromycin for 2 weeks. Five surviving single colonies were picked from each transfection and cultured for an additional 2 weeks. The cells secreting fewer EVs among the transfectants were considered to be stable cell lines.
Plasmid constructs and luciferase reporter assay
The following annealed oligonucleotides (Thermo Fisher Scientific) were used for constructing 3′UTR-reporter vectors: PFDN4_3UTR_S, 5′-TCGAGACATTTTATAATACTTTTTTTATTTGTTTAATAAACTTGAATATTGTTTAAAATGATAATTTTCCTTCTTCAAATGACATGGAGC-3′; PFDN4_3UTR_AS, 5′-GGCCGCTCCATGTCATTTGAAGAAGGAAAATTATCATTTTAAACAATATTCAAGTTTATTAAACAAATAAAAAAAGTATTATAAAATGTC-3′; SHC4_3UTR_S, 5′-TCGAGAAAAGCACAACTAAAATTTCACATGCTAACGACAACTTGAATGAACTGCTGGGGCAGTGGTATGTGCCTTTCAACTTGATAATTGC-3′; SHC4_3UTR_AS, 5′-GGCCGCAATTATCAAGTTGAAAGGCACATACCACTGCCCCAGCAGTTCATTCAAGTTGTCGTTAGCATGTGAAATTTTAGTTGTGCTTTTC-3′; CHORDC1_1_3UTR_S, 5′-TCGAGTTCTCCCTACTGGTAGGAACCATAGTTGTGTCCTATACTTGAAGAGGCTGGAAAGTAGCCCATAACCATAATTGCAGTATTTCTTGC-3′; CHORDC1_1_3UTR_AS, 5′-GGCCGCAAGAAATACTGCAATTATGGTTATGGGCTACTTTCCAGCCTCTTCAAGTATAGGACACAACTATGGTTCCTACCAGTAGGGAGAAC-3′.
The annealed oligonucleotide for the 3′UTR of PFDN4, SHC4, or CHORDC1 was subcloned into psiCHECK2 that had been digested with Xho I and Not I. To mutate miR-26a recognition sites in PFDN4, SHC4, or CHORDC1, the following annealed oligos were used: PFDN4_3UTR_mut_S, 5′-TCGAGACATTTTATAATACTTTTTTTATTTGTTTAATATTGAACTTTATTGTTTAAAATGATAATTTTCCTTCTTCAAATGACATGGAGC-3′; PFDN4_3UTR_mut_AS, 5′-GGCCGCTCCATGTCATTTGAAGAAGGAAAATTATCATTTTAAACAATAAAGTTCAATATTAAACAAATAAAAAAAGTATTATAAAATGTC-3′; SHC4_3UTR_mut_S, 5′-TCGAGAAAAGCACAACTAAAATTTCACATGCTAACGACATGAACTTTGAACTGCTGGGGCAGTGGTATGTGCCTTTCAACTTGATAATTGC-3′; SHC4_3UTR_mut_AS, 5′-GGCCGCAATTATCAAGTTGAAAGGCACATACCACTGCCCCAGCAGTTCAAAGTTCATGTCGTTAGCATGTGAAATTTTAGTTGTGCTTTTC-3′; CHORDC1_1_3UTR_mut_S, 5′-TCGAGTTCTCCCTACTGGTAGGAACCATAGTTGTGTCCTAATGAACTTGAGGCTGGAAAGTAGCCCATAACCATAATTGCAGTATTTCTTGC-3′; CHORDC1_1_3UTR_mut_AS, 5′-GGCCGCAAGAAATACTGCAATTATGGTTATGGGCTACTTTCCAGCCTCAAGTTCATTAGGACACAACTATGGTTCCTACCAGTAGGGAGAAC-3′.
Luciferase reporter assays were performed by cotransfecting HEK293 cells with 8 μl of 5 μM miR-26a mimic or negative control mimic with 500 ng of psiCHECK2 reporter plasmids using Lipofectamine 3000 (Thermo Fisher Scientific) and performing the Dual-Luciferase Reporter Assay System (Promega) after 48 hours.
Mouse studies
Animal experiments in this study were performed in compliance with the guidelines of the Institute for Laboratory Animal Research, National Cancer Center Research Institute (number T19-003). Five-week-old male BALB/C nude mice (Charles River Laboratories, Kanagawa, Japan) were used for animal experiments. PC3M-control, SHC4-, PFDN4-, or CHORDC1-modified cells (1 × 106 cells were injected in 50 μl of Matrigel diluted with PBS) were subcutaneously injected into anesthetized mice. Mice were carefully monitored, and the size of their tumors was measured using Vernier calipers. For the rescue experiment, PC3M-SHC4, PC3M-PFDN4, or PC3M-CHORDC1-KD cells (1 × 106 cells suspended in 50-μl volume Matrigel diluted with PBS) were subcutaneously injected. After 7 days of implantation, 3 μg of EVs from PC3M control cells was injected intratumorally (50 μl in PBS) every other day for up to 28 days.
Mice were monitored carefully, and the size of their tumors was measured using Vernier calipers. Tumors were harvested 31 days after the inoculation of cancer cells, and tumor weight was measured.
Statistical analysis
Unless otherwise described, the data are presented as the means ± SE, and statistical significance was determined by Student’s t test. In the dot plot, the bars indicate the median and interquartile range, and statistical significance was determined by Student’s t test. P < 0.05 was considered statistically significant. A correlation matrix was computed and visualized using R version 3.1.2 (R Foundation for Statistical Computing, www.R-project.org) and the R corrplot package.
Supplementary Material
Acknowledgments
We thank A. Inoue for the support on this work. Funding: This study was supported by the Practical Research for Innovative Cancer Control (18ck0106366h0001) from the Japan Agency for Medical Research and Development (AMED) and the “Development of Diagnostic Technology for Detection of miRNA in Body Fluids” grant from AMED; the Princess Takamatsu Cancer Research Fund; the Kobayashi Foundation for Cancer Research; Novartis Research Grants; and the Foundation for Promotion of Cancer Research. Department of Extracellular Vesicle Science, Industrial-Academic Collaboration, Tokyo Medical University, supported by Theoria Science, Inc. Japan. Author contributions: F.U., Y.Y., K.I., T.Y., T.K., S.E., T.O., and N.K. designed the study. F.U., Y.Y., K.I., T.Y., and N.K. performed the experiments. F.U., Y.S., Y.Y., and N.K. analyzed the data. F.U. and N.K. wrote the manuscript with contribution from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaay3051/DC1
REFERENCES AND NOTES
- 1.Denzer K., Kleijmeer M. J., Heijnen H. F., Stoorvogel W., Geuze H. J., Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 113, 3365–3374 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Fauré J., Lachenal G., Court M., Hirrlinger J., Chatellard-Causse C., Blot B., Grange J., Schoehn G., Goldberg Y., Boyer V., Kirchhoff F., Raposo G., Garin J., Sadoul R., Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 31, 642–648 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Kosaka N., Yoshioka Y., Fujita Y., Ochiya T., Versatile roles of extracellular vesicles in cancer. J. Clin. Invest. 126, 1163–1172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hosseini-Beheshti E., Choi W., Weiswald L.-B., Kharmate G., Ghaffari M., Roshan-Moniri M., Hassona M. D., Chan L., Chin M. Y., Tai I. T., Rennie P. S., Fazli L., Tomlinson Guns E. S., Exosomes confer pro-survival signals to alter the phenotype of prostate cells in their surrounding environment. Oncotarget 7, 14639–14658 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundholm M., Schröder M., Nagaeva O., Baranov V., Widmark A., Mincheva-Nilsson L., Wikström P., Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLOS ONE 9, e108925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye Y., Li S.-L., Ma Y.-Y., Diao Y.-J., Yang L., Su M.-Q., Li Z., Ji Y., Wang J., Lei L., Fan W.-X., Li L.-X., Xu Y., Hao X.-K., Exosomal miR-141-3p regulates osteoblast activity to promote the osteoblastic metastasis of prostate cancer. Oncotarget 8, 94834–94849 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciravolo V., Huber V., Ghedini G. C., Venturelli E., Bianchi F., Campiglio M., Morelli D., Villa A., Mina P. D., Menard S., Filipazzi P., Rivoltini L., Tagliabue E., Pupa S. M., Potential role of HER2-overexpressing exosomes in countering trastuzumab-based therapy. J. Cell. Physiol. 227, 658–667 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Marleau A. M., Chen C.-S., Joyce J. A., Tullis R. H., Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 10, 134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nishida-Aoki N., Tominaga N., Takeshita F., Sonoda H., Yoshioka Y., Ochiya T., Disruption of circulating extracellular vesicles as a novel therapeutic strategy against cancer metastasis. Mol. Ther. 25, 181–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kosaka N., Iguchi H., Hagiwara K., Yoshioka Y., Takeshita F., Ochiya T., Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J. Biol. Chem. 288, 10849–10859 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pritchard C. C., Cheng H. H., Tewari M., MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 13, 358–369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rupaimoole R., Calin G. A., Lopez-Berestein G., Sood A. K., miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discov. 6, 235–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshioka Y., Kosaka N., Konishi Y., Ohta H., Okamoto H., Sonoda H., Nonaka R., Yamamoto H., Ishii H., Mori M., Furuta K., Nakajima T., Hayashi H., Sugisaki H., Higashimoto H., Kato T., Takeshita F., Ochiya T., Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 5, 3591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato M., Goto Y., Matsushita R., Kurozumi A., Fukumoto I., Nishikawa R., Sakamoto S., Enokida H., Nakagawa M., Ichikawa T., Seki N., MicroRNA-26a/b directly regulate La-related protein 1 and inhibit cancer cell invasion in prostate cancer. Int. J. Oncol. 47, 710–718 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Théry C., Witwer K. W., Aikawa E., Alcaraz M. J., Anderson J. D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G. K., Ayre D. C., Bach J.-M., Bachurski D., Baharvand H., Balaj L., Baldacchino S., Bauer N. N., Baxter A. A., Bebawy M., Beckham C., Zavec A. B., Benmoussa A., Berardi A. C., Bergese P., Bielska E., Blenkiron C., Bobis-Wozowicz S., Boilard E., Boireau W., Bongiovanni A., Borràs F. E., Bosch S., Boulanger C. M., Breakefield X., Breglio A. M., Brennan M. Á., Brigstock D. R., Brisson A., Broekman M. L., Bromberg J. F., Bryl-Górecka P., Buch S., Buck A. H., Burger D., Busatto S., Buschmann D., Bussolati B., Buzás E. I., Byrd J. B., Camussi G., Carter D. R., Caruso S., Chamley L. W., Chang Y.-T., Chen C., Chen S., Cheng L., Chin A. R., Clayton A., Clerici S. P., Cocks A., Cocucci E., Coffey R. J., Cordeiro-da-Silva A., Couch Y., Coumans F. A., Coyle B., Crescitelli R., Criado M. F., D’Souza-Schorey C., Das S., Chaudhuri A. D., de Candia P., De Santana E. F., De Wever O., Del Portillo H. A., Demaret T., Deville S., Devitt A., Dhondt B., Vizio D. D., Dieterich L. C., Dolo V., Rubio A. P. D., Dominici M., Dourado M. R., Driedonks T. A., Duarte F. V., Duncan H. M., Eichenberger R. M., Ekström K., Andaloussi S. E., Elie-Caille C., Erdbrügger U., Falcón-Pérez J. M., Fatima F., Fish J. E., Flores-Bellver M., Försönits A., Frelet-Barrand A., Fricke F., Fuhrmann G., Gabrielsson S., Gámez-Valero A., Gardiner C., Gärtner K., Gaudin R., Gho Y. S., Giebel B., Gilbert C., Gimona M., Giusti I., Goberdhan D. C., Görgens A., Gorski S. M., Greening D. W., Gross J. C., Gualerzi A., Gupta G. N., Gustafson D., Handberg A., Haraszti R. A., Harrison P., Hegyesi H., Hendrix A., Hill A. F., Hochberg F. H., Hoffmann K. F., Holder B., Holthofer H., Hosseinkhani B., Hu G., Huang Y., Huber V., Hunt S., Ibrahim A. G.-E., Ikezu T., Inal J. M., Isin M., Ivanova A., Jackson H. K., Jacobsen S., Jay S. M., Jayachandran M., Jenster G., Jiang L., Johnson S. M., Jones J. C., Jong A., Jovanovic-Talisman T., Jung S., Kalluri R., Kano S.-I., Kaur S., Kawamura Y., Keller E. T., Khamari D., Khomyakova E., Khvorova A., Kierulf P., Kim K. P., Kislinger T., Klingeborn M., Klinke D. J. II, Kornek M., Kosanović M. M., Kovács Á. F., Krämer-Albers E.-M., Krasemann S., Krause M., Kurochkin I. V., Kusuma G. D., Kuypers S., Laitinen S., Langevin S. M., Languino L. R., Lannigan J., Lässer C., Laurent L. C., Lavieu G., Lázaro-Ibáñez E., Lay S. L., Lee M.-S., Lee Y. X. F., Lemos D. S., Lenassi M., Leszczynska A., Li I. T., Liao K., Libregts S. F., Ligeti E., Lim R., Lim S. K., Linē A., Linnemannstöns K., Llorente A., Lombard C. A., Lorenowicz M. J., Lörincz Á. M., Lötvall J., Lovett J., Lowry M. C., Loyer X., Lu Q., Lukomska B., Lunavat T. R., Maas S. L., Malhi H., Marcilla A., Mariani J., Mariscal J., Martens-Uzunova E. S., Martin-Jaular L., Martinez M. C., Martins V. R., Mathieu M., Mathivanan S., Maugeri M., McGinnis L. K., McVey M. J., Meckes D. G. Jr, Meehan K. L., Mertens I., Minciacchi V. R., Möller A., Jørgensen M. M., Morales-Kastresana A., Morhayim J., Mullier F., Muraca M., Musante L., Mussack V., Muth D. C., Myburgh K. H., Najrana T., Nawaz M., Nazarenko I., Nejsum P., Neri C., Neri T., Nieuwland R., Nimrichter L., Nolan J. P., Nolte-’t Hoen E. N., Hooten N. N., O’Driscoll L., O’Grady T., O’Loghlen A., Ochiya T., Olivier M., Ortiz A., Ortiz L. A., Osteikoetxea X., Østergaard O., Ostrowski M., Park J., Pegtel D. M., Peinado H., Perut F., Pfaffl M. W., Phinney D. G., Pieters B. C., Pink R. C., Pisetsky D. S., von Strandmann E. P., Polakovicova I., Poon I. K., Powell B. H., Prada I., Pulliam L., Quesenberry P., Radeghieri A., Raffai R. L., Raimondo S., Rak J., Ramirez M. I., Raposo G., Rayyan M. S., Regev-Rudzki N., Ricklefs F. L., Robbins P. D., Roberts D. D., Rodrigues S. C., Rohde E., Rome S., Rouschop K. M., Rughetti A., Russell A. E., Saá P., Sahoo S., Salas-Huenuleo E., Sánchez C., Saugstad J. A., Saul M. J., Schiffelers R. M., Schneider R., Schøyen T. H., Scott A., Shahaj E., Sharma S., Shatnyeva O., Shekari F., Shelke G. V., Shetty A. K., Shiba K., Siljander P. R.-M., Silva A. M., Skowronek A., Snyder O. L. II, Soares R. P., Sódar B. W., Soekmadji C., Sotillo J., Stahl P. D., Stoorvogel W., Stott S. L., Strasser E. F., Swift S., Tahara H., Tewari M., Timms K., Tiwari S., Tixeira R., Tkach M., Toh W. S., Tomasini R., Torrecilhas A. C., Tosar J. P., Toxavidis V., Urbanelli L., Vader P., van Balkom B. W., van der Grein S. G., Van Deun J., van Herwijnen M. J., Van Keuren-Jensen K., van Niel G., van Royen M. E., van Wijnen A. J., Vasconcelos M. H., Vechetti I. J. Jr, Veit T. D., Vella L. J., Velot É., Verweij F. J., Vestad B., Viñas J. L., Visnovitz T., Vukman K. V., Wahlgren J., Watson D. C., Wauben M. H., Weaver A., Webber J. P., Weber V., Wehman A. M., Weiss D. J., Welsh J. A., Wendt S., Wheelock A. M., Wiener Z., Witte L., Wolfram J., Xagorari A., Xander P., Xu J., Yan X., Yáñez-Mó M., Yin H., Yuana Y., Zappulli V., Zarubova J., Žėkas V., Zhang J.-Y., Zhao Z., Zheng L., Zheutlin A. R., Zickler A. M., Zimmermann P., Zivkovic A. M., Zocco D., Zuba-Surma E. K., Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colombo M., Moita C., van Niel G., Kowal J., Vigneron J., Benaroch P., Manel N., Moita L. F., Théry C., Raposo G., Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 126, 5553–5565 (2013). [DOI] [PubMed] [Google Scholar]
- 17.He L., Hannon G. J., MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Fu X., Meng Z., Liang W., Tian Y., Wang X., Han W., Lou G., Wang X., Lou F., Yen Y., Yu H., Jove R., Huang W., miR-26a enhances miRNA biogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth and metastasis. Oncogene 33, 4296–4306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J., Liang J., Huang J., Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncol. Lett. 12, 3397–3402 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang B., Tang X., Wang Z., Sun D., Wei X., Ding Y., TUG1 promotes prostate cancer progression by acting as a ceRNA of miR-26a. Biosci. Rep. 38, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosaka N., Iguchi H., Yoshioka Y., Takeshita F., Matsuki Y., Ochiya T., Secretory mechanisms and intercellular transfer of microRNAs in living cells. J. Biol. Chem. 285, 17442–17452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baietti M. F., Zhang Z., Mortier E., Melchior A., Degeest G., Geeraerts A., Ivarsson Y., Depoortere F., Coomans C., Vermeiren E., Zimmermann P., David G., Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 14, 677–685 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Jackson C. E., Scruggs B. S., Schaffer J. E., Hanson P. I., Effects of Inhibiting VPS4 support a general role for ESCRTs in extracellular vesicle biogenesis. Biophys. J. 113, 1342–1352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Phuyal S., Hessvik N. P., Skotland T., Sandvig K., Llorente A., Regulation of exosome release by glycosphingolipids and flotillins. FEBS J. 281, 2214–2227 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., Amigorena S., Eradication of established murine tumors using a novel cell-free vaccine: Dendritic cell derived exosomes. Nat. Med. 4, 594–600 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Kharait S., Dhir R., Lauffenburger D., Wells A., Protein kinase Cδ signaling downstream of the EGF receptor mediates migration and invasiveness of prostate cancer cells. Biochem. Biophys. Res. Commun. 343, 848–856 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Sakaguchi K., Okabayashi Y., Kasuga M., Shc mediates ligand-induced internalization of epidermal growth factor receptors. Biochem. Biophys. Res. Commun. 282, 1154–1160 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Wills M. K. B., Lau H. R., Jones N., The ShcD phosphotyrosine adaptor subverts canonical EGF receptor trafficking. J. Cell Sci. 130, 2808–2820 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Kelly E. E., Horgan C. P., McCaffrey M. W., Rab11 proteins in health and disease. Biochem. Soc. Trans. 40, 1360–1367 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Savina A., Vidal M., Colombo M. I., The exosome pathway in K562 cells is regulated by Rab11. J. Cell Sci. 115, 2505–2515 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Michowski W., Ferretti R., Wisniewska M. B., Ambrozkiewicz M., Beresewicz M., Fusella F., Skibinska-Kijek A., Zablocka B., Brancaccio M., Tarone G., Kuznicki J., Morgana/CHP-1 is a novel chaperone able to protect cells from stress. Biochim. Biophys. Acta 1803, 1043–1049 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Lauwers E., Wang Y.-C., Gallardo R., Van der Kant R., Michiels E., Swerts J., Baatsen P., Zaiter S. S., McAlpine S. R., Gounko N. V., Rousseau F., Schymkowitz J., Verstreken P., Hsp90 mediates membrane deformation and exosome release. Mol. Cell 71, 689–702.e9 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Vainberg I. E., Lewis S. A., Rommelaere H., Ampe C., Vandekerckhove J., Klein H. L., Cowan N. J., Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Collins C., Volik S., Kowbel D., Ginzinger D., Ylstra B., Cloutier T., Hawkins T., Predki P., Martin C., Wernick M., Kuo W. L., Alberts A., Gray J. W., Comprehensive genome sequence analysis of a breast cancer amplicon. Genome Res. 11, 1034–1042 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshioka Y., Konishi Y., Kosaka N., Katsuda T., Kato T., Ochiya T., Comparative marker analysis of extracellular vesicles in different human cancer types. J. Extracell. Vesicles 2, 10.3402/jev.v2i0.20424 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yokoi A., Yoshioka Y., Yamamoto Y., Ishikawa M., Ikeda S.-I., Kato T., Kiyono T., Takeshita F., Kajiyama H., Kikkawa F., Ochiya T., Malignant extracellular vesicles carrying MMP1 mRNA facilitate peritoneal dissemination in ovarian cancer. Nat. Commun. 8, 14470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaay3051/DC1