

Structural mass spectrometry enables dissection of a transient activated ensemble in influenza hemagglutinin fusion machinery.
Abstract
The influenza virus hemagglutinin (HA) fusion protein has long been viewed as a “spring-loaded” fusion machine whereby activation at low pH initiates a rapid and irreversible cascade of conformational changes that drives the membrane fusion reaction. This mechanism has shaped our understanding of how type 1 viral fusion proteins function as a whole. Experimental limitations have hindered efforts to expand our mechanistic and structural understanding of viral membrane fusion. Here, we used pulse-labeling hydrogen/deuterium exchange mass spectrometry and cryo–electron tomography to monitor and characterize the structural dynamics of HA during fusion activation on intact virions. Our data reveal how concurrent reorganizations at the HA1 receptor binding domain interface and HA2 fusion subunit produce a dynamic fusion intermediate ensemble in full-length HA. The soluble HA ectodomain transitions directly to the postfusion state with no observable intermediate.
INTRODUCTION
Infection by influenza virus and all enveloped viruses requires fusion of the viral and host membranes. Enveloped viruses have evolved specialized fusion protein machinery that undergoes major conformational changes to drive the membrane fusion reaction to completion (1). For influenza virus, the hemagglutinin (HA) fusion glycoprotein trimer mediates entry into host cells by its receptor binding and membrane fusion activities (1, 2). HA is first synthesized as an inactive, single-chain polypeptide precursor (HA0) that trimerizes and is activated by proteolytic processing (3, 4). The resulting functional HA assembly is a homotrimer of disulfide-linked heterodimers consisting of a receptor binding subunit, HA1, and a membrane fusion subunit, HA2 (Fig. 1A). The N terminus of HA2 includes the hydrophobic fusion peptide, which, in the prefusion configuration, is sequestered away from membranes in a pocket that extends into the central core of the HA trimer, while the C terminus of the HA2 subunit is anchored in the virus membrane by a transmembrane domain. Influenza virus infection begins when HA binds sialic acid on cell surface receptors through a low-affinity, high-avidity interaction that initiates internalization through receptor-mediated endocytosis. The HA fusion machinery is activated by low pH in the maturing endosome, triggering a cascade of large-scale conformational changes where the refolding of HA to the postfusion state is coupled to overcoming the activation energy barrier that separates pre- and postfused membranes (Fig. 1B) (5–8). The formation of the resulting fusion pore enables transfer of the viral genome into the cell (9).
Fig. 1. Monitoring HA’s structure and the low pH–induced reorganization by pulse-labeling HDX-MS.
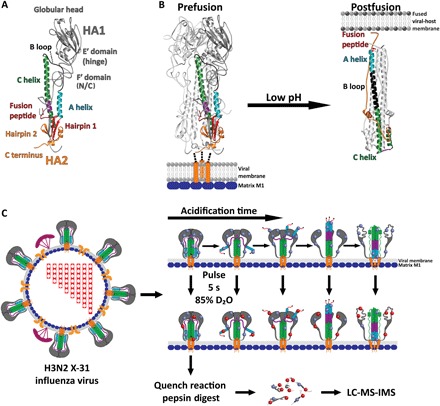
(A) Core HA structural features and regions colored and mapped onto a single HA heterodimer protomer [HA numbering according to Wilson et al. (5)] [Protein Data Bank (PDB): 2HMG]. (B) Comparison of the pre- and postfusion crystal structures for the HA ectodomain reveals the marked structural reorganization that occurs in HA2 following activation by low pH; currently, the structure of HA1 in the postfusion state is unknown (PDB: 2HMG and 1QU1). (C) Schematic depicting pulse-labeling HDX-MS of whole infectious H3N2 X-31 virions. Cartoon model of HA fusion activation during pulse labeling with amide hydrogens (blue spheres) exchanging with deuterium (red spheres) and analysis by LC-MS with ion mobility spectrometry (IMS).
Crystal structures of the prefusion HA ectodomain and an HA2 fragment of the postfusion state illustrate the beginning and end points of the membrane fusion process (Fig. 1B). Comparison of the pre- and postfusion structures reveals the marked structural re-organizations that occur in the fusion domain during membrane fusion, but they do not reveal the structural calisthenics occurring between these end points (5–8). The mechanism by which a fusion protein activates and mediates membrane fusion remains poorly understood. The conventional mechanistic model for HA fusion activation and membrane fusion describes prefusion HA as a spring-loaded fusion machine (7, 10, 11). The salient features of the spring-loaded model are that the prefusion structure represents a high-energy metastable configuration of the HA trimer, which, upon exposure to acidic pH, rapidly and irreversibly refolds to a new postfusion hairpin conformation. In this postfusion state, the fusion peptide is relocated ~100 Å toward the target membrane at the apex of an extended coiled coil, where it is colocalized with the relocated transmembrane anchor. Key interactions that maintain HA in the metastable state have been suggested to center on a “clamp” in HA1 that constrains a key HA2 loop and a “hook” formed by the fusion peptide that lashes two helices together in the prefusion trimer (7). The “uncaging” model for HA activation suggests that HA1 acts as a cage or clamp containing the spring-loaded HA2 fusion machine; whereupon low pH releases the HA1 cage, allowing rapid and irreversible activation of the spring-loaded HA2 fusion machine (7, 11–13). By contrast, a “fusion peptide release” model suggests that low pH first stimulates the release of the HA2 N-terminal fusion peptide hook from its sequestered pocket in the core of HA2 before HA1 head domains dissociating from each other (12, 14–21). In this model, the exposed fusion peptide, tethered to a highly dynamic HA2 subunit, is deployed and available to engage with the target membrane before the large-scale refolding of HA2 into the postfusion, hairpin conformation (22).
To date, only piecemeal biophysical evidence has been available to support one or the other of these models of HA activation. No detailed structural analysis describing how different regions of HA1 and HA2 respond to acidic pH once the fusion machinery is triggered has been available. Hydrogen/deuterium exchange mass spectrometry (HDX-MS) analysis of the soluble bromelain-released HA (BHA) ectodomain at pH values approaching the threshold of activation revealed dynamic structural changes throughout HA, suggesting that it is primed for fusion peptide release (12). Increased dynamics were observed at and around the fusion peptide, whereas the HA1 receptor binding domain (RBD) interface became more protected before acid activation (12). Crystal structures of BHA under similar conditions revealed a structural relaxation in the HA2 B loop at the HA1-HA2 interface, suggesting that exposure to mildly acidic conditions loosens structural constraints around the fusion domain and primes HA for activation (23). By cryo–electron tomography (cryo-ET), Fontana et al. (15) observed morphological changes in HA at low resolution on the virus surface, which led them to infer that fusion peptide release precedes dissociation of the HA1 globular head and that both of these events are reversible. Recently, single-molecule Förster resonance energy transfer (sm-FRET) imaging enabled the first observation of a dynamic HA intermediate state during membrane fusion (14). To gain a mechanistic understanding of fusion protein activation and function, we need detailed resolution of structural reorganization throughout the fusion protein and sequencing of conformational events traversed by HA.
Here, we present a pulse-labeling HDX-MS method that enables us to capture snapshots of the HA fusion machinery undergoing its complex conformational changes during activation (Fig. 1C). Pulse-labeling HDX-MS is sensitive to changes in a protein’s local secondary structure, conformation, and global quaternary organization. It enables one to monitor accessibility of amide hydrogens across the protein backbone (24). Changes in the local structure of a given peptide segment can be distinguished from the initial state by measuring changes in the extent of amide exchange. In our pulse deuteration approach using intact infectious virions, we trigger HA’s activation and then introduce a rapid deuteration pulse that labels the HA ensemble immediately before quenching the exchange reaction. Using binomial fitting and bimodal deconvolution, we were able to resolve and characterize distinct, often coexisting structural states, measure their relative abundances, and monitor the transition between conformational states (24, 25).
The transitions observed throughout the protein reveal the nature and sequence of the local conformational changes that occur throughout HA during fusion activation on intact infectious virions. Our data provide new insight into the conformational changes traversed by HA and allow us to probe the on-pathway dynamic intermediate ensemble conformation populated by HA during its activation. This intermediate ensemble exhibits a structurally flexible fusion domain before the formation of the postfusion state. These changes coincide with reorganization of the N-terminal HA2 fusion domain, the HA1-HA2 interface, as well as between HA1 subunits, but are clearly distinguished from the transition to a stable postfusion conformation. These data thus provide a detailed structural, dynamic, and temporal characterization of a type 1 fusion protein’s activation in the native context on the surface of infectious virions.
RESULTS
HA presented on whole influenza virions remains in the prefusion conformation at neutral pH
The prefusion conformation of HA on intact X-31 H3N2 influenza virions was first examined under neutral pH (preactivation) conditions (pH 7.5 at 37°C) from our pulse deuteration series to assess the presence of any conformational heterogeneity among HA spikes before activation. Our experimental conditions allowed peptides spanning most of HA1 to be monitored, including the F′ domain, hinge region, RBD, and trimeric interface (Fig. 1A) [HA numbering follows Wilson et al. (5)]. We obtained greater than 93% sequence coverage for HA1 on whole virus resulting from more than 30 unique peptides (fig. S1). The coverage throughout HA2 on whole virus was more limited (34% sequence coverage); however, we were able to monitor key peptides involved in the structural reorganization from the pre- to postfusion state, including the N-terminal fusion peptide, A helix, B loop, and portions of the C helix. Only those peptides present and trackable throughout the acidified reactions were included in analysis. Under neutral pH conditions, we observed unimodal HDX profiles for all identifiable HA peptides, indicating that the HA on the virus surface was in a conformationally uniform state and that no detectable subpopulations (e.g., misfolded or unprocessed HA) were present. These pulse deuteration results are consistent with our previously reported continuous deuteration-labeling HDX-MS studies of prefusion H3 Aichi/68 HA on intact virions (12). Western blot analysis of HA on whole virions and BHA further confirms that no unprocessed HA is present (fig. S2). Correct HA processing was also confirmed by MS as no peptides unique to HA0 were found. Analysis of the internal exchange standard Pro-Pro-Pro-Ile (PPPI) showed consistent labeling times for each reaction (fig. S3).
The HA2 fusion machinery becomes highly solvent accessible and dynamic in a transient intermediate state following low pH activation
To monitor the conformational pathway traversed by HA, we next activated influenza virions from the same stock as above by lowering the pH to 5.1. At time points ranging from 0 s to 360 min, the virus was exposed to a 5-s pulse of D2O that transiently raised the pH* to 8.0 (Fig. 1C). Following this brief pulse deuteration, samples were rapidly quenched, digested, and frozen for subsequent analysis by MS. Pulse-labeling HDX-MS enables us to monitor structural changes as they occur during protein motion; however, we are limited to monitoring only those structural changes substantial enough to produce a new, unique, and resolvable HDX state. It remains possible that early structural changes that are too subtle to alter amide protection occur too quickly, are reversed by the brief elevated pH pulse (pH* 8.0), or go undetected by this approach. For example, it has been suggested that some early acid-induced structural changes in HA are reversible upon reneutralization, as recently demonstrated for fusion peptide release by Das et al. (14) using sm-FRET (15, 18). We note, however, that under our pulse conditions (pH* 8.0 at 22°C), exposed amide hydrogens will exchange fully in 20 ms (five half-lives for an exposed amide), meaning that in our approach, HA becomes labeled very rapidly in most cases before large-scale refolding events that can take place (26). Our data show that the rate of exchange for exposed amides across HA is faster than the rate of refolding as the alternative scenarios would produce markedly different data. If the rate of refolding was greater than the rate of exchange, then this would result in all peptides being in the refolded state before they become labeled (24, 27). Similarly, if the rate of refolding was comparable to the rate of exchange, then we would be able to detect a population with an intermediate level of deuteration as the peptide transitions to a more protected state and is simultaneously labeled (24, 27). As we do not observe either of these phenomena in our data, we can assume with confidence that refolding does not have an appreciable effect on the structural transitions observed here. Hence, we can monitor all but the fastest refolding events in HA.
Notably, in contrast to the ubiquitous unimodal spectra for all HA peptide segments that were observed before low pH–induced activation, once exposed to pH 5.1 conditions, bimodal spectra began to emerge for numerous peptides throughout the HA trimer. From the pool of peptides showing clear bimodal isotopic distributions throughout the acidification process, it was possible to apply bimodal deconvolution to quantify the contribution and relative abundance of each conformational state throughout the full time course (25). Examples of peptide-specific population shift data are shown in Figs. 2, 3, and 5 and figs. S5 to S7. The shift in the relative populations across the complete time course revealed two distinct, sequential conformational transitions occurring throughout HA1 and at least three conformational transitions in HA2. Below, we describe these changes sequentially as they appeared following acidification.
Fig. 2. Formation of a dynamic intermediate ensemble in full-length HA.
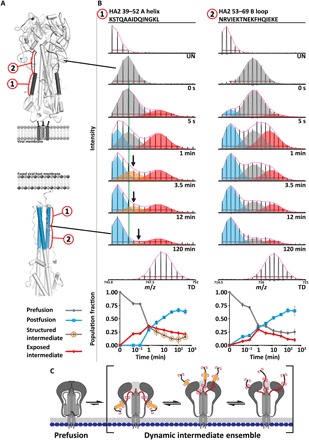
Fusion subunit triggering and reorganization of the HA1 RBD interface directly result in formation of a dynamic fusion-active intermediate state in HA2. (A) The HA2 A helix and B loop segments are highlighted on ribbon diagrams for the prefusion (top; PDB: 3HMG) and postfusion (bottom; PDB: 1QU1) crystal structures. (B) The HA2 A helix (segment 1) adopts a relatively unstructured intermediate state (red spectral envelope) following acid activation that is distinct from the prefusion (gray) and postfusion (light blue) helical states. The neutral pH prefusion conformation is tracked by the centroid (green vertical line). The presence of a fourth coexisting state by 3.5 min that is slightly more exposed than the neutral pH prefusion state is denoted by the orange envelopes. Formation of the postfusion helical bundle is indicated by the formation of the low mass/charge ratio (m/z), minimally deuterated population that increases in relative abundance over time (light blue). The HA2 B loop (segment 2) likewise reveals the formation of a highly exposed, dynamic intermediate state (red envelopes) on the same time scale as the A helix. Undeuterated (UN) and totally deuterated (TD) controls are shown for each peptide. The red horizontal bar in each spectrum corresponds to the distribution width, and the magenta curve above each spectrum displays the summation of each binomial fit. (C) Schematic illustrating the nature of the dynamic intermediate ensemble.
Fig. 3. Concurrent fusion subunit triggering and HA1 RBD interface reorganization in full-length HA.
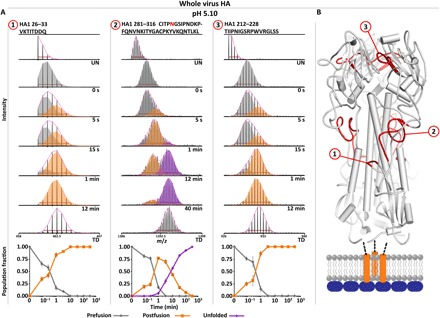
(A) N-terminal HA1 peptides between HA1 and HA2 fusion domain (peptide 1, F′ region) display a transition to a more exposed state (orange envelope and traces) soon after acidification, indicative of a loss of quaternary contacts. The HA1 hinge region (peptide 2, hinge and F′ region), which forms quaternary contacts with the HA2 fusion domain, displays two distinct structural changes, with the first corresponding to a loosening of the HA1-HA2 quaternary contacts (orange) and the second to unfolding of the HA1 globular head (purple). The HA1 RBD interface (peptide 3, HA1-HA1 trimeric interface) transitions to a more exposed state on the same time scale as the fusion peptide release event. Undeuterated and totally deuterated controls are shown for each peptide. The red horizontal bar in each spectrum corresponds to the distribution width, and the magenta curve above each spectrum displays the summation of each binomial fit. (B) Peptide segments highlighted in 1 to 3 mapped onto ribbon diagram (PDB: 3HMG). Error bars indicate SDs from triplicate measurements.
Fig. 5. The soluble BHA ectodomain transitions directly to the postfusion state following acid activation.
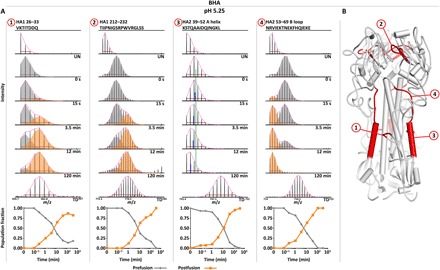
(A) Activation of the soluble BHA ectodomain at pH 5.25 reveals that fusion peptide release (segment 1) and reorganization of the HA1 RBD interface (segment 2) occur similarly to full-length HA. However, both the HA2 A helix and B loop display a rapid transition from the prefusion state directly to the postfusion state upon acid activation with no observable intermediate state (A helix segment 3 and B loop segment 4). The A helix is fit with a single binomial, and the centroid of the 0-s prefusion state (green line) is plotted for comparison with the centroid of each time point (blue line). Undeuterated and totally deuterated controls are shown for each peptide. The red horizontal bar in each spectrum corresponds to the distribution width, and the magenta curve above each spectrum displays the summation of each binomial fit. (B) Peptide segments highlighted in 1 to 4 mapped onto ribbon diagram (PDB: 3HMG).
The first observable conformational changes following exposure of the virus to acidic conditions (pH 5.10) occurred rapidly after acidification. Analysis of key peptides in the HA2 fusion machinery subunit reveals the concurrent formation of a dynamic fusion peptide–released intermediate ensemble state that emerges as soon as 5 s after activation of HA on intact influenza virions (Fig. 2B). As seen in the mass spectra, a transient intermediate conformation reported by the A helix is highly solvent accessible (red envelopes and traces in Fig. 2B, peptide 1) compared to the initial and final protected states. This exposed conformational population shows a clear increase and subsequent decrease in abundance over the course of HA’s conformational change, peaking at 1 min after acid activation. As the exposed population is almost maximally deuterated, we infer that this segment is highly exposed and likely sampling relatively disordered conformations (Fig. 2B, peptide 1). At 1 min, a highly protected conformational state, consistent with the proposed extended helical intermediate and known postfusion helical bundle conformation that incorporate the A helix into the bundle, also begins to appear and grows monotonically over the course of the transition as the exposed, dynamic intermediate decreases in abundance (Fig. 2B, peptide 1).
Analysis of the A helix required constraining the binomial fits for the exposed intermediate and postfusion states (Fig. 2B, peptide 1, red and blue envelopes, respectively), which were well defined and consistent throughout the time course. This revealed an additional transition where the prefusion neutral pH conformation shifts to a slightly more exposed state, emerging by 3.5 min after acidification (Fig. 2B, peptide 1, see peak shift of orange envelopes relative to vertical green line). Described in more detail below, this likely reflects the structural relaxation of adjacent stem regions of HA1 that pack against the A helix in prefusion HA. It is likely that this population is present at earlier time points; however, we are unable to clearly resolve its presence as the prefusion, neutral pH–like state is more abundant, and we are limited to monitoring at most three states concurrently. When we tried instead to constrain the mass/charge ratio (m/z) centroid position of the initial prefusion, neutral pH conformation (gray envelopes) throughout the time course, the deconvolution of the multimodal spectra produced a poorer overall fit for each state but did not change the overall kinetics of the population changes (Δχ2 = −4.3 × 107). This indicates that, at some of the intermediate time points, there are at least four coexisting states sampled by the A helix (Fig. 2, B and C): the ordered and solvent-protected helical prefusion neutral pH state (gray); a slightly more dynamic, exposed but structured fusion peptide–released intermediate state (orange); a highly exposed unstructured fusion peptide–released intermediate state (red); and the highly protected helical postfusion state (blue). Together, these two resolvable intermediate populations contribute to the dynamic intermediate ensemble state. It is highly likely that there exist a number of interconverting structures that contribute to these mass envelopes that we are unable to resolve. While quantitative analysis of the HA2 N-terminal fusion peptide itself was confounded by a significant loss of signal intensity over time, perhaps because of the significant hydrophobicity of this segment, peak width analysis revealed an increase in conformational heterogeneity and formation of an exposed state at 5 s after acidification (fig. S4). The peptide’s behavior was thus consistent with the formation of a fusion peptide–released state as early as 5 s after acidification.
We observed parallel behavior starting as early as 5 s after acidification for the formation of an intermediate state involving the HA2 B loop (Fig. 2B, peptide 2). In this intermediate state, the B loop is more exposed than the structured loop observed in the prefusion state. The prefusion B loop is defined by ordered but exposed amides, whereas the intermediate state appears fully exposed (red envelopes and traces in Fig. 2B, peptide 2). As with the A helix, the B loop subsequently reports on the transition from the dynamic intermediate state to the ordered postfusion helical bundle conformation (8). We note that a small subpopulation of highly protected B helices (approximately 15%) began to appear starting at 5 s after acidification. This minor fraction’s behavior may be consistent with previous sm-FRET observations that some fraction of HA can transition relatively directly to the postfusion state without traversing the dynamic intermediate state that is observed (14). Analysis of the B loop required constraining the prefusion neutral pH state (gray) throughout the time course. It is possible that the broad multimodal distribution represented by the gray and red fits at intermediate time points contains more than two distinct structural states that are indistinguishable by HDX and cannot be resolved in our data.
Together, these data indicate that the dynamic intermediate ensemble state reported by the A helix (HA2 39–52) and B loop (HA2 53–69) segments appears rapidly, starting within 5 s after acidification. In this state, the critical fusion peptide–associated subdomains of HA2 are structurally dynamic and distinct from the known pre- and postfusion structures of HA2 (Fig. 2C). The intermediate ensemble becomes populated well before formation of the postfusion state, where these segments form highly protected, stable helices as part of the trimeric core helical bundle (Fig. 2) (6, 8).
Concomitant with HA2 activation, the HA1-HA2 and HA1-HA1 interfaces reorganize
The HA1 subunit has been described as serving as a clamp around the HA2 fusion machinery subunit; however, as our data above reveal, key portions of HA2 become highly dynamic and exposed soon after activation conditions were introduced (7). To understand how and on what time scale HA1 reorganizes, we examined peptide segments that first form contacts with HA2 in the prefusion state; these peptides include the HA1 F′ domain, which packs against the HA2 N-terminal fusion peptide proximal subdomain and A helix (Fig. 1A). We then examined peptides involved in maintaining the trimerized HA1-HA1 head, which must eventually dissociate in order for HA2 to be able to adopt the postfusion state.
In the same early time frame in which HA2 began to populate a dynamic intermediate state, we observe that within the HA1 subunit, the fusion peptide proximal HA1 F′ region (HA1 26–33 and 281–316) began to transition to a more exposed state (Fig. 3A, peptides 1 and 2) and, by 3.5 min after acidification, almost completely populated this new state (figs. S5, A and B, and S6, A1 and A6, and table S1). The increase in exposure is likely due to a loss of quaternary packing interactions between the F′ domain and the N-terminal segment of HA2 that are present in prefusion HA. In regions of HA1 that span the HA1-HA2 interface including the HA1 hinge and F′ region (Fig. 3, figs. S5 and S6, and table S1), peptides show a distinct initial increase in deuterium uptake and broadening of the mass envelope from 5 s up until 1 min, where the mass envelope then narrows to a single, more uniform population that reflects a conformational state with an intermediate level of exposure between the prefusion and fully exposed states (Fig. 3 and fig. S5A). These changes occur in concert with the early changes in HA2 in the fusion peptide–associated regions. Together, these peptide segments cover nearly the entire HA1-HA2 quaternary interface and show the large-scale structural reorganization that occurs following fusion peptide release and the coinciding reorganization of the N-terminal HA2 fusion domain (Figs. 2 and 3 and figs. S5 and S6).
In contrast, HA1 peptides that do not form quaternary contacts with HA2 or adjacent HA1 protomers do not show any increased deuterium uptake until 3.5 min after acidification (figs. S5D and S6). These peptides, in a sense, serve as internal standards, demonstrating that the changes we monitor in other sites are significant and a result of structural reorganization.
In the first, early conformational transition, we also observed reorganization of the protomeric interface between HA1 RBSs that reveals a shift to a more exposed state (HA1 212–228) (Fig. 3A, peptide 3, and fig. S5A). This shift takes place on the same time scale as fusion peptide release, occurring between 15 s and 1 min. Reorganization of the HA1-HA1 interface thus coincides with changes that take place proximal to the fusion peptide subdomain in the HA stem region and across the HA1-HA2 interface.
A large-scale unfolding in HA1 is seen at late time points following fusion activation
In a late stage of structural reorganization within HA1, beginning at 3.5 min, we observed widespread unfolding of the HA1 globular head, marked by transitions to nearly fully deuterated states (Fig. 3A, peptide 2; figs. S5, C and D, and S6; and table S1). Peptides spanning the C-terminal segment of the F′ domain (HA1 281–316), which form quaternary contacts with the HA2 fusion domain, show an initial conformational change and increase in exposure associated with fusion peptide release (Fig. 3A, peptide 2, gray to orange coloring) and subsequently a distinct shift to a maximally exposed state (Fig. 3A, peptide 2, orange to purple coloring). HA1 peptides that do not form quaternary contacts with HA2 or adjacent HA1 protomers do not show any increased deuterium uptake until 3.5 min after acidification, where they transition to fully exposed state (figs. S5D and S6). Certain HA1 peptides with strong secondary and tertiary structural elements, including portions of the 110 helix and peptides spanning the receptor-binding site, do not fully unfold but nevertheless transition to a significantly more exposed state in concert with the unfolding event (HA1 109–119, 175–194, and 234–243) (figs. S5A and S6). These data suggest that receptor binding HA1 head domain transitions to a molten globule–like state following extended exposure to endosomal pH conditions.
pH dependence of HA fusion activation
To determine whether HA fusion activation follows a similar trajectory across a range of fusogenic pH conditions, we performed pulse-labeling HDX-MS on HA acidified to pH 5.25 at 37°C exactly as described above. The overall nature and sequencing of events mirror the trajectory described at pH 5.10; however, the onset of each conformational event was delayed (Fig. 4). Both fusion subunit triggering and reorganization of the HA1 RBD at the protomeric interface commenced between 15 s and 1 min after acidification and reached completion by 12 min (Fig. 4, A to D, orange coloring). The same form of dynamic intermediate state was observed in both the A helix and B loop with HDX profiles; however, this state was slightly less abundant at each time point when compared to activation at pH 5.10 (Figs. 2 and 4 and fig. S7). The late stage unfolding of the globular head was also observed with delayed onset and kinetics compared to the pH 5.10 condition (Fig. 4, C to F, purple coloring). These data suggest that shifting the pH of activation can accelerate or slow the onset and rate of change for each conformational event but does not change the structural or dynamic nature or sequence of those conformational changes.
Fig. 4. Global kinetic comparison and pH dependence of HA fusion activation for full-length HA.
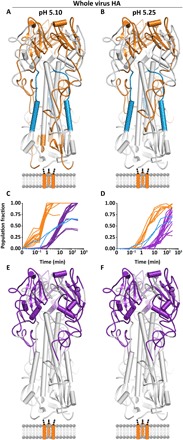
The specific pH of activation did not alter the nature (A and B) or sequence of any observed structural changes in full-length HA but simply accelerated the onset and rate of change of each conformational event (C and D). Conformational transitions that take place before 3.5 min are colored in orange. The last transition reported by HA1 peptides is to a largely unfolded, highly flexible state, which begins at 3.5 min [purple; (C) and (D) and (E and F)]. Formation of the postfusion helical bundle in full-length HA is delayed because of the formation of the intermediate state [(A) to (D); blue peptides and traces].
Soluble BHA ectodomain follows a two-state transition from pre- to postfusion forms
The soluble BHA ectodomain or analogous trimeric ectodomain constructs are commonly used for the study of HA’s structure and function in place of the native full-length membrane-anchored HA. However, it has been suggested that significant differences exist between how BHA and full-length HA behave during fusion activation (28). Detailed structural and functional analysis of HA activation for intact trimeric spikes versus soluble ectodomain has not been possible with previously used methods. Here, we sought to use the pulse deuteration approach to characterizing the conformational changes in BHA and to comparing them with the behavior of intact HA on virions under identical fusion activation conditions at pH 5.25.
Pulse-labeling HDX-MS demonstrated that fusion peptide release and head-opening events occurred similarly between BHA and HA on virions both in terms of the structural changes that occur and the kinetics of each conformational change (Fig. 5A, peptides 1 and 2, and fig. S8). Most notably, in contrast to what we observed for HA on intact virions, in soluble BHA, the A helix and B loop display unambiguous two-state behavior transitioning directly from the pre- to postfusion states with no observable intermediate (Fig. 5A, peptides 3 and 4). Both the A helix and B loop transition to their respective protected postfusion states in concert with the fusion peptide release and head-opening events (Fig. 5A, peptides 3 and 4, and fig. S8). The two coexisting populations in the A helix data could not be resolved by bimodal deconvolution, as the centers of each population were too close. Analysis using a single binomial fit reveals an increase and subsequent decrease in the peak width over time. Most notably, there are no changes in the mass envelope that indicate the presence of a more exposed state, and the increase in width can be unambiguously attributed to the formation of the postfusion state (Fig. 5A, peptide 3). BHA thus transitions from pre- to postfusion conformations in a two-state fashion, whereas intact HA on whole virus particles exhibits the presence of a transient but highly populated, dynamic conformational intermediate state.
Visualization of low pH–induced HA structures by cryo-ET
To obtain complementary structural characterization of HA under neutral pH and fusion activating conditions, we performed cryo-ET of virions treated identically to the HDX-MS experiments. At neutral pH, the HA trimeric spikes are closely spaced and well ordered on the virion surface, consistent with previous cryo-ET imaging of influenza virus (figs. S9, A to C, and S10A) (29, 30). By contrast, influenza virion tomograms of the virus treated for 1 min at pH 5.10 show a high degree of variability in the HA structure and organization (figs. S9, D and E, and S10B). Here, individual HA appears to populate a heterogenous mixture of low pH–induced configurations and has, in part, lost the characteristic hour glass–shaped structure of neutral pH HA (fig. S9, A, B, D, and E). Even HA spikes on the same virion have variability in their relative conformations. In these acid-triggered states, HA still maintains its overall trimeric nature as seen from closely clustered HA1 subunits in the top view cross sections (fig. S9F). From these types of images, the low pH HA appears to be sampling different conformation states, some of which show variable degrees of separation of the HA1 interface or overall thinning of HA (fig. S9), but there does not seem to be a singular dominant, discrete intermediate state.
DISCUSSION
Viral fusion proteins face a unique challenge in terms of staying poised in a prefusion state on the surface of the virus until exposed to the necessary triggering events that activate them into a fusogenic state. Once activated, they undergo structural calisthenics that are required to draw two opposing membranes together and induce them to fuse. This refolding process is believed to be critical to driving fusion forward. To date, however, we have had a very limited view into how these dynamic fusion machines carry out their function and the structural dynamic nature of the activated fusogenic forms of the machinery.
HA populates an ensemble of highly dynamic intermediate states during activation
The influenza HA membrane fusion protein has been viewed as a “spring-loaded” fusion machine that rapidly and irreversibly activates upon exposure to the low pH environment of the maturing endosome (7, 10, 11, 13, 31). This view was proposed on the basis of experiments, indicating that the prefusion state is in a trapped, high-energy metastable state, which can be induced to transition irreversibly to the stable, low-energy postfusion state through a range of destabilizing conditions (7, 8, 11). Recent methodological advances have made it possible to study the transitions between viral fusion protein conformational states under native activation conditions (14, 15). For example, an sm-FRET imaging of HA displayed on engineered lentivirus particles enabled the conformational change of the HA2 subunit to be monitored during fusion activation and membrane fusion (14). Using HA with two fluorescent dyes that served as FRET pairs engineered into HA2, Das et al. (14) show that acid-activated HA reversibly samples multiple intermediate conformations and proceeds through a highly flexible obligate intermediate state during activation and membrane fusion. While sm-FRET provides a powerful means to probe the relative disposition of the two fluorescent probe molecules that were covalently attached to HA, the behavior of most of the trimer remained uncharacterized. No information has been available for sites such as HA1 receptor binding subunits, which have been hypothesized to cage the HA2 fusion subunit modulating its activation, as well as regions of HA2 that undergo marked refolding that is key to the fusion protein’s ability to deploy the fusion peptide and draw the two membranes together.
Using HDX-MS and cryo-ET, we investigated the nature and sequence of the conformational changes that occur throughout the HA fusion machinery activation on intact virions and in soluble isolated ectodomain forms. Our data reveal concurrent reorganization of the HA1-HA2 interface centered around the HA2 N-terminal fusion peptide proximal subdomain (segments spanning from the B loop to the fusion peptide) and HA1 protomeric interface (Figs. 2 to 4 and figs. S4 to S7). The loss of quaternary contacts between HA1 and HA2 is also seen across the HA1-HA2 interface at the HA2 apex. As a result of the loosening of these key restraints, the HA2 A helix and B loop are freed to populate at least two structurally distinct states in the intermediate ensemble (Fig. 2). We propose that, in this intermediate state, the A helix and B loop flexibility enables the released fusion peptide to sample space and grapple to a membrane (Fig. 6). Furthermore, the A helix clearly adopts two structurally distinct states in the intermediate ensemble corresponding to a slightly more dynamic, exposed but structured fusion peptide–released intermediate state and a highly exposed, unstructured fusion peptide–released intermediate state (Figs. 2 and 6). The structural dynamics monitored by HDX-MS are consistent with recent molecular dynamics simulations, indicating that during the transition to the postfusion helical bundle, the A helix and B loop are capable of sampling diverse secondary structure elements and can become relatively unstructured (20). The sm-FRET studies by Das et al. (14) likewise support the conclusion that a high degree of conformational dynamics contribute to the observed intermediate states.
Fig. 6. Proposed model for HA fusion activation.
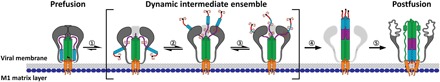
Full-length HA on whole influenza activates in response to low pH conditions with (1) concurrent reorganization of the HA2 fusion domain and HA1 RBD interface, resulting in formation of a dynamic, fusion peptide–released intermediate state. This intermediate state is not a single discrete or static state but rather a dynamic intermediate ensemble where the exposed fusion peptide is sampling conformational space in search of the target membrane (2 and 3). In the dynamic intermediate ensemble, the HA2 fusion peptide proximal subdomain is structurally dynamic. Formation of the extended helical intermediate (4) and the subsequent refolding to the postfusion state (5) in the absence of a target membrane is a relatively slow process for most of HAs. Last, HA1 unfolds after extended exposure to low pH.
Previously reported low-resolution cryo-ET images of HA on influenza particles at acidic pH were interpreted in terms of two intermediate conformations including one in which the HA stalk was narrowed because of HA2 fusion peptide release and another in which the globular head domains had dissociated from each other (15). It was not clear in the past study whether the subtomogram-averaged states reflected the behavior of most of HAs or a subset that was most amenable to classification in those tomograms. On the basis of cryo-ET data we gathered under conditions identical to those used for HDX-MS, we found that the HA spikes to be considerably more conformationally heterogeneous than one might expect for two discrete states (figs. S9 and S10). We conclude that this is most likely due to the flexibility and dynamic nature of key regions in the fusion machinery in HA1 and HA2. Subtomogram averaging attempts to isolate different states and obtain a high-resolution model were not successful; the small fraction of particles that could be averaged gave a low-resolution map similar to that of Fontana et al., but this could not be improved, again most likely due to heterogeneity among the HA spikes (15).
The apparent structural heterogeneity of HA supports the idea that HA populates a structurally dynamic intermediate ensemble during activation rather than one or two discrete, well-ordered intermediate conformations. The highly dynamic nature throughout the HA spike has important implications for efforts that might seek to target the fusion-active intermediate with inhibitors (32) and may suggest that stabilization of the prefusion state or prefusion structural elements (33) is a more fruitful aim than attempting to target intermediate conformations directly.
Our findings and the recent sm-FRET results reframe our understanding of how HA functions as a biomechanical fusion machine. The data do not necessarily challenge the characterization of prefusion HA as existing in a metastable state or the fact that, in the course of mediating fusion, it transitions irreversibly to the true low-energy postfusion state. Rather, the data now clearly indicate that the activated forms of these fusion machines transiently populate highly dynamic ensembles of intermediate states that can persist for minutes before completing the final irreversible transition to the stable postfusion state.
Presentation on virions yields a more sequential HA activation pathway than in isolated HA ectodomain
These data provide the most detailed portrait to date of a viral fusion protein intermediate state and the traversal of fusion-active HA trimers across the conformational pathway. While the conventional view of type 1 fusion proteins as “mouse traps” would suggest that once triggered or destabilized, the fusion protein will snap to the postfusion conformation, our data demonstrate that HA on the surface of intact virions populates an ensemble of highly dynamic intermediate states that are not observed for soluble ectodomain. A marked, two-state change is exhibited by soluble BHA ectodomain, which does not appear to populate any discernable intermediate states, instead transitioning directly between the prefusion and highly stable postfusion state (Figs. 4 and 5). This comparison of full-length HA and BHA during fusion activation underscores the significant differences in functional behavior that result from excising the HA ectodomain from the context of the intact virion (28). Stabilizing interactions present in full-length HA in virus particles appear to encourage the formation of the transiently populated intermediate state (9, 12, 14, 15, 34, 35). Additional interactions include that those involving the viral membrane, HA transmembrane domain, HA cytoplasmic tail, and internal viral components such as the M1 matrix protein are absent with soluble BHA (34, 36). We note that by cryo-ET, it is clear that the M1 matrix layer remains assembled and membrane associated under the pH 5.1 conditions used in the HDX-MS pulse-labeling experiments (fig. S9). Past studies have suggested an interaction between the HA cytoplasmic tail and the matrix layer (9, 34, 35, 37–40). These data indicate that those interactions with an intact matrix layer could persist during activation. In addition, trimerization of the transmembrane domain or presentation on the membrane serve as brakes on the HA2 C-terminal reorganization (34) These interactions borne by HA2 on the surface of influenza virions likely regulate that subdomain’s ability to re-organize, allowing the fusion peptides to be released and insert into a target membrane before complete refolding to the postfusion state.
In addition, studies have suggested that there exists a long-range connection between the HA1 RBD and the HA2 fusion peptide proximal subdomain. Mutations in and around the HA2 fusion peptide were found to significantly alter the dynamic behavior of the HA1 RBD and protomeric interface (41). Recent studies have shown that “breathing” motions in the HA1 RBD and protomeric interface are linked to flexibility of the HA2 fusion peptide (42, 43). Das et al. (14) demonstrated that receptor binding markedly increased dynamics in HA2 and promoted formation of a fusion peptide–released state at neutral pH. We previously demonstrated that while a neutralizing antibody that binds to the HA1 subunit stabilized the prefusion or prefusion-like configuration for the trimerized HA head, its binding did not prevent fusion peptides from being released such that they could disrupt liposomal membranes (33). In some circumstances, it appears that the various structural elements of the HA spike respond to acidic pH in relatively independent rather than concerted fashion, meaning that HA does not function as one cooperative unit but rather each domain does appear to be linked in some manner. While the present data do not directly probe the allosteric linkage between spike apex and fusion peptide–associated regions, the reorganizations observed throughout the HA2 fusion peptide proximal subdomain and the HA1 RBD indicate a concurrent, if not necessarily concerted, reorganization of distal regions.
Mechanistic differences between influenza subtypes
Our observations are based on an H3N2 influenza virus strain. Different influenza virus strains vary widely in their acid stabilities and fusion kinetics and may exhibit different mechanisms of fusion activation (44–47). In the sm-FRET study, H5 HA was examined. In one notable difference, substantial sampling of conformational states reported by the fluorescent probes in HA2 was reported even under neutral pH prefusion conditions. The HDX-MS data for H3 HA examined here and in past continuous deuterium-labeling experiments did not show signatures of conformational sampling before triggering (12). We do not yet understand the structural basis for these functional variations. It is not clear how different HAs, with varying acid stabilities, would influence or alter the mechanism of fusion activation (44). Our results show that, in the absence of a target membrane, the early conformational changes in HA that produce the fusion-active intermediate ensemble occur rapidly upon acidification and that refolding to the postfusion state is relatively slow. When a target membrane is present, the rate of formation for the intermediate is unperturbed, while the transition to the postfusion state is rapidly accelerated, meaning that formation of the fusion-active intermediate is the rate-limiting step for fusion (14). It is possible that by modulating the acid stability of its HA, a virus can control when and how quickly fusion will occur during infection, ensuring that the virus does not prematurely and spontaneously inactivate before reaching the correct subcellular location. In vitro membrane fusion experiments, including our own, initiate fusion by rapid acidification to a single fusogenic pH (12, 14, 15, 17–19, 35, 44). Evidence suggests that during infection, the modified endosomal acidification pathway proceeds through distinct pH stages with varying rates of acidification between them (37, 48). This staged acidification pathway may have an effect on HA fusion activation or other viral components involved in the membrane fusion process, including acidification of the viral interior by the matrix M2 proton channel and reorganization of the matrix M1 layer (16, 35, 37, 48, 49). It is also possible that this stepwise acidic priming might accelerate the formation of the fusion-active intermediate ensemble by gradually increasing the dynamics across HA as the pH approaches the activation threshold.
Powerful, new complementary biophysical and structural techniques enable us to develop a more complete mechanistic model for protein-membrane fusion in an enveloped virus. Future experiments examining pathways of activation in other membrane fusion systems will enable us to test the universality of fusion protein activation and function. The time-resolved, pulse deuteration HDX-MS approach we used opens the door to analysis of highly complex biological assemblies, enabling one to probe intact functional complexes, including whole virions. As the data with influenza virus demonstrate, investigating the complete functional system provides key insights into its behavior that are lost when components are examined in isolation. This approach thus provides a step toward realizing a long-standing goal of performing structural analysis of intact biological systems as they carry out their functions.
MATERIALS AND METHODS
Influenza virus and BHA purification
Purified influenza virus A X-31 A/Aichi/68 (H3N2) was purchased from Charles River Laboratories and stored at −80°C before use. Virus was purified by centrifugation concentrated in HDX HBS buffer [150 mM NaCl, 10 mM Hepes (pH 7.50), and 0.02% NaN3]. Whole-virus HA concentration was determined by Western blotting as previously described (fig. S2) (33). The soluble BHA ectodomain was produced and purified from influenza virus as previously described and concentrated in HDX HBS (pH 7.50) buffer (12).
Pulse-labeling HDX of whole influenza virus
For each reaction, a stock of purified influenza virus containing 4.0 μg of HA in HDX HBS (pH 7.50) buffer was rapidly diluted 1:1 with HDX HBS acidification buffer (150 mM NaCl, 10 mM Hepes, 80 mM citrate, and 0.02% NaN3) to a final pH of either 5.10 or 5.25 at 37°C. Virus was acidified for 5 and 15 s, 1, 3.50, 12, and 40 min, and 2 and 6 hours. Following acidification, each reaction was rapidly pulse-labeled with deuterium for 5 s at room temperature (22°C) by dilution into pulse deuteration buffer [25 mM phosphate, 150 mM NaCl, 0.02% NaN3, and 85% D2O (Cambridge Isotope Laboratories)] to a final pH* 8.0. The pulse-labeling reactions were then rapidly quenched by dilution 1:1 with ice-cold quench buffer {200 mM TCEP [tris(2-carboxyethyl) phosphine] and 0.2% formic acid (FA)} with porcine pepsin (30 μg/ml; Worthington Labs) to a final pH of 2.50 and digested on ice for 5 min. The labeled HA peptides were separated from intact virions by high-speed centrifugation at 25,000 relative centrifugal force (rcf) and 0°C for 2 min. The supernatant was collected and immediately flash-frozen in liquid nitrogen and then stored at −80°C until analysis. Undeuterated samples were prepared identically to the deuterated samples with water in place of D2O. A no-acidification or “0-s” control was also prepared, where the acidification buffer was included with the pulse deuteration buffer and was otherwise treated identically to acidified reactions. All samples were prepared containing 0.25 μg/ml of each PPPI and Pro-Pro-Pro-Phe (PPPF) tetrapeptides (AnaSpec) as internal exchange standards (50). Pulse-labeling reactions were performed in triplicate by manual pipetting with the assistance of a metronome to ensure consistent labeling times. Labeling consistency was verified by analyzing the PPPI and PPPF internal exchange standards (fig. S3). All HDX buffers were prepared with liquid chromatography–MS (LC-MS) grade Optima Water (Thermo Fisher Scientific). Totally deuterated samples were prepared by collecting purified peptide eluent following reverse-phase LC separation of a pepsin-digested undeuterated sample. Following evaporation of the LC elution buffer, the peptides were resuspended in HDX HBS (pH 7.50) buffer, deuterated in pulse deuteration buffer for 2 hours at 37°C, and quenched and frozen as described above.
Pulse-labeling HDX of BHA
Pulse-labeling HDX of the soluble BHA ectodomain was performed identically to the whole influenza virus reactions at pH 5.25. Samples containing 2.5 μg of BHA each were acidified to pH 5.25 at 37°C and pulse-labeled with deuterium at pH 8.0 and 22°C following the same time course outlined above. The pulse-labeled reactions were quenched by dilution 1:1 with ice-cold quench buffer (200 mM TCEP and 0.2% FA) and immediately flash-frozen in liquid nitrogen and stored at −80°C until analysis. The 0-s, totally deuterated, and undeuterated samples were prepared as stated above, and all reactions contained the PPPI and PPPF tetrapeptides (AnaSpec) as internal exchange standards.
Reverse-phase LC-MS
Samples were thawed for 5 min on ice and manually injected into a Waters HDX Manager kept at 1°C. Whole-virus HA samples were trapped on a Waters ACQUITY UPLC CSH C18 VanGuard, 130 Å, 1.7 μm, 2.1 mm by 5 mm trap column for 3 min with a flow of solvent A (2% acetonitrile, 0.1% FA, and 0.025% trifluoroacetic acid) at a rate of 150 μl/min. BHA samples were digested online with immobilized pepsin for 5 min and trapped as described previously (51). Peptides were resolved over a Waters ACQUITY UPLC CSH C18, 130 Å, 1.7 μm, 1 mm by 100 mm column using a 10-min linear gradient of 3 to 50% solvent B (solvent B: 100% acetonitrile and 0.1% FA) and analyzed using a Waters Synapt G2-Si Q-TOF with ion mobility enabled. Source and desolvation temperatures were 70° and 130°C, respectively. The StepWave ion guide settings were tuned to prevent nonuniform gas-phase proton exchange in the source (52). A series of trap column wash steps were implemented between each injection to minimize carryover (53).
HDX-MS data analysis
Peptic HA peptides were identified previously (12). For glycopeptides, only the most abundant glycoforms were examined. Ion mobility and LC retention time filtered spectra were extracted from the raw MS files using CDCReader (54). Spectra were analyzed using HX-Express V2 with binomial fitting and bimodal deconvolution (25, 55). Fitting with more than one binomial distribution was only performed when a single binomial was not sufficient to encompass the mass envelope, and the two populations could be separated with confidence. The relative areas of each population were plotted as a function of time in acidification buffer to track the kinetics of each structural transition. When necessary, the 0-s neutral pH, or prefusion, control HDX population was used as a fitting constraint for bimodal deconvolution. In cases where a bimodal distribution is indicated by broadened isotopic distributions, but cannot be deconvoluted with confidence, peak width analysis was used to support the population analysis (56). Peptides showing more than 10% chromatographic carryover were not used for bimodal deconvolution. Peptides whose signal intensity dropped significantly over the course of the acidification reaction, presumably due to the formation of the proteolytically resistant postfusion HA2 structure, were also excluded from bimodal analysis. HDX quality data can be found in table S2.
Cryo–electron tomography
Purified influenza virus in HBS (pH 7.50) buffer was rapidly diluted 1:1 with HBS acidification buffer (150 mM NaCl, 10 mM Hepes, 80 mM citrate, and 0.02% NaN3) to a final pH of 5.10 at 37°C and incubated for 1 min. Bovine serum albumin gold beads (10 nm) were then added to sample. Using a Vitrobot Mark IV (FEI Co.), 3 μl of this sample was applied to C-flat 2/2 200-mesh grid, blotted, and plunge-frozen in liquid ethane. Frozen grids were imaged using a 300-kV Titan Krios with a Gatan K2 direct electron detector. Tilt series were collected from −48° to +48° with a 3° step size at a magnification of ×53,000, which corresponds to 2.58 Å per pixel. The total dose per tilt series was ~64 e/Å2. Thirty tilt series were collected and further processed.
Tilt series image frames were corrected for electron beam–induced motion using motioncor2 (57). These were then processed for tomogram generation using batch tomography in IMOD using standard procedures (58). Briefly, the tilt series images were aligned using the gold bead markers. The aligned images were then reconstructed to give a three-dimensional volume using weighted back-projection. The final tomograms were rotated, binned, and low pass–filtered for visualization using IMOD and ImageJ (59, 60).
Statistical analysis
Whole-virus HDX-MS experiments were performed in triplicate. The means and SDs of these replicates were used to determine the significance of changes in a peptide deuteration level or population fractions when bimodal distributions were present. When bimodal distributions were present but could not be resolved into clear isotopic distributions, the peak width and deuterium uptake values of the beginning and end points of the reaction were used to calculate the population shift.
Supplementary Material
Acknowledgments
We thank D. Whittington and S. Edgar from the University of Washington School of Pharmacy Mass Spectrometer Center for help and support. We thank the members of the Lee Lab for helpful discussions. We thank D. H. Ross for assistance in developing code for analysis of HDX-MS data. Funding: This work was supported by NIH R01-GM099989 and T32GM008268-27. Author contributions: M.A.B., N.K.G., and K.K.L. designed the HDX-MS experiments. M.A.B. performed the HDX-MS experiments and analyzed the data. N.K.G. and M.G. provided guidance during analysis of the HDX-MS data. V.M.P. designed and conducted the cryo-ET experiments and analyzed the data. M.A.B. and K.K.L. wrote the manuscript with contributions from all other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data and code related to this paper may be requested from the authors.
SUPPLEMENTAY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8822/DC1
REFERENCES AND NOTES
- 1.White J. M., Delos S. E., Brecher M., Schornberg K., Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Skehel J. J., Wiley D. C., Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 69, 531–569 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Chen J., Lee K. H., Steinhauer D. A., Stevens D. J., Skehel J. J., Wiley D. C., Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95, 409–417 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Lazarowitz S. G., Choppin P. W., Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 68, 440–454 (1975). [DOI] [PubMed] [Google Scholar]
- 5.Wilson I. A., Skehel J. J., Wiley D. C., Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 289, 366–373 (1981). [DOI] [PubMed] [Google Scholar]
- 6.Chen J., Skehel J. J., Wiley D. C., N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. U.S.A. 96, 8967–8972 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carr C. M., Kim P. S., A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823–832 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Bullough P. A., Hughson F. M., Skehel J. J., Wiley D. C., Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37–43 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Lee K. K., Architecture of a nascent viral fusion pore. EMBO J. 29, 1299–1311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boonstra S., Blijleven J. S., Roos W. H., Onck P. R., van der Giessen E., van Oijen A. M., Hemagglutinin-mediated membrane fusion: A biophysical perspective. Annu. Rev. Biophys. 47, 153–173 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Carr C. M., Chaudhry C., Kim P. S., Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. U.S.A. 94, 14306–14313 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia N. K., Guttman M., Ebner J. L., Lee K. K., Dynamic changes during acid-induced activation of influenza hemagglutinin. Structure 23, 665–676 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kemble G. W., Bodian D. L., Rosé J., Wilson I. A., White J. M., Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 66, 4940–4950 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das D. K., Govindan R., Nikić-Spiegel I., Krammer F., Lemke E. A., Munro J. B., Direct visualization of the conformational dynamics of single influenza hemagglutinin trimers. Cell 174, 926–937.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontana J., Cardone G., Heymann J. B., Winkler D. C., Steven A. C., Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 86, 2919–2929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fontana J., Steven A. C., Influenza virus-mediated membrane fusion: Structural insights from electron microscopy. Arch. Biochem. Biophys. 581, 86–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Q., Sivaramakrishna R. P., Ludwig K., Korte T., Böttcher C., Herrmann A., Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: Stability and energetics of the hemagglutinin. Biochim. Biophys. Acta 1614, 3–13 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Leikina E., Ramos C., Markovic I., Zimmerberg J., Chernomordik L. V., Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 21, 5701–5710 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin X., Noel J. K., Wang Q., Ma J., Onuchic J. N., Lowered pH leads to fusion peptide release and a highly dynamic intermediate of influenza hemagglutinin. J. Phys. Chem. B 120, 9654–9660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin X., Noel J. K., Wang Q., Ma J., Onuchic J. N., Atomistic simulations indicate the functional loop-to-coiled-coil transition in influenza hemagglutinin is not downhill. Proc. Natl. Acad. Sci. U.S.A. 115, E7905–E7913 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mair C. M., Meyer T., Schneider K., Huang Q., Veit M., Herrmann A., A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J. Virol. 88, 13189–13200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park H. E., Gruenke J. A., White J. M., Leash in the groove mechanism of membrane fusion. Nat. Struct. Biol. 10, 1048–1053 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Xu R., Wilson I. A., Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 85, 5172–5182 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walters B. T., Mayne L., Hinshaw J. R., Sosnick T. R., Englander S. W., Folding of a large protein at high structural resolution. Proc. Natl. Acad. Sci. U.S.A. 110, 18898–18903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guttman M., Weis D. D., Engen J. R., Lee K. K., Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J. Am. Soc. Mass Spectrom. 24, 1906–1912 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai Y., Milne J. S., Mayne L., Englander S. W., Primary structure effects on peptide group hydrogen exchange. Proteins 17, 75–86 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malhotra P., Jethva P. N., Udgaonkar J. B., Chemical denaturants smoothen ruggedness on the free energy landscape of protein folding. Biochemistry 56, 4053–4063 (2017). [DOI] [PubMed] [Google Scholar]
- 28.White J. M., Wilson I. A., Anti-peptide antibodies detect steps in a protein conformational change: Low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 105, 2887–2896 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris A., Cardone G., Winkler D. C., Heymann J. B., Brecher M., White J. M., Steven A. C., Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. U.S.A. 103, 19123–19127 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calder L. J., Wasilewski S., Berriman J. A., Rosenthal P. B., Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. U.S.A. 107, 10685–10690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godley L., Pfeifer J., Steinhauer D., Ely B., Shaw G., Kaufmann R., Suchanek E., Pabo C., Skehel J. J., Wiley D. C., Wharton S., Introduction of intersubunit disulfide bonds in the membrane-distal region of the influenza hemagglutinin abolishes membrane fusion activity. Cell 68, 635–645 (1992). [DOI] [PubMed] [Google Scholar]
- 32.Kilby J. M., Hopkins S., Venetta T. M., DiMassimo B., Cloud G. A., Lee J. Y., Alldredge L., Hunter E., Lambert D., Bolognesi D., Matthews T., Johnson M. R., Nowak M. A., Shaw G. M., Saag M. S., Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4, 1302–1307 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Williams J. A., Gui L., Hom N., Mileant A., Lee K. K., Dissection of epitope-specific mechanisms of neutralization of influenza virus by intact IgG and Fab fragments. J. Virol. 92, e02006-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benton D. J., Nans A., Calder L. J., Turner J., Neu U., Lin Y. P., Ketelaars E., Kallewaard N. L., Corti D., Lanzavecchia A., Gamblin S. J., Rosenthal P. B., Skehel J. J., Influenza hemagglutinin membrane anchor. Proc. Natl. Acad. Sci. U.S.A. 115, 10112–10117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gui L., Ebner J. L., Mileant A., Williams J. A., Lee K. K., Visualization and sequencing of membrane remodeling leading to influenza virus fusion. J. Virol. 90, 6948–6962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruigrok R. W. H., Barge A., Durrer P., Brunner J., Ma K., Whittaker G. R., Membrane interaction of influenza virus M1 protein. Virology 267, 289–298 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Stauffer S., Feng Y., Nebioglu F., Heilig R., Picotti P., Helenius A., Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza A virus cores after penetration. J. Virol. 88, 13029–13046 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ali A., Avalos R. T., Ponimaskin E., Nayak D. P., Influenza virus assembly: Effect of influenza virus glycoproteins on the membrane association of M1 protein. J. Virol. 74, 8709–8719 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin H., Leser G. P., Zhang J., Lamb R. A., Influenza virus hemagglutinin and neuraminidase cytoplasmic tails control particle shape. EMBO J. 16, 1236–1247 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enami M., Enami K., Influenza virus hemagglutinin and neuraminidase glycoproteins stimulate the membrane association of the matrix protein. J. Virol. 70, 6653–6657 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yewdell J. W., Taylor A., Yellen A., Caton A., Gerhard W., Bachi T., Mutations in or near the fusion peptide of the influenza virus hemagglutinin affect an antigenic site in the globular region. J. Virol. 67, 933–942 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bangaru S., Lang S., Schotsaert M., Vanderven H. A., Zhu X., Kose N., Bombardi R., Finn J. A., Kent S. J., Gilchuk P., Gilchuk I., Turner H. L., Garcia-Sastre A., Li S., Ward A. B., Wilson I. A., Crowe J. E. Jr., A site of vulnerability on the influenza virus hemagglutinin head domain trimer interface. Cell 177, 1136–1152.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner H. L., Pallesen J., Lang S., Bangaru S., Urata S., Li S., Cottrell C. A., Bowman C. A., Crowe J. E. Jr., Wilson I. A., Ward A. B., Potent anti-influenza H7 human monoclonal antibody induces separation of hemagglutinin receptor-binding head domains. PLOS Biol. 17, e3000139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costello D. A., Whittaker G. R., Daniel S., Variations in pH sensitivity, acid stability, and fusogenicity of three influenza virus H3 subtypes. J. Virol. 89, 350–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Lella S., Herrmann A., Mair C. M., Modulation of the pH stability of influenza virus hemagglutinin: A host cell adaptation strategy. Biophys. J. 110, 2293–2301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mair C. M., Ludwig K., Herrmann A., Sieben C., Receptor binding and pH stability — How influenza A virus hemagglutinin affects host-specific virus infection. Biochim. Biophys. Acta 1838, 1153–1168 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Schrauwen E. J. A., Richard M., Burke D. F., Rimmelzwaan G. F., Herfst S., Fouchier R. A. M., Amino acid substitutions that affect receptor binding and stability of the hemagglutinin of influenza A/H7N9 virus. J. Virol. 90, 3794–3799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakadamyali M., Rust M. J., Babcock H. P., Zhuang X., Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. U.S.A. 100, 9280–9285 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puri A., Booy F. P., Doms R. W., White J. M., Blumenthal R., Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: Effects of acid pretreatment. J. Virol. 64, 3824–3832 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z., Zhang A., Xiao G., Improved protein hydrogen/deuterium exchange mass spectrometry platform with fully automated data processing. Anal. Chem. 84, 4942–4949 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Verkerke H. P., Williams J. A., Guttman M., Simonich C. A., Liang Y., Filipavicius M., Hu S.-L., Overbaugh J., Lee K. K., Epitope-independent purification of native-like envelope trimers from diverse HIV-1 isolates. J. Virol. 90, 9471–9482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guttman M., Wales T. E., Whittington D., Engen J. R., Brown J. M., Lee K. K., Tuning a high transmission ion guide to prevent gas-phase proton exchange during H/D exchange MS analysis. J. Am. Soc. Mass Spectrom. 27, 662–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang J., Rand K. D., Beuning P. J., Engen J. R., False EX1 signatures caused by sample carryover during HX MS analyses. Int. J. Mass Spectrom. 302, 19–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marty M. T., Baldwin A. J., Marklund E. G., Hochberg G. K., Benesch J. L., Robinson C. V., Bayesian deconvolution of mass and ion mobility spectra: From binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weis D. D., Engen J. R., Kass I. J., Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Weis D. D., Wales T. E., Engen J. R., Hotchko M., Ten Eyck L. F., Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17, 1498–1509 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mastronarde D. N., Held S. R., Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 197, 102–113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 60.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kavan D., Man P., MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 302, 53–58 (2011). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8822/DC1