

Abstract
Objectives
Disease‐modifying therapies (DMTs) targeting B cells are amongst the most effective for preventing multiple sclerosis (MS) progression. IgG3 antibodies and their uncharacterised B‐cell clones are predicted to play a pathogenic role in MS. Identifying subsets of IgG3 + B cells involved in MS progression could improve diagnosis, could inform timely disease intervention and may lead to new DMTs that target B cells more specifically.
Methods
We designed a 31‐parameter B‐cell‐focused mass cytometry panel to interrogate the role of peripheral blood IgG3 + B cells in MS progression of two different patient cohorts: one to investigate the B‐cell subsets involved in conversion from clinically isolated syndrome (CIS) to MS; and another to compare MS patients with inactive or active stages of disease. Each independent cohort included a group of non‐MS controls.
Results
Nine distinct CD20+IgD−IgG3 + B‐cell subsets were identified. Significant changes in the proportion of CD21+CD24+CD27−CD38− and CD27+CD38hiCD71hi memory B‐cell subsets correlated with changes in serum IgG3 levels and time to conversion from CIS to MS. The same CD38− double‐negative B‐cell subset was significantly elevated in MS patients with active forms of the disease. A third CD21+CD24+CD27+CD38− subset was elevated in patients with active MS, whilst narrowband UVB significantly reduced the proportion of this switched‐memory B‐cell subset.
Conclusion
We have identified previously uncharacterised subsets of IgG3 + B cells and shown them to correlate with autoimmune attacks on the central nervous system (CNS). These results highlight the potential for therapies that specifically target IgG3 + B cells to impact MS progression.
Keywords: B cells, clinically isolated syndrome, mass cytometry, multiple sclerosis, phototherapy
Mass cytometry has allowed us to identify nine unique IgG3+ B‐cell subsets. Using two independent cohorts of multiple sclerosis (MS) patients, we show that a number of these IgG3+ subsets are not only associated with MS progression but also affected by disease‐modifying therapies. These studies highlight the potential for therapies that specifically target IgG3+ B cells to impact MS progression.

Introduction
Relapsing–remitting multiple sclerosis (RRMS) is an autoimmune disease caused by the destruction of the myelin‐producing cells in the central nervous system (CNS). As a consequence of this immune attack, nerve impulses cannot be transmitted efficiently and uninterrupted from the CNS to the periphery. The only successful disease‐modifying therapies (DMTs) limit the damage caused to the CNS by targeting the cells and molecules of the immune system. DMTs that target B cells are proving to be highly effective at halting MS, not only in RRMS but also notably in progressive forms of the disease. 1 The success of some B‐cell‐targeting DMTs such as the anti‐CD20 monoclonal antibodies, rituximab and ocrelizumab, but not others such as atacicept, 2 suggests that not all B cells are pathogenic in the context of MS. DMTs targeting specific B‐cell subsets that are involved in MS pathogenesis are likely to be more effective in the treatment of this CNS disease.
The immunoglobulin subclasses IgG1 and IgG3 have long been associated with autoimmunity, 3 , 4 particularly in MS. 5 We recently showed that, compared with baseline, IgG3 serum levels were higher in clinically isolated syndrome (CIS) patients who were close to converting to MS. 6 Identification of the IgG3 B‐cell subsets dysregulated by MS will allow for the design of more targeted therapeutics. To that end, using mass cytometry to interrogate circulating IgG3 + B‐cell subsets in two different MS cohorts, we have discovered nine previously unidentified subsets of IgG3 + B cells. CD21+CD24+CD27−CD38− and CD27+CD38hiCD71hi memory IgG3 + B cells were found to be significantly increased as CIS patients progress to MS, which correlated with increased serum levels of IgG3, and in patients with active disease. Finally, we show that phototherapy, which delays progression of CIS to MS in a subset of individuals, 7 is associated with a significant decrease in CD21+CD24+CD27+CD38−IgG3 + B‐cell subsets mirroring the lower proportion of IgG3 + B cells we found in MS patients with inactive or quiescent disease. Our study provides evidence that specific IgG3 + B‐cell subsets are associated with autoimmune attack on the CNS and that DMTs targeting these subsets may have an impact on disease progression.
Results
Serum IgG3 levels correlate with the proportion of IgG3 + B‐cell subsets
Consistent with serum levels of individual IgG subclasses correlating with IgG+ B cells, 8 there was a statistically significant positive correlation between IgG3 serum levels and total IgG3 + B cells (as a proportion of all B cells, across cohort 1 irrespective of phototherapy status; Figure 1a). IgG3 + B cells could be manually subdivided into nine distinct subsets based on their expression of CD21, CD20, CD24, CD27 and CD38 (Figure 1b). The nine IgG3 + subsets were IgD− (Figure 1b) and differed in their expression of CD71 (transferrin receptor), CD80, CD185 (CXCR5), CD210 (IL‐10 receptor), CD360 (IL‐21 receptor) and HLA‐DR (Figure 1c). No other markers were able to differentiate the nine IgG3 + subsets (Supplementary figure 1b). Subset 9 had the most activated phenotype, expressing the highest amount of HLA‐DR, CD71 and CD80. B‐cell subset 4, which resembled double‐negative (DN)‐1 B cells 9 in that it was IgD−CD21+CD24+CD27−CXCR5+ but lacked CD38, showed a statistically significant positive correlation with IgG3 serum levels (Figure 1d). The CD27+ memory B‐cell subset 9, which was CD21+ and expressed high levels of CD38 but not CD24, also showed a statistically significant positive correlation with IgG3 serum levels (Figure 1e). This suggests that in the PhoCIS cohort, two B‐cell subsets, one with a unique CD38− DN‐1 phenotype 9 and the other with a subset of CD27+ memory cells, were primarily responsible for changes in serum IgG3 levels.
Figure 1.
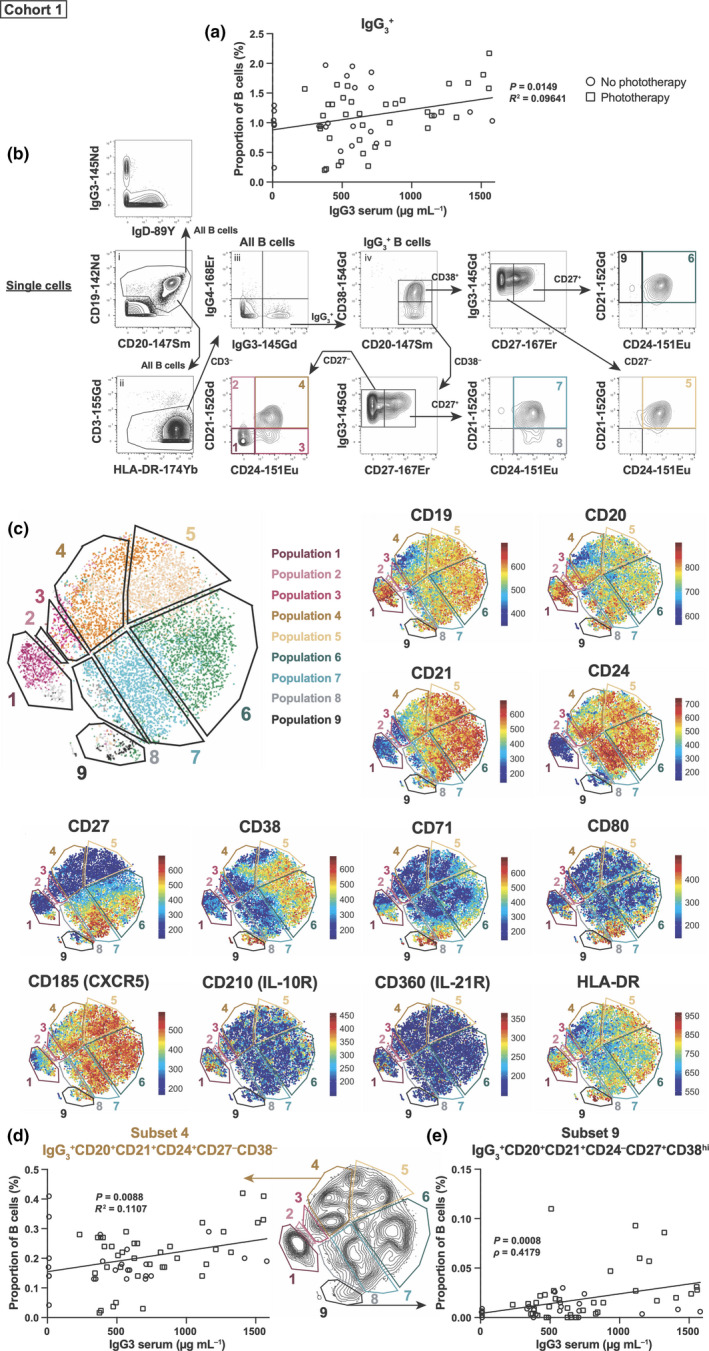
Serum IgG3 levels correlate with IgG3 + B‐cell subsets. (a) IgG3 serum levels were compared with IgG3 + B‐cell levels (as a proportion of total B cells). (b) Manual gating strategy progressing from (i) to (iv) and beyond as indicated by the arrows to identify IgG3 +IgD−B cells and nine subsets using CD20, CD21, CD24, CD27 and CD38. (c) tSNE plots showing the position of the nine subsets and heatmap of markers used for gating and differentiating between these nine subsets. The proportions of subsets (d) 4 and (e) 9 had statistically significant correlations with IgG3 serum levels. Open black circles represent clinically isolated syndrome (CIS) patients who did not receive phototherapy (n = 8 patients, 25 time points), whilst open black squares represent patients who did receive phototherapy (n = 8 patients, 36 time points). A linear regression was done (with line shown), with reported P‐values and R 2‐values or ρ (rho)‐value. Mass cytometry data were generated from seven independent experiments.
Changes in the proportion of IgG3 + B cells correlate with time to convert from CIS to MS and success of DMT
IgG3 serum levels are higher in CIS patients close to converting to MS. 6 The same correlation held true for IgG3 + B‐cell subsets as there was a significant increase in total IgG3 + B cells as CIS patients went on to develop MS (day 0 being MS diagnosis) (Figure 2a). B‐cell subsets 4 and 9 again showed a significant correlation as did the CD38+ B‐cell subset 5 and an additional CD24− B‐cell subset (subset 2; Figure 2b). For all subsets, MS disease progression increased the proportion of IgG3 + B cells to levels that were comparable to that of non‐MS controls (Figure 2; red line and shaded areas represent median and interquartile range of the non‐MS controls, respectively; individual values for non‐MS controls are shown in Supplementary figure 2). Whilst the correlation between time and relative proportion of IgG3 + B cells was lost after conversion to MS (day 0, dashed vertical line in Figure 2), commencement of DMTs, dimethyl fumarate (n = 4), natalizumab (n = 5) or fingolimod (n = 2), reversed the rising trend in IgG3 + B cells. This negative correlation over time was statistically significant for subset 4 (green line, Figure 2b). Indeed, in the absence of DMT this subset of CD21+CD24+CD27−CD38− B cells continued to rise over time. Together, these data suggest that there may be an inherent defect in the IgG3 + B‐cell compartment that is affected by the course of MS disease and that DMT can correct this defect. Furthermore, this IgG3 + B‐cell defect does not seem to be present in non‐MS controls.
Figure 2.
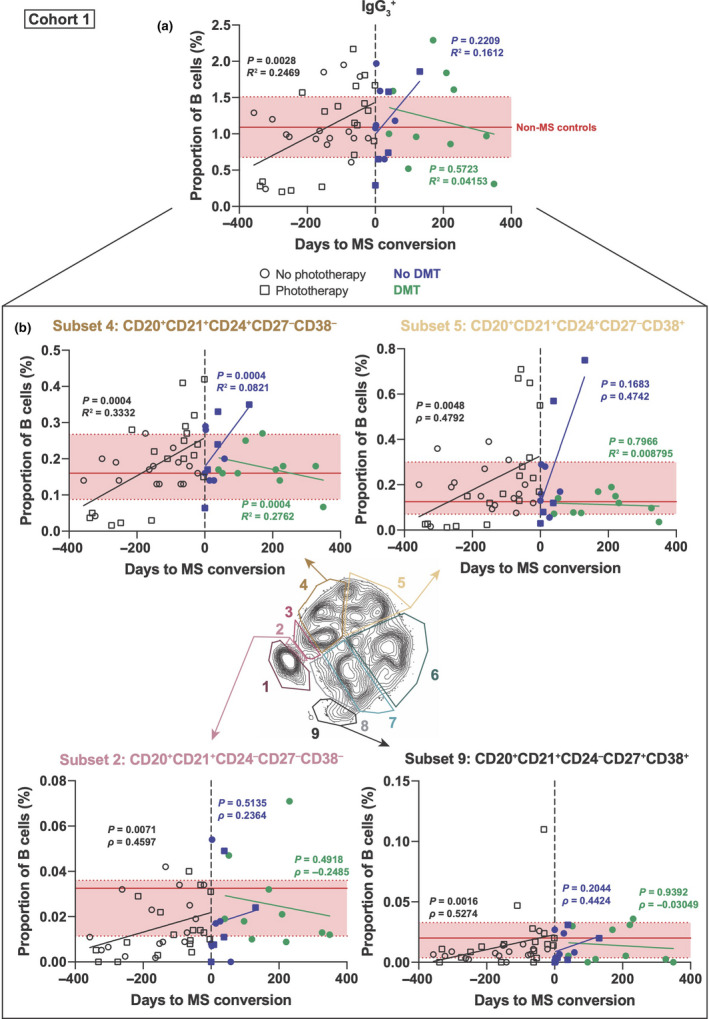
IgG3 + B‐cell levels positively correlate with CIS conversion to MS. (a) IgG3 + B‐cell levels (as a proportion of total B cells) are shown for clinically isolated syndrome (CIS) patients as they go on to convert to multiple sclerosis (MS). At time point 0 (vertical dotted line), patients were diagnosed with MS (prior to this, they had CIS). Open black squares represent CIS patients who received phototherapy (n = 7 patients, 28 time points), whilst open black circles did not receive phototherapy (n = 5 patients, 27 time points). A linear regression was done, with the line shown for CIS/prior (black, n = 12 patients, 34 time points) and MS/after with (green, n = 6 patients, 10 time points) or without (purple, n = 9 patients, 11 time points) disease‐modifying therapies (DMTs), with the reported P‐value and R 2‐value or ρ (rho)‐value. (b) Subsets of IgG3 + B cells that had significant correlations either CIS/prior or MS/after. Solid horizontal red line represents median proportion for non‐MS controls, with interquartile range highlighted in red between dashed horizontal red lines (n = 14 patients). Mass cytometry data were generated from seven independent experiments.
Subsets of IgG3 + B cells increase during active MS disease
In a second independent cohort of MS patients, we observed no significant difference in the proportion of total IgG3 + B cells comparing iMS, aMS and non‐MS controls (Figure 3a). However, when we interrogated the nine different subsets of IgG3 + B cells, we found that, compared with inactive disease states, there was a significant increase in the proportion of B‐cell subset 4 in those with aMS (Figure 3b). This was the same subset found to be affected by MS progression and DMT in the PhoCIS cohort (cohort 1; Figure 2b). CD27+ B‐cell subsets 7 and 8 that were CD21+ and CD21−, respectively, were also significantly higher in patients with aMS (Figure 3b). Similar to what we observed in the first cohort, the proportion of all IgG3 + B‐cell subsets in those with aMS disease was comparable to that of non‐MS controls.
Figure 3.
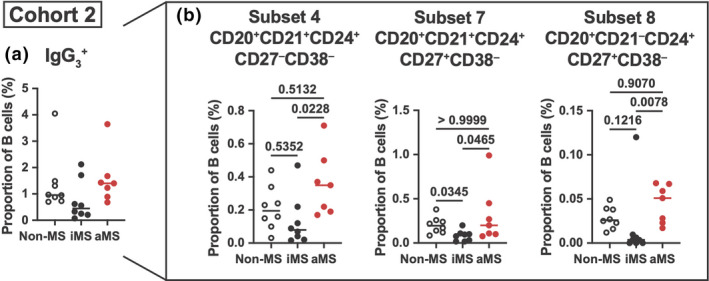
IgG3 + B cells are decreased during inactive MS. (a) IgG3 + B‐cell levels as a proportion of total B cells between non‐MS controls, inactive MS (iMS) and active MS (aMS) patients. (b) IgG3 + B‐cell subsets 4, 7 and 8 that had significant differences between disease and control groups are shown as a proportion of total B cells. As no groups were normally distributed, a non‐parametric Kruskal–Wallis test with Dunn's multiple comparisons test (median shown) was used with P‐values shown. n = 7 or 8 individual patients/controls. Mass cytometry data were generated from two independent experiments.
Phototherapy decreases the levels of circulating IgG3 + B cells
In the PhoCIS trial, 100% of CIS patients who did not receive phototherapy converted to MS within 12 months. In contrast, only 70% of CIS patients who were given phototherapy three times per week for 8 weeks converted to MS in the 12 months of follow‐up. 7 Changes in IgG3 + B‐cell levels (as a proportion of all B cells) were calculated as the difference in their baseline level. Compared with control CIS patients, those receiving phototherapy over the 2‐month period showed a significant reduction in total IgG3 + B cells (Figure 4a) with subsets 5 and 7 specifically affected (Figure 4b). Of note, the proportions of subsets 4 and 9, both of which correlated with serum IgG3 (Figure 1d), and in the case of subset 4, it was affected by active MS disease (Figure 3b), were not significantly affected by phototherapy.
Figure 4.
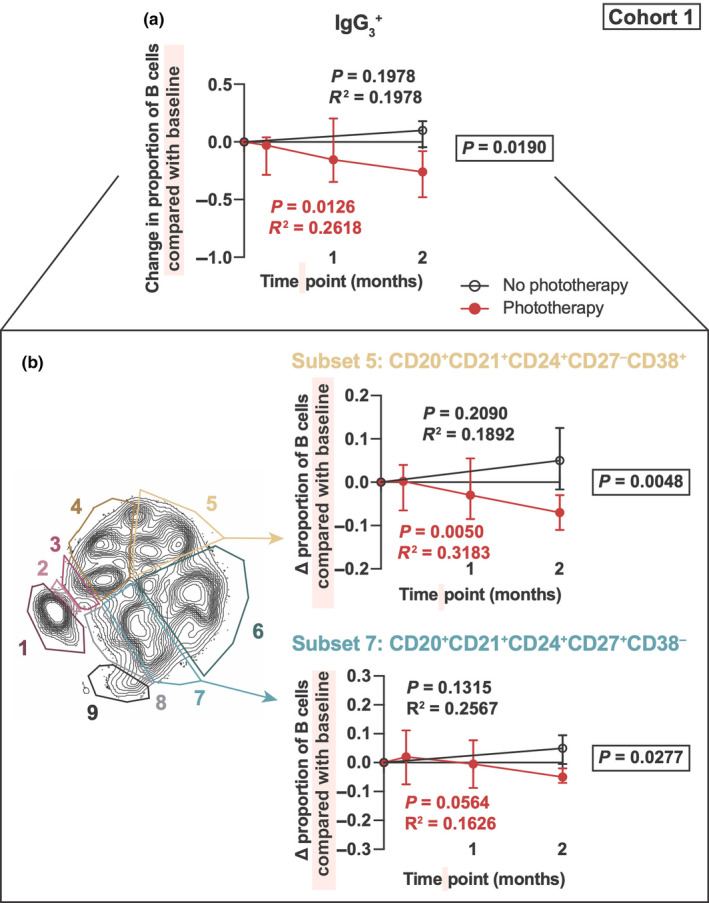
Phototherapy decreases IgG3 + B‐cell proportions in CIS patients. IgG3 + B‐cell levels (as a proportion of total B cells) were compared between clinically isolated syndrome (CIS) patients who either received phototherapy for 2 months and CIS patients who did not. (a) The change in IgG3 + B cells compared with baseline prior to phototherapy. CIS patients who received phototherapy are indicated in red, whilst CIS patients who did not are indicated in black open circles. A linear regression was done with shown P‐values and R 2‐values for CIS patients who did not receive phototherapy (black) or did receive phototherapy (red). Slopes of linear regression lines were compared between groups, by a comparable analysis of covariance (ANCOVA) with the P‐value reported in a black box. (b) Subsets of IgG3 + B cells that had significant differences between CIS patients who did or did not receive phototherapy. n = 8 individual patients. Mass cytometry data were generated from seven independent experiments.
Discussion
Using two separate cohorts, we have uncovered a previously undescribed association for subsets of IgG3 + B cells in the conversion of CIS to MS and in MS patients with a more aggressive disease course. The serum concentration of IgG3 positively correlated with the proportion of these peripheral IgG3 + B cells (Figure 1a), suggesting that the source of the elevated IgG3 levels that predict conversion from CIS to MS are these same B‐cell subsets. Compared with other B cells (Supplementary figure 1c), almost all IgG3 + B cells expressed high levels of CXCR5 (CD185), which is encoded by a known MS risk gene. 10 CXCR5 is the receptor for CXCL13, a chemokine that mediates B‐cell trafficking into germinal centres, and is expressed at significantly higher levels in the cerebrospinal fluid of RRMS patients. 11 Whether CXCR5‐expressing IgG3 + B cells find their way into the CNS, or exert their effects in lymphoid follicles remains to be determined.
Two specific subsets that differed in CD27 expression showed the most striking correlation with serum IgG3 concentrations. Subset 4, which accounts for 15% of all IgG3 + B cells in non‐MS controls, expressed CD21 and CD24 but not IgD, CD27 or CD38, which together with their expression of CXCR5 makes them phenotypically similar, but not identical to DN1 B cells (which are CD38+). 9 Whether this difference in CD38 expression represents abnormal class switching and/or germinal centre B‐cell maturation in MS is not known. A decrease in CD38 expression on double‐negative B cells has been described previously, 12 suggesting that subset 4 may be at a more mature stage of development. IgG3 class switching, at least in vitro, can be induced by IL‐21, 13 and although the IL‐21 receptor (CD360) is associated with MS, 14 its expression was absent from all IgG3 + subsets except for subset 1. This lack of IL‐21R expression suggests that the IgG3 + B cells we found to be associated with MS progression are likely to be switched‐memory B cells. 15 Whether upstream factors such as IL‐21 influence the course of MS through IgG3 B‐cell subsets remains to be determined, although both IL‐21 and its receptor are expressed by immune cells and neurons at greater levels within MS brains compared with controls. 16
Double‐negative B cells are broadly defined by their lack of both IgD and CD27. Supporting our findings, Fraussen and colleagues recently showed an increase in IgD−CD27− DN‐B cells in people with MS. 12 DN1 B cells are thought to represent an early switched‐memory B‐cell that still expresses CD21, CD24 and CD38 but has not yet acquired CD27. 9 In our MS patients, DN1 cells would correspond to subset 5 (Figure 1c) which in cohort 1 we found to be positively associated with conversion of CIS to MS (Figure 2b), and were one of the only two IgG3 + B‐cell subsets significantly reduced by phototherapy (Figure 4b). IgD−CD27− DN2 B cells are CD19hiCD21−CD24−CD38lowCD185low and would correspond to subset 1 in our analysis (Figure 1c). However, despite producing the highest amounts of IgG3 in systemic lupus erythematosus, 17 we found no role for IgG3 + DN2 B cells in the progression of MS.
We also observed changes in the CD27+ memory B‐cell subset 9, which expressed the highest levels of CD38, suggesting that it may be a precursor population to plasmablasts. 18 , 19 Indeed, CD20+CD27hiCD38hi blood‐derived B cells, but not traditional memory B cells, spontaneously produce antibodies in vitro. 18 Changes in subset 9 would be consistent with a suspected pathogenic role for memory B cells in MS. 20 Indeed, the proportion of this IgG3 + B‐cell subset not only correlated with IgG3 serum levels, but also correlated with progression from CIS to MS. Memory B‐cell subset 9 expressed the highest levels of CD80, HLA‐DR and CD71, the latter being a recently described early activation marker of proliferating antigen‐specific B cells. 21 The functional significance of high CD71 expression on activated B cells is unclear. Although we did not find a significant change in subset 9 within cohort 2, others have previously shown that CD80+CD71+ leucocytes (which included B cells) are increased during active MS. 22 CD71, otherwise known as the transferrin receptor, plays a key role in iron metabolism, which may be dysregulated in MS. 23 Whilst it remains to be seen if targeting transferrin‐bound iron in serum modulates these CD71‐expressing B cells, our data strongly support the idea that activated IgG3 + memory B cells are involved in MS pathogenesis.
Supporting a role for IgG3 + B‐cell subsets in MS pathogenesis is our observation in a second cohort that MS patients with clinically confirmed active forms of the disease also had significantly elevated levels of specific IgG3 + subsets. IgG3 + B‐cell subset 4 was again implicated in this cohort, and management of MS disease in cohort 1 through administration of DMTs known to affect B cells including dimethyl fumarate, 24 , 25 natalizumab 26 and fingolimod 27 , 28 was associated with lower proportions of this IgG3‐expressing subset. Although phototherapy did not significantly suppress the conversion of CIS to MS, 7 exposure to narrowband UVB does lead to a significant but short‐lived decrease in circulating CD20+CD27+IgD− B cells as a proportion of total B cells. 29 Here, we have discovered using mass cytometry that these UVB‐targeted ‘switched‐memory’ B cells are likely to include a population of IgG3 + B cells (subset 7; Figure 4b). This same population of IgG3 + B cells were significantly elevated in those with active forms of MS that subsequently have been found to be more aggressive (Figure 3b), suggesting that this subset is also involved in MS progression.
It is not entirely clear what role IgG3 plays in MS pathogenesis. IgG3 has the highest affinity for the complement protein C1q 30 and is consequently a potent activator of the classical complement pathway. 31 , 32 Complement proteins C1q and C3 are present in demyelinated MS lesions, 33 , 34 , 35 , 36 , 37 and the membrane attack complex, which represents the end stage of complement activation, has been found surrounding myelin. 33 Thus, IgG3 and the B cells that produce it are likely to be involved in complement‐mediated MS pathogenesis. Our findings suggest that reducing circulating IgG3 or specific subsets of B cells that produce IgG3 may inhibit MS disease activity. CIS patients receiving phototherapy had an overall decrease in circulating IgG3 + B cells, as well as select IgG3 + B‐cell subsets. Phototherapy delays the onset of MS in a subset of CIS patients, 7 and exposure to UV radiation (UVR) decreases the expression of classical complement genes including C1q. 38 These two UVR‐driven events may work synergistically to lower IgG3 complement‐mediated damage in MS, which may explain the observations from epidemiological 39 and animal studies 40 that UVR protects from MS.
In the PhoCIS cohort, we noticed, somewhat paradoxically, that as CIS patients progressed to MS, the proportion of circulating IgG3 + B cells rose to levels that were similar to that found in non‐MS controls. This was also observed in the second cohort whereby proportions of IgG3 + B cells in those with iMS were significantly lower than those of non‐MS controls despite not being on any DMT at the time of blood sampling. However, in untreated MS patients with active disease, their proportions were no longer different from non‐MS controls. This suggests that whilst people without CIS or MS do have IgG3 + B cells, they are presumably not producing IgG3 antibodies either to autoantigens or pathogens, which may have implications for viral defence. IgG3 has the most flexible and extended hinge region of all the IgG isotypes. 41 It contains multiple glycosylation sites and has a high affinity for Fcγ receptors, particularly CD16a, 42 which is highly expressed by NK cells and monocytes/macrophages, and IgG3 is the most potent activator of classical complement. 31 , 32 This makes IgG3, despite only accounting for 5–8% of total IgG in the circulation, an important immunological player, especially in response to infectious diseases. 43 What our data show is that in those susceptible to MS, subsets of IgG3 + B cells are abnormally low during inactive disease states. However, during periods of progression or relapse, these B cells are activated to clonally expand from a low level and differentiate to secrete IgG3 into the circulation. This may indicate that there is an inherent defect in the IgG3 + B‐cell compartment in those at risk of MS with abnormally low levels of IgG3 increasing the susceptibility to certain infections and/or autoreactive IgG3 B‐cell clones being activated to secrete autoantibodies.
In conclusion, mass cytometry has provided us with unprecedented insight into the changes that occur in IgG3 + B cells as CIS patients convert to MS, and when MS patients develop active forms of the disease. Our results highlight that interventions (e.g. phototherapy) that reduce these populations of IgG3 + B cells protect CIS patients from transitioning to MS. These findings justify consideration for subsets of IgG3 + B cells as biomarkers and/or mediators of disease progression in MS, as well as future targets of novel immunotherapies.
Methods
Study participants: cohort 1 – CIS/MS/phototherapy patients
Recruitment of these patients was conducted in Perth, Western Australia. The Phototherapy for Clinically Isolated Syndrome (PhoCIS) trial design has been published. 7 Briefly, 16 of the 20 individuals (10 females: range 29–54, median 43 years old; and six males: range 27–42, median 36 years old) presenting with CIS within 120 days from their first demyelinating event at study enrolment, and meeting PatyA or PatyB criteria based on magnetic resonance imaging (MRI) were included. Insufficient biobanked cells were available from the other 4 trial participants. All participants with CIS were drug‐naïve, but if they did not have 25(OH)D levels > 80 nmol L−1 at enrolment, they were supplemented with oral vitamin D. 7 MRI scans and clinical assessments were performed at 3, 6 and 12 months after enrolment to detect conversion to MS, the diagnosis of which was based on the appearance of new MRI‐confirmed lesions. Following the detection of new lesions and confirmation of a diagnosis of MS, patients were offered one of three DMTs: dimethyl fumarate, natalizumab or fingolimod. Non‐MS controls (n = 14) were age‐ and sex‐matched to the patient group and were free of neurological signs/symptoms.
The PhoCIS study was carried out in accordance with the recommendations of the National Health and Medical Research Council of Australia's National Statement on Ethical Conduct in Human Research. The PhoCIS study protocol was approved by the Bellberry Human Research Ethics Committee (2014‐02‐083) and endorsed by the Human Research Ethics Office of the University of Western Australia (RA/4/1/6796), and the study of MS participants was approved by Sir Charles Gairdner Hospital Human Research Ethics Committee (2006‐073). All participants gave written informed consent in accordance with the Declaration of Helsinki prior to study procedures being performed.
Phototherapy intervention
In the PhoCIS trial, CIS participants were randomised to receive (n = 8), or not receive (controls, n = 8), narrowband UVB phototherapy three times per week for the first 8 weeks (24 sessions in total). Phototherapy was delivered according to the Dundee protocol, based on patient skin type as previously described. 7
Blood sampling
Peripheral blood mononuclear cells (PBMC) were isolated from heparinised blood collected in lithium heparin vacutainers (BD) using a Lymphoprep (Axis‐Shield, Oslo, Norway) density separation gradient. The CIS patients (n = 16) from the PhoCIS trial had their peripheral venous blood taken at 1 week, 1 month, 2 months, 3 months, 6 months and 12 months post‐baseline. PBMC were cryopreserved in 5–10% DMSO/FBS for storage in liquid nitrogen prior to mass cytometry staining.
Cytometric bead array for IgG3 serum levels
IgG3 serum levels were measured using cytometric bead arrays (BD Biosciences, North Ryde, NSW, Australia), with data captured using the BD LSRFortessa flow cytometer, according to the manufacturer's instructions and as previously described. 6
Study participants: cohort 2 – inactive and active MS patients and non‐MS controls
Ethical consent for the study was obtained from the Research Integrity and Ethics Administration of the University of Sydney (project number 2012/1851). Multiple sclerosis was defined by McDonald 2010 criteria, and disease activity was defined by neurological signs/symptoms and the presence of new T2 or gadolinium‐enhancing MRI lesions. No patients were on DMTs at the time of blood sampling. All patients had low disability (EDSS range 0–2.5). Cohort 2 patient data are shown in Table 1.
Table 1.
Cohort 2 patient data
| Non‐MS controls (n = 8) | iMS (n = 8) | aMS (n = 7) | |
|---|---|---|---|
| Median age (range) | 40 (25–59) | 50 (27–60) | 32 (25–42) |
| Female sex | 5 (62.5%) | 7 (87.5%) | 6 (85.7%) |
| Median EDSS (range) | |||
| Prior treatment | –– | 0 (0–1) | 0 (0–1) |
| Current | –– | 0 (0–0) | 0 (0–2.5) |
| Previous treatment prior to blood sampling | |||
| Time since last dose | |||
| Untreated | –– | 6 | 6 |
| 1 month | –– | 1 | 0 |
| 2 years | –– | 0 | 1 |
| 10 years | –– | 1 | 0 |
| Disease‐modifying therapy | |||
| Untreated | –– | 6 | 6 |
| IFN‐β | –– | 2 | 1 |
| Clinical/MRI activity prior to blood collection | |||
| Sensory symptoms > 1 year | –– | 3 | 0 |
| No symptoms > 9 months | –– | 3 | 0 |
| No disease activity > 4 years | –– | 2 | 0 |
| Activity < 6 months | –– | 0 | 7 |
| Lesion load | |||
| Brain | |||
| Low | –– | 7 | 4 |
| Moderate | –– | 1 | 1 |
| High | –– | 0 | 2 |
| Spinal | |||
| No | –– | 2 | 1 |
| Yes | –– | 6 | 3 |
| Several | –– | 0 | 1 |
| Enhancing | –– | 0 | 1 |
| Large + thoracic cord lesion | –– | 0 | 1 |
| Other characteristics | |||
| Pars planitis | –– | 1 | 0 |
Inactive MS (iMS, n = 8) was defined as having no disease activity in the previous 6 months of blood sampling, a low lesion load, and in follow‐up, these patients have had a benign clinical course, a number never having been started on DMTs. Six of eight iMS patients had not been on DMTs prior to blood collection. Active MS (aMS, n = 7) was defined as having disease activity within 6 months of blood sampling and subsequently an aggressive clinical course; all patients having ended up on high‐efficacy DMT since the blood collection. Six of seven aMS patients had not been on therapeutics prior to blood collection. Non‐MS controls (n = 8) were age‐ and sex‐matched to the patient cohort and were free of neurological signs/symptoms.
Blood sampling
PBMC were isolated from blood and collected in EDTA vacuette tubes (Greiner Bio‐One International, Kremsmünster, Austria) using a Ficoll‐Paque Plus (GE Healthcare, Chicago, IL, USA) density separation gradient. Samples were cryopreserved in 5–10% DMSO/FBS for storage in liquid nitrogen prior to mass cytometry staining.
Cell staining and analysis by mass cytometry
A total of 2.5–5 × 106 cells were resuscitated by thawing in a 37°C water bath and washed in RPMI medium. Individual patient/time point samples were first barcoded with anti‐human CD45 (on three different metal isotopes) for 30 min, such that three independent samples could be combined for further staining as described. 44 Samples were combined (for 7.5–15 × 106 cells) and stained with cisplatin (Fluidigm, South San Francisco, CA, USA) for 5 min as a live/dead marker. PBMC were stained with antibodies specific for the markers in Table 2 for 30 min. These antibodies were purchased unlabelled in a carrier‐protein‐free medium and conjugated with the indicated metal isotope using the x8 MaxPAR conjugation kit (Fluidigm) according to the manufacturer's protocol. Conjugations done by the Ramaciotti Facility for Human Systems Biology are indicated in Table 2. Cells were fixed in 4% paraformaldehyde (PFA) for 20 min prior to being incubated in Foxp3 permeabilisation buffer (eBioscience Inc., San Diego, CA, USA) for 15 min. Cells were finally resuspended in DNA intercalator mix [1/1000 iridium intercalator (Fluidigm) in 4% PFA] and washed with Milli‐Q water before being resuspended in 10% EQ four element beads (Fluidigm) in Milli‐Q water at a concentration of 0.8 × 106 cells mL−1, to be run on a CyTOF 2 Helios mass cytometer (Fluidigm).
Table 2.
Antibodies used for human mass cytometry staining
| Target | Isotope | Clone | Company | tSNE a | IgG3 gating b |
|---|---|---|---|---|---|
| CD3 | 155Gd | UCHT1 | BioLegend | ||
| CD19 | 142Nd c | HIB19 | BD Biosciences | ✓ | |
| CD20 | 147Sm c | 2H7 | BioLegend | ✓ | ✓ |
| CD21 | 152Sm c | BU32 | BioLegend | ✓ | ✓ |
| CD23 | 144Nd | M‐L23.4 | Miltenyi Biotec | ✓ | |
| CD24 | 151Eu | ML5 | BioLegend | ✓ | ✓ |
| CD25 | 149Sm c | 2A3 | BD Biosciences | ✓ | |
| CD27 | 167Er c | M‐T271 | BioLegend | ✓ | ✓ |
| CD38 | 154Sm | HIT2 | BioLegend | ✓ | ✓ |
| CD45 d | 104Pd c | HI30 | BD Biosciences | ||
| 115In c | |||||
| 209Bi c | |||||
| CD71 | 164Er | CY1G4 | BioLegend | ✓ | |
| CD79b | 153Eu | CB3‐2 | BioLegend | ✓ | |
| CD80 | 162Dy c | L307.4 | BD Biosciences | ✓ | |
| CD86 | 156Gd c | IT2.2 | BD Biosciences | ✓ | |
| CD120a | 158Gd | REA252 | Miltenyi Biotec | ✓ | |
| CD120b | 160Gd | 3G7A02 | BioLegend | ✓ | |
| CD138 | 150Nd c | DL‐101 | BioLegend | ✓ | |
| CD184 (CXCR4) | 175Lu c | 12G5 | BD Biosciences | ✓ | |
| CD185 (CXCR5) | 146Nd | J252D4 | BioLegend | ✓ | |
| CD210 (IL‐10 receptor) | 169Tm | REA239 | Miltenyi Biotec | ✓ | |
| CD267 (TACI) | 159Gd | 1A1 | BioLegend | ✓ | |
| CD274 (PD‐L1) | 161Dy | 29E.2A3 | BioLegend | ✓ | |
| CD360 (IL‐21 receptor) | 165Ho | REA233 | Miltenyi Biotec | ✓ | |
| HLA‐DR | 174Yb c | L243 | BD Biosciences | ✓ | |
| IgD | 89Y c | IA6‐2 | BD Biosciences | ||
| IgE | 148Nd | MHE‐18 | BioLegend | ||
| IgG3 | 145Nd | HP6047 | BioLegend | ||
| IgG4 | 168Er | HP6023 | BioLegend | ||
| 5‐HT2A receptor | 166Er | Polyclonal | Novus Biologicals | ✓ | |
| PAF receptor | 163Dy | AA14 (clone21) | Cayman Chemicals | ✓ | |
| S1P1 | 170Er | MM0045‐21L9 | Novus Biologicals | ✓ | |
| TNF (membrane) | 141Pr c | Mab11 | BioLegend | ✓ |
Markers used to run tSNE plots.
Markers used for gating IgG3 + B‐cell subsets.
Isotopes conjugated by the Ramaciotti Facility for Human Systems Biology, The University of Sydney, Australia.
CD45 was used for barcoding to allow up to three samples to be stained together.
Samples were initially gated as shown in Supplementary figure 1a using FlowJo v10.4 (Becton Dickinson, Ashland, OR, USA). All analyses were done on CD3−, HLA‐DR+, CD19dim/hi, IgG4− and IgG3+ B cells. tSNE plots were generated using the following markers: CD19, CD20, CD21, CD23, CD24, CD25, CD27, CD38, CD71, CD79b, CD80, CD86, CD120a, CD120b, CD138, CD184, CD185, CD210, CD267, CD274, CD360, HLA‐DR, 5 HT2A, PAF‐R, S1P1 and membrane TNF (Table 2). Markers not expressed on IgG3 + B cells (CD3, IgD, IgE and IgG4) and CD45 (used as a barcode) were not included in generating the tSNE plot. Although IgM was included in the panel design, it needed to be excluded from the final analysis because of quality control issues with staining in this channel. tSNE plot was generated using the script Cytometry Analysis Pipeline for large and complex data sets (CAPX) 45 in R, after downsampling for a total of 10 000 cells. tSNE parameters included 1000 iterations with a perplexity of 30 and theta of 0.5. Subsets of IgG3 + B cells were gated manually in FlowJo.
Statistical analysis: cohort 1
Statistical analysis was performed using GraphPad Prism software (GraphPad Software, San Diego, CA, USA) version 8.2.1. A P‐value < 0.05 was considered statistically significant.
IgG3 serum analysis
Patient samples were combined for analysis purposes regardless of phototherapy treatment. For IgG3 serum comparison, eight control patients (with 25 time points) and eight phototherapy patients (with 36 time points) were included in the analyses. If data passed a Shapiro–Wilk normality test, then a parametric Pearson correlation coefficient was used (and a R 2‐value is shown). If the data were not normally distributed, a non‐parametric Spearman correlation was done and a ρ (rho)‐value is shown.
Days to conversion analysis
IgG3 + B‐cell levels in CIS patients who converted to MS irrespective of phototherapy were compared. Three phototherapy patients did not go on to develop MS during the 12‐month study so were not included in the analysis, whilst one control patient (no phototherapy) dropped out of the study so disease status is not known. This left seven control patients (with 28 time points) and 5 phototherapy patients (with 27 time points). Once again, a Shapiro–Wilk normality test was done across all time points (either 34, 11 after without DMT or 10 after with DMT) to determine distribution of data. If data passed, a parametric Pearson correlation coefficient was used (and R 2‐value was shown). If the data were not normally distributed, a non‐parametric Spearman correlation was done (and a ρ (rho)‐value is shown).
Phototherapy analysis
CIS patients received phototherapy for the first 8 weeks (2 months) of the study with blood being analysed at baseline, 1 week (phototherapy only), 1 month (phototherapy only) and 2 months post‐treatment. Five no phototherapy (with 10 time points) and seven phototherapy (with 23 time points) patients were included for analysis. A linear regression was done to compare changes in proportion of IgG3 + B cells compared with baseline over time for each group. Phototherapy and no phototherapy groups were then compared with a comparable analysis of covariance (ANCOVA).
Statistical analysis: cohort 2 – iMS, aMS and non‐MS analysis
A Shapiro–Wilk normality test was first done across all groups to determine the distribution of data. Only if all three groups (non‐MS, iMS and aMS) passed was a parametric one‐way ANOVA (with Tukey's multiple comparisons test) used. If any one group failed the normality test, a non‐parametric Kruskal–Wallis test (with Dunn's multiple comparisons test) was performed.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Felix Marsh‐Wakefield: Data curation; Formal analysis; Investigation; Methodology; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Thomas Ashhurst: Data curation; Formal analysis; Investigation; Methodology; Resources; Software; Writing‐review & editing. Stephanie Trend: Data curation; Formal analysis; Investigation; Resources; Writing‐review & editing. Helen McGuire: Data curation; Formal analysis; Investigation; Methodology; Writing‐review & editing. Pierre Juillard: Data curation; Resources; Writing‐review & editing. Anna Zinger: Data curation; Resources; Writing‐review & editing. Anderson P Jones: Data curation; Resources; Writing‐review & editing. Allan G Kermode: Resources; Writing‐review & editing. Simon Hawke: Conceptualization; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Writing‐review & editing. Georges E Grau: Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Writing‐review & editing. Prue H Hart: Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing. Scott N Byrne: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Supporting information
Supplementary figure S1
{kind=link}
Supplementary figure S2
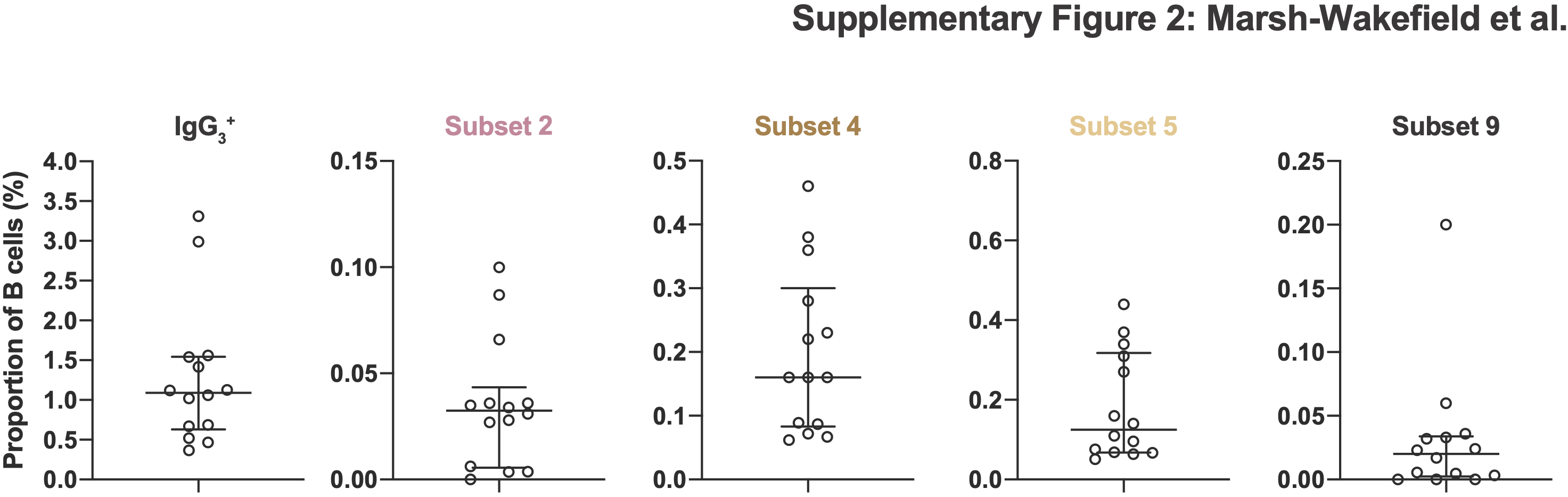{kind=link}
Supplementary Material
Acknowledgments
This work was supported by a Sydney Medical School Foundation Grant and a Multiple Sclerosis Research Australia Project Grant (#17‐014). The trial recruiting the CIS/MS/phototherapy patients of cohort 1 was supported by the National Health and Medical Research Council of Australia (ID 1067209) and MS Western Australia. We acknowledge and thank Professor Martyn French for his expert and critical review of this manuscript. We also acknowledge and thank the support staff at Sydney Cytometry and the Ramaciotti Facility for Human Systems Biology for their assistance with the mass cytometry studies. TA and HMM are International Society for Advancement of Cytometry Marylou Ingram Scholars.
References
- 1. Montalban X, Hauser SL, Kappos L et al Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017; 376: 209–220. [DOI] [PubMed] [Google Scholar]
- 2. Kappos L, Hartung H‐P, Freedman MS et al Atacicept in multiple sclerosis (atams): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol 2014; 13: 353–363. [DOI] [PubMed] [Google Scholar]
- 3. Rubin RL, Tang FL, Chan EK, Pollard KM, Tsay G, Tan EM. IgG subclasses of autoantibodies in systemic lupus erythematosus, Sjogren's syndrome, and drug‐induced autoimmunity. J Immunol 1986; 137: 2528–2534. [PubMed] [Google Scholar]
- 4. Gharavi AE, Harris EN, Lockshin MD, Hughes GR, Elkon KB. IgG subclass and light chain distribution of anticardiolipin and anti‐DNA antibodies in systemic lupus erythematosus. Ann Rheum Dis 1988; 47: 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Losy J, Mehta PD, Wisniewski HM. Identification of IgG subclasses' oligoclonal bands in multiple sclerosis CSF. Acta Neurol Scand 1990; 82: 4–8. [DOI] [PubMed] [Google Scholar]
- 6. Trend S, Jones AP, Cha L et al Higher serum immunoglobulin G3 levels may predict the development of multiple sclerosis in individuals with clinically isolated syndrome. Front Immunol. 2018; 9: 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hart PH, Jones AP, Trend S et al A randomised, controlled clinical trial of narrowband UVB phototherapy for clinically isolated syndrome. The phocis study. Mult Scler J Exp Transl Clin 2018; 4: 2055217318773112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lighaam LC, Vermeulen E, Bleker T et al Phenotypic differences between IgG4 + And IgG1 + B cells point to distinct regulation of the IgG4 response. J Allergy Clin Immunol 2014; 133: 267–270. [DOI] [PubMed] [Google Scholar]
- 9. Sanz I, Wei C, Jenks SA et al Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front Immunol 2019; 10: 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. International Multiple Sclerosis Genetics Consortium , Lill CM, Schjeide BM et al MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 are genetic risk loci for multiple sclerosis. Brain 2013; 136: 1778–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalinowska‐Łyszczarz A, Szczuciński A, Pawlak MA, Losy J. Clinical study on CXCL13, CCL17, CCL20 and IL‐17 as immune cell migration navigators in relapsing‐remitting multiple sclerosis patients. J Neurol Sci 2011; 300: 81–85. [DOI] [PubMed] [Google Scholar]
- 12. Fraussen J, Marquez S, Takata K et al Phenotypic and Ig repertoire analyses indicate a common origin of IgD–CD27– double negative B cells in healthy individuals and multiple sclerosis patients. J Immunol 2019; 203: 1650–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pene J, Gauchat JF, Lecart S, et al IL‐21 is a switch factor for the production of IgG1 and IgG3 by human B cells. J Immunol 2004; 172: 5154–5157. [DOI] [PubMed] [Google Scholar]
- 14. Nohra R, Beyeen Ad, Guo JP et al RGMA and IL21R show association with experimental inflammation and multiple sclerosis. Genes Immun 2010; 11: 279–293. [DOI] [PubMed] [Google Scholar]
- 15. Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL‐21. J Immunol 2006; 177: 5236–5247. [DOI] [PubMed] [Google Scholar]
- 16. Tzartos JS, Craner MJ, Friese Manuel A et al IL‐21 and IL‐21 receptor expression in lymphocytes and neurons in multiple sclerosis brain. Am J Pathol 2011; 178: 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jenks SA, Cashman KS, Zumaquero E et al Distinct effector B cells induced by unregulated toll‐like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 2018; 49: 725–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Quach TD, Rodriguez‐Zhurbenko N, Hopkins TJ et al Distinctions among circulating antibody‐secreting cell populations, including B‐1 cells, in human adult peripheral blood. J Immunol 2016; 196: 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Clavarino G, Delouche N, Vettier C et al Novel strategy for phenotypic characterization of human B lymphocytes from precursors to effector cells by flow cytometry. PLoS One 2016; 11: e0162209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K. Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. Ebiomedicine 2017; 16: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ellebedy AH, Jackson KJL, Kissick HT et al Defining antigen‐specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat Immunol 2016; 17: 1226–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Genc K, Dona DL, Reder AT. Increased CD80+ B cells in active multiple sclerosis and reversal by interferon β‐1B therapy. J Clin Invest 1997; 99: 2664–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valberg LS, Flanagan PR, Kertesz A, Ebers GC. Abnormalities in iron metabolism in multiple sclerosis. Can J Neurol Sci 1989; 16: 184–186. [DOI] [PubMed] [Google Scholar]
- 24. Traub J, Traffehn S, Ochs J et al Dimethyl fumarate impairs differentiated B cells and fosters central nervous system integrity in treatment of multiple sclerosis. Brain Pathol 2019; 29: 640–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Montes Diaz G, Fraussen J, Van Wijmeersch B, Hupperts R, Somers V. Dimethyl fumarate induces a persistent change in the composition of the innate and adaptive immune system in multiple sclerosis patients. Sci Rep 2018; 8: 8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Putzki N, Baranwal MK, Tettenborn B, Limmroth V, Kreuzfelder E. Effects of natalizumab on circulating B cells, T regulatory cells and natural killer cells. Eur Neurol 2010; 63: 311–317. [DOI] [PubMed] [Google Scholar]
- 27. Nakamura M, Matsuoka T, Chihara N et al Differential effects of fingolimod on B‐cell populations in multiple sclerosis. Mult Scler 2014; 20: 1371–1380. [DOI] [PubMed] [Google Scholar]
- 28. Zinger A, Latham SL, Combes V et al Plasma levels of endothelial and B‐cell‐derived microparticles are restored by fingolimod treatment in multiple sclerosis patients. Mult Scl J 2016; 22: 1883–1887. [DOI] [PubMed] [Google Scholar]
- 29. Trend S, Jones AP, Cha L et al Short‐term changes in frequencies of circulating leukocytes associated with narrowband UVB phototherapy in people with clinically isolated syndrome. Sci Rep 2019; 9: 7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu Y, Harding SE, Michaelsen TE et al Solution conformation of wild‐type and mutant IgG3 and IgG4 immunoglobulins using crystallohydrodynamics: possible implications for complement activation. Biophys J 2007; 93: 3733–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruggemann M, Williams GT, Bindon CI et al Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987; 166: 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype‐specific differences in complement activation. J Exp Med 1993; 178: 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prineas JW, Kwon EE, Cho E‐S et al Immunopathology of secondary‐progressive multiple sclerosis. Annals Neurol. 2001; 50: 646–657. [DOI] [PubMed] [Google Scholar]
- 34. Breij ECW, Brink BP, Veerhuis R et al Homogeneity of active demyelinating lesions in established multiple sclerosis. Annals Neurol 2008; 63: 16–25. [DOI] [PubMed] [Google Scholar]
- 35. Barnett MH, Parratt JD, Cho E‐S, Prineas JW. Immunoglobulins and complement in postmortem multiple sclerosis tissue. Annals Neurol 2009; 65: 32–46. [DOI] [PubMed] [Google Scholar]
- 36. Michailidou I, Willems JGP, Kooi E‐J et al Complement C1q‐C3‐associated synaptic changes in multiple sclerosis hippocampus. Annals Neurol 2015; 77: 1007–1026. [DOI] [PubMed] [Google Scholar]
- 37. Lm Watkins, Neal JW, Loveless S et al Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflam 2016; 13: 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Damian DL, Patterson CRS, Stapelberg M, Park J, Barnetson RSTC, Halliday GM. UV radiation‐induced immunosuppression is greater in men and prevented by topical nicotinamide. J Invest Dermatol 2008; 128: 447–454. [DOI] [PubMed] [Google Scholar]
- 39. Marsh‐Wakefield F, Byrne SN. Photoimmunology and multiple sclerosis In: La Flamme AC, Orian JM (Eds). Current Topics in Behavioral Neurosciences. Cham, Switzerland: Springer International Publishing; 2015;117–141. [DOI] [PubMed] [Google Scholar]
- 40. Kok LF, Marsh‐Wakefield F, Marshall JE, Gillis C, Halliday GM, Byrne SN. B cells are required for sunlight protection of mice from a CNS‐targeted autoimmune attack. J Autoimmunity 2016; 73: 10–23. [DOI] [PubMed] [Google Scholar]
- 41. Carrasco B, Garcia De La Torre J, Davis KG et al Crystallohydrodynamics for solving the hydration problem for multi‐domain proteins: open physiological conformations for human IgG. Biophys Chem 2001; 93: 181–196. [DOI] [PubMed] [Google Scholar]
- 42. Bruhns P, Iannascoli B, England P et al Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113: 3716–3725. [DOI] [PubMed] [Google Scholar]
- 43. Damelang T, Rogerson SJ, Kent SJ, Chung AW. Role of IgG3 in infectious diseases. Trends Immunol 2019; 40: 197–211. [DOI] [PubMed] [Google Scholar]
- 44. Wagar LE. Live cell barcoding for efficient analysis of small samples by mass cytometry. Methods Mol Biol 2019; 1989: 125–135. [DOI] [PubMed] [Google Scholar]
- 45. Ashhurst TM, Cox DA, Smith AL, King NJC. Analysis of the murine bone marrow hematopoietic system using mass and flow cytometry. Methods Mol Biol 2019; 1989: 159–192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure S1
Supplementary figure S2
Supplementary Material