

Abstract
CRISPR-Cas systems are adaptive immune systems that protect bacteria from bacteriophage (phage) infection1. To provide immunity, RNA-guided protein surveillance complexes recognize foreign nucleic acids, triggering their destruction by Cas nucleases2. While the essential requirements for immune activity are well understood, the physiological cues that regulate CRISPR-Cas expression are not. Here, a forward genetic screen identifies a two-component system (KinB/AlgB), previously characterized in regulating Pseudomonas aeruginosa alginate biosynthesis3,4, as a regulator of the expression and activity of the P. aeruginosa Type I-F CRISPR-Cas system. Downstream of KinB/AlgB, activators of alginate production AlgU (a σE orthologue) and AlgR, repress CRISPR-Cas activity during planktonic and surface-associated growth5. AmrZ, another alginate regulator6, is triggered to repress CRISPR-Cas immunity during surface-association. Pseudomonas phages and plasmids have taken advantage of this regulatory scheme, and carry hijacked homologs of AmrZ that repress CRISPR-Cas expression and activity. This suggests that while CRISPR-Cas regulation may be important to limit self-toxicity, endogenous repressive pathways represent a vulnerability for parasite manipulation.
Type I CRISPR-Cas systems are comprised of a multi-subunit RNA-guided surveillance complex, a trans-acting nuclease (Cas3)2,7,8, and proteins dedicated to spacer acquisition, Cas1 and Cas29. Pseudomonas aeruginosa has become a powerful model organism for studying Type I CRISPR-Cas mechanisms10–15, functions16–19, evolution20–22, and interactions with phages utilizing anti-CRISPR proteins23–26. The P. aeruginosa strain PA14 possesses a naturally active Type I-F CRISPR-Cas immune system, comprised of two CRISPR arrays, an operon encoding surveillance complex subunits Csy1–412, and a separate operon encoding Cas1 and a Cas2–3 fusion protein. Quorum sensing has been shown to activate CRISPR-Cas expression in Pseudomonas aeruginosa27, as well as other species of bacteria28. However, little is known regarding the factors that temper CRISPR-Cas activity and mitigate the risk of acquiring and expressing a nucleolytic immune system.
To discover new CRISPR-Cas regulators in P. aeruginosa, we utilized a P. aeruginosa strain PA14 engineered to express lacZ in place of the csy3 gene (csy3::lacZ) 17. This strain was subjected to C9 mariner transposon mutagenesis and ~40,000 colonies screened on X-gal plates. Multiple independent insertions were identified within lacZ and upstream genes (csy1 and csy2), and thirty mutants with transposon insertions outside of this region were isolated and mapped (Extended Data Fig. 1). Four independent insertions were identified in a single gene, kinB, which resulted in decreased β-galactosidase production on solid plates (Extended Data Fig. 2a) and ~30% less csy3::lacZ activity in liquid culture compared to the unmutagenized parent (Extended Data Fig. 2b). We selected kinB (a sensor kinase/phosphatase) for follow-up study as it was the only gene with >1 independent transposon insertion and displayed the largest β-galactosidase activity change.
We measured the ability of kinB::Tn insertions to limit the survival and replication of phage when introduced into the wild-type (CRISPR active) strain. Phages used to assay activity are: DMS3acrIE3 which is an untargeted control phage, DMS3macrIE318, which is fully targeted by the PA14 Type I-F CRISPR-Cas system, and phage DMS3macrIF4, which is partially targeted, by virtue of encoding a “weak” anti-CRISPR, acrIF4, that binds to the surveillance complex to inhibit CRISPR-Cas function23,25,29. The kinB::Tn strains remained resistant to DMS3macrIE3 infection, but DMS3macrIF4 formed 10-fold more plaques relative to WT, demonstrating attenuated CRISPR-Cas activity (Figure 1a, Extended Data Fig. 2c). This defect was complemented by expression of kinB in trans (Extended Data Fig. 2c). Growth of control phage DMS3acrIE3 was not impacted in the absence of kinB (Figure 1a, Extended Data Fig. 2c). Furthermore, two other phages that are partially targeted, JBD26 (naturally possessing acrIF4) and JBD25 (a phage with no Acr that is targeted by a weak spacer that provides incomplete immunity) also showed increased survival in the kinB:Tn strain (Extended Data Fig. 2d) relative to WT PA14. Survival of a phage with a weak anti-CRISPR or one that is targeted by a less active spacer is therefore a sensitive barometer for perturbations in CRISPR-Cas levels. Together, these data confirm that in the absence of kinB, csy gene expression and phage targeting are decreased.
Figure 1: A forward genetic screen identifies a role for an alginate-activating pathway in repressing CRISPR-Cas immunity.
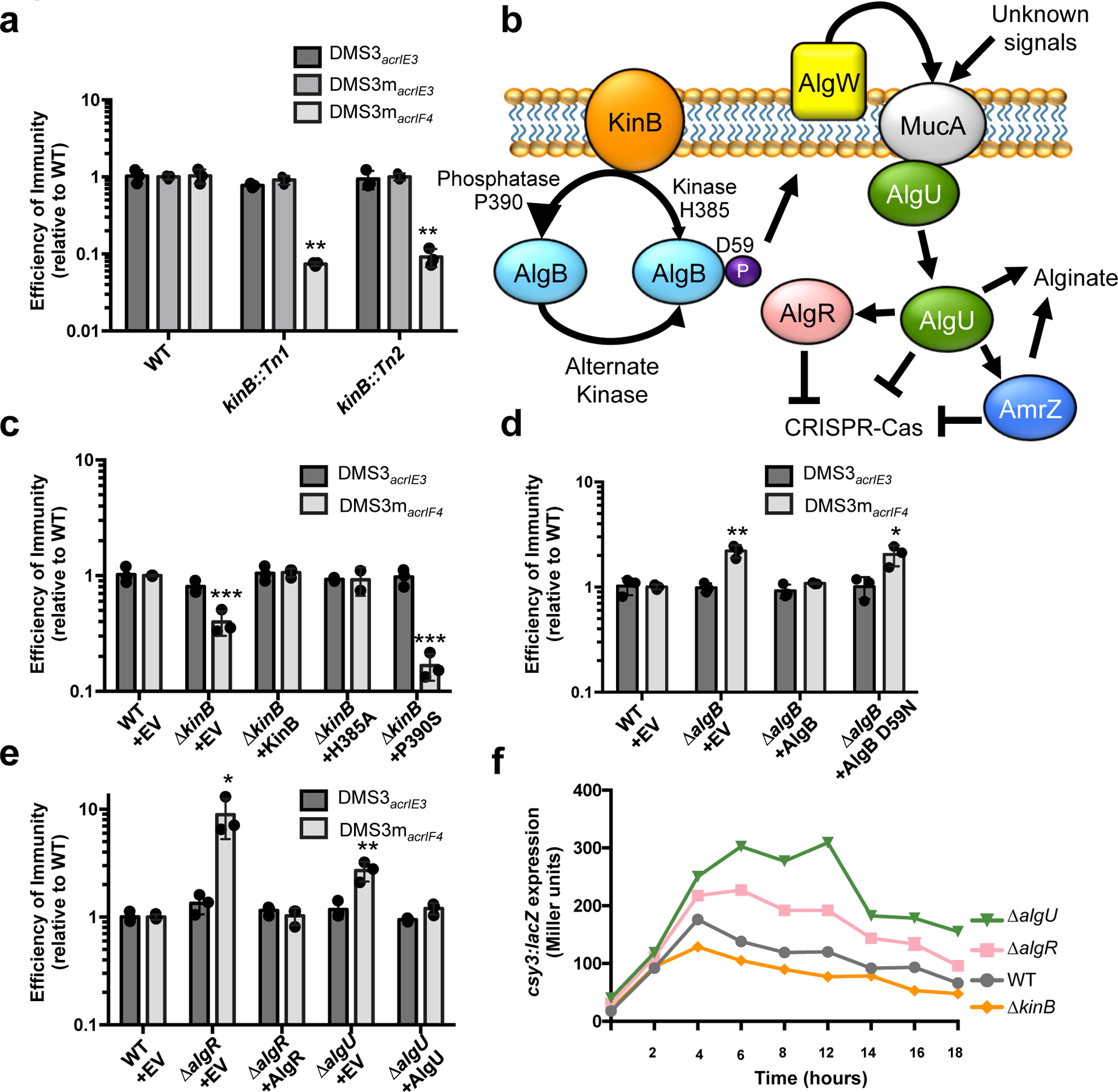
a. Efficiency of immunity (EOI) against isogenic phages DMS3acrIE3 (non-targeted), DMS3macrIE3 (no I-F anti-CRISPR, CRISPR- targeted), and DMS3macrIF4 (weak I-F anti-CRISPR, CRISPR-targeted). Plaque forming units (PFUs) are presented as a ratio relative to the number of PFUs measured on WT PA14, quantified on two independent kinB transposon mutants (kinB::Tn1 and kinB::Tn2). Tn mutants show altered EOI against DMS3macrIF4 relative to WT (Tn1, P = 2.9 × 10−3, Tn2, P = 3.2 × 10−3) b. A cartoon summarizing the KinB/AlgB two component system and downstream effects, based on prior work46 (see text) with CRISPR-Cas regulation added. c,d,e. EOI measurements for indicated ΔalgB, ΔkinB, ΔalgR, and ΔalgU strains with complementation. Mutants show altered EOI against DMS3macrIF4 relative to WT (ΔkinB + EV, P = 4.30 × 10−4, ΔkinB + P390S, P = 5.6 × 10−6 ΔalgB + EV, P = 2.8 × 10−3, ΔalgB + D59N, P = 1.8 × 10−2 ΔalgR + EV, P = 1.9 × 10−2, ΔalgU + EV, P = 6.6 × 10−3). f. csy3::lacZ β-galactosidase activity over time in the indicated strain backgrounds. Experiment was replicated twice with fewer timepoints and consistent results seen. All EOI data are represented as the mean of 3 biological replicates +/− SD and β-galactosidase reporter activity is represented as the mean of 3 technical replicates. Two-tailed unpaired Student’s T-test was used to calculate P values, *p < 0.05, **p < 0.01, ***p < 0.001.
KinB is a sensor kinase/phosphatase in a two-component system with response regulator AlgB. The KinB/AlgB system has a large regulon within P. aeruginosa, and controls the biosynthesis of the extracellular polysaccharide alginate4. This pathway is well-studied due to the recurrent isolation of alginate-overproducing (mucoid) P. aeruginosa from the lungs of cystic fibrosis patients, where alginate plays an important role in the formation of antibiotic resistant biofilms during chronic infection. The absence of KinB function results in the accumulation of the phosphorylated form of the response regulator AlgB (P-AlgB), while the phosphorylation of AlgB has been attributed to unknown kinases30,31 (Fig. 1b). P-AlgB activates the periplasmic protease AlgW (a DegS homolog), which degrades MucA, liberating sigma factor AlgU3,32,33 (Fig. 1b). AlgU positively regulates many factors involved in alginate production, including AlgR, AlgD, AlgB, and AmrZ5,6,34.
WT kinB or kinase inactive H385A kinB complemented an in-frame ΔkinB deletion, restoring CRISPR targeting of DMS3macrIF4 (Fig. 1c). However, a P390S kinB mutant incapable of dephosphorylating the response regulator AlgB did not complement, and in fact decreased CRISPR-Cas activity further (Fig. 1c). A ΔkinBΔalgB double mutant restored CRISPR-Cas targeting to levels two-fold above WT (Extended Data Fig. 3b), confirming the role of this signaling pathway. A strain lacking algB (ΔalgB) or possessing a D59N mutant that cannot be phosphorylated also elevated CRISPR-Cas activity two-fold, supporting the repressive role of P-AlgB (Fig. 1d). These data show that accumulation of high levels of P-AlgB (achieved in kinB::Tn, ΔkinB, or kinB P390S) leads to CRISPR-Cas repression.
We next assayed anti-phage immunity in ΔalgU and ΔalgR backgrounds, revealing increased targeting of DMS3macrIF4 but not control phage DMS3acrIE3 in both knockouts (Fig. 1e). Complementation restored CRISPR-Cas levels (Fig. 1e), demonstrating that AlgU and AlgR repress CRISPR-Cas immunity. Double knockouts of each gene combined with ΔkinB also demonstrated increased CRISPR-Cas immunity, consistent with these factors acting as repressors downstream of KinB (Extended Data Fig. 3a). All changes in DMS3macrIF4 phage replication and survival are CRISPR-dependent, as double knockouts (kinB, algB, algU, algR mutants combined with csy3::lacZ, a loss of function mutation) revealed plaquing equivalent to csy3::lacZ alone (Extended Data Fig. 3b). β-galactosidase activity was measured in these strains during growth in liquid culture, revealing a peak in csy expression around 8 h, with repression of this operon during entry into stationary phase (Fig. 1f). As suggested by the phage targeting experiments, a marked increase in expression of the csy operon was noted for both ΔalgR and ΔalgU strains, with a decrease in csy expression for ΔkinB.
Next, we performed RT-qPCR of the cas3 and the csy3 transcripts in the mutant strains. We measured the relative abundance of Cas3 and Csy complex protein by fusing a sfCherry tag to the endogenous cas3 or csy1 gene in the mutant backgrounds, and used fluorescence as a proxy for protein abundance. We found that kinB loss decreased expression of both the cas3 and csy operon, resulting in lower cas3 and csy3 transcripts and Cas3-sfCherry and Csy1-sfCherry levels relative to WT (Fig. 2a,b). Conversely, we observed increased levels of cas3 and csy3 transcripts and Cas3-sfCherry and Csy1-sfcherry in the ΔalgR and ΔalgU mutants relative to WT (Fig. 2a,b). These data demonstrate that this pathway controls the levels of both Cas3 and the Csy complex in the bacterial cell by transcriptionally controlling the cas3 and csy operons.
Figure 2. The KinB/AlgB pathway modulates Cas3 and Csy protein and RNA levels.
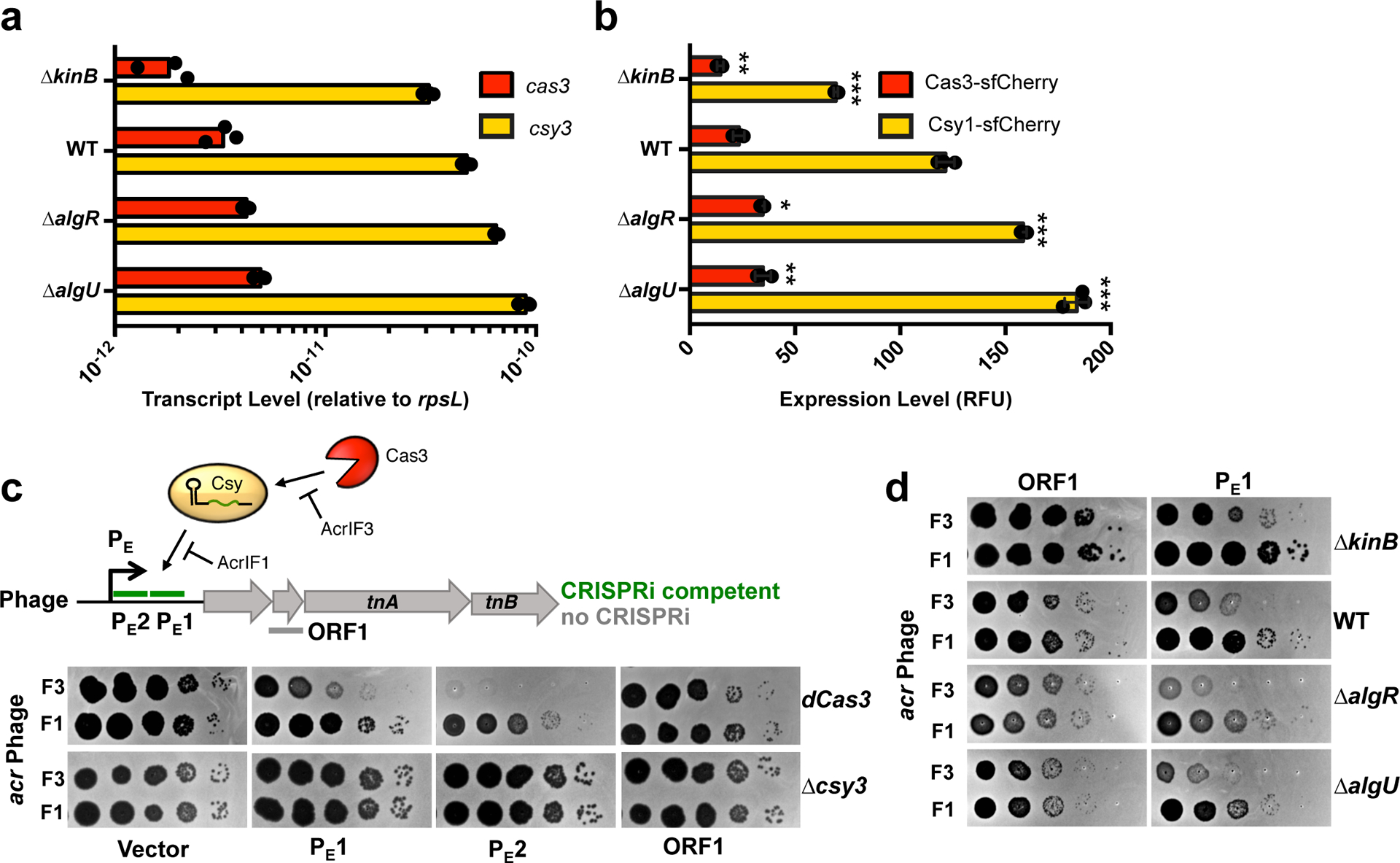
a. qRT-PCR measurements of transcript levels of cas3 (red) and csy3 (yellow) normalized to the housekeeping gene rpsL after 8 h of growth in liquid culture. Measurements are represented as the mean of 3 technical replicates. b. Measurement of the fluorescence levels of Cas3-sfCherry (red) or Csy1-sfCherry (yellow) reporter strains after 10 h of growth in liquid culture. Fluorescence measurements are represented as the mean of 3 biological replicates +/− SD. Mutants show altered Cas3-sfCherry levels (ΔkinB, P = 7.8 × 10−3 ΔalgR, P = 1.5 × 10−4 ΔalgU, P = 1.1 × 10−4) and Csy1-sfCherry levels relative to WT (ΔkinB, P = 3.3 × 10−5, ΔalgR, P = 1.5 × 10−4, ΔalgU, P = 1.1 × 10−4). Two-tailed unpaired Student’s T-test was used to calculate P values,*p < 0.05, **p < 0.01, ***p < 0.001. c. Spot titration of F3 (DMS3macrIF3) or F1 (DMS3macrIF1) on dCas3 (dead Cas3) or Δcsy3 (active Cas3, no Csy complex) strains. Phages are targeted by natural spacer CR2_sp1, as well as crRNAs designed to target DMS3m genome in positions designated on ORF map. d. Spot titration of DMS3macrIF3 and DMS3macrIF1 phages on WT PA14 or deletion mutants expressing the indicated crRNA. Plaquing experiments were replicated 3 times and consistent results seen.
As Cas3-sfCherry was expressed at low levels relative to Csy1-sfCherry, and is also known to be subject to post-translational control by Cas114, we sought to dissect the relative contribution of nuclease versus surveillance complex disregulation in driving the immune phenotypes of the KinB/AlgB pathway mutants. To specifically measure the anti-phage activity of the Csy complex, we developed a Cas3-indepedent bioassay to read out the activity of the surveillance complex in the cell. Through the rational design of crRNAs to target an early phage promoter (PE1, PE2), we observed inhibition of phage survival in a P. aeruginosa strain with a nuclease dead Cas3 (dCas3), while an ORF-targeting crRNA (ORF1) was ineffective (Fig. 2c). This CRISPR-based transcriptional interference (CRISPRi) effect was remarkably strong enough to completely limit phage replication in the absence of Cas3 nuclease activity for crRNA PE2. Phage inhibition via CRISPRi occurred when infecting with a phage that expressed the inhibitor of Cas3 recruitment, AcrIF3, but not an inhibitor that blocks Csy complex-phage DNA binding25, AcrIF1 (Fig. 2c). We selected PE1 as a moderately-functional CRISPRi spacer and expressed it in KinB/AlgB pathway mutants. We observed decreased CRISPRi activity against phage DMS3macrIF3 in the ΔkinB background, but increased CRISPRi in ΔalgR and ΔalgU, (Fig. 2d, compare F3 and F1 phage). This demonstrates modulation of csy gene expression is sufficient to impact phage targeting in a Cas3-independent manner. We conclude that the KinB/AlgB pathway regulates Cas3 and Csy complex levels, and repression of Csy complex levels has a large impact on anti-phage immunity.
To identify downstream CRISPR-Cas regulators in the AlgU regulon35, we focused on another factor involved in alginate production, the alginate and motility and regulator Z, amrZ6,36. We generated a knockout of amrZ and observed a CRISPR-dependent increase in efficiency of immunity (EOI) against phage DMS3macrIF4 (Fig. 3a, Extended Data Fig. 3b). This was complemented when amrZ was expressed in trans (Fig. 3a). A ΔkinBΔamrZ double knockout also showed increased CRISPR-Cas activity, consistent with its role as a repressor downstream of KinB (Extended Data Fig. 3a). However, when we measured cas3 and csy3 transcript levels and Cas3-sfCherry and Csy1-sfCherry levels in ΔamrZ, neither transcript or protein levels differed from WT (Extended Data Fig. 4a,b). In considering these discrepant results, we realized that the anti-phage plaque assay is performed on solid plates whereas RNA quantification and sfCherry fluorescence measurements were conducted on liquid culture samples. To measure anti-phage activity of ΔamrZ in planktonic growth, we challenged WT and ΔamrZ with 106 PFU (MOI = 0.2) of virulent DMS3macrIF4 in liquid culture. Both strains succumbed to phage infection with similar kinetics (Fig. 3b), and phage replication did not differ significantly between the two strains (Fig. 3c). Phage replication in the absence of CRISPR-Cas immunity also did not differ between the two strains (Fig. 3c). This demonstrates that under our conditions, AmrZ does not control CRISPR-Cas during planktonic growth.
Figure 3: AmrZ is a surface-activated repressor of CRISPR-Cas immunity.
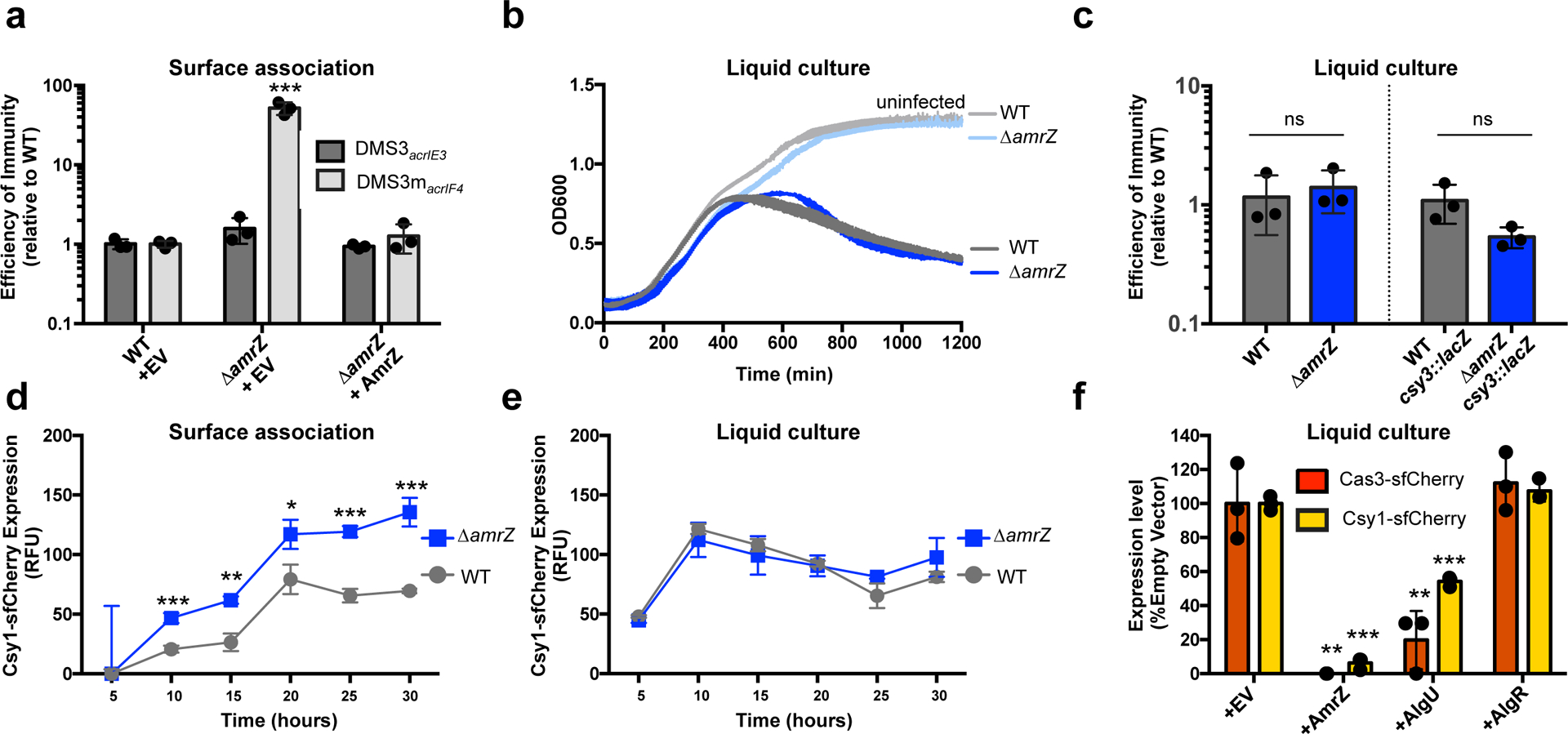
a. Efficiency of immunity (EOI) against phages DMS3acrIE3 (non-targeted) and DMS3macrIF4 (CRISPR-targeted). Plaque forming units (PFUs) were quantified on ΔamrZ or the complemented strain, then represented as a ratio of the number of PFUs measured on WT PA14. ΔamrZ + EV shows increased EOI against DMS3macrIF4 relative to WT (P = 7.3 × 10−4). EOI measurements are represented as the mean of 3 biological replicates +/− SD. b. Growth curves of PA14 WT and ΔamrZ infected with 106 PFU of virulent DMS3macrIF4 alongside uninfected controls. c. EOI against virulent DMS3macrIF4 in liquid culture of WT and ΔamrZ strains (CRISPR active) or WT csy3::lacZ and ΔamrZ csy3::lacZ (CRISPR inactive). PFUs were quantified after 24 h from ΔamrZ or amrZ csy3::lacZ, then represented as a ratio of PFUs from WT or WT csy3::lacZ, respectively. OD600 and EOI measurements are represented as the mean of 3 biological replicates +/− SD. ΔamrZ and ΔamrZ csy3::lacZ show no significant difference of EOI relative to WT and WT csy3::lacZ, respectively (ΔamrZ, P = 0.6 ΔamrZ csy3::lacZ, P = 0.08). d, e. Timecourse of the fluorescence levels of Csy1-sfCherry reporter strains during surface-association (d) or planktonic growth (e). ΔamrZ has increased Csy1-sfCherry levels during surface association relative to WT (10 h, P = , 8.9 × 10−4, 15 h, P = 1.5 × 10−3, 20 h, P = 2.0 × 10−2, 25 h, P = 2.2 × 10−4 , 30 h , P = 7.0 × 10−4) f. Normalized fluorescence measurements of WT Cas3-sfCherry (red) or Csy1-sfCherry (yellow) overexpressing the indicated transcription factor after 10 h growth in liquid culture. AmrZ and AlgU overexpression reduced Cas3-sfCherry (AmrZ, P = 1.5 × 10−3, AlgU, P = 7.8 × 10−3) and Csy1-sfCherry (AmrZ, P = 7.5 × 10−6, AlgU, P = 9.9 × 10−5) levels relative to WT. Fluorescence measurements are represented as the mean of 3 biological replicates +/− SD. Two-tailed unpaired Student’s T-test was used to calculate P values, ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
To test the hypothesis that AmrZ is a surface-activated repressor of CRISPR-Cas, we measured the levels of Csy complex during surface association and planktonic growth in WT and ΔamrZ cells using an endogenous Csy1-sfCherry reporter over a period of 30 h. In WT cells, the levels of Csy complex were attenuated during surface-association relative to planktonic growth (~50% reduction of peak Csy1-cherry levels, Fig. 3d), but in the absence of AmrZ, Csy complex levels during surface association increased to levels comparable to those in planktonic growth (Fig. 3d). Deletion of amrZ did not impact Csy complex levels in liquid culture at any timepoint (Fig. 3e). To increase the levels of AmrZ during planktonic growth, we ectopically expressed AmrZ from a high copy plasmid, and measured the impact on the our transcriptional reporter csy3::lacZ and our translational reporters Csy1-sfCherry and Cas3-sfCherry. Here, high levels of AmrZ in liquid growth reduced β-galactosidase activity of the csy3::lacZ reporter (Extended Data Fig. 4c) and strongly limited expression of Csy1-sfCherry and Cas3-sfCherry (Fig. 3f). These results suggest that low AmrZ activity in planktonic growth underlies its surface-activated control of CRISPR-Cas. In contrast to AmrZ, overexpression of AlgU only moderately impacted Csy complex and Cas3 levels and AlgR did not impact the levels of either reporter when overexpressed (Fig. 3f).
We next considered if phages and other mobile genetic elements had evolved mechanisms to manipulate this CRISPR-Cas repressive pathway. Inspired by the discovery of a Paraburkholderia phage that carried a distant homolog of AmrZ37, we searched the NCBI database for AmrZ homologs on Pseudomonas mobile genetic elements (MGE). Excitingly, we identified 14 diverse Pseudomonas mobile elements carrying AmrZ homologs (Extended Data Fig. 5). These MGEs included obligately lytic and temperate Myophages, temperate Siphophages, and plasmids. AmrZ has been structurally characterized in complex with operator DNA38, and these mobile AmrZ homologs showed perfect conservation of critical DNA-interacting residues in the ribbon-helix-helix domain, suggesting conserved binding specificity (Fig. 4a, b, red residues/arrowheads). To test if these mobilized AmrZ variants were capable of regulating CRISPR-Cas activity in Pseudomonas aeruginosa, we assayed the ability of 6 MGE-encoded AmrZ homologs to complement the ΔamrZ strain. Five out of six homologs complemented the ΔamrZ mutant to various degrees, indicating they were active in the PA14 transcriptional network and were bona fide CRISPR-Cas regulators (Fig. 4c). Next, each gene was expressed in WT cells, revealing 3 P. aeruginosa phage AmrZ homologs (AmrZPaBG, AmrZphi3, AmrZJBD68) inhibited Csy complex biogenesis (Fig. 4d).
Figure 4. Phage-derived AmrZ homologs control CRISPR-Cas immunity.
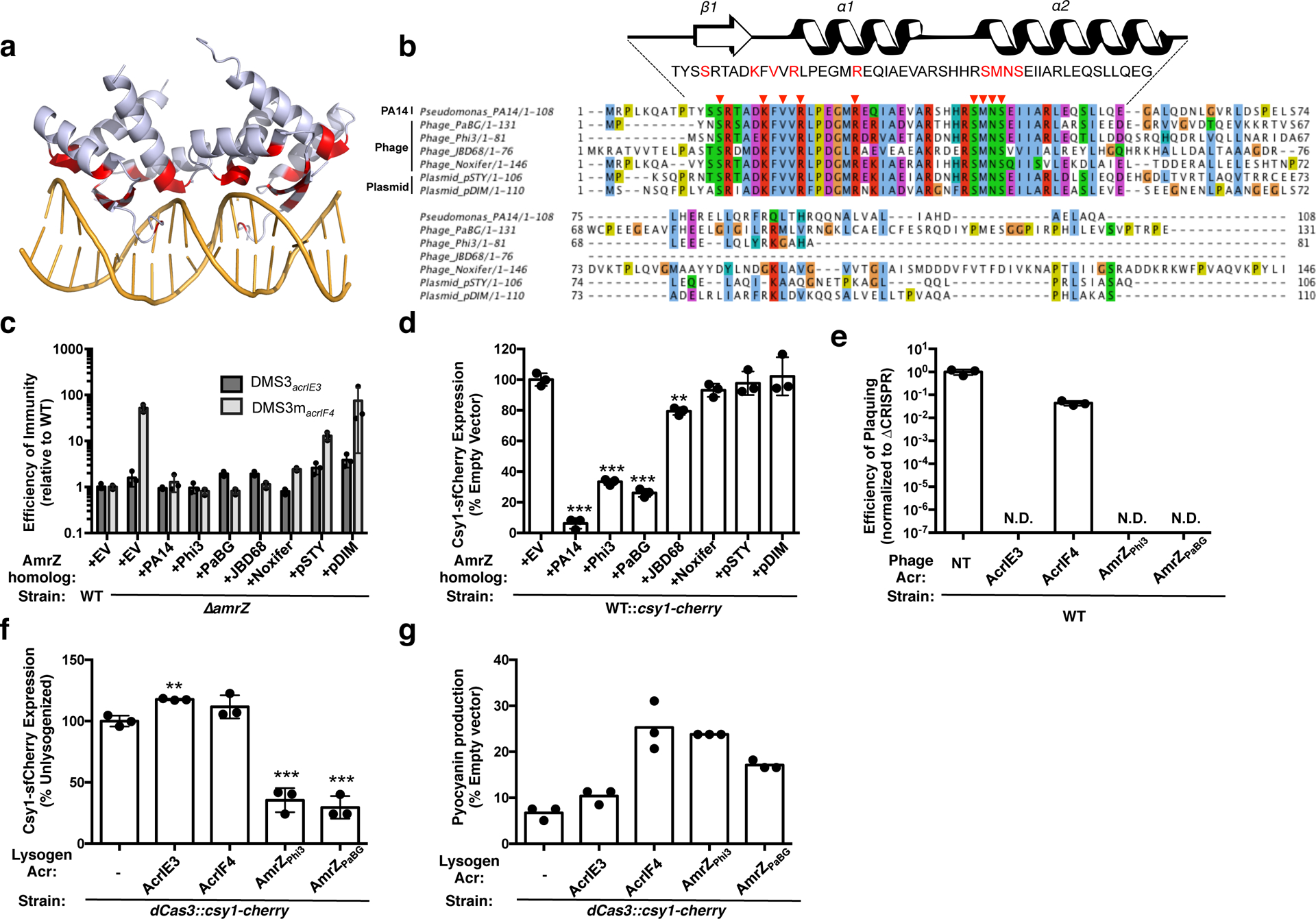
a. Structure of an AmrZ tetramer bound to 18bp of operator DNA38 with DNA-contacting residues highlighted in red. b. Alignment of six mobile AmrZ homologs and the native PA14 AmrZ homolog, with the ribbon-helix-helix DNA binding domain schematized and DNA-contacting residues indicated with red arrows and text. c. Efficiency of immunity (EOI) against DMS3acrIE3 (non-targeted) and DMS3macrIF4 (CRISPR-targeted). Plaque forming units (PFUs) were quantified on ΔamrZ or the strains complemented with AmrZ homologs, and represented as a ratio to the number of PFUs measured on WT PA14. Measurements are represented as the mean of 3 biological replicates +/− SD. d. Normalized fluorescence levels of Csy1-sfCherry reporter strains expressing AmrZ homologs after 10 h of growth in liquid culture, shown as mean of 3 biological replicates, +/− SD. AmrZ homologs from PA14, Phi3, PaBG, and JBD68 repressed Csy1-sfCherry relative to WT (PA14, P = 7.5 × 10−6, Phi3, P = 1.5 × 10−5, PaBG, P = 1.3 × 10−5, JBD68, P = 1.9 × 10−3). e. Efficiency of plaquing (EOP) of non-targeted DMS3acrIE3 phage (NT) or targeted DMS3macr phages. EOP is the ratio of PFUs on PA14 WT over PFUs formed on PA14 ΔCRISPR, represented as the mean of 3 biological replicates, +/− S.D. N.D. = not detectable. f. Fluorescence levels of dCas3::csy1-sfCherry after 16 h liquid growth lysogenized with the indicated DMS3macr phage, normalized to the unlysogenized control (−), and represented as the mean of 3 biological replicates +/− SD. Expression of AmrZPhi3 (P = 4.9 × 10−4) and AmrZPaBG (P = 2.8 × 10−4) from a prophage repressed Csy1-sfCherry expression relative to an unlysogenized control. g. Pyocyanin production from dCas3::csy1-sfCherry reporter strains lysogenized with the indicated DMS3macr phage or the unlysogenized control (−) after 16 h of growth in liquid culture. Pyocyanin levels during phzM-targeting are shown as a percentage of pyocyanin levels in an empty vector control, and represented as the mean of 3 technical replicates. Experiment was replicated three times and consistent results seen. Two-tailed unpaired Student’s T-test was used to calculate P values, ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
We next studied the anti-CRISPR function of these mobilized AmrZ homologs in the context of the phage life cycle. By inserting the two most potent phage AmrZ homologs, amrZphi3 and amrZPaBG into the anti-CRISPR locus of phage DMS3m, we compared the anti-CRISPR capacity of these repressors relative to bona-fide Type I-F inhibitor AcrIF4 and the negative control inhibitor AcrIE3. While the AmrZ homologs provided no protection during lytic growth (likely because they cannot act on previously synthesized CRISPR-Cas complexes) (Fig. 4e), they were able to significantly reduce the expression and activity of the CRISPR-Cas complex during lysogeny (Fig. 4f,g). By lysogenizing a strain of PA14 with a catalytically dead Cas3 and an endogenously tagged copy of Csy1-sfCherry, we demonstrated that the presence of AmrZPhi3 or AmrZPaBG reduced Csy complex levels more than 50% of an unlysogenized control, while AcrIF3 and AcrIF4 did not reduce Csy complex levels (Fig. 4f). To measure the activity of the Csy complex in these lysogens, we programmed the Csy complex to transcriptionally repress the phzM gene, which is responsible for the generation of the green pigment pyocyanin. De-repression of phzM expression can be quantified by measuring accumulation of the pyocyanin pigment in an overnight culture. We found that AmrZPhi3 or AmrZPaBG de-repressed phzM to a similar extent as AcrIF4 (Fig. 4g), demonstrating anti-CRISPR activity for these hijacked CRISPR-Cas repressors.
Regulation of bacterial processes is highly variable across species, reflecting niche-specific adaptations. Here, a genetic screen reveals that the KinB/AlgB two-component system regulates CRISPR-Cas in P. aeruginosa. Removal of KinB or inactivation of its phosphatase activity leads to the accumulation of P-AlgB, activating CRISPR-Cas repressors AlgU, AlgR, and AmrZ. This pathway also drives alginate production, which is responsible for the formation of the characteristic mucoid biofilms of cystic fibrosis P. aeruginosa isolates3,39,40. We show that P-AlgB (via kinB deletion), AlgU, and AlgR repress CRISPR-Cas activity during surface-association and planktonic growth, and AmrZ is triggered to further repress CRISPR-Cas during surface-association. Some Pseudomonas genetic parasites encode hijacked AmrZ homologs, which retain their ability to repress CRISPR-Cas expression and inhibit CRISPR-Cas biogenesis during lysogeny. Strikingly, we have identified multiply-lysogenized strains of Pseudomonas aeruginosa with as many as 4 independent copies of AmrZ on mobile elements in addition to host AmrZ (Extended Data Fig. 6). The evolutionary success of AmrZ in the Pseudomonas mobilome and core genome suggests a “guns for hire41” role for this gene in the arms race between bacteria and their parasites.
We and others observe CRISPR-Cas activation27,28 during exponential growth, where phage infection risk is high (i.e. metabolically active, well-mixed planktonic culture21). Surface-association lessens infection risk, as spatial structure limits phage dispersal and prevents a phage bloom from overtaking the entire bacterial population42. Though not measured here, spatial stratification and polysaccharide secretion in a mucoid biofilm likely also provide high levels of intrinsic phage resistance.
The observation that CRISPR-Cas expression and surface-association/biofilm formation are inversely regulated is supported by our analysis of a previously published PA14 RNAseq data set43 and proteomic data set44, which show activation of CRISPR-Cas expression in exponential phase, and repression during stationary phase and biofilm growth at 24 and 48 h (Extended Data Fig. 7a). Cas proteins are still detected in stationary phase and biofilm growth, suggesting the cells retain some immunity after transcriptional shutdown (Extended Data Fig. 7b). Furthermore, previous studies show that the P. aeruginosa genome is hyper-sensitive to CRISPR-induced DNA damage during surface-association and biofilm formation, leading to cell death when a mismatched prophage sequence target is present in the chromosome16,17. This suggests that CRISPR auto-immunity costs are also dependent on the growth state and physical environment of the cell.
Here, we identify a CRISPR-Cas repressive pathway in P. aeruginosa. We speculate that the ability to control CRISPR-Cas activity during lifestyle transitions may be essential for P. aeruginosa to safely maintain a CRISPR-Cas system by limiting self-toxicity. In our discovery of MGE-encoded CRISPR-Cas repressors we reveal an unexpected cost to CRISPR-Cas regulation: the evolution of CRISPR-Cas repression has created an Achilles Heel that is exploited by genetic parasites.
Methods
Bacterial strains and bacteriophages.
P. aeruginosa UCBPP-PA14 (PA14) strains and E. Coli strains (Supplementary Table 1) were grown on lysogeny broth (LB) agar or liquid at 37 °C. Media was supplemented with gentamicin (50 μg ml−1 for P. aeruginosa and 30 μg ml−1 for E. Coli) to maintain the pHERD30T plasmid or carbenicillin (250 μg ml−1) for P. aeruginosa or ampicillin (100 μg ml−1) for E. coli containing the pHERD20T plasmids. pHERD plasmids were induced with 0.1% arabinose. Bacteriophage stocks (Supplementary Table 1) were prepared as described previously18. In brief, 3 ml of SM buffer was added to plate lysates of the desired purified bacteriophage and incubated at room temperature for 15 minutes. SM buffer containing phages was collected and 100 μl of chloroform was added. This was centrifuged at 10,000 × g for 5 minutes and supernatant containing phages was transferred to a screw cap storage tube and incubated at 4 °C.
Transposon mutagenesis screen.
The csy::lacZ reporter strain was subjected to transposon mutagenesis and colonies were isolated on plates containing X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside). ~50,000 colonies were visually examined for increased or decreased levels of β-galactosidase and insertions mapped by semi-random PCR. To conduct transposon mutagenesis, overnights of PA14 csy3::lacZ and E. coli containing the pBTK30 Tn suicide vector were mixed in a 1:2 ratio (donor : recipient) for conjugation. Mixed cells were centrifuged at 4,000 × g for 10 minutes to pellet cells. 100 μl of resuspended conjugation pellet was then spotted on LB agar plates and incubated at 37 °C for 6h. Conjugation spots were collected and resuspended in LB liquid media. Conjugation was then plated on an LB agar plates supplemented with nalidixic acid (30 μg ml−1) and gentamicin (50 μg ml−1). Surviving colonies containing Tn insertions were collected into 1ml of LB liquid media. Serial dilution of were prepared and plated on LB agar plates supplemented with x-gal (200 μg ml−1) and gentamicin (50 μg ml−1) and nalidixic acid (30 μg ml−1). Plates were incubated at 37 °C for 24 h to allow for colonies to change color. Colonies displaying changed expression levels as compared to the unmutagenized parental strain (PA14 csy3::lacZ no pBTK30) were then isolated onto secondary LB agar plates with X-gal, gentamicin, and nalidixic acid at the stated concentrations. Genomic DNA (gDNA) was collected from isolated single colonies by resuspending bacterial colonies in 0.02% SDS and boiling the sample for 15 minutes. Samples were then centrifuged at 10,000 × g and supernatants containing gDNA were collected. Transposon insertion junctions were mapped using semi-random PCR (Supplementary Table 1). PCR samples were sequenced and reads were then mapped to the P. aeruginosa UCBPP-PA14 genome using BLAST. Expression changes were then verified via modified β-galactosidase assay in liquid culture.
Plaque assays.
Plaque assays were performed on LB agar plates (1.5% agar) with LB top agar (0.7% agar), supplemented MgSO4 (10 mM final concentration) and gentamicin (50 μg ml−1) and arabinose (0.1%) as needed for plasmid maintenance and induction. Spot titrations were done by mixing 150 μl of a P. aeruginosa overnight culture with 3 ml of top agar, which was dispersed evenly on a LB MgSO4 plate. 3 μl of 10-fold phage dilutions were then spotted on the surface. Plates were incubated overnight at 30 °C. To count plaques, full plate assays were used, except when CRISPR-targeting was so strong that discrete plaques could not be accurately measured. In this case, spot titrations are shown. For full plate assays, 10 μl of the phage dilution giving single plaques was incubated with 150 μl of P. aeruginosa overnight for 10 minutes at 37 °C. 3 ml of top agar was then added and the mixture was dispersed evenly on a LB MgSO4 plate. Individual plaques were then counted to assess differences in efficiency of bacterial immunity and phage. Efficiency of immunity (EOI) of a bacterial mutant relative to WT was calculated by dividing the number of plaque-forming units (PFUs) formed on WT by the number of PFUs formed on the mutant strain. EOI>1 means less plaques formed on the mutant than on WT, so the mutant was more immune to phage infection than WT. EOI<1 means more plaques formed on the mutant, so the mutant was less immune to phage infection than WT. Efficiency of plaquing (EOP) of a phage (Fig. 4e) was calculated by dividing the number of PFUs formed on WT (CRISPR+) by the number of PFUs formed on ΔCRISPR. An EOP of 1 means that CRISPR does not impact phage replication, and EOP of 0 means that the phage cannot replicate in the presence of CRISPR.
β-galactosidase assay.
A β-galactosidase assay described previously45 was used to measure lacZ activity in transcriptional fusions. Bacterial cultures were grown overnight at 37 °C. Cultures were then diluted 1:100 into LB liquid medium supplemented with the desired antibiotic, and incubated at 37 °C with shaking until the desired time point was reached. Culture density was measured with a spectrophotometer (OD600) and 200 μl of the sample was added to 800 μl of permeabilization solution. Cells were mixed via inversion and vortexed for 1 minute to permeabilize the cells. 200 μl of ONPG (4 mg ml−1) was added and samples were incubated at 30 °C until sample turned yellow. Enzymatic reaction was stopped by addition of 300 μl of 1M Na2CO3. Samples were centrifuged at 13,000 × g for 5 minutes to remove debris and 200 μl of supernatant was moved to a 96-well plate to read absorbance at 420 nm and at 550 nm. Miller units were calculated using the Miller equation: (1000*OD420 nm −1.75*OD550 nm)/(Tmin* VmLs *OD600nm).
Phage transduction of kinB::Tn alleles.
Transposon insertions in kinB from a csy3::lacZ background were transduced into WT PA14 to enable testing of CRISPR-Cas function with the same transposon insertion. Phage phiPA3 was used to infect the donor strain (kinB::Tn), on plates with top agar overlays, using ~104 PFU to generate near confluent lysis. Plates were soaked in 3–4 mL of phage SM buffer and 2 mL collected over chloroform, vortexed, and pelleted to isolate transducing phage in the supernatant. Lysates were used to infect recipient strains (WT PA14). ~108 PFU were used to infect a culture at an MOI of 1. After 30 minutes of static incubation on the bench, cultures were gently shaken at 37 °C for 20 min and then pelleted at 5000 × g. Cells were washed twice with LB, and subsequently incubated at 37 °C for 1 hour to allow recombination and gentamicin resistance outgrowth. Cultures were pelleted and resuspended in 200 μL of LB, and plated on LB plates containing gentamicin. Controls included uninfected cells and cells infected with phages not propagated on a gentamicin resistant donor strain. Additionally, phage lysate was directly plated under selection to confirm no residual donor strain in the phage preparation. Plates were incubated overnight at 37 °C and their identity (i.e. CRISPR-Cas intact) confirmed with a plaque assay using DMS3macrIE3 as the target phage and PCR of the kinB locus.
Introduction of csy3::lacZ P. aeruginosa UCBPP-PA14 strains.
The lacZ gene was introduced into PA14 strains of interest via allelic replacement. Recombination vector pMQ30 containing lacZ flanked by homology arms matching csy2 and csy4 was introduced via conjugation. PA14 strains and E. coli containing vector were mixed at a ratio of 1:2 (recipient:donor). Mixture was heat shocked at 42 °C for 10 min. Mating spot was then plated on a LB agar plate and incubated overnight for 30 °C. Mating spot was then collected, resuspended in 1 ml of LB liquid media and plated on VBMM plates supplemented with 50 ug/mL gentamicin to select for colonies with the integrated homology plasmid. Colonies were cultured overnight in LB in the absence of selection at 37 °C, and were then diluted and counterselected on no salt LB (NSLB) agar plates supplemented with 15% sucrose. Surviving colonies were then grown on LB agar plates supplemented with gentamicin and X-gal to check for lacZ insertion via color change and lacZ insertion was further verified via PCR.
Reverse transcriptase quantitative PCR (RT-PCR).
Total RNA extracts were harvested using an acid-phenol chloroform extraction from liquid cultures subcultured 1:100 and grown for 8 h in LB media. RNA treated with DNAse (Ambion) to remove DNA and 1ng of total RNA was used in a series of RT-qPCR reactions. Reactions were conducted in a BioRad CFX connect qPCR cycler, using clear BioRad plates with the Luna Universal One-Step Reaction Mix (NEB). A standard curve for each primer set was generated using pooled RNA samples. The housekeeping gene, rpsL, was used for normalization, and gene specific primers against cas3 and csy3 (Supplementary Table 1) were used to quantify expression from the cas and csy operons. For RT-qPCR reactions, 1 ng of total RNA was used in each reaction, performed in triplicate. Reverse transcription was conducted using to generate cDNA using Luna WarmStart® RT Enzyme Mix (NEB). Standard curves were used to calculate the relative abundance of target transcripts, and cas3 and csy3 transcript levels were then normalized to rpsL levels.
Generation of endogenous Csy1-sfCherry and Cas3-sfCherry reporters
Endogenous Csy1-sfCherry and Cas3-sfCherry reporters were constructed similar to the construction of csy3::lacZ. We initially verified that tagging of sfCherry at the N-terminus of Csy1 and Cas3 are functional, when expressed from a plasmid. pMQ30-sfCherry-Csy1, which contains sfCherry sequence flanked by 657 bp upstream of csy1 and 701 bp downstream of csy1 start codon, was cloned in pMQ30 plasmid between HindIII and BamHI sites using Gibson assembly. pMQ30-sfCherry-Cas3, which contains sfCherry sequence flanked by 353 bp upstream of cas3 and 350 bp downstream of the cas3 start codon, was cloned in pMQ30 plasmid between HindIII and BamHI sites using Gibson assembly. The 4 bp that overlap between the end of cas1 and the beginning of cas3 were duplicated in the final construct. Both pMQ30-sfCherry-Csy1 and pMQ30-sfCherry-Cas3 contains ggaggcggtggagcc sequence (encoding GGGGA) as linker between sfCherry and the respective tagged proteins. The Csy1-sfCherry and Cas3-sfCherry construct were introduced into PA14 strains of interest via allelic replacement. Strains containing appropriate insertion were verified via PCR.
sfCherry reporter profiling.
Liquid:
Cells were diluted 1:100 from an overnight culture into fresh LB (with 0.1% arabinose and 50 ug/mL gentimicin if required for plasmid induction and maintenance), and grown for the indicated number of hours in biological triplicate. 500 μl of each sample was then spun down at 8,000xg for 2 minutes, and resuspended in 500 μl of M9 media. Samples were loaded in to a 96 well plate (150 μl/well) in technical triplicate and red fluorescence and OD600 were measured using a Biotek H4 Synergy 96 well plate reader. M9 media alone was measured to obtain a background fluorescence and absorbance reading.
Solid:
Cells were diluted 1:100 from an overnight culture into fresh LB and 20 μl plated onto individual wells in biological triplicate in a 24 well plate with each well containing solidified 1.5% LB Agar (with 0.1% arabinose and 50 ug/mL gentimicin if required for plasmid induction and maintenance). The 24 well plate was then covered with a breathable Aeraseal, and incubated at 37 °C with no shaking. At the indicated timepoint, cells were harvested by flooding each well with 500 μl of M9 buffer, and were spun down at 8,000xg for 2 minutes, and resuspended in 500 μl of M9 media. Samples were loaded in to a 96 well plate (150 μl/well) in technical triplicate and red fluorescence (excitation 580 nm, emission 610 nm) and OD600 were measured using a Biotek H4 Synergy 96 well plate reader. M9 media alone was measured to obtain a background fluorescence and absorbance reading. To calculate the relative fluorescence units for each sample, the background fluorescence and background OD600 values obtained were subtracted from the sample values, and the sample fluorescence was then normalized to the sample OD600.
Generation of PA14 ΔamrZ using the endogenous I-F CRISPR-Cas system.
Complementary oligonucleotides encoding a crRNA targeting the amrZ gene of PA14 were annealed and ligated into the multiple cloning site of the pHERD30T vector. A fragment possessing homology arms flanking the desired mutation (500 bp upstream and 500 bp downstream) around amrZ was cloned into a distinct location (NheI site) of the same vector via Gibson assembly. The new plasmid containing both a crRNA and homology region was introduced into WT PA14 via electroporation. Transformation efficiency dropped dramatically in the presence of the crRNA due to the toxicity caused by self-targeting. All surviving colonies had the desired clean deletion of the amrZ gene. Deletions were confirmed by PCR of the region of interest and subsequent Sanger sequencing of the amplicon. A 2000 bp region flanking amrZ was PCR amplified and sequencing primers were designed to sequence both the deletion junction and outside of the original 500 bp flanking regions to confirm the removal of the amrZ gene.
Liquid phage infection assay.
Liquid phage infections were performed as described in29. In brief, an overnight culture of cells was diluted 1:100 into fresh media, and infected with 106 PFU virulent phage DMS3macrIF4 in biological triplicate in a 96 well Costar plate. Cells were incubated at 37 °C with constant rotation and OD600 measured every 5 minutes in a Biotek H4 Synergy plate reader. Phage were harvested from each well and quantified by plaque assay after 24 h. In these experiments, all strains used in the assay carried 2 spacers against the DMS3macrIF4 phage to prevent phage escape: one endogenous spacer (CRISPR2_sp1), and the other spacer was provided on a pHERD30T plasmid.
AmrZ homolog discovery and characterization.
BLASTp was used to search the nonredundant protein database for AmrZ homologs (accession: ABJ12639.1) in Pseudmonas sp. (taxid: 286) in May 2019. This homolog list (e value > 0.001) was then examined for homologs found on phage or plasmid genomes. Representative homologs were aligned using Clustal and the alignment visualized in Jalview, and key conserved residues were mapped onto the structure in Pymol (PDB ID: 3QOQ). Select homologs were synthesized (TWIST Biosciences) and cloned into the SacI/PstI site of the arabinose-inducible plasmid pHERD30T using Gibson assembly. Vectors were electroporated into Pseudomonas aeruginosa strains for functional testing, where they were induced with 0.1% arabinose and maintained with 50 ug/mL gentimicin.
Construction of recombinant DMS3m AmrZ phages.
Phages were generated as previously described29. Briefly, Gibson assembly was used to generate a recombination plasmid on pHERD 30T with amrZphi3 or amrZPaBG flanked by homology arms up and downstream of the DMS3m Acr locus. This plasmid was transformed into PA14 ΔCRISPR, and infected with phage DMS3macr-gent – a phage that is sick as the result of the insertion of a large gentimicin resistance cassette into its anti-CRISPR locus. Healthy plaques resulting from the recombination were screened for their incorporation of amrZphi3 or amrZPaBG into the anti-CRISPR locus with PCR.
Construction of PA14 lysogens.
Lysogens were obtained by first spotting phage onto a bacterial lawn, then streaking out surviving colonies from phage spots. These colonies were screened for phage resistance using a cross streak method, and lysogeny verified by prophage induction.
Pyocyanin Repression Assay.
The pyocyanin repression assay was performed as previously described25. Lysogens were transformed with a plasmid encoding a Type I-F crRNA targeting the promoter region of the gene phzM, which is required for the synthesis of the green pigment pyocyanin. As a control, each lysogen was also transformed with the empty vector plasmid. These strains were grown overnight (~16 h) in 5 mLs of LB media supplemented with 50 μg ml−1 gentamicin and 0.01% arabinose, to induce expression of the crRNA. Pyocyanin was extracted with an equal volume of chloroform, and then mixed with a half-volume of 0.2 M HCl, which produces a pink color proportional to the amount of pyocyanin and can be quantified by measuring absorbance at 520 nm. The absorbance value of each crRNA-expressing lysogen was expressed as a percentage of the pyocyanin level measured in the empty vector control lysogen. Samples were measured in technical triplicate.
Statistical Testing.
This study used a two-tailed unpaired Student’s T-test for statistical testing. In all cases, sample size is n = 3, degrees of freedom is n-1, confidence interval is 95%. In all plots, bar or data point height is equivalent to the mean and error bars are shown as +/−1*standard deviation (SD).
Data Availability
Source data and statistics used to generate figures are provided with the paper. Additional data supporting the findings of this paper will be made available from the corresponding author by request.
Extended Data
Extended Data Figure 1. Mapped insertions from transposon mutagenesis screen.
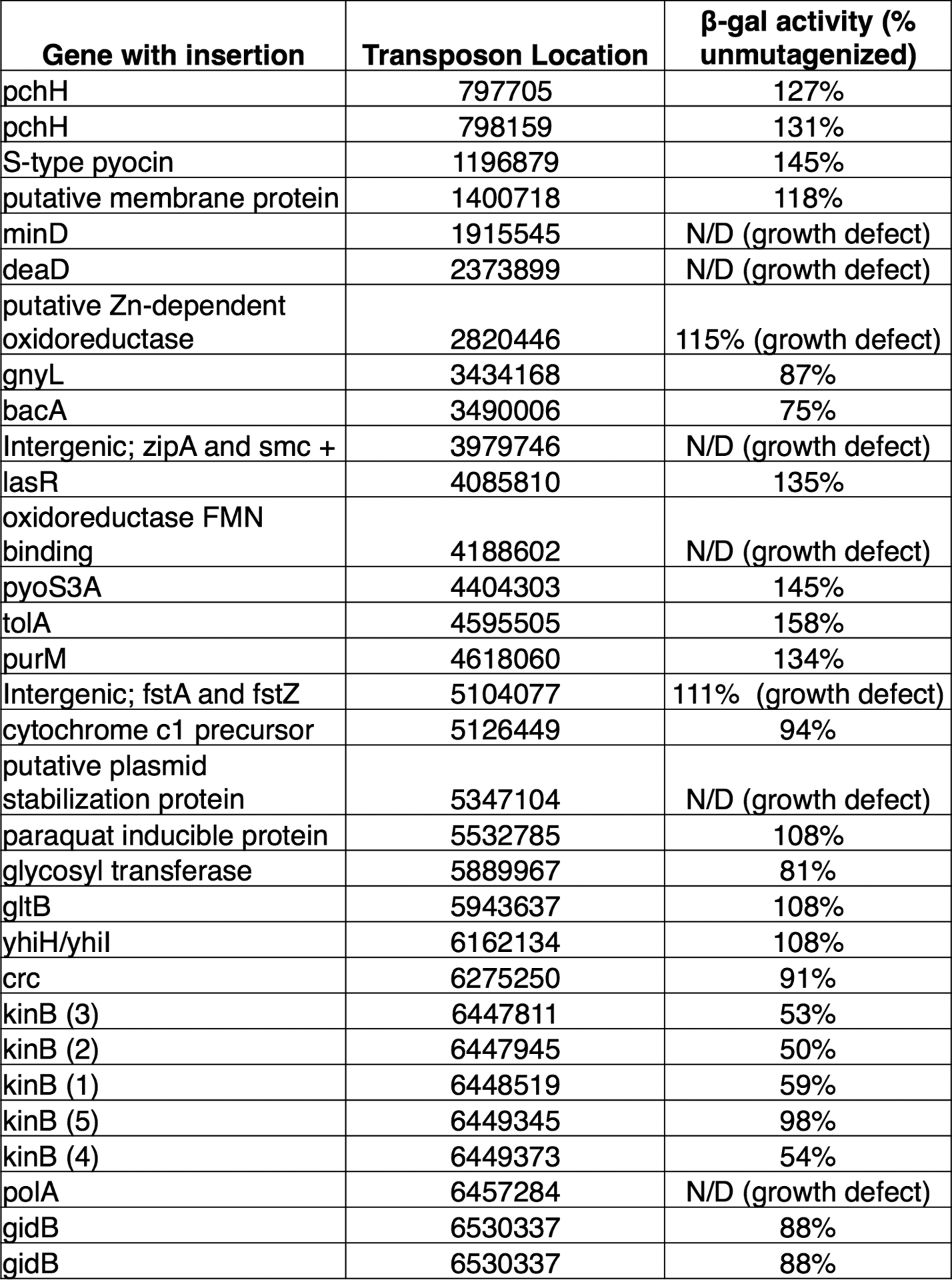
All independent transposon insertions identified and mapped by visual screening with increased or decreased csy3::lacZ β-galactosidase activity. β-galactosidase activity is expressed as a percentage of the unmutagenized parent strain, and measurements were taken at a single timepoint after 8 h of growth in liquid culture. The insertion location in the PA14 genome is shown, along with the measured level of β-galactosidase enzyme at the 8 hour timepoint. These measurements were not determined (N/D) for strains with a growth defect.
Extended Data Figure 2. Characterization of kinB::Tn mutants.
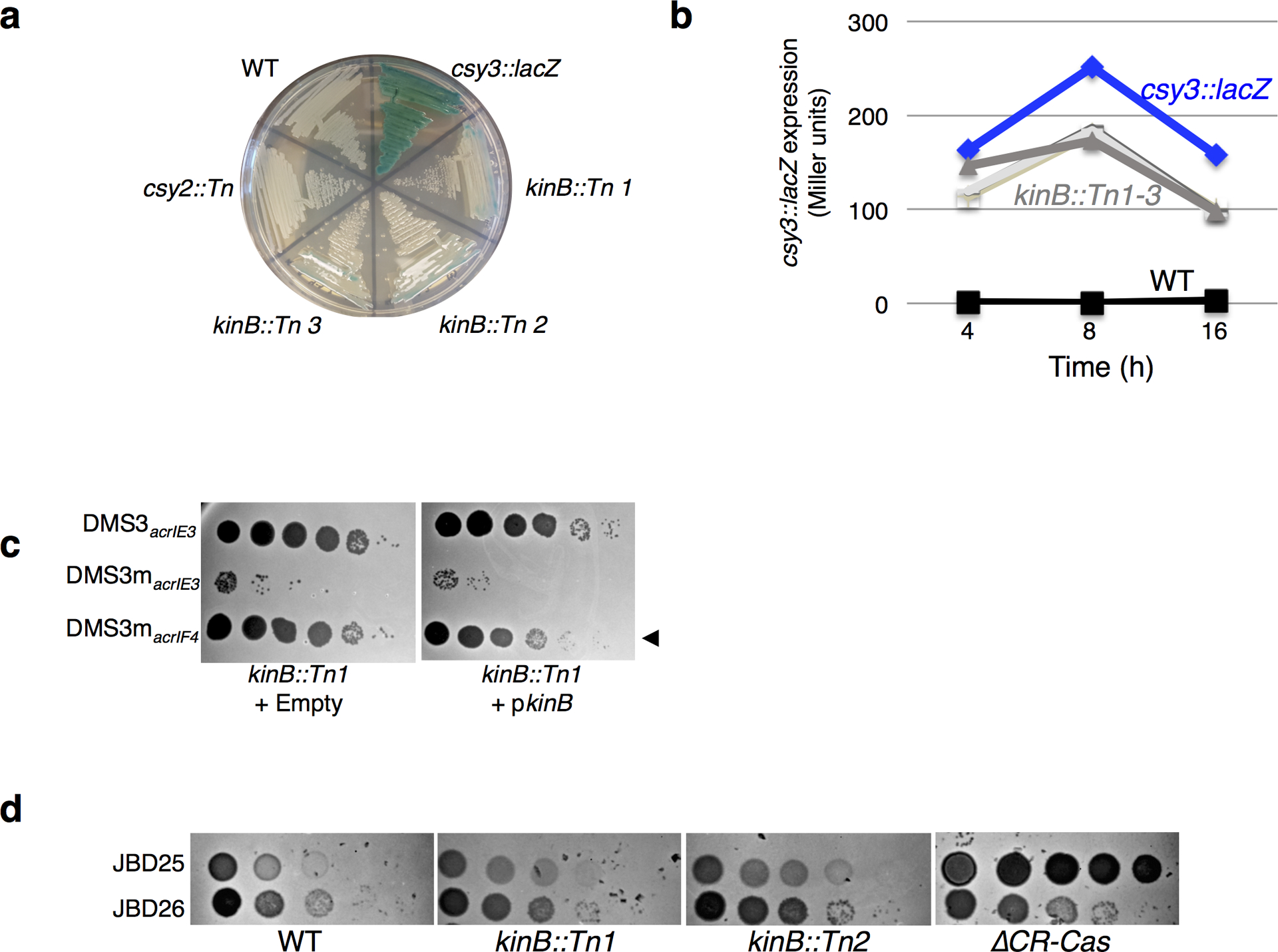
a. A streak plate on X-gal plates, showing strains involved in this study and isolated transposon (Tn) insertions. csy3::lacZ is a derivative of WT PA14, and is the unmutagenized parent of kinB::Tn 1–3. b. β-galactosidase measurements of strains grown in liquid culture for the indicuated time. Measurements for the unmutagenized (csy3::lacZ) parent strain and three isolated kinB transposon mutants (kinB::Tn1–3) are shown, as well as a control PA14 culture with no lacZ insertion. c. Phage titration on lawns of the kinB::Tn1 mutant transformed with empty vector or kinB. d. Spot titration of phages JBD26 (CR2_sp17, sp20-targeted, possessing acrIF4), JBD25 (CR1_sp1 targeted) on kinB::Tn mutants and ΔCRISPR-Cas. These experiments have been replicated at least 2 times with consistent results.
Extended Data Figure 3. Double knockouts of pathway members.
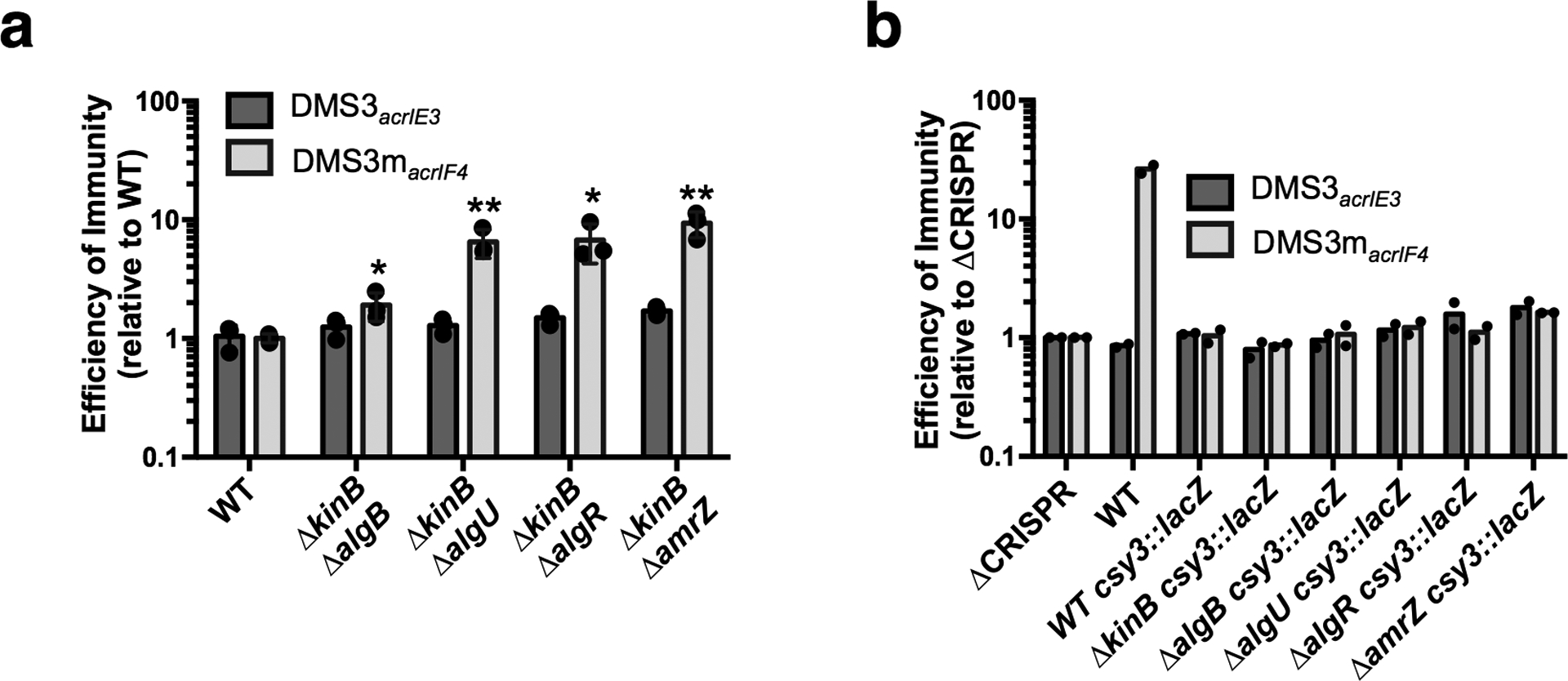
a-b. Efficiency of immunity measurements for indicated mutants relative to WT. a. Double knockouts show ΔkinB combined with algB, algU, algR, or amrZ. EOI measurements are shown as the mean of 3 biological replicates, +/− S.D. Mutants show increased EOI against DMS3macrIF4 relative to WT (ΔkinBΔalgB, P = 3.8 × 10−2, ΔkinBΔalgU, P = 5.9 × 10−3, ΔkinBΔalgR , P = 1.5 × 10−2 , ΔkinBΔamrZ, P = 3.2 × 10−3) Two-tailed unpaired Student’s T-test was used to calculate P value, *p < 0.05, **p <0.01. b. Indicated knockouts were combined with csy3::lacZ, EOI shown as the mean of two biological replicates.. These experiments have been replicated at least 2 times with consistent results.
Extended Data Figure 4. AmrZ activity in liquid growth.
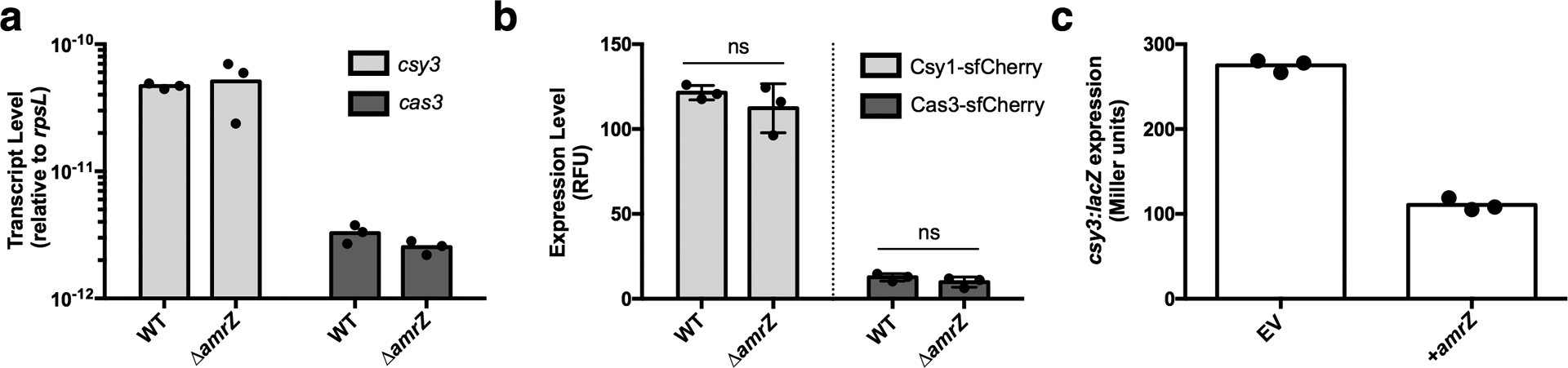
a. qRT-PCR measurements of transcript levels of csy3 (light grey) and cas3 (dark grey) normalized to the housekeeping gene rpsL after 8 h of growth in liquid culture. Measurements are represented as the mean of 3 technical replicates. b. Measurement of the fluorescence levels of Csy1-sfCherry (light grey) or Cas3-sfCherry (dark grey) reporter strains after 10 h of growth in liquid culture. Fluorescence measurements are represented as the mean of 3 biological replicates +/− SD. Cas3-sfCherry (P = 0.26) and Csy1-sfCherry levels (P = 0.35) in ΔamrZ did not differ significantly from WT. Two-tailed unpaired Student’s T-test was used to calculate P value, ns = not significant c. csy3::lacZ β-galactosidase activity from PA14 WT csy3::lacZ transformed with either empty vector (EV) or a plasmid overexpressing AmrZ (+AmrZ). β-galactosidase reporter activity was measured after 8 h in liquid growth and is represented as the mean of 3 technical replicates. Experiment was replicated two times with consistent results.
Extended Data Figure 5. Mobile AmrZ homologs.
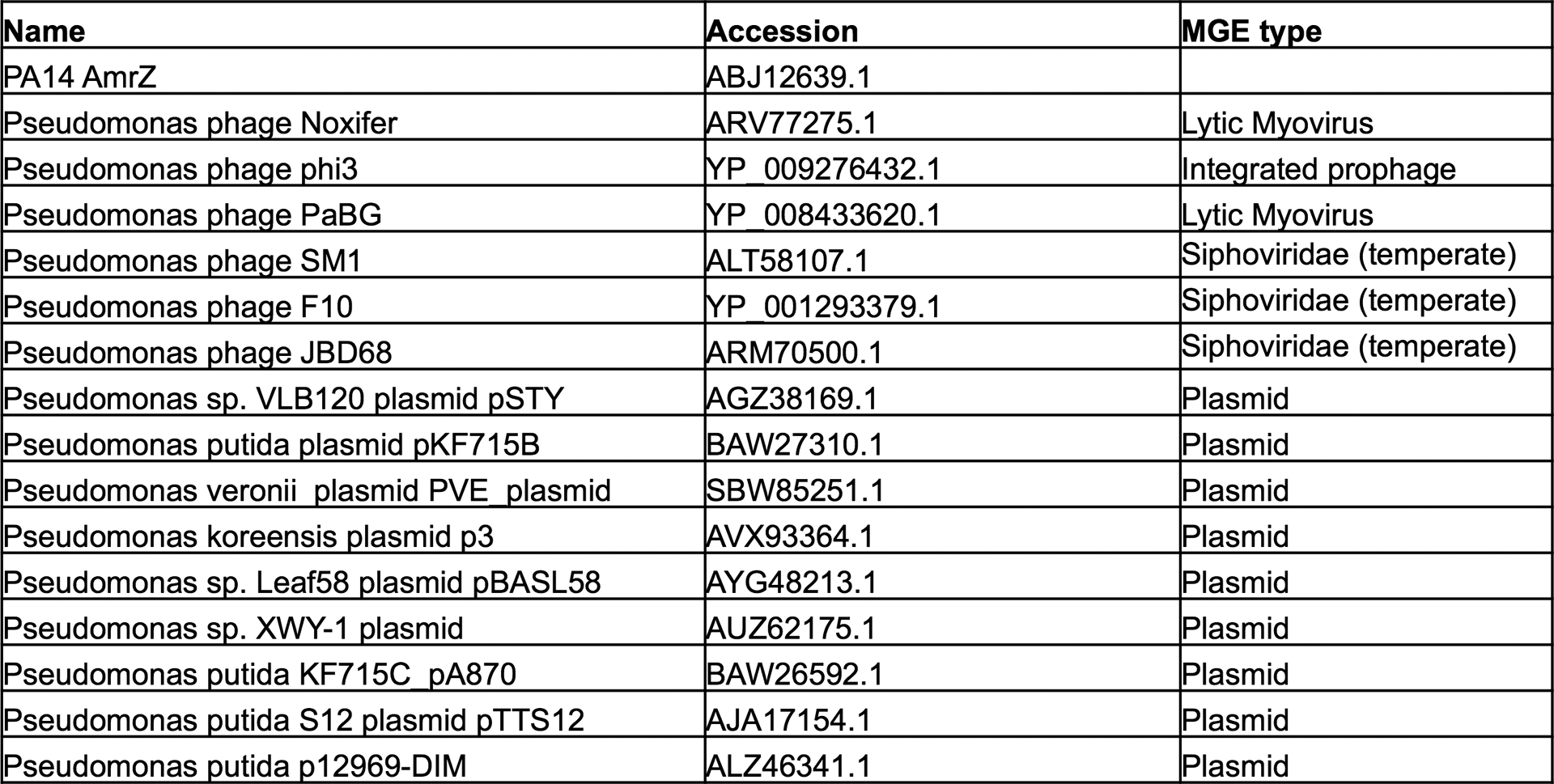
AmrZ homologs listed by the genome that encodes them, the accession number, and the mobile genetic element type.
Extended Data Figure 6. AmrZ copy number analysis of two Pseudomonas aeruginosa strains.
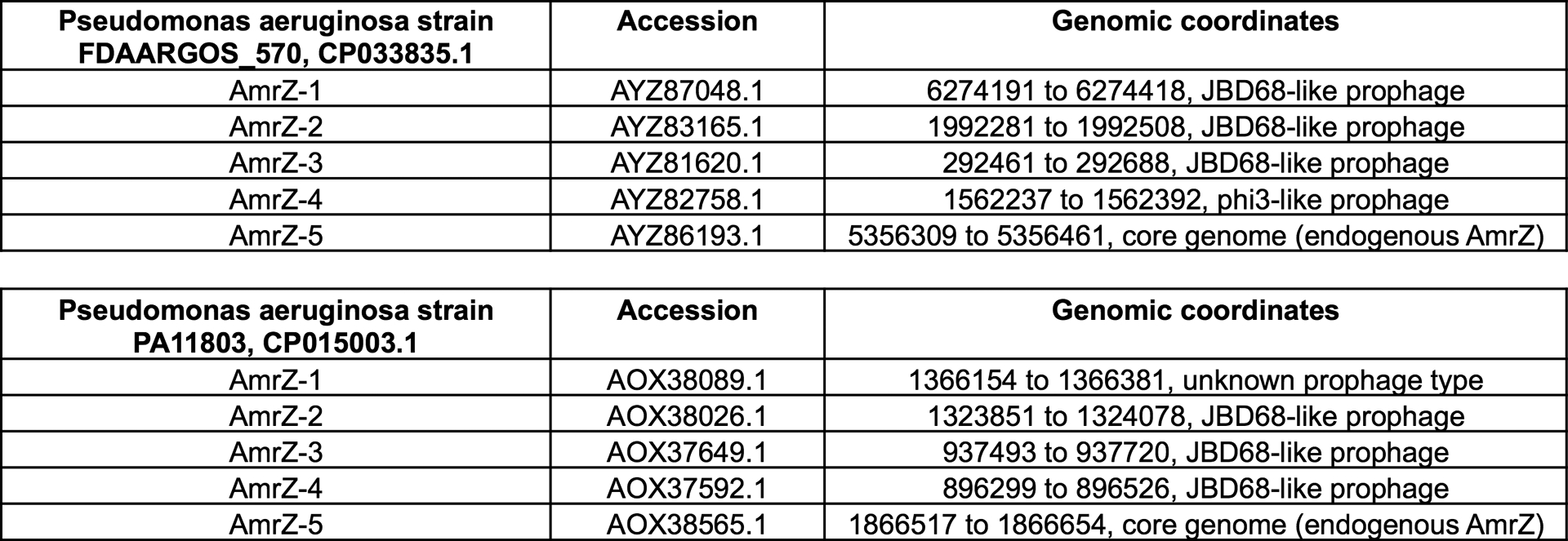
AmrZ copy number analysis of two different strains of Pseudomonas aeruginosa. AmrZ homologs listed by accession number and their genomic coordinates. Phaster46 was used to identify the prophages encoding mobile AmrZ copies.
Extended Data Figure 7. Cas and Csy RNA and protein levels across growth conditions.
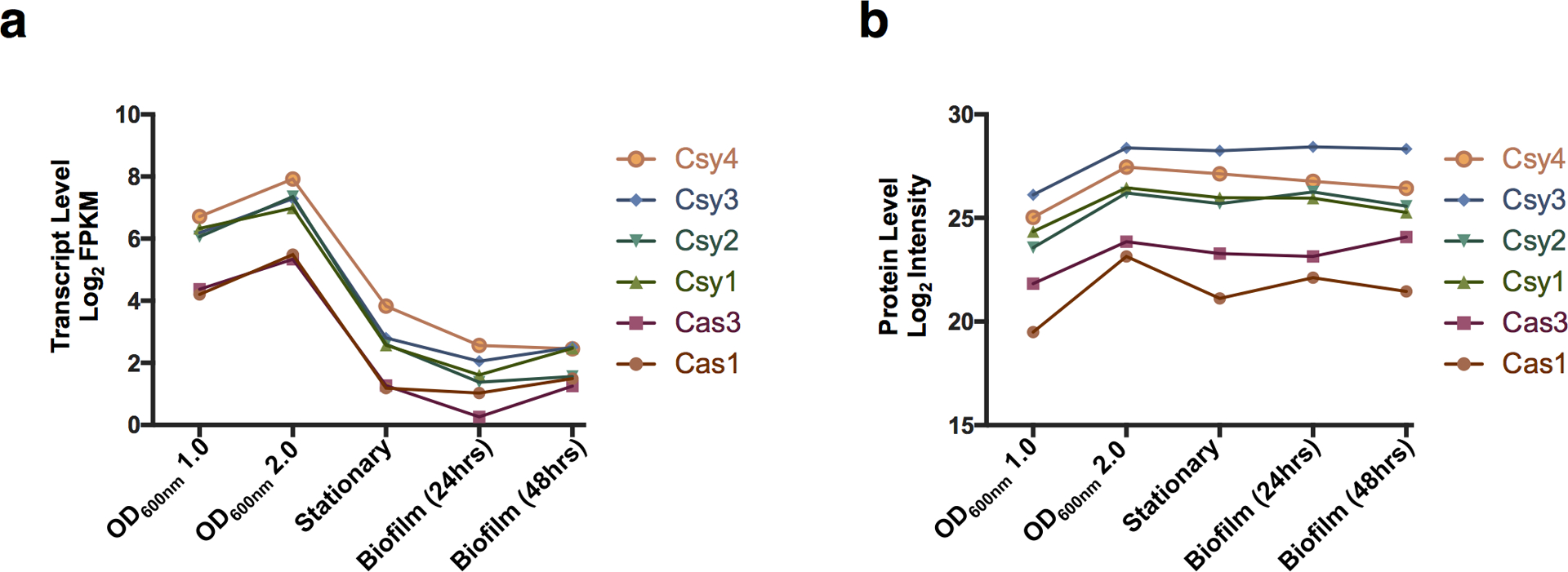
a. Log2 of Fragments Per Kilobase of transcript per Million mapped reads (FPKM) shown for each I-F cas gene in PA14 in the indicated growth condition43. b. Log2 of protein levels for each of the I-F Cas proteins in PA14 in the indicated growth condition44.
Supplementary Material
Supplementary Table 1. Strains, plasmids, and primers.
Acknowledgements
We thank Deborah Hung’s lab for providing ΔkinB, ΔalgB, ΔalgR, ΔalgU mutants, and George O’Toole’s lab providing the csy3::lacZ PA14 strain. We thank Kristine Trotta (Seemay Chou’s lab, UCSF) for advice in the development of fluorescent assays, and Andrew Santiago-Frangos (Blake Wiedenheft’s lab, MSU) for advice and consultation. The Bondy-Denomy lab was supported by the University of California, San Francisco Program for Breakthrough in Biomedical Research, funded in part by the Sandler Foundation, and an NIH Office of the Director Early Independence Award (DP5-OD021344), and R01GM127489..
Footnotes
Competing Interests
J.B.-D. is a scientific advisory board member of SNIPR Biome and Excision Biotherapeutics and a scientific advisory board member and co-founder of Acrigen Biosciences.
REFERENCES
- 1.Barrangou R et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Brouns SJJ et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damron FH, Qiu D & Yu HD The Pseudomonas aeruginosa sensor kinase KinB negatively controls alginate production through AlgW-dependent MucA proteolysis. J. Bacteriol 191, 2285–2295 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Damron FH et al. Analysis of the Pseudomonas aeruginosa Regulon Controlled by the Sensor Kinase KinB and Sigma Factor RpoN. J. Bacteriol 194, 1317–1330 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wozniak DJ & Ohman DE Transcriptional analysis of the Pseudomonas aeruginosa genes algR, algB, and algD reveals a hierarchy of alginate gene expression which is modulated by algT. J. Bacteriol 176, 6007–6014 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baynham PJ & Wozniak DJ Identification and characterization of AlgZ, an AlgT-dependent DNA-binding protein required for Pseudomonas aeruginosa algD transcription. Molecular Microbiology 22, 97–108 (1996). [DOI] [PubMed] [Google Scholar]
- 7.Hochstrasser ML et al. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proceedings of the National Academy of Sciences 111, 6618–6623 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westra ER et al. CRISPR Immunity Relies on the Consecutive Binding and Degradation of Negatively Supercoiled Invader DNA by Cascade and Cas3. Mol Cell 46, 595–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy A et al. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520, 505–510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haurwitz RE, Jinek M, Wiedenheft B, Zhou K & Doudna JA Sequence- and Structure-Specific RNA Processing by a CRISPR Endonuclease. Science 329, 1355–1358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiedenheft B et al. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 17, 904–912 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Wiedenheft B et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proceedings of the National Academy of Sciences 108, 10092–10097 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rollins MF, Schuman JT, Paulus K, Bukhari HST & Wiedenheft B Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Research (2015). doi: 10.1093/nar/gkv094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rollins MF et al. Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proceedings of the National Academy of Sciences 23, 201616395–E5121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowdhury S et al. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169, 47–57.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zegans ME et al. Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol 191, 210–219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cady KC & O’Toole GA Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol 193, 3433–3445 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR & O’Toole GA The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol 194, 5728–5738 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vorontsova D et al. Foreign DNA acquisition by the I-F CRISPR-Cas system requires all components of the interference machinery. Nucleic Acids Research 43, 10848–10860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Belkum A et al. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio 6, e01796–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westra ER et al. Parasite Exposure Drives Selective Evolution of Constitutive versus Inducible Defense. Curr. Biol 25, 1043–1049 (2015). [DOI] [PubMed] [Google Scholar]
- 22.van Houte S et al. The diversity-generating benefits of a prokaryotic adaptive immune system. Nature 532, 385–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bondy-Denomy J, Pawluk A, Maxwell KL & Davidson AR Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pawluk A, Bondy-Denomy J, Cheung VHW, Maxwell KL & Davidson AR A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. mBio 5, e00896–e00896–14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bondy-Denomy J et al. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawluk A et al. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nature Microbiology 1, 1–6 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Høyland-Kroghsbo NM et al. Quorum sensing controls the Pseudomonas aeruginosa CRISPR-Cas adaptive immune system. Proceedings of the National Academy of Sciences 114, 201617415–135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patterson AG et al. Quorum Sensing Controls Adaptive Immunity through the Regulation of Multiple CRISPR-Cas Systems. Mol Cell 64, 1102–1108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borges AL et al. Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity. Cell 174, 917–925.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chand NS et al. The Sensor Kinase KinB Regulates Virulence in Acute Pseudomonas aeruginosa Infection. J. Bacteriol 193, 2989–2999 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chand NS, Clatworthy AE & Hung DT The Two-Component Sensor KinB Acts as a Phosphatase To Regulate Pseudomonas aeruginosa Virulence. J. Bacteriol 194, 6537–6547 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cezairliyan BO & Sauer RT Control of Pseudomonas aeruginosa AlgW protease cleavage of MucA by peptide signals and MucB. Molecular Microbiology 72, 368–379 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schurr MJ, Yu H, Martinez-Salazar JM, Boucher JC & Deretic V Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol 178, 4997–5004 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tart AH, Blanks MJ & Wozniak DJ The AlgT-dependent transcriptional regulator AmrZ (AlgZ) inhibits flagellum biosynthesis in mucoid, nonmotile Pseudomonas aeruginosa cystic fibrosis isolates. J. Bacteriol 188, 6483–6489 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schulz S et al. Elucidation of sigma factor-associated networks in Pseudomonas aeruginosa reveals a modular architecture with limited and function-specific crosstalk. PLoS Pathog 11, e1004744 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wozniak DJ et al. Alginate is not a significant component of the extracellular polysaccharide matrix of PA14 and PAO1 Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci USA 100, 7907–7912 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pratama AA & van Elsas JD A novel inducible prophage from the mycosphere inhabitant Paraburkholderia terrae BS437. Sci Rep 7, 9156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pryor EE et al. The transcription factor AmrZ utilizes multiple DNA binding modes to recognize activator and repressor sequences of Pseudomonas aeruginosa virulence genes. PLoS Pathog 8, e1002648 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin DW et al. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc Natl Acad Sci USA 90, 8377–8381 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones AK et al. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J. Bacteriol 192, 5709–5717 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koonin EV & Makarova KS Mobile Genetic Elements and Evolution of CRISPR-Cas Systems: All the Way There and Back. Genome Biol Evol 9, 2812–2825 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heilmann S, Sneppen K & Krishna S Sustainability of virulence in a phage-bacterial ecosystem. Journal of Virology 84, 3016–3022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dötsch A et al. The Pseudomonas aeruginosaTranscriptional Landscape Is Shaped by Environmental Heterogeneity and Genetic Variation. mBio 6, e00749–15–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erdmann J, Preusse M, Khaledi A, Pich A & Häussler S Environment-driven changes of mRNA and protein levels in Pseudomonas aeruginosa. Environmental Microbiology 20, 3952–3963 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Smale ST β-Galactosidase Assay. Cold Spring Harb Protoc 2010, pdb.prot5423 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Arndt D et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research 44, W16–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Strains, plasmids, and primers.
Data Availability Statement
Source data and statistics used to generate figures are provided with the paper. Additional data supporting the findings of this paper will be made available from the corresponding author by request.