

Targeting Vps34 makes melanoma and CRC eligible to anti–PD-1/PD-L1 blockade.
Abstract
One of the major challenges limiting the efficacy of anti–PD-1/PD-L1 therapy in nonresponding patients is the failure of T cells to penetrate the tumor microenvironment. We showed that genetic or pharmacological inhibition of Vps34 kinase activity using SB02024 or SAR405 (Vps34i) decreased the tumor growth and improved mice survival in multiple tumor models by inducing an infiltration of NK, CD8+, and CD4+ T effector cells in melanoma and CRC tumors. Such infiltration resulted in the establishment of a T cell−inflamed tumor microenvironment, characterized by the up-regulation of pro-inflammatory chemokines and cytokines, CCL5, CXCL10, and IFNγ. Vps34i treatment induced STAT1 and IRF7, involved in the up-regulation of CCL5 and CXCL10. Combining Vps34i improved the therapeutic benefit of anti–PD-L1/PD-1 in melanoma and CRC and prolonged mice survival. Our study revealed that targeting Vps34 turns cold into hot inflamed tumors, thus enhancing the efficacy of anti–PD-L1/PD-1 blockade.
INTRODUCTION
Immune checkpoint inhibitors (ICIs), exemplified by antibodies targeting programmed death 1 (PD-1) or programmed death ligand 1 (PD-L1), are groundbreaking treatment for many cancers, including melanoma (1). However, the clinical responses are only observed in a limited number of cancer patients treated with these drugs. The majority of patients treated with ICIs as single agents reap limited or no benefits at all (2). Therefore, there is a strong clinical need to improve the efficacy of ICI by designing synergistic combinations with other rational therapies to increase response rates and extend their use to a larger number of patients and tumor types. In many cases, one of the major causes of nonresponsiveness to these agents is the limited tumor penetrance of effector T cells. Durable clinical responses using anti–PD-1/PD-L1–based therapy have been associated with T cell–inflamed tumor microenvironment (TME) favoring the infiltration of functional cytotoxic T lymphocytes (CTLs) (3). It is now generally appreciated that driving cytotoxic immune cells into the tumor bed could significantly improve the efficacy of ICI, notably in patients displaying an immune cold phenotype. This statement is supported by clinical data showing that the presence of preexisting T cells close to the invasive tumor margin is a key determinant to achieving a long-lasting response to anti–PD-1 therapy in melanoma (4). On the basis of these studies, significant efforts to identify therapeutic approaches able to turn immune cold tumors hot by enhancing immune cell infiltration into the tumor bed are currently undertaken (4).
Autophagy is a cellular process involved in the degradation and recycling of cytoplasmic contents in well-defined structures called autophagosomes. The fusion of autophagosomes with lysosomes leads to the degradation of sequestered materials by lysosomal hydrolases (5). Several reports have suggested that autophagy inhibition can enhance the antitumor efficacy of chemotherapy or targeted therapies (6). We have reported extensive data showing that autophagy activation in tumor cells plays a major role in impairing the antitumor immune response, providing evidence that targeting autophagy inhibits tumor growth and improves CTL and natural killer (NK)–mediated killing (7, 8). More recently, we showed that targeting the autophagy-related gene Beclin1 inhibited melanoma tumor growth by inducing the infiltration of functional NK cells into the TME by a mechanism involving the release of CCL5/RANTES by tumor cells (9). These results point toward autophagy activation in tumor cells as a major hijacker of the antitumor immune response and reinforce the inhibition of autophagy as a new approach in the field of cancer immunotherapy. Developing selective drugs inhibiting autophagy is challenging, as most autophagy-related proteins also display nonautophagic functions (10). Recently, vacuolar protein sorting 34 (Vps34) or class III phosphoinositide 3-kinase (PIK3C3) has attracted immense interest as a potential druggable protein to inhibit autophagy (11, 12). Vps34 forms two distinct complexes involved either in the initiation of autophagy by interaction with Vps15/Atg14/UVRAG/Beclin1 or in the process of endocytosis where it interacts with Vps15/UVRAG/Rab5/EEA1 (13).
Here, we show that either genetic targeting of the PIK3C3 gene Vps34 or pharmacologically inhibiting its kinase activity, using two selective drugs, had a broad and marked impact on the immune landscape of melanoma and colorectal cancer (CRC) by inducing the infiltration of not only NK cells but also CD8+ and CD4+ T effector cells into the tumor bed. We found that such infiltration is mechanistically related to the reprogramming of immune cold desert TME into a hot inflamed immune cell–infiltrated TME. We showed that such reprogramming is the result of the establishment of a pro-inflammatory cytokine signature in the TME and in the blood of tumor-bearing mice treated with Vps34 inhibitors (Vps34i). Treatment of melanoma or CRC tumor–bearing mice with Vps34i improves the therapeutic benefit of targeting PD-1 and PD-L1. This study provides evidence that Vps34 inhibition makes melanoma and CRC tumors more susceptible to ICI-based immunotherapies, providing the preclinical rationale for clinical trials using selective Vps34i in combination with various ICIs.
RESULTS
Targeting Vps34 inhibits tumor growth and improves mice survival in multiple cancer models
We first evaluated the impact of targeting Vps34 (both genetically and pharmacologically) on tumor growth and tumor weight in different cancer models. Genetic targeting of Vps34 was achieved by stable transfection of B16-F10 and CT26 cells with a vector encoding Vps34 short hairpin RNA (shVps34). The efficient knockdown of Vps34 protein resulted in complete inhibition of autophagy flux in B16-F10 and CT26 cells (fig. S1, A and B). After inoculation into the left flank of immunocompetent mice, the growth of tumors, transfected with control vector (shCT), and shVps34 B16-F10 and CT26 cells was monitored. Our results in Fig. 1 (A and B) and fig. S1C show that genetic targeting of Vps34 significantly decreased tumor growth and tumor weight and improved mice survival. We next assessed whether, similar to genetic targeting of Vps34, pharmacological inhibition of Vps34 kinase activity also affects the tumor growth, tumor weight, and mice survival of several tumor types. Two diverse and selective Vps34 kinase inhibitors (Vps34i) were used: SB02024 developed by Sprint Bioscience (12) and SAR405 developed by Sanofi Pharma (11). In vitro, Vps34i efficiently inhibited the autophagy flux in B16-F10 melanoma, CT26 CRC, Renca renal cell carcinoma, and genetically engineered melanoma mouse (GEMM) YUMM cells, derived from a spontaneous mouse melanomas driven by Braf activation, Pten inactivation, and Cdkn2a inactivation (fig. S1D) (14). Systemic treatment (oral gavage) of tumor-bearing mice with Vps34i resulted in significant inhibition of tumor growth, reduction of tumor weight, and significant improvement of mice survival in B16-F10 (Fig. 1C and fig. S1E), CT26 (Fig. 1D and fig. S1F), YUMM (Fig. 1E and fig. S1G), and Renca (Fig. 1F and fig. S1H). These results demonstrate that the inhibition of tumor growth, the decrease in tumor weight, and the improvement of mice survival resulting from SB02024 or SAR405 treatment are not restricted to a specific tumor histology or mouse genetic background, and they could be broadly applied to multiple tumor models, including melanoma and CRC.
Fig. 1. Targeting Vps34 decreases tumor growth and tumor weight and improves mice survival in multiple tumor types.
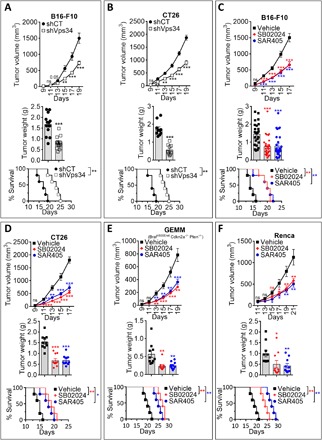
(A and B) Growth curves, weight in grams, and mice survival curves of control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (A) and CT26 CRC (B) tumors. Each curve represents three independent experiments of four or five mice per group for B16-F10 and two independent experiments of five mice per group for CT26. (C to F) Growth curves, weight in grams, and mice survival curves of B16-F10 melanoma (C); CT26 colorectal (D), GEMM (YUMM) melanoma (E); or Renca renal carcinoma (F) tumors in mice treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). For B16-F10, each curve represents three independent experiments of eight or nine mice per group. For CT26, YUMM, or Renca, each curve represents two independent experiments of five mice per group. All results are shown as mean ± SEM (error bars). Statistically significant differences (indicated by asterisks) are calculated compared to control conditions using an unpaired two-tailed Student’s t test. Not significant (ns) = P > 0.05; *P < 0.05; **P < 0.005; and ***P < 0.0005. Mice survival curves (five mice per group for all tumor models) were generated from tumor-bearing mice. Lack of survival was defined as death or tumor size >1000 mm3. Mice survival percentage was defined using GraphPad Prism, and P values were calculated using the log-rank (Mantel-Cox) test (*P ≤ 0.05 and **P ≤ 0.01).
Vps34 targeting enhances the infiltration of various antitumor immune effector cells
We next investigated whether the Vps34-dependent antitumor activity was associated with a modulation of the tumor immune landscape. We showed that the percentage of live CD45+ cells was significantly increased in shVps34 B16-F10 tumors as compared to shCT B16-F10 tumors (Fig. 2A, top left). Similarly, Vps34i treatment significantly increased the percentage of live CD45+ cells in both B16-F10 and CT26 tumors (Fig. 2A, top middle and right). The increased infiltration of CD45+ cells into B16-F10 melanoma tumors treated with Vps34i was further confirmed by immunohistochemistry staining on tumor sections (Fig. 2A, bottom). We next performed comprehensive immune phenotyping of different immune cell subpopulations by flow cytometry to identify and quantify both immune effector and immune suppressor cell subsets infiltrating B16-F10 tumors genetically defective in Vps34 or pharmacologically treated with Vps34i. The gating strategies used for immune phenotyping are reported in fig. S2. We observed a significant increase in the infiltration of immune effectors NK, CD8+ T cells, CD4 T effector cells, dendritic cells (DCs), and M1 macrophages in shVps34- and Vps34i-treated B16-F10 tumors as compared to shCT- and vehicle-treated controls (Fig. 2B, top and middle). The increased infiltration of CD8+ T cells into B16-F10 melanoma tumors treated with SB02024 or SAR405 was also confirmed by immunohistochemistry on three different tumor sections (Fig. 2B, middle). Enlarged tumor sections showing the infiltration of CD45+ cells (fig. S3A) and CD8+ cells (fig. S3B) into three different B16-F10 tumors are shown. Similar to B16-F10 tumors, an increased infiltration of NK, CD8+ T cells, DCs, and M1 macrophages, but not CD4+ T effector cells, was also observed in Vps34i-treated CT26 tumors as compared to vehicle-treated control tumors (Fig. 2B, bottom). An increase in the expression level of the activation marker CD69 was observed on NK, CD8+, and CD4+ T effector cells infiltrating shVps34 B16-F10 tumors and Vps34i-treated B16-F10 and CT26 tumors as compared to shCT and vehicle-treated control tumors (Fig. 2C). No significant difference in the infiltration of M2 macrophages, regulatory T (Treg) cells, and myeloid-derived suppressor cells (MDSCs) was observed in shVps34 B16-F10 tumors and in Vps34i-treated B16-F10 and CT26 tumors as compared to controls (Fig. 2D). However, CD8/Treg ratio was significantly increased in shVps34 and Vps34i-treated B16-F10 tumors (Fig. 2E).
Fig. 2. Targeting Vps34 induces profound changes in the immune landscape of tumors.
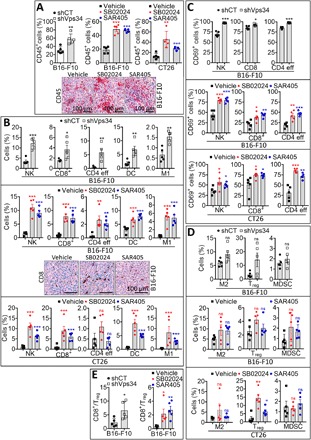
(A) Top: Flow cytometry quantification of CD45+ leukocytes (gated in live cells) infiltrating the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (left) and B16-F10 melanoma (middle) or CT26 colorectal (right) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). Bottom: Representative images of immunohistochemical staining of CD45 performed on B16-F10 tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). Scale bars, 100 μm. (B) Flow cytometry quantification of NK cells (NK), CD8+ T cells (CD8), CD4+ effector T cells (CD4 eff), DCs, and type 1 macrophages (M1) infiltrating the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (top) and B16-F10 melanoma (middle) or CT26 colorectal (bottom) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). Representative images of immunohistochemical staining of CD8+ performed on B16-F10 tumors treated with vehicle (vehicle) or Vps34i (SB02024 or SAR405) are included in the bottom of the middle panel. Scale bars, 100 μm. (C) Quantification of the percent of CD69+ activated NK cells, CD8+ T cells, and CD4+ effector T cells infiltrating the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (top) and B16-F10 melanoma (middle) or CT26 colorectal (bottom) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). (D) Quantification of the percent of type 2 macrophages (M2), Treg, and MDSCs infiltrating the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (top) and B16-F10 melanoma (middle) or CT26 colorectal (bottom) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). (E) CD8+–to–Treg cell ratio (CD8/Treg) in control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (left) and in B16-F10 melanoma tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405) (right). Experiments described in (A) to (E) were conducted on well-established tumors on day 15. The defined subpopulations were gated and quantified in live CD45+ cells. Each dot represents one tumor. The data are reported as the average of five mice per group (each dot represents one mouse). Results are shown as mean ± SEM (error bars). Statistically significant differences (indicated by asterisks) are calculated compared to control conditions using an unpaired two-tailed Student’s t test (*P < 0.05, **P < 0.005, and ***P < 0.0005).
To evaluate the involvement of adaptive immunity in Vps34-mediated decrease in tumor growth and tumor weight, we investigated the effect of shVps34 and Vps34i treatment on immunocompromised NOD scid gamma (NSG) mice, lacking mature B, T, and NK cells. We found that both shVps34 (Fig. 3A) and Vps34i (Fig. 3B) treatment had no effect on tumor growth and tumor weight in B16-F10 tumor–bearing NSG mice. We next assessed to what extent the inhibition of tumor growth and weight were related to the Vps34-dependent increase in infiltration of NK or CD8+ T cells. NK or CD8+ T cells were depleted in shCT or shVps34 and in vehicle- or Vps34i-treated B16-F10 tumor–bearing mice. The strategy and the efficacy of NK and CD8+ T cell depletions are shown in fig. S4 (A to C). While the depletion of NK or CD8+ T cells had moderate or no impact on the growth of weakly infiltrated shCT or vehicle-treated tumors, such depletion significantly increased the volume and the weight of shVps34 (Fig. 3C) or Vps34i-treated (Fig. 3, D and E) B16-F10 melanoma tumors. The impact of depleting NK cells on the rescue of tumor growth inhibition of B16-F10 was higher than that observed by depleting CD8+ T cells. This result suggests that although both NK and CD8+ T effector cells are the major immune cells controlling the growth of Vps34-defective B16-F10 tumors, NK cells seem to be more important than CD8+ T cells.
Fig. 3. Tumor growth inhibition in Vps34-targeted tumors is dependent on NK and CD8+ T cells.
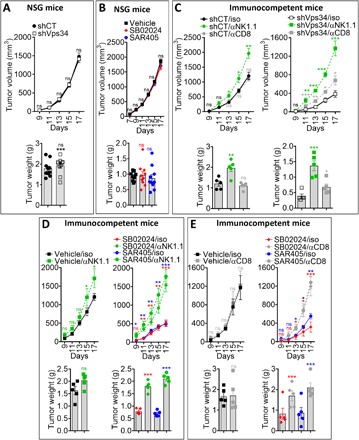
(A) Growth curve (top) and weight in grams (bottom) in immunodeficient NSG mice of shCT and shVps34 B16-F10 tumors. (B) Growth curve (top) and weight in grams (bottom) of B16-F10 tumors in immunodeficient NSG mice treated with vehicle or Vps34i (SB02024 or SAR405). (C) Growth curves (top) and weight in grams (bottom) of shCT and shVps34 B16-F10 tumors in control (iso), NK-depleted (αNK1.1), and CD8-depleted (αCD8) mice. (D) Growth curves (top) and weight in grams (bottom) of B16-F10 tumors in control (iso) or NK-depleted (αNK1.1) mice treated with vehicle or Vps34i (SB02024 or SAR405). (E) Growth curves (top) and weight in grams (bottom) of B16-F10 tumors in control (iso) or CD8-depleted (αCD8) mice treated with vehicle or Vps34i (SB02024 or SAR405). Data described in (A) to (E) are reported as the average of 10 (A and B) and 5 (C to E) mice per group. Each dot represents one mouse. Results are shown as mean ± SEM (error bars). Tumor weights were assessed at day 17. Statistically significant differences (indicated by asterisks) are calculated compared to control conditions using an unpaired two-tailed Student’s t test (*P < 0.05, **P < 0.005, and ***P < 0.0005).
Inhibition of Vps34 establishes a pro-inflammatory TME responsible for the recruitment of NK and CD8+ cells
A mechanistic link between autophagy and the secretion of proteins, including cytokines, in the TME has been described (15). Moreover, it has been shown that autophagy promotes cancer cell invasion by coordinating the production of multiple secreted factors (16). We next evaluated the expression of a subset of six chemokines (CCL3, CCL4, CCL5, CXCL9, CXCL10, and CXCL11), previously described to be involved in the recruitment of CD8+ T cells into human melanomas (17). Both Ccl5/Rantes and Cxcl10/IP10 mRNA and their corresponding secreted protein levels were significantly up-regulated in Vps34-silenced (shRNA and si-RNA Vps34) B16-F10 melanoma (fig. S5, A and B) and in Vps34i-treated B16-F10 and CT26 tumor cells as compared to controls (fig. S5, C and D). Similarly, CCL5 and CXCL10 mRNA and their corresponding secreted protein levels were significantly increased in YUMM cells treated with Vps34i (fig. S5E). The increase in CCL5 and CXCL10 was further confirmed in different types of human and mouse cancer cell lines treated with SB02024 or SAR405 in vitro (fig. S6, A to C). Genetic or pharmacological Vps34 inhibition resulted in significantly higher levels of CCL5, CXCL10, and interferon-γ (IFNγ) in the tumor plasma within the TME of B16-F10 and CT26 tumors as compared to controls (Fig. 4A). We next evaluated whether the increased levels of CCL5 and CXCL10 could also be observed in the blood of shVps34 tumor–bearing mice. We did not observe any difference in CCL5 and CXCL10 in the blood plasma of shVps34 compared to control (shCT) B16-F10 melanoma (Fig. 4B, top). However, the levels of CCL5 and CXCL10 were significantly higher in the blood plasma of B16-F10 and CT26 tumor–bearing mice treated with SB02024, but not in the blood plasma of mice treated with SAR405 (Fig. 4B, middle and bottom). Such an increase in CCL5 and CXCL10 is most likely generated from the tumor, because in SB02024 treatment did not increase CCL5 or CXCL10 levels in the blood of non–tumor-bearing mice (fig. S6D). Together, our data indicate that Vps34 inhibition induced the secretion of pro-inflammatory chemokines and cytokines (CCL5, CXCL10, and IFNγ) mostly by the tumor cells within the TME. CCL5, CXCL10, and IFNγ are all known to promote recruitment of cytotoxic T cells into the TME (18), and this is likely the explanation for why we observed enhanced recruitment of NK, CD8+ T, and CD4+ T effector cells into the tumor bed following Vps34 inhibition. To experimentally validate this statement, we blocked CCL5 by using anti–CCL5-neutralizing antibody in Vps34i-treated B16-F10 and CT26 tumor–bearing mice and investigated whether the increased infiltration of NK and CD8+ T cells was abrogated. Our results showed that neutralizing CCL5 in vehicle-treated B16-F10 and CT26 tumor–bearing mice has no impact on tumor growth and tumor weight of poorly infiltrated control tumors. However, blocking CCL5 in Vps34i-treated B16-F10 and CT26 tumor–bearing mice significantly rescued the inhibition of tumor growth and weight. This rescue of the tumor growth was associated with a significant decrease in the infiltration of both NK and CD8+ into tumors. On the basis of these data, we conclude that Vps34 treatment induces CCL5 and CXCL10 in tumor cells, which resulted in attracting more NK and CD8+ T cells into tumors (Fig. 4, C and D).
Fig. 4. Targeting of Vps34 increases pro-inflammatory CCL5 and CXCL10 chemokine secretion in tumors.
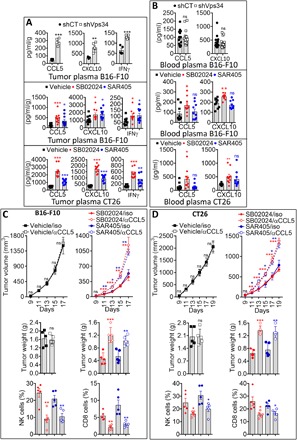
(A) Enzyme-linked immunosorbent assay (ELISA) quantification of the CCL5, CXCL10, and IFNγ secreted in the microenvironment of the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (top) and B16-F10 melanoma (middle) and CT26 colorectal (bottom) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). Data are reported in pg/ml standardized to excised tumor weight (in grams) and represented as an average of 5 or 10 tumors per group (each dot represents one mouse). (B) ELISA quantification of CCL5 and CXCL10 protein levels in the blood of mice bearing the following tumors: control (shCT) or Vps34-targeted (shVps34) B16-F10 melanoma (top) and B16-F10 melanoma (middle) and CT26 colorectal (bottom) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405). Data are reported in pg/ml and represented as an average of 10 or 15 blood samples per group collected from different mice (each dot represents one mouse). (C and D) Growth curves (top), weight in grams (middle), and flow cytometry quantification of NK cells and CD8+ T cells (bottom) infiltrating B16-F10 melanoma (C) and CT26 colorectal (D) tumors treated with control vehicle (vehicle) or Vps34i (SB02024 or SAR405) and injected with either control (iso) or CCL5-neutralizing antibody (αCCL5). Results are reported as the average of five mice per group. All results are shown as mean ± SEM (error bars). Statistically significant differences (indicated by asterisks) are calculated compared to control conditions using an unpaired two-tailed Student’s t test (*P < 0.05, **P < 0.005, and ***P < 0.0005).
Vps34 targeting induces CCL5 and CXCL10 via STAT1/IRF7
We next explored in vitro the underlying mechanisms involved in the up-regulation of CCL5 and CXCL10 following Vps34 targeting using siRNA or Vps34i. As shown in Fig. 5 (A and B) and fig. S6E, we observed an induction of phospho–signal transducer and activator of transcription 1 (pSTAT1) and increased STAT1 mRNA and protein levels in B16-F10 melanoma cells transfected with si-RNA targeting Vps34 (si-Vps34) and in Vps34i-treated B16-F10 and CT26 cells as compared to controls. We next assessed whether the activation of IFNγ/Janus kinase 2 (JAK2)/STAT1 signaling pathway was associated with increased expression of IFN regulatory factor (IRF) family of transcription factors. We analyzed the expression of five IRF family members (IRF1, IRF3, IRF4, IRF7, and IRF8) and found that IRF1, IRF7, and IRF8 mRNA levels were significantly up-regulated in si-Vps34–transfected B16-F10 cells as compared to cells transfected with si-CT (Fig. 5C, left). The increased mRNA expression of IRF1 and IRF7 was also observed in Vps34i-treated B16-F10 and CT26 tumor cells compared to controls (Fig. 5C, middle and right). Silencing Vps34 (siRNA Vps34) or in vitro treatment of B16-F10 and CT26 cells with Vps34i induced the protein expression of both IRF1 and IRF7 (Fig. 5D and fig. S6F).
Fig. 5. STAT1/IRF7 axis is involved in the up-regulation of CCL5 and CXCL10 in Vps34i (SB02024 or SAR405)–treated tumor cells.
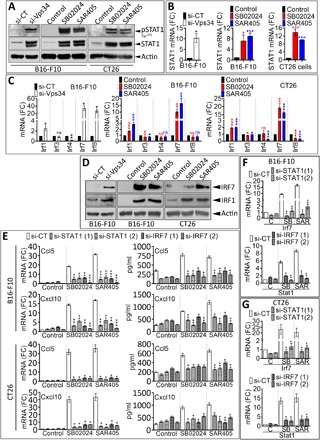
(A) Expression of pSTAT1 (Tyr701) and total STAT1 protein levels by Western blot in control (si-CT) or Vps34-targeted (si-Vps34) B16-F10 melanoma cells (left) and B16-F10 melanoma cells (middle) and CT26 CRC cells (right) treated with control dimethyl sulfoxide (DMSO) (control) or Vps34i (SB02024 or SAR405). β-Actin was used as a loading control. (B) Expression of STAT1 mRNA by real-time quantitative polymerase chain reaction (RT-qPCR) in control (si-CT) or Vps34-targeted (si-Vps34) B16-F10 melanoma cells (left) and B16-F10 melanoma cells (middle) and CT26 CRC cells (right) treated with control DMSO (control) or Vps34i (SB02024 or SAR405). Results are reported as fold change (FC) compared to control conditions. (C) The expression of Irf1, Irf3, Irf4, Irf7, and Irf8 mRNA by RT-qPCR in control (si-CT) or Vps34-targeted (si-Vps34) B16-F10 melanoma cells (left) and B16-F10 melanoma cells (middle) and CT26 CRC cells (right) treated with control DMSO (control) or Vps34i (SB02024 or SAR405). Results are reported as fold change compared to control conditions. (D) The expression of IRF7 and IRF1 protein levels by Western blot in control (si-CT) or Vps34-targeted (si-Vps34) B16-F10 melanoma cells (left) and B16-F10 melanoma cells (middle) and CT26 CRC cells (right) treated with control DMSO (control) or Vps34i (SB02024 or SAR405). β-Actin was used as a loading control. (E) Expression of CCL5 and CXCL10 mRNA by RT-qPCR (left) and ELISA quantification of CCL5 and CXCL10 protein levels in the supernatants (right) of B16-F10 melanoma cells and CT26 CRC cells pretransfected with control siRNA (si-CT) or two different siRNA sequences (1 or 2) targeting either STAT1 (si-STAT1) or IRF7 (si-IRF7) and then treated with control DMSO (C), SB02024 (SB), or SAR405 (SAR). Results in left panels are reported as fold change compared to control conditions. Results in right panels are reported in pg/ml. (F and G) Expression of Irf7 and Stat1 mRNA by RT-qPCR in B16-F10 melanoma (F) and CT26 CRC (G) cells described in (E). C, control DMSO; SB, SB02024; SAR, SAR405. Results represent the average of three independent experiments (A to D) and two independent experiments (E to G). All results are shown as mean ± SEM (error bars). Statistically significant differences (indicated by asterisks) are calculated compared to control conditions using an unpaired two-tailed Student’s t test (*P < 0.05, **P < 0.005, and ***P < 0.0005).
Many genes encoding the inflammatory signature, including CCL5 and CXCL10, are STAT1 targets (19). To establish a direct relationship between the activation of STAT1/IRF7 and the expression of pro-inflammatory cytokines CCL5 and CXCL10, we targeted STAT1 or IRF7 in cells pretreated with Vps34i and analyzed CCL5 and CXCL10 expression. We focused on IRF7 as it is the only transcription factor that was highly up-regulated using both genetic and pharmacological approaches targeting Vps34. We transfected B16-F10 cells with siRNA targeting either STAT1 or IRF7, treated them with Vps34i, and assessed the CCL5 and CXCL10 expression levels. Our results demonstrated that, in the absence of STAT1 or IRF7, Vps34i failed to induce CCL5 and CXCL10 mRNA and protein expression (Fig. 5E and fig. S7, A and B) in B16-F10 and CT26 cells in vitro. Our results revealed that IRF-7 induction was no longer observed in both Vps34i-treated B16-F10 (Fig. 5F, top) and CT26 (Fig. 5G, top) cells pretransfected with STAT1 siRNA. Similarly, Vps34i treatment failed to induce STAT1 in both B16-F10 (Fig. 5F, bottom) and CT26 (Fig. 5G, bottom) cells pretransfected with IRF7 siRNA. We next used fludarabine, which has been described as STAT1 inhibitor in addition to its effect on DNA synthesis. The induction of pSTAT1 and STAT1 was no longer observed in B16-F10 and CT26 cells when cultured with fludarabine combined with Vps34i (fig. S7, C and D). The release of CCL5 and CXCL10 was significantly decreased in B16-F10 and CT26 cells treated with fludarabine combined with Vps34i (fig. S7E). Fludarabine also abrogated the Vps34i-mediated induction of mRNA levels of STAT1, IRF1, IRF7, CCL5, and CXCL10 in B16-F10 and CT26 cells (fig. S7F). Together, our results provide strong evidence that the pharmacological inhibition of Vps34 induced both STAT1 and IRF7, which independently up-regulates CCL5 and CXCL10 pro-inflammatory cytokines.
Vps34i improves the therapeutic benefit of anti–PD-L1/PD-1
In addition to their increased infiltration into Vps34i-treated B16-F10 and CT26 tumors, we observed that NK, CD8+, and CD4+ T effector cells expressed high levels of early exhaustion marker PD-1 (fig. S8, A and B, left). Similar results were obtained in shVps34 B16-F10 tumors (fig. S8C, left). Furthermore, Vps34i-treated B16-F10 and CT26 tumors showed increased expression of PD-L1 on the surface of CD45− cells, CD11b+ myeloid cells, F4/80 macrophages (only in B16-F10), DCs (only in CT26), and MDSCs (only in CT26) (fig. S8, A and B, right). In shVps34 B16-F10 tumors, the increased expression of PD-L1 was only observed on the surface of CD45− cells and DCs (fig. S8C, right). The response to anti–PD-1/PD-L1–based therapy can be predicted by several biomarkers, including the expression of PD-L1 on the surface of tumor cells [reviewed in (20)]. Cancer patients expressing PD-L1 expression have been considered as responders to anti–PD-1/PD-L1–based therapies (1). We next evaluated whether Vps34i treatment could improve the response to anti–PD-L1 or anti–PD-1 therapy by assessing the impact of this combined treatment on tumor growth, tumor weight, and mice survival. We used melanoma B16-F10, previously described as immune hot tumors but not responding to anti–PD-L1/PD-1 (21), and CRC CT26, reported as immune cold tumors and partially responding to anti–PD-L1/PD-1 (22). The treatment schedule of B16-F10 and CT26 tumor–bearing mice is reported in fig. S8D. Our results reported in Fig. 6 (A and B) confirm that Vps34i (SB02024 or SAR405) treatment alone decreased the tumor growth and weight of B16-F10 and CT26 and prolonged the survival of tumor-bearing mice. We also showed, as expected, that anti–PD-L1 or anti–PD-1 monotherapy had no, or marginal, effect on B16-F10 and CT26 tumor volume, tumor weight, and mice survival. Combining Vps34i with either anti–PD-L1 or anti–PD-1 significantly improved their therapeutic benefit as compared to anti–PD-L1/PD-1 monotherapy in both B16-F10 (Fig. 6A) and CT26 (Fig. 6B) tumor models. Such improvement was translated into a significant delay in the tumor growth, decrease in the tumor weight, and large survival benefit of both B16-F10 and CT26 tumor–bearing mice. We observed a remarkable delay of almost 9 days in the survival of mice treated with a combination of Vps34i (SB02024 or SAR405) with either anti–PD-1 or anti–PD-L1 compared to monotherapy (anti–PD-1/PD-L1 alone). Together, our results provided strong evidence that combining Vps34i makes B16-F10 and CT26 tumor–bearing mice, who were nonresponders, responders to anti–PD-1/PD-L1.
Fig. 6. Treatment of tumor-bearing mice with Vps34i (SB02024 or SAR405) improves the therapeutic benefit of anti–PD-1/PD-L1 immunotherapy.
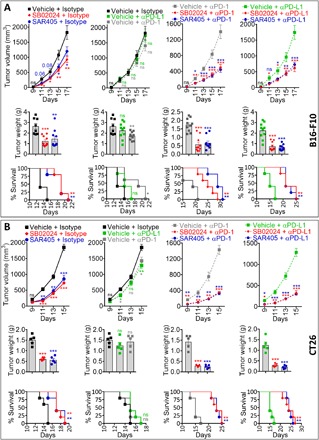
(A and B) Tumor growth curves of B16-F10 melanoma (A) (top) or CT26 CRC (B) (top) in mice treated with control (vehicle) or Vps34i (SB02024 or SAR405) combined with either control isotype, anti–PD-L1 (αPD-L1), or anti–PD-1 (αPD-1). Tumor weight (in grams) of B16-F10 (A) (middle) on day 17 and CT26 (B) (middle) on day 15. Results are reported for B16-F10 as the average of 8 or 10 mice per group from two independent experiments conducted with four to five mice per group and for CT26 as the average of five mice per group. Results are shown as mean ± SEM (error bars). Statistically significant differences (indicated by asterisks) are calculated using an unpaired two-tailed Student’s t test (*P < 0.05, **P < 0.005, and ***P < 0.0005). Mice survival curves (five mice per group) were generated from B16-F10 (A) (bottom) or CT26 (B) (bottom) tumor–bearing mice treated with control (vehicle) or Vps34i (SB02024 or SAR405) alone or in combination with either control isotype, anti–PD-L1 (αPD-L1), or anti–PD-1 (αPD-1). Lack of survival was defined as death or tumor size >1000 mm3. Mice survival percentage was defined using GraphPad Prism, and P values were calculated using the log-rank (Mantel-Cox) test (*P ≤ 0.05 and **P ≤ 0.01).
DISCUSSION
The major significance of this work relies on addressing the unmet clinical needs to find innovative strategies to improve the efficacy of immune checkpoint inhibition. In this study, we demonstrate that Vps34 inhibition converts immune cold (poorly infiltrated) tumors into inflamed (highly infiltrated) ones. Vps34 inhibition leads to STAT1-mediated up-regulation of IFN-regulated genes, which, in turn, results in the expression and secretion of chemokines CCL5 and CXCL10, recruiting T cells and NK into the TME. This finding was then capitalized upon by combining Vps34 inhibition and anti–PD-1 or anti–PD-L1 antibodies, resulting in enhanced antitumor efficacy.
While the impact of blocking autophagy on the inhibition of tumor growth has been well documented by several studies (7, 8, 14, 23–26), the effect of targeting autophagy on the immune landscape of tumors is still only partially defined. New evidence points to a critical role of autophagy in regulating the infiltration of immune cells. Recently, it has been shown that pancreatic tumor–bearing mice carrying a dominant-negative ATG4B mutant displayed significantly more CD68+ macrophages in their tumors compared to those carrying a wild-type ATG4B (27). Moreover, whole-body deletion of Atg7 in mice significantly decreased the tumor growth of YUMM melanoma through the release of Arginase I from the liver, which is responsible for the degradation of Arginine required for the tumor growth (14). We have recently reported that genetic targeting of Beclin1 (BECN1) in melanoma cells induced a massive infiltration of NK cells into tumors (9). Here, we demonstrated that, in addition to improving the infiltration of NK cells, genetic targeting of Vps34 or pharmacological inhibition of its kinase activity has a broader impact on the immune landscape of melanoma and CRC. The data presented here provide evidence that the microenvironment of melanoma and CRC tumors treated with Vps34i became highly inflamed compared with that of vehicle-treated tumors. It is likely that the establishment of an inflamed signature in Vps34i-treated tumors resulted in a reprogramming of the immunosuppressive TME and an increase in the infiltration of immune effector cells that elicit potent antitumor immune responses including NK, CD4+, and CD8+ T lymphocytes and those displaying pro-inflammatory and antitumor properties such as M1 macrophages.
A recent study reported that targeting autophagy had no impact neither on tumor growth nor on the infiltration and function of T lymphocytes, still suggesting that autophagy inhibitors could be combined to chemotherapies or immunotherapies (28). However, the role of targeting autophagy on the inhibition of tumor growth and the infiltration of cytotoxic immune cells is well supported by several studies (8, 9, 14, 23–25, 27, 29). This raises an important issue regarding which autophagy protein would be the best druggable target in tumor cells to elicit a major clinical impact in cancer immunotherapy. Currently, there is a major interest in developing selective autophagy inhibitors for various tumor types. Druggable autophagy target proteins have been recently proposed, including Beclin1 and Vps34, members of the phosphatidylinositol 3-kinase (PI3K) class III complex involved in the early steps of autophagy (30). Previous studies have found that complete genetic ablation of Vps34 in liver or heart led to the development of hepatomegaly and hepatic steatosis or cardiomegaly and decreased contractility, respectively (31). However, in our experimental settings, we did not observe any major failure in liver or heart functions in Vps34i-treated tumor-bearing mice, indicating that systemic (oral administration) treatment with drugs inhibiting the kinase activity of Vps34 could be well tolerated and effective in inducing a potent inflammatory signature in the TME. It has been proposed that the six cytokines constituting the inflammatory signature defined in melanoma and CRC (17, 32) may have partially redundant roles for the recruitment of CD8+ T cells (17). It is now well established that efficient trafficking of CTL to the metastatic site in patients with melanoma correlated better with the expression of CXCL9 and CXCL10 (17). Notably, our cytokine profiling data identified CCL5 and CXCL10 as major chemokines up-regulated by Vps34i treatment in both melanoma and CRC tumors. Furthermore, we assume that CCL5 and CXCL10 are released, at least in part, from the tumor in response to Vps34i treatment. This assumption is supported by our data showing that (i) in addition to their presence in the TME, CCL5 and CXCL10 are also released in vitro by Vps34i-treated B16-F10 and CT26 cells, and (ii) CCL5 and CXCL10 are increased in the blood of tumor-bearing mice treated with SB02024 but not in naïve (tumor-free) mice treated with Vps34i as compared to control. However, it would be important to determine whether increased blood levels of CCL5 and CXCL10 in response to Vps34i are sustained or are a general response to systemic inflammation caused by tumor lysis. While the Vps34-related mechanism(s) underlying the release of high amount of CCL5 and CXCL10 in melanoma and CRC tumors is (are) not fully understood, our study highlights the involvement of activated STAT1/IRF pathway in the transcriptional regulation of these chemokines. Among the five IRFs assessed, we found that IRF7 and IRF1 are the major factors induced in Vps34i-treated B16-F10 and CT26 cells. We provided circumstantial evidence indicating that STAT1 acts upstream and is the major inducer of IRF7 and IRF1; as such, induction was significantly abrogated in Vps34i-treated cells pretransfected with either STAT1 or IRF7 siRNA or incubated with the STAT1 inhibitor fludarabine. Nevertheless, it is important to highlight that Vps34i treatment was still able to induce the expression of CCL5 and CXCL10 in STAT1- or IRF7-defective cells, but to a lesser extent (Fig. 5E). These results suggest that, in addition to STAT1/IRF7, there are additional non–mutually exclusive mechanisms, such as nuclear factor κB (NFκB) and mitogen-activated protein kinase (MAPK) pathways, by which Vps34i or, to a larger extent, autophagy inhibition could be able to induce the expression of CCL5 and CXCL10.
Overall, our data imply that both CCL5 and CXCL10 constitute the key chemokines involved in the recruitment of major cytotoxic immune cells into melanoma and CRC tumors in response to Vps34i treatment. These data are further supported by other studies showing that CCL5 and CXCL10 induced the recruitment of CD8+ T cells, activated CD4+ T cells, activated NK cells, and M1 macrophages into triple-negative breast cancer (33). The immune phenotyping of tumors combined with the cytokine network results reported in this study strongly predicts that the immune-supportive characteristic of Vps34i-treated TME relies on the establishment of an inflamed TME supported by the presence of tumor-infiltrating lymphocytes, high levels of IFNγ-producing CD8+ T cells, and higher expression of PD-L1 in tumor-infiltrating immune cells as previously reported (34). It should be underlined that autophagy was previously described to suppress inflammation (35) and regulate NFκB signaling by several mechanisms, including the suppression of inflammasome activation or the degradation of pro-inflammatory signaling factors. Autophagy can elicit both anti- and pro-inflammatory effects in the TME via its cell-specific effects on NFκB signaling (36). In keeping with this, we strongly believe that inhibiting Vps34 in B16-F10 and CT26 tumors leads to the establishment of an inflamed microenvironment, which facilitates the infiltration of major cytotoxic effector cells, including NK and T cells. In addition to CCL5 and CXCL10, we showed an increase in the level of IFNγ in both Vps34i-treated melanoma and CT26 tumors. Although the precise molecular mechanism linking Vps34 kinase inhibition to IFNγ is not yet fully understood, it has been proposed that several autophagy proteins such as Beclin1-hVps34–interacting protein Rubicon, Atg5-Atg12, and Atg9 directly inhibit the formation or suppress the activation of pro-inflammatory protein complexes, including type I IFN signaling. Nevertheless, our data imply that the mechanism whereby Vps34 inhibition induces a massive infiltration of cytotoxic immune cells, including NK and CD8+ T cells into B16-F10 and CT26 tumors, is primarily related to the establishment of characteristic T cell–inflamed microenvironment with sustained type I IFN signature. In this regard, it is tempting to speculate that type I IFN signature could be responsible for triggering the release of more CCL5 and CXCL10 in the TME, as previously reported (37), to attract more NK and CD8+ T cells into the tumor bed. In addition to such a role, it is generally appreciated that IFNγ released in the TME plays a major role in activating tumor killing. Accumulating evidence points also to the involvement of IFNγ in the up-regulation of PD-L1 and therefore in driving T cell exhaustion and tumor immune escape (1). Despite these apparently contradictory effects of IFNγ in the TME, it has been proposed that, in contrast to expectations based on the well-known immunosuppressive function of PD-L1, its intratumoral expression has been generally associated with improved survival in melanoma patients (1) and increased response to anti–PD-1/PD-L1 therapy. Therefore, it stands to reason that the overexpression of PD-L1 on CD45− cells and PD-1 on immune effector cells in the microenvironment of Vps34i-treated tumors most likely resulted from the increased level of IFNγ in these tumors. In this context, PD-L1/PD-1 expression associated with tumor-infiltrating immune cells is therefore considered to be a marker of an active host antitumor immune response and positive prognostic feature (1). Our data showing that Vps34i-treated tumors displayed (i) T cell−inflamed characteristic, (ii) increased infiltration of CD8+ T cells, and (iii) increased expression of PD-L1 on CD45− cells have inspired significant interest to combine anti−PD-L1/PD-1−based therapy. Our results showed that the resistance to anti−PD-L1 or PD-1 observed in control B16-F10 and CRC tumors could be overcome by combining the treatment with Vps34i. These results are in line with a previous report showing that creating a T cell–inflamed TME by targeting tumors with tumor necrosis factor superfamily member LIGHT overcomes tumor resistance to checkpoint blockades (3). It would be interesting to evaluate the impact of Vps34i on the quality and the functional impact of different immune cells infiltrating tumors, notably NK and CD8+ T cells. Atg5-depleted T cells have been shown to display a marked shift to an effector memory phenotype and produce higher level of IFNγ and tumor necrosis factor–α (TNF-α). The mechanism underlying this shift seems to be related to an enhanced glucose metabolism (38). Furthermore, the impact of targeting autophagy in the immune cells on the tumor development has also been investigated. It has been recently reported that the impairment of the LC3-associated phagocytosis (LAP) in the myeloid compartment, rather than canonical autophagy, leads to tumor growth control by tumor-associated macrophages (TAMs) upon phagocytosis of dying tumor cells. This effect was related to the impact of LAP deficiency on inducing a pro-inflammatory gene expression and triggering STING-mediated type I IFN responses in TAM (39).
The clinical relevance of the results presented in this study is supported by The Cancer Genome Atlas (TCGA) data showing that our “Vps34i response signature,” consisting of CCL5/CXCL10/IFNγ/PD-1/PD-L1/CD8A/CD8B, is associated with an improved melanoma patient survival. Vps34i response signature was defined on the basis of proteins whose expression levels were increased in vivo upon Vps34i treatment. Proteins involved in the direct signaling pathway activated in response to Vps34i treatment, such as IRF7 and STAT1, were not included. Using Vps34i response signature, we were able to cluster 470 melanoma patients into three groups expressing high, intermediate, and low expression levels of Vps34i response signature, which are respectively denoted as hot, intermediate, and cold tumors (fig. S9A). Kaplan-Meier survival curve showed that the median overall survival and disease-free survival of patients displaying hot tumors (high Vps34 response signature) are significantly improved compared to those bearing cold tumors (low Vps34 response signature) (fig. S9B). Information about the TCGA patient ID, vital status, overall survival, and disease-free survival of patients can be found in table S1.
While overcoming resistance to immune checkpoint blockade–based therapies and increasing their objective response rates are still unmet clinical needs and have become an urgent challenge, our study provides a cutting edge approach and establishes the first proof of concept to design innovative and rational clinical trials using Vps34i in combination with immune checkpoint blockades. Such combination therapy will benefit from the tremendous progress of the cancer immunotherapies and extend their use to nonresponder cancer patients.
MATERIALS AND METHODS
Cells and reagents
B16-F10, CT26, YUMM GEMM, DLD-1, and NCI-H1299 cell lines were purchased from ATCC (American Type Culture Collection). T47D, Kasumi-1, THP-1, OCI-M1, and U937 were purchased from DSMZ (German Collection of Microorganisms and Cell Cultures). Renca cell line (440321) was purchased from CLS (cell lines service), and PC9 was purchased from Sigma-Aldrich. Me30966 (human metastatic melanoma) was from the Istituto Nazionale dei Tumori, Milan, Italy. RPMI 1640, Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), and antibiotics were obtained from Life Technologies. Cell lines were cultured in RPMI 1640 or DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2. The cell lines were mycoplasma-free based on tests with a MycoAlert kit (Lonza). The B16-F10 cells and CT26 cells were transfected according to the manufacturer’s protocol with either Control shRNA Lentiviral Particles (sc-108080) or Pik3c3 (Vps34) Lentiviral Particles (sc-62803-V) purchased from Santa Cruz Biotechnology. Bafilomycin A1 (S1413) and fludarabine (S1491) were purchased from Selleckchem.
In vivo study approval
Mice were treated in accordance with the European Union guidelines, and the in vivo experimentation protocols were approved by the Luxembourg Institute of Health (LIH) ethical committee, Animal Welfare Society Luxembourg (agreement nos. LECR-2016-02, LECR-2017-02, LECR-2017-03, and LECR-2018-12).
In vivo tumor growth and Vps34i’s treatments in mice
C57BL/6, BALB/C, immunodeficient NSG mice (7 weeks old) were purchased from Janvier and housed under pathogen-free conditions for 1 week before the experiments. Mice were injected subcutaneously in the right flank with different cell lines diluted in 100 μl of phosphate-buffered saline. Vps34i SAR405 (HY-12481, Bio-Connect) was administered to the mice at 10 mg/kg by oral gavage. Vps34i SB02024 was provided by Sprint Bioscience (Sweden) and administered to the mice at 20 mg/kg by oral gavage. Vehicle treatment was performed using methylcellulose (0.5% in water) supplemented with 1% polysorbate 80. Tumor volume was measured using a caliper every other day and estimated as follows: volume (cm3) = (width) 2 × length × 0.5. Mice that did not develop tumors or developed tumors larger than the threshold defined in the in vivo experimentation protocols approved by the animal welfare committee of LIH (volume, ≥2000 mm3) were excluded.
Tumor immune phenotyping and flow cytometry analysis
Tumors were harvested and mechanically dissociated into fragments (<4 mm) and then enzymatically digested using a mouse tumor dissociation kit (Miltenyi Biotec) for 45 min at 37°C. Single-cell suspensions were prepared, and red blood cells were lysed by ACK (10-548E, Lonza). The cells were next counted using a Countess Automated Cell Counter (Invitrogen) and blocked for 30 min on ice with Fc block [TruStain fcX (anti-mouse CD16/32) Antibody 101320, BioLegend]. The samples were first stained for surface markers for lymphoid and myeloid immune populations followed by intracellular staining. For FoxP3 and intracellular staining, True-Nuclear Transcription Factor Buffer Set 424401 (BioLegend) was used according to the manufacturer’s recommended protocol. The following antibodies were purchased from BioLegend: fluorescein isothiocyanate (FITC) anti-mouse CD45, Brilliant Violet 785 anti-mouse CD3, allophycocyanin (APC) anti-mouse CD8a, APC/Fire 750 anti-mouse CD4, phycoerythrin (PE)/Cy7 anti-mouse CD49b (pan-NK cells), PE/Cy7 anti-mouse NK-1.1 antibody, Brilliant Violet 605 anti-mouse CD69, PE/Cy5 anti-mouse CD25, Brilliant Violet 421 anti-mouse FOXP3, PE/Dazzle 594 anti-mouse CD279 (PD-1), Brilliant Violet 785 anti-mouse/human CD11b, APC anti-mouse F4/80, PE/Cy5 anti-mouse CD11c, PE/Cy7 anti-mouse Ly-6G, APC/Fire 750 anti-mouse Ly-6C, Brilliant Violet 605 anti-mouse CD206 macrophage mannose receptor (MMR), and Brilliant Violet 421 anti-mouse CD274 (B7-H1, PD-L1). LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Thermo Fisher Scientific) was used as a viability dye. For compensation controls, single dye stains were performed, and fluorescence spread was checked by carrying out Fluorescence Minus One FMO (Fluorescence Minus One) controls. The level of nonspecific binding was evaluated on isotype controls.
In vivo blocking antibodies
The InVivoMab anti-mouse CD8alpha (BE0061), InVivoMab anti-mouse NK1.1 (BE0036), InVivoMab anti-mouse PD-1 (CD279) (BE0273), InVivoMab anti-mouse PD-L1 (B7-H1) (BE0101), InVivoMab rat IgG2a isotype control (BE0089), and InVivoMab rat IgG2b isotype control (BE0090) were purchased from Bio X Cell, diluted in InVivoPure pH 7.0 Dilution Buffer (IP0070), and administered as indicated in the corresponding figures. Rabbit anti-murine RANTES (CCL5) (500-P118) was purchased from PeproTech.
SYBR Green RT-qPCR and Western blot
SYBR Green real-time quantitative polymerase chain reaction (RT-qPCR) and Western blot were performed as described previously (40). Primer sequences are available upon request. The following antibodies were used for Western blots: Vps34 (4263; Cell Signaling), p62 (GP62-C; Progen), LC3B (2775; Cell Signaling), phospho-STAT1 (Tyr701) (7649; Cell Signaling), STAT1 (9172; Cell Signaling), IRF1 (8478; Cell Signaling), IRF7 (696402; Cell Signaling), and actin (A5441; Sigma-Aldrich).
Immunohistochemistry on tumors
Tumors were fixed in 4% paraformaldehyde for 48 hours and then embedded in paraffin. Formalin-fixed, paraffin-embedded tumor sections (5 μm thick) from different tumors were stained using anti-CD45 and anti-CD8 antibodies. Tumor sections and staining were performed by HistoWiz Company (NY, USA).
Enzyme-linked immunosorbent assay
For tumor plasma preparation, tumors were dissociated in DMEM and then centrifuged to collect the supernatant. The supernatant was concentrated with Protein Concentrator PES, 3K MWCO (88526, Thermo Fisher Scientific) according to the manufacturer’s protocol. For blood plasma preparation, blood was collected by cardiac puncture and centrifuged at 1500g for 10 min. The supernatant was collected for enzyme-linked immunosorbent assay (ELISA). For in vitro cell supernatants, cells were plated in six-well dishes, either transfected with scrambled or siRNA Vps34, Stat1, and Irf7 (Qiagen) or treated with SB02024 (5 μM) and SAR405 (10 μM) for 48 hours. The past 24 hours, cells were deprived from serum. CCL5, CXCL10, and IFNγ were quantified using mouse CCL5/RANTES DuoSet ELISA (DY478-05, R&D Systems), mouse CXCL10/IP-10/CRG-2 DuoSet ELISA (DY466-05, R&D Systems), cxcl10/IP-10/CRG-2 ELISA kit (NBP1-92665, Novus Biologicals), and Mouse IFN-gamma DuoSet ELISA (DY485-05, R&D Systems).
Statistical analysis
Statistical analyses were conducted using GraphPad Prism software (version 7.00). The error bars represent the SEM. The data are represented as the average ± SEM. P values were calculated using an unpaired, two-tailed Student’s t test to compare the two groups. A P value less than 0.05 was considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001; ns: not significant.
Melanoma patient clustering strategy
TCGA skin cutaneous melanoma (SKCM) gene expression data were downloaded from the Broad Institute Morpheus software (https://software.broadinstitute.org/morpheus/). The expression of genes of interest (GOIs) was extracted, and unsupervised hierarchical clustering was performed with Euclidean distance. The mRNAseq expectation maximization (RSEM) value was defined at 0.5 for all genes. Melanoma patients were segregated into three clusters displaying high, moderate, and low expression levels of GOI. The cluster with high GOI expression level was termed hot tumors (comprising 211 patients), and that with low GOI expression level was termed immune cold tumors (comprising 131 patients). The vital status, overall survival, and disease-free survival of each patient segregated into the hot tumor and the cold tumor clusters were uploaded from TCGA database. The percent overall survival and disease-free survival curves, the median survival, and the P value were calculated by using log-rank (Mantel-Cox) test in GraphPad software.
Supplementary Material
Acknowledgments
We thank U. Nehrbass and F. Glod (LIH) and R. Bjerkvig (Department of Biomedicine, University of Bergen, Norway) for their helpful discussions and critical reading of the manuscript. Funding: This work was supported by grants from the Luxembourg National Research Fund C18/BM/12670304/COMBATIC and PRIDE15/10675146/CANBIO; FNRS Televie (grants 7.4664.15, 7.6503.16, and 7.4606.18); Fondation Cancer, Luxembourg (FC/2016/01 and FC/2018/06); Kriibskrank Kanner Foundation; Luxembourg (2016-08-15); Janssen Cilag Pharma; and Action LIONS Vaincre le Cancer Luxembourg. Y.Y. was supported by the Swedish Foundation for Strategic Research (grant ID16-0034). Author contributions: Study concept and experimental design: M.Z.N., and B.J.; conducting experiments: M.Z.N., K.V.M., M.X., M.H., Y.Y., J.V., and S.P.; data acquisition: M.Z.N., K.V.M., M.X., and Y.Y.; data analysis and interpretation: M.Z.N., S.P., J.V., A.D.M., M.A., R.K.A., and B.J.; study supervision: J.M., G.B., and B.J.; writing the manuscript: M.Z.N. and B.J. Competing interests: S.P., Y.Y., J.V., A.D.M., M.A., and J.M. are employees and shareholders at Sprint Bioscience. R.K.A. has equity in Pinpoint Therapeutics and is part of the scientific advisory board for Sprint Biosciences and Immunaccel Inc. M.Z.N., G.B., and B.J. are co-inventors of a patent application filed pertaining to the results presented in the paper. The other authors declare that they have no competing interests. Data and materials availability: SB02024 compound can be provided by Sprint Bioscience pending scientific review and completed material transfer agreement. SB02024 request should be submitted to S.P., principal scientist at Sprint Bioscience AB. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaax7881/DC1
REFERENCES AND NOTES
- 1.Topalian S. L., Taube J. M., Anders R. A., Pardoll D. M., Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 16, 275–287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smyth M. J., Ngiow S. F., Ribas A., Teng M. W., Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 13, 143–158 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Tang H., Wang Y., Chlewicki L. K., Zhang Y., Guo J., Liang W., Wang J., Wang X., Fu Y. X., Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell 29, 285–296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tumeh P. C., Harview C. L., Yearley J. H., Shintaku I. P., Taylor E. J., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., West A. N., Carmona M., Kivork C., Seja E., Cherry G., Gutierrez A. J., Grogan T. R., Mateus C., Tomasic G., Glaspy J. A., Emerson R. O., Robins H., Pierce R. H., Elashoff D. A., Robert C., Ribas A., PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N., Komatsu M., Autophagy: Renovation of cells and tissues. Cell 147, 728–741 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Galluzzi L., Bravo-San Pedro J. M., Levine B., Green D. R., Kroemer G., Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 16, 487–511 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baginska J., Viry E., Berchem G., Poli A., Noman M. Z., van Moer K., Medves S., Zimmer J., Oudin A., Niclou S. P., Bleackley R. C., Goping I. S., Chouaib S., Janji B., Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. U.S.A. 110, 17450–17455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noman M. Z., Janji B., Kaminska B., Van Moer K., Pierson S., Przanowski P., Buart S., Berchem G., Romero P., Mami-Chouaib F., Chouaib S., Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 71, 5976–5986 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Mgrditchian T., Arakelian T., Paggetti J., Noman M. Z., Viry E., Moussay E., Van Moer K., Kreis S., Guerin C., Buart S., Robert C., Borg C., Vielh P., Chouaib S., Berchem G., Janji B., Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 114, E9271–E9279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao M., Noman M. Z., Menard L., Chevigne A., Szpakowska M., Bosseler M., Ollert M., Berchem G., Janji B., Driving cytotoxic natural killer cells into melanoma: If CCL5 plays the music, autophagy calls the shots. Crit. Rev. Oncog. 23, 321–332 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Ronan B., Flamand O., Vescovi L., Dureuil C., Durand L., Fassy F., Bachelot M. F., Lamberton A., Mathieu M., Bertrand T., Marquette J. P., El-Ahmad Y., Filoche-Romme B., Schio L., Garcia-Echeverria C., Goulaouic H., Pasquier B., A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 10, 1013–1019 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Dyczynski M., Yu Y., Otrocka M., Parpal S., Braga T., Henley A. B., Zazzi H., Lerner M., Wennerberg K., Viklund J., Martinsson J., Grandér D., De Milito A., Pokrovskaja Tamm K., Targeting autophagy by small molecule inhibitors of vacuolar protein sorting 34 (Vps34) improves the sensitivity of breast cancer cells to Sunitinib. Cancer Lett. 435, 32–43 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Marsh T., Debnath J., Ironing out VPS34 inhibition. Nat. Cell Biol. 17, 1–3 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Poillet-Perez L., Xie X., Zhan L., Yang Y., Sharp D. W., Hu Z. S., Su X., Maganti A., Jiang C., Lu W., Zheng H., Bosenberg M. W., Mehnert J. M., Guo J. Y., Lattime E., Rabinowitz J. D., White E., Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kraya A. A., Piao S., Xu X., Zhang G., Herlyn M., Gimotty P., Levine B., Amaravadi R. K., Speicher D. W., Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 11, 60–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lock R., Kenific C. M., Leidal A. M., Salas E., Debnath J., Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 4, 466–479 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harlin H., Meng Y., Peterson A. C., Zha Y., Tretiakova M., Slingluff C., McKee M., Gajewski T. F., Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 69, 3077–3085 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagarsheth N., Wicha M. S., Zou W., Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 17, 559–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sikorski K., Wesoly J., Bluyssen H. A., Data mining of atherosclerotic plaque transcriptomes predicts STAT1-dependent inflammatory signal integration in vascular disease. Int. J. Mol. Sci. 15, 14313–14331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maleki Vareki S., Garrigós C., Duran I., Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 116, 116–124 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Curran M. A., Montalvo W., Yagita H., Allison J. P., PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl. Acad. Sci. U.S.A. 107, 4275–4280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim K., Skora A. D., Li Z., Liu Q., Tam A. J., Blosser R. L., Diaz L. A. Jr., Papadopoulos N., Kinzler K. W., Vogelstein B., Zhou S., Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. U.S.A. 111, 11774–11779 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maes H., Kuchnio A., Peric A., Moens S., Nys K., De Bock K., Quaegebeur A., Schoors S., Georgiadou M., Wouters J., Vinckier S., Vankelecom H., Garmyn M., Vion A. C., Radtke F., Boulanger C., Gerhardt H., Dejana E., Dewerchin M., Ghesquière B., Annaert W., Agostinis P., Carmeliet P., Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26, 190–206 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Rao S., Tortola L., Perlot T., Wirnsberger G., Novatchkova M., Nitsch R., Sykacek P., Frank L., Schramek D., Komnenovic V., Sigl V., Aumayr K., Schmauss G., Fellner N., Handschuh S., Glösmann M., Pasierbek P., Schlederer M., Resch G. P., Ma Y., Yang H., Popper H., Kenner L., Kroemer G., Penninger J. M., A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 5, 3056 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Rosenfeldt M. T., O’Prey J., Morton J. P., Nixon C., MacKay G., Mrowinska A., Au A., Rai T. S., Zheng L., Ridgway R., Adams P. D., Anderson K. I., Gottlieb E., Sansom O. J., Ryan K. M., p53 status determines the role of autophagy in pancreatic tumour development. Nature 504, 296–300 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Yang S., Wang X., Contino G., Liesa M., Sahin E., Ying H., Bause A., Li Y., Stommel J. M., Dell’antonio G., Mautner J., Tonon G., Haigis M., Shirihai O. S., Doglioni C., Bardeesy N., Kimmelman A. C., Pancreatic cancers require autophagy for tumor growth. Genes Dev. 25, 717–729 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang A., Herter-Sprie G., Zhang H., Lin E. Y., Biancur D., Wang X., Deng J., Hai J., Yang S., Wong K. K., Kimmelman A. C., Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 8, 276–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Starobinets H., Ye J., Broz M., Barry K., Goldsmith J., Marsh T., Rostker F., Krummel M., Debnath J., Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J. Clin. Invest. 126, 4417–4429 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei H., Guan J. L., Blocking tumor growth by targeting autophagy and SQSTM1 in vivo. Autophagy 11, 854–855 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rebecca V. W., Amaravadi R. K., Emerging strategies to effectively target autophagy in cancer. Oncogene 35, 1–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaber N., Dou Z., Chen J. S., Catanzaro J., Jiang Y. P., Ballou L. M., Selinger E., Ouyang X., Lin R. Z., Zhang J., Zong W. X., Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. U.S.A. 109, 2003–2008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sweis R. F., Spranger S., Bao R., Paner G. P., Stadler W. M., Steinberg G., Gajewski T. F., Molecular drivers of the non-T-cell-inflamed tumor microenvironment in urothelial bladder cancer. Cancer Immunol. Res. 4, 563–568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pfirschke C., Siwicki M., Liao H. W., Pittet M. J., Tumor microenvironment: No effector T cells without dendritic cells. Cancer Cell 31, 614–615 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Hegde P. S., Karanikas V., Evers S., The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 22, 1865–1874 (2016). [DOI] [PubMed] [Google Scholar]
- 35.White E., Karp C., Strohecker A. M., Guo Y., Mathew R., Role of autophagy in suppression of inflammation and cancer. Curr. Opin. Cell Biol. 22, 212–217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monkkonen T., Debnath J., Inflammatory signaling cascades and autophagy in cancer. Autophagy 14, 190–198 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sistigu A., Yamazaki T., Vacchelli E., Chaba K., Enot D. P., Adam J., Vitale I., Goubar A., Baracco E. E., Remédios C., Fend L., Hannani D., Aymeric L., Ma Y., Niso-Santano M., Kepp O., Schultze J. L., Tüting T., Belardelli F., Bracci L., La Sorsa V., Ziccheddu G., Sestili P., Urbani F., Delorenzi M., Lacroix-Triki M., Quidville V., Conforti R., Spano J. P., Pusztai L., Poirier-Colame V., Delaloge S., Penault-Llorca F., Ladoire S., Arnould L., Cyrta J., Dessoliers M. C., Eggermont A., Bianchi M. E., Pittet M., Engblom C., Pfirschke C., Préville X., Uzè G., Schreiber R. D., Chow M. T., Smyth M. J., Proietti E., André F., Kroemer G., Zitvogel L., Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014). [DOI] [PubMed] [Google Scholar]
- 38.DeVorkin L., Pavey N., Carleton G., Comber A., Ho C., Lim J., McNamara E., Huang H., Kim P., Zacharias L. G., Mizushima N., Saitoh T., Akira S., Beckham W., Lorzadeh A., Moksa M., Cao Q., Murthy A., Hirst M., De Berardinis R. J., Lum J. J., Autophagy regulation of metabolism is required for CD8+ T cell anti-tumor immunity. Cell Rep. 27, 502–513.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Cunha L. D., Yang M., Carter R., Guy C., Harris L., Crawford J. C., Quarato G., Boada-Romero E., Kalkavan H., Johnson M. D. L., Natarajan S., Turnis M. E., Finkelstein D., Opferman J. T., Gawad C., Green D. R., LC3-associated phagocytosis in myeloid cells promotes tumor immune tolerance. Cell 175, 429–441.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noman M. Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P., Bronte V., Chouaib S., PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 211, 781–790 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaax7881/DC1