

Abstract
Caspases are a family of conserved cysteine proteases that play key roles in programmed cell death and inflammation. In multicellular organisms, caspases are activated via macromolecular signaling complexes that bring inactive procaspases together and promote their proximity-induced autoactivation and proteolytic processing. Activation of caspases ultimately results in programmed execution of cell death, and the nature of this cell death is determined by the specific caspases involved. Pioneering new research has unraveled distinct roles and cross talk of caspases in the regulation of programmed cell death, inflammation, and innate immune responses. In-depth understanding of these mechanisms is essential to foster the development of precise therapeutic targets to treat autoinflammatory disorders, infectious diseases, and cancer. This review focuses on mechanisms governing caspase activation and programmed cell death with special emphasis on the recent progress in caspase cross talk and caspase-driven gasdermin D–induced pyroptosis.
Keywords: inflammatory caspase, inflammasome, pyroptosis, gasdermin, infection, innate immunity
INTRODUCTION
In physiologic settings, intrinsic genetic programs allow eukaryotic cells to undergo cell death in a regulated manner. This regulated cell death is conventionally called programmed cell death because of its execution through a defined set of genetically encoded mechanisms. Apoptosis is the most studied form of programmed cell death and is essential for organismal development. In apoptosis, death is executed by intrinsic molecular programs in the cell to ensure there is no impact on surrounding, living cells. Necrosis, which is phenotypically different from apoptosis, is considered a lytic form of unregulated, accidental cell death (1). However, this was contradicted by recent findings that some types of necrosis are molecularly controlled and termed programmed necrosis (1, 2). In programmed necrosis, cell death is executed in association with the induction of inflammation; therefore, programmed necrosis falls into the category of processes termed inflammatory cell death. Programmed necrosis releases intracellular damage-associated molecular patterns (DAMPs) to further amplify inflammation. Whereas apoptosis primarily orchestrates cellular homeostasis and development and often acts to suppress inflammation, inflammatory cell death pathways are emerging as a central component of the innate immune response in infectious and inflammatory diseases (1–3).
Programmed cell death is facilitated by caspases, a highly conserved set of intracellular proteases (4–6). Caspases are cysteine proteases that cleave their targeted proteins after aspartic acid residues. They are expressed in both immune and nonimmune cells, and the majority of caspases are constitutively expressed during homeostasis. The caspases are produced as enzymatically inactive zymogens called procaspases (4, 5) that undergo dimerization or oligomerization for activation. The protease effector domain of procaspases (caspase domain) is cleaved into large and small subunits during the activation process, further forming complexes for enzymatic activity (7–9). Caspases were first characterized as key biological players in inflammation and apoptotic cell death. The molecular pathways controlled by caspase-dependent substrate cleavage are evolutionarily conserved in eukaryotes and have evolved to regulate fundamental cellular processes by inducing apoptosis or triggering inflammatory cell death. Identification of new mechanisms of caspase activation and the discovery of novel substrates provide opportunities to understand their functional regulation in health and disease.
CLASSIFICATION OF CASPASES
Based on function, mammalian caspases are categorized into two major groups: apoptotic and inflammatory caspases (Table 1). Apoptotic caspases function predominantly to initiate and execute apoptosis, which is mostly immunologically silent (4). These apoptotic caspases are further subgrouped as initiator and effector caspases based on their order of function in the execution of apoptosis (4, 5, 9). Initiator caspases, including caspase-2, -8, -9, and -10, act as proteolytic signal amplifiers to activate effector caspases. Effector caspases such as caspase-3, -6, and -7 proteolytically cleave several cellular proteins at their target sites to facilitate apoptosis. Caspase-1, -4, -5, -11, and -12 belong to the inflammatory caspase family and are functionally distinct from apoptotic caspases (5, 10). Caspase-2, -6, -12, and -14 are poorly characterized and need further investigation (Table 1). Caspase-2 is activated in PIDDosome in response to genotoxic stress and failure of cytokinesis to inhibit cell proliferation (11, 12). Caspase-6 was shown to cleave Lamin A to promote chromosomal condensation during apoptotic execution, and its deregulation was observed in inflammatory neurodegenerative diseases such as Huntington disease and Alzheimer disease; however, its role in infection-induced cell death and the specific upstream signaling pathway responsible for the activation remain obscure (13–15). Caspase-14 is not categorized as an apoptotic or inflammatory caspase. It is known to participate in epidermal differentiation (16, 17). Inflammatory caspases are activated by the assembly of a macromolecular, oligomeric complex called the inflammasome (10, 18). Inflammatory caspase activation induces the activation of a lytic form of inflammatory cell death called pyroptosis and also triggers activation and secretion of DAMPs to promote inflammation.
Table 1.
Classifification of human and mouse caspases and their functions in eliciting cell death and inflammation
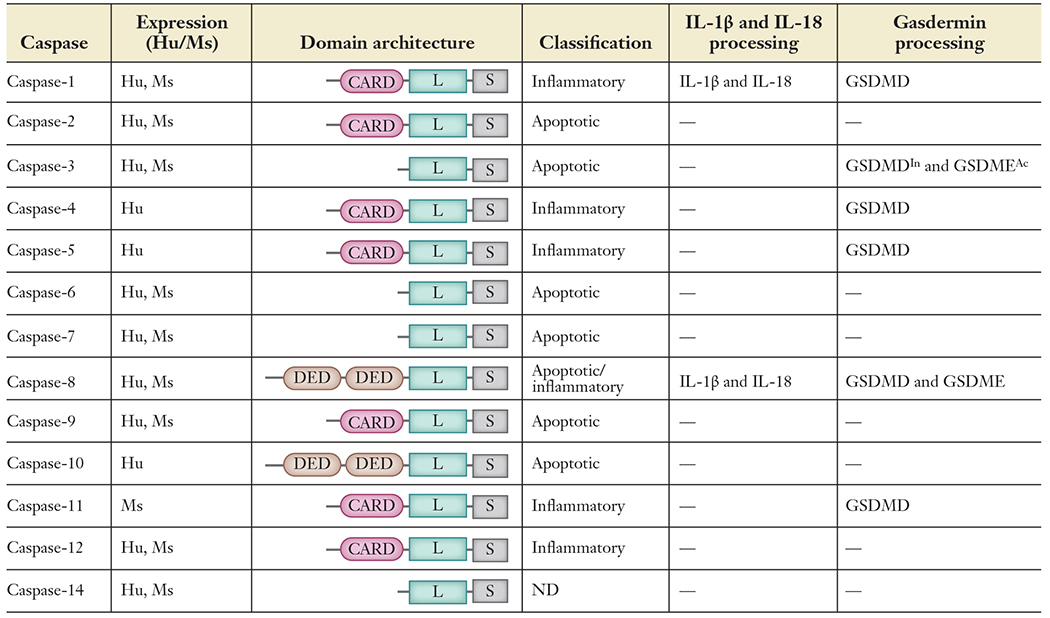 |
Abbreviations: Ac, activation; CARD, caspase activation and recruitment domain; DED, death effector domain; GSDMD, gasdermin D; GSDME, gasdermin E; Hu, human; In, inactivation; L, large domain; Ms, mouse; S, small domain, ND, not defined; —, unknown/no data available.
All caspases contain a C-terminal caspase domain that proteolytically processes the target protein (Table 1). In addition to this caspase domain, some of the caspases contain nonenzymatic domains such as the death effector domain (DED) or caspase activation and recruitment domain (CARD) at their N terminus (Table 1). Several caspases (both apoptotic and inflammatory) contain CARD domains, and only caspase-8 and -10 contain DED domains. The DED and CARD domains belong to a death domain (DD) superfamily (19) and are known to facilitate the assembly of large oligomeric signaling complexes through self-association (homotypic interactions) and protein-protein interactions with DED/CARD-containing proteins. DED/CARD-dependent oligomerization can trigger proximity-dependent activation of caspases or activate caspase-independent functions (19, 20). Inactivation of the enzymatic activity of some of these DED/CARD-containing caspases compromises their cell death function but preserves their ability to engage in other biological processes, like inflammatory responses (19–23). These additional nonenzymatic functions may be mediated via the assembly of oligomeric signaling scaffolds, thus allowing further categorization of DED/CARD-containing caspases as scaffolding (e.g., caspase-8) and non-scaffold-forming caspases. Scaffolding caspases act as a molecular framework to enable the nonenzymatic assembly of a variety of signaling complexes and perform distinct biological functions. In some cases, caspases in scaffolding complexes still retain enzymatic activity to regulate inflammatory cell death pathways (21, 24–26). This suggests that caspases have evolved to perform proteolytic processing-dependent and -independent functions to regulate biological functions.
MECHANISMS OF APOPTOSIS
As discussed above, apoptosis is an intrinsic mechanism of silent cell death evolved to carry out the programmed dismantling of cellular components for cellular homeostasis. Caspases are central to this process and systematically hydrolyze key cellular proteins in a complex cascade of cellular events. Because caspase-mediated proteolysis is an irreversible event, caspases are synthesized as inactive monomeric precursors. Pro-apoptotic stimuli prime the cells to initiate the caspase cascade, resulting in the proximity-induced activation of initiator caspases (27–29). This in turn proteolytically activates effector caspases, which facilitate the systematic dismantling of cellular components, leading to characteristic intracellular changes including DNA degradation, nuclear shrinkage (pyknosis) and fragmentation (karyorrhexis), membrane blebbing, and release of apoptotic bodies (7, 30, 31), and ultimately resulting in apoptosis and clearance of the dead cells by phagocytosis (32). Diverse upstream signaling events such as death receptor (DR) engagement (extrinsic) or cellular stress (intrinsic) lead to distinct sets of death-inducing mechanisms of apoptosis (Figure 1). Therefore, the apoptotic pathways can generally be classified as extrinsic or intrinsic based on the mode of initiation and the specific adapters and initiator caspases involved in the process (33, 34).
Figure 1.
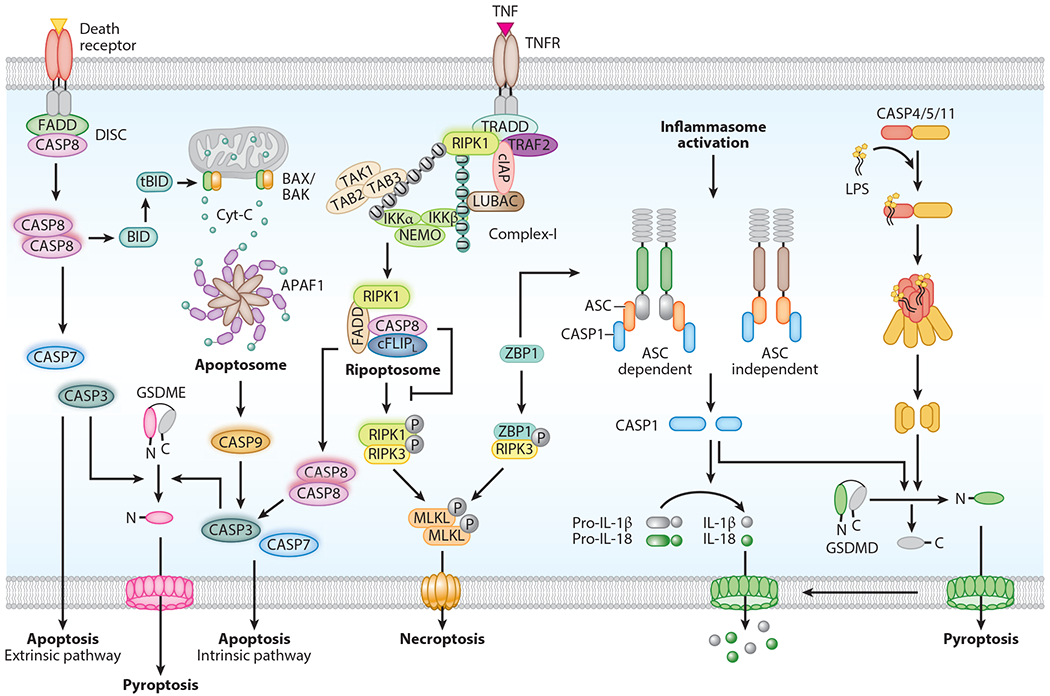
Mechanism of caspase activation and execution of cell death. Caspases trigger activation of apoptosis, a noninflammatory form of cell death. In extrinsic apoptosis, death receptor activation facilitates recruitment of FADD and caspase-8 to form the death-inducing signaling complex (DISC). Activated caspase-8 in the DISC further processes executioner caspases (caspase-3, -7) to engage apoptosis. In intrinsic apoptosis, intracellular stress stimuli induce mitochondrial outer membrane permeabilization (MOMP). This leads to the release of cytochrome c into the cytosol. Binding of cytochrome c to APAF1 triggers assembly of the apoptosome complex, which facilitates activation of caspase-9. This in turn promotes activation of caspase-3 and -7 to execute apoptosis. Complex-I assembly is initiated by TNF binding to TNFR on the membrane. This promotes prosurvival signaling via receptor-interacting serine/threonine protein kinase 1 (RIPK1), TAK-TAB, and NEMO proteins. Loss of RIPK1 ubiquitination promotes formation of the intracellular ripoptosome complex. Inactivation of the catalytic activity of caspase-8 in the ripoptosome complex induces RIPK1-RIPK3 association and phosphorylation of MLKL to form pores on the membrane and engage necroptosis. ZBP1 also activates necroptosis via RIPK3. Activation of inflammatory caspases induces pyroptosis. Inflammasome assembly activates caspase-1 enzymatic function. ASC in the inflammasome complex recruits caspase-1. Some inflammasome-forming innate immune receptors recruit caspase-1 without the involvement of an ASC protein. ZBP1 activates the NLRP3 inflammasome in response to influenza infection. LPS binds to caspase-11 (or human caspase-4/-5) and triggers its activation through oligomerization and cleavage. Activation of caspase-1 and -11 proteolytically processes gasdermin D (GSDMD) to release the N-terminal domain, which forms pores on the membrane to further induce pyroptosis. Caspase-1 also cleaves pro-IL-1β and pro-IL-18 into IL-1β and IL-18, which are released through GSDMD pores. Caspase-3-mediated gasdermin E (GSDME) cleavage also drives pyroptosis.
Intrinsic Apoptosis Pathway
The intrinsic apoptosis pathway is triggered when mitochondrial integrity is compromised in response to stressors (Figure 1). These stressors include a wide range of exogenous and endogenous triggers, such as DNA damage, endoplasmic reticulum (ER) stress, and reactive oxygen species. The stressors induce activation of the proapoptotic BCL-2 antagonist or killer (BAK) and/or BCL-2-associated X (BAX) protein, which results in mitochondrial outer membrane permeabilization (MOMP) (35) (Figure 1). The loss of mitochondrial integrity leads to the release of its inner membrane proteins, such as cytochrome c and second mitochondria-derived activator of caspase (SMAC; also known as DIABLO), which directly promote caspase activation and intrinsic apoptosis (35). Cytoplasmic cytochrome c binds the WD domain of apoptotic protease-activating factor 1 (APAF1), inducing a conformational change and exposing the nucleotide-binding site of APAF1 (Figure 1). The nucleotide-bound form of APAF1 undergoes further structural changes resulting in the assembly of a heptameric complex with the initiator caspase-9, called the apoptosome (36–38). During this process, caspase-9 initiates the caspase cascade, driving the activation of caspase-3/-6/-7/-9/-10 to cause the robust execution of apoptosis. This process is further amplified by SMAC, which blocks the caspase-inhibitory function of inhibitor of apoptosis proteins (IAPs) by targeting them for proteasomal degradation (39).
Extrinsic Apoptosis Pathway
The extrinsic apoptosis pathway is central to extracellular ligand-induced DR signaling (Figure 1) (33, 34). Members of the well-characterized DR family include the tumor necrosis factor (TNF) superfamily members TNF receptor 1 (TNFR1/CD120a), apoptosis antigen 1 (APO-1, Fas/CD95), death receptor 3 (DR3), DR4 [also called TNF-related apoptosis-inducing ligand receptor 1 (TRAIL-R1)], and DR5 (TRAIL-R2/APO-2). Upon binding to their cognate ligands, Fas, TRAIL-R1, and TRAIL-R2 induce apoptosis. TNFR1 engagement often results in inflammatory signaling that drives the expression of antiapoptotic molecules and inflammatory cytokines, except when the prosurvival cellular checkpoints are blocked (40–42). Binding of the DR to its cognate ligand leads to receptor oligomerization and recruitment of the downstream adaptors FAS-associated death domain (FADD) or TNFR1-associated death domain (TRADD) protein. These adaptors bridge the initiator caspases, such as caspase-8 or -10, to form a macromolecular death-initiating signaling complex (DISC) (7). Caspase-10 is presumed to be proapoptotic; however, it was shown to promote cell survival by altering DISC signaling to NF-κB activation or by cleaving the BCL2-interacting protein BCLAF1 to block autophagic cell death (43, 44). Caspase-10 contains N-terminal DED domains and is expressed in humans but not in mice. Caspase-8 is the only other caspase that contains DED domains similar to those in caspase-10, suggesting a possible similarity in their function. Loss-of-function genetic mutations in human caspase-8 and caspase-10 are associated with autoimmune lymphoproliferative syndrome (ALPS) due to defective caspase activity and apoptosis (45, 46). DISC acts as a tailored molecular platform to promote dimerization and proximity-induced activation of initiator caspase-8/10 (Figure 1). Therefore, not surprisingly, cells with a genetic deficiency for DISC components fail to undergo apoptosis in response to DRs engaging their corresponding ligands (47–49).
There exists an intimate connection between extrinsic and intrinsic apoptosis pathways through the BCL-2 homology 3 (BH3)-interacting domain death agonist (BID) (50–52). Caspases activated downstream of the extrinsic pathway cleave BID to produce tBID, which acts on the proapoptotic BAX-BAK molecular switch, resulting in MOMP and intrinsic apoptosis (35) (Figure 1). Cells are broadly categorized as type I or type II based on their critical requirement for MOMP to undergo apoptosis. Type I cells show robust DR-mediated activation of caspase-8 and -3, which is sufficient to drive apoptosis, whereas type II cells need MOMP-mediated antagonism to counteract IAP- and Bcl-2-driven prosurvival mechanisms to ensure efficient execution of apoptosis.
Activation of Apoptotic Caspases via the Ripoptosome Complex
In immune cells, the TNF-TNFR1 complex at the cell membrane facilitates the formation of a membrane-associated signaling complex called complex-I to engage prosurvival responses (1, 53). Upon TNF stimulation, receptor-interacting serine/threonine protein kinase 1 (RIPK1) and cellular IAPs (cIAPs) are recruited to TNFR via TNFR-associated factor 2 (TRAF2) and TRADD (1, 54, 55). Here, cIAPs modify RIPK1 by Lys63-linked ubiquitination (1, 54, 55) to promote the scaffolding function, but not kinase activity, of RIPK1 to assemble a multiprotein signaling complex that activates prosurvival NF-κB transcriptional responses. However, functional attenuation of some of the complex-I components such as cIAP1, transforming growth factor β–activated kinase 1 (TAK1), or TANK-binding kinase 1 (TBK1) facilitates formation of cytosolic complex-II, also called the ripoptosome complex (Figure 1). The ripoptosome is a large (≈2 MDa) protein complex containing RIPK1, FADD, caspase-8, and cellular FLICE-like inhibitory protein (cFLIP) as core components associated through their conserved DD interactions (56, 57). The ripoptosome functions to further recruit either prosurvival or cell death–inducing proteins to decide cell fate. The ripoptosome complex promotes caspase-8-FADD-dependent apoptosis through the activation of executioner caspase-3 and -7 (Figure 1). Recruitment of RIPK3 to the ripoptosome complex promotes MLKL-mediated necroptosis (1, 53, 56, 57). However, recruitment of cFLIP to the ripoptosome complex diverts the apoptotic activity of caspase-8 to prosurvival inflammatory signaling (1, 21, 25, 56, 57). This complex also inhibits RIPK3-dependent necroptosis to promote cell survival.
CASPASE-8 INHIBITION, SWITCHING EXTRINSIC APOPTOSIS TO NECROPTOSIS
Genetic and biochemical inactivation of caspases has revealed caspase-independent forms of cell death and a more complex relationship between apoptosis and inflammation (33, 58, 59). Inactivation of caspases blocks apoptosis but switches the cell fate to a form of programmed necrotic death called necroptosis. Deletion of the components of the extrinsic apoptosis pathway such as caspase-8 or FADD results in TNF-induced necrotic cell death (60). Accordingly, deletion of FADD, caspase-8, or its regulator cFLIP is lethal in mouse embryos (48, 61, 62). Furthermore, cell-specific studies showed that FADD and caspase-8 play nonapoptotic roles in cell death and inflammation (63–66). In vivo genetic evidence demonstrated that elimination of FADD and caspase-8 or cFLIP results in spontaneous activation of RIPK3 and mixed lineage kinase domain-like pseudokinase (MLKL) to execute necroptosis (25, 67, 68) (Figure 1). Caspase-8 is also reported to cleave and inactivate cylindromatosis (CYLD), a deubiquitinase known to activate RIPK1 to drive RIPK3-dependent necroptosis (69). During viral infections, Z-DNA binding protein 1 (ZBP1), also known as DNA-dependent activator of interferon regulatory factors (DAI) or DLM-1, engages inflammatory cell death via caspase-8 and RIPK3 (70–72) (Figure 1). Together, these studies unequivocally establish that extrinsic apoptosis and caspase-8-dependent suppression of necroptosis are critical for cellular homeostasis and normal embryonic development.
INFLAMMATORY CASPASES
In sharp contrast to the apoptotic caspases, inflammatory caspases play central roles in the innate immune response and promote pyroptosis, an immunologically active, proinflammatory form of cell death. Myeloid cells of the immune system strategically express high levels of the inflammatory caspases and their corresponding signaling platforms for programmed activation in response to infection or injury. Inflammatory caspase activation is one of the key aspects of the innate immune system and provides the first line of defense against invading pathogens. While the human genome encodes the inflammatory caspases caspase-1, -4, -5, and -12, caspase-11 represents both caspase-4 and -5 in mice. Although, it is now well established that caspase-1 is activated in a multiprotein oligomeric complex called the inflammasome (discussed below), the inflammatory caspase-4, -5, and -11 do not need such molecular platforms for their activation (Figure 1). These inflammatory caspases act as direct receptors to sense pathogen-encoded molecules such as lipopolysaccharide (LPS) and undergo self-oligomerization and proximity-induced autoactivation. Caspase-11 was also shown to directly bind self-encoded oxidized phospholipids (oxPAPC), although different functional consequences were reported (73, 74).
Inflammasomes and Inflammatory Caspase Activation Mechanisms
Inflammasomes are heterologous oligomeric protein complexes that are assembled after an innate immune receptor senses pathogen-associated molecular patterns (PAMPs) or danger signals (3, 10, 18) (Figure 1). The inflammasome complex acts as a scaffold to recruit caspase-1 and promote its proximity-induced activation to induce pyroptosis (3, 5, 10, 75). Various innate immune receptors assemble inflammasome complexes. The best-studied inflammasome complexes are Nod-like receptor (NLR) family pyrin domain (PYD)-containing 1 (NLRP1), NLRP3, NLR family CARD domain-containing 4 (NLRC4), absent in melanoma 2 (AIM2), and the pyrin inflammasome; these inflammasomes are activated upon sensing bacterial toxins and secretion system components, nucleic acids, pathogenic crystals, and altered cellular components (5, 10, 18, 76–90). Many inflammasome proteins use their CARD or PYD to recruit the adapter protein ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain) through homotypic protein-protein interactions (5, 10). ASC contains both a PYD and a CARD, which can promote further homotypic interactions with different innate immune receptors and promote ASC polymerization into large, filamentous structures that are characteristic of cytosolic membraneless compartments. This exposes the CARDs to recruit caspase-1 through CARD-CARD homotypic interactions. Some inflammasomes (i.e., NLRP1 and NLRC4) can directly engage caspase-1 through CARD-CARD interactions and do not require ASC for activation (Figure 1). Inflammasome assembly is a crucial step to activate caspase-1 to induce pyroptosis and release proinflammatory cytokines.
The inflammatory cytokines IL-1β and IL-18 are expressed in inactive proforms that require maturation into active cytokines by inflammatory caspase-mediated processing (5, 10) (Figure 1; Table 1). More importantly, these cytokines lack signal peptides and hence do not enter into the conventional secretion pathway. Instead, they are secreted via inflammatory caspase-induced membrane pores or lytic cell death (75, 91). In response to various infections, inflammatory caspase-1, -4, -5, and -11 induce pyroptosis of innate immune cells, macrophages, and monocytes. Caspase-1 was identified initially as an important factor for the maturation of proinflammatory cytokines lacking signal peptides for secretion (IL-1β and IL-18) (3, 92, 93).
The gene encoding caspase-11 is adjacent to the gene encoding caspase-1 on the same chromosome. Mice lacking Casp1 are resistant to endotoxic shock and lethality induced by bacterial cell wall component LPS. However, Caspl knockout mice also do not express Casp11 (3, 75, 94). Deleting only Casp11 revealed that loss of Casp11 is sufficient to confer resistance to LPS-induced septic shock (94). Caspase-11 induces macrophage pyroptotic death during various gram-negative bacterial infections (3, 94, 95). Unlike caspase-1, which requires assembly of inflammasome complexes for its activation, caspase-11 can be directly activated by sensing LPS to induce pyroptosis and the indirect activation of the noncanonical NLRP3 inflammasome and caspase-1 to promote activation of IL-1β and IL-18 (95). Human caspase-4 and -5 behave similarly to mouse caspase-11 and induce pyroptosis upon LPS sensing (3, 94–96).
Caspase-12 is also an inflammatory caspase, but its function is poorly understood (5, 97, 98). Caspase-12 seems to mediate ER-stress-induced apoptosis but is dispensable for apoptosis induced by classical stimuli (97, 99, 100). Caspase-12 undergoes self-cleavage but does not cleave other caspases. It was shown to interact with caspase-1 and -5 in macrophages and inhibit caspase-1 activity and the release of IL-1β (101). Although this study demonstrated enhanced caspase-1 activity in Casp12-deficient mice, later studies contested these findings by revealing caspase-12 does not suppress caspase-1 activity (101–103). In humans, a truncated version of caspase-12 with only the CARD domain is the predominant allele, while the full-length form with a functional caspase domain is less common (5). The precise cellular and biological functions of caspase-12 are still enigmatic.
PYROPTOSIS—AN INFLAMMATORY CASPASE-INDUCED LYTIC CELL DEATH
During infection, death of infected cells and induction of immune response are critical for maintaining organismal homeostasis. Pyroptosis (pyro meaning fire and ptosis meaning falling) emerged as a major form of programmed cell death in vertebrates that is important to fight infections (2, 3, 104). Pyroptosis is specifically induced in infected cells to eliminate the pathogen niches and engage inflammatory responses to potentiate protective host immunity (2, 105–108). Pyroptosis also plays an important role in eliciting proinflammatory responses to bring innate immune cells to the site of injury or infection. Pyroptosis is activated by various bacterial infections, such as Francisella, Legionella, Shigella, and Salmonella, and viral infections, such as influenza A virus, HIV, and hepatitis C virus (2, 3, 70, 71, 105–107). Although pyroptosis has long been considered a monocytic cell death pathway driven by caspase-1, it was later identified that caspase-4, -5, and -11 also drive pyroptosis efficiently, but are ineffective in direct cytokine processing (3, 104, 107, 109, 110) (Figure 1). Recent studies suggest that the induction of pyroptosis also occurs in epithelial cells (111). Caspase-1-induced pyroptotic cell death in HIV-infected lymphoid CD4+ T cells is a major cause of depletion of CD4+ T cells in HIV infection (112, 113). Thus, pyroptosis plays a critical role in diverse cell types.
Gasdermin D, an Executioner of Pyroptosis
Although inflammatory caspases were known to trigger pyroptosis and cleave multiple substrates during this process, the identity of the key effector substrate was not known until recently, when cleavage of gasdermin D (GSDMD) by inflammatory caspases was identified as the key event triggering pyroptosis in immune cells (114, 115). Initially, GSDMD was known to be one of the substrates of caspase-1, but its functional significance was not determined (116). Two independent approaches by Kayagaki et al. (114) and Shi et al. (115) identified the role of GSDMD in the caspase-1- and caspase-11-mediated activation of pyroptosis (Figures 1 and 2). Additionally, lack of Gsdmd inhibits the secretion of IL-1β and confers resistance to LPS-induced septic shock (114). Mechanistically, inflammatory caspases cleave the linker region between the N- and C-terminal domains of GSDMD, which liberates the active N terminus from the autoinhibiting C-terminal domain to execute pyroptosis (114, 115) (Figure 2a–c). Lack of interferon regulatory transcription factor 2 (IRF2) expression inhibits both canonical and noncanonical inflammasome-induced pyroptosis, as IRF2 is essential for the transcriptional upregulation of Gsdmd (117) (Figure 2c). Furthermore, gasdermin family members are predominantly expressed in the skin, gastrointestinal tract, and immune cells, which suggests they are strategically located to function at physical and mucosal barrier systems to actively eliminate infected cells through pyroptosis (75, 118).
Figure 2.
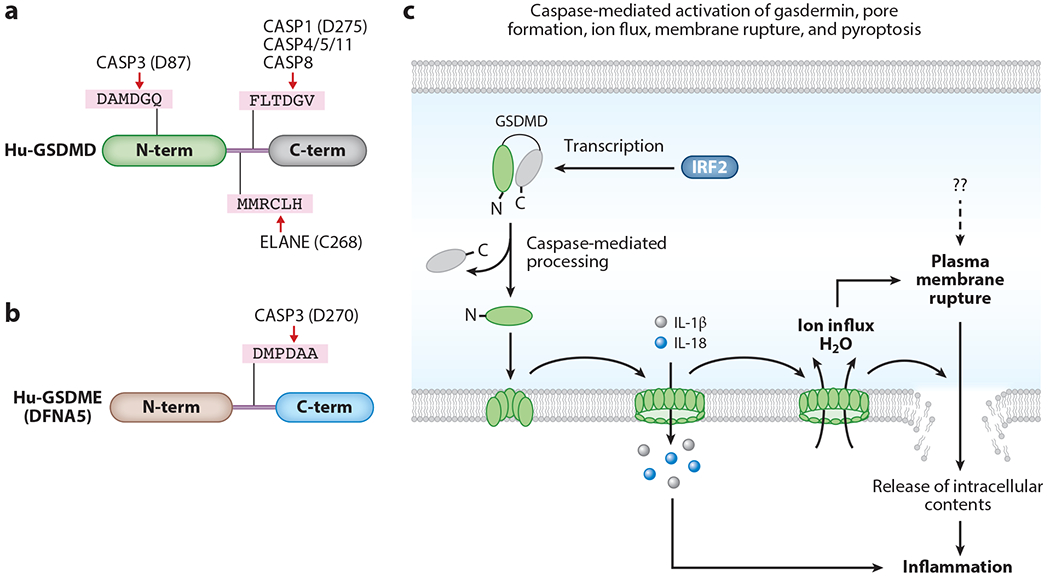
Caspase-mediated processing of gasdermin proteins and execution of pyroptosis. (a) Gasdermin D (GSDMD) is cleaved by several caspases. Cleavage of GSDMD at D275 of the linker region by caspase-1, caspase-4/-5/-11, and caspase-8 or at C268 by ELANE liberates the functionally active N terminus of GSDMD. Caspase-3 cleavage of GSDMD at D87 in the N-terminal domain inactivates its pyroptotic function. (b) Caspase-3 cleaves gasdermin E (GSDME) (DFNA5) at D270 of the linker region to release its active N-terminal domain. (c) Inflammatory caspases cleave at the linker region connecting the N and C termini of GSDMD to facilitate the induction of pyroptosis. Cleavage of GSDMD liberates the pore-forming N-terminal domain from the autoinhibitory C-terminal domain. The N-terminal domain of GSDMD is transported to the membrane to assemble into a pore structure. The inner region of the pore is hollow with a large diameter that is sufficient to enable passage of small ions and proteins such as mature IL-1β and IL-18. The pore formation by GSDMD is essential for the induction of pyroptosis. IRF2 transcriptionally upregulates GSDMD expression. Once the GSDMD pore is formed on the cell membrane, it facilitates ion and water flux from the extracellular space into the cell. GSDMD pore formation, ion and water flux, and loss of cytoskeletal integrity facilitate plasma membrane rupture and pyroptosis of cells. Pyroptosis releases intracellular components, which are immunostimulatory and cause inflammation. Abbreviations: N-term, N-terminal domain; C-term, C-terminal domain.
The gasdermin protein family consists of GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5), and DFNB59 (75, 118, 119). These proteins share similar N- and C-terminal domain architecture in which the C-terminal domain functions as an inhibitory domain to restrict N-terminal function. However, the caspase cleavage site residues in the linker region vary among the gasdermin family members, and not all gasdermin proteins are cleaved by caspases. Only the GSDMD linker region is cleaved by inflammatory caspases (114, 115) (Figure 2a,c). Although not all gasdermins are cleaved by caspases, they contain defined C-terminal inhibitory domain and N-terminal functional domain structures. Further studies identified that caspase-3 cleaves GSDME (DFNA5) in the linker region to liberate an N-terminal fragment, which activates pyroptosis in a similar mechanism to that of the GSDMD N terminus (120, 121) (Figure 2b). Caspase-3 also cleaves GSDMD; however, this cleavage occurs within the N-terminal domain, rather than at the linker region, which inhibits its function (121).
Pore-Forming Activity of GSDMD Executes Pyroptosis
Proteolytic cleavage of GSDMD at its linker region by an inflammatory caspase separates its N-terminal (p30) and C-terminal (p20) fragments, and the N-terminal fragment (hereafter called GN-term) is sufficient to induce pyroptosis (114, 115). The GN-term directly binds to membrane lipids, such as phosphatidylinositide, phosphatidylserine, and cardiolipin, which are primarily present in the inner leaflet of the cell membrane (122–125). Binding of the GN-term of GSDMD or GSDMA can disrupt membrane structures, causing cytotoxicity in mammalian cells (122–124). The GN-term forms pore structures on cell membranes through oligomerization (Figure 2c). The pore-forming activity of the GN-term is critical for initiating pyroptosis by inflammatory caspases (122–124) (Figure 2c). In addition, proteolytic cleavage of GSDME by caspase-3 switches TNF- or chemotherapeutic-induced apoptosis to pyroptosis by releasing the GN-term, which also exhibits pore-forming activity (120, 121).
Studies using atomic force microscopy revealed that GN-term protomers assemble arc- or slit-shaped oligomers in the membrane, which farther recruit additional protomers to build a stable ring-shaped pore in the membrane (126). Formation of a stable pore structure facilitates osmotic cell swelling, rupture of the plasma membrane, and permeability of cellular contents (114, 115, 127, 128). Calpain enzyme-dependent cleavage of vimentin proteins leads to the loss of intermediary filament (cytoskeletal components) structures, which causes the rupture of cells by extrinsic forces (127). Several caspases are also shown to cleave vimentin to disrupt intermediate filaments (129, 130). Calpain activity is associated with Ca2+ influx, perhaps through GSDMD pores, to promote loss of cellular architecture. Furthermore, cell rupture contributes to the release of several immune-stimulatory DAMPs, particularly molecules of large size, that trigger elevated immune stimulation. Overall, gasdermin-associated pore formation in the cell membrane leads to pyroptosis and the subsequent release of intracellular contents into the extracellular environment, which in turn exacerbates inflammatory responses.
In contrast to the cleavage mechanism in macrophages, cleavage of GSDMD is independent of inflammatory caspases in neutrophils (131). GSDMD is cleaved by the neutrophil-specific serine protease elastase ELANE at a site distinct from the inflammatory caspase cleavage site (131) (Figure 2a). Interestingly, this study indicated that GSDMD-induced lytic cell death in neutrophils is anti-inflammatory, in contrast with proinflammatory pyroptotic death in macrophages. Although neutrophils are known to exhibit pyroptosis downstream of inflammasome activation, they also drive NETosis-mediated antimicrobial functions at the site of infection (132, 133). Perhaps neutrophils express low levels of inflammasome components compared to macrophages, resulting in a relatively reduced frequency of pyroptosis (134–136).
Structural Insights into the Activation of Gasdermin-Induced Pores
The fundamental role of the pore-forming activity of gasdermin proteins in pyroptosis execution is an important framework for therapeutic targeting. Structural knowledge of gasdermin-induced pores is necessary to understand, at a molecular level, the formation of ring-like structures via oligomerization of GN-term protomers and also the regions that facilitate membrane insertion. Ding et al. (123) crystallized the full-length GSDMA3 protein and showed that it exists in a two-domain (N-terminal and C-terminal) architecture, which is considered an autoinhibitory conformation (75). This study also visualized GSDMA3 and GSDMD pores formed by GN-term protomers in lipid monolayers. They identified that gasdermin pores are typically made of 16 protomers, with a diameter of approximately 10–16 nm (123). A recent study by Ruan et al. (137) solved a cryo–electron microscopy (cryo-EM) structure of the GSDMA3 pore present in lipid bilayers with high resolution and showed that the gasdermin pore contains 27–28 protomers and typically attains an antiparallel β-barrel structure with an inner diameter of 180 Å (18 nm); this is distinct from the structure reported by Ding et al. Furthermore, Ruan et al. suggest that the differences observed in the pore structures are due to the lipid layers used to form the pores and that the pore structure solved in lipid bilayers is more physiologically relevant (137). Nevertheless, both structures indicate that the pores formed by gasdermin proteins contain a large inner space that is sufficient to facilitate the passage of ions and even cytokines such as IL-1β and IL-18, whose diameter is approximately 4–8 nm. The high-resolution structure proposed by Ruan et al. showed that the α1 helix of the GN-term of GSDMA3 provides a basic (i.e., positively charged) surface to bind to the acidic face of lipids, whereas its elongated β-hairpin structures contribute to the oligomeric interface of the pore structure. Once cleavage liberates the autoinhibitory conformation of GSDMA3, its active GN-term further undergoes extensive conformation transitions to become inserted into the membrane (137, 138). Thus, gasdermin proteins are conformationally dynamic and undergo an inflammatory caspase-driven sequential transition from a monomeric autoinhibitory state to an oligomeric pore structure for executing pyroptosis (75, 122–125, 137, 139, 140).
The GN-term of gasdermin proteins exhibits bacterial killing activity (123, 140). In particular, a specific region of the GN-term (residues ≈70–90) exhibits antibacterial activity against Escherichia coli and Mycobacterium smegmatis (140). On the basis of the cryo-EM structure of the GSDMA3 pore, the approximately 70–90 residues of the GN-term correspond with the region that is important for the oligomerization of gasdermin protomers in the pore structure (i.e., the β3 strand of the elongated hairpin structure). Some regions of GSDMD (residues 120–125 and 211–216) also attain higher-order oligomers or fibril-like structures (140), suggesting that oligomerization of the GN-term of gasdermin proteins is structurally crucial to induce pyroptosis and membrane-disrupting cytotoxicity. A recent study solved crystal structures of full-length murine and human GSDMD that define specific features required for intramolecular gasdermin domain association that restricts autoactivation (141). In addition, the new structural insights from this study demonstrate that mutations disrupting lipid binding and oligomerization of gasdermins inhibit cell lysis (141). Because the lipid-binding surface and oligomerization interface of the GN-term are essential for pore formation, these regions provide new attractive targets for structure-based design of therapeutic molecules. While new mechanisms are emerging that unravel the intricate regulation of gasdermin function, the full picture of the intracellular factors that regulate the pore-forming activity of gasdermins and pyroptosis is not yet available.
Consequences of Gasdermin Pore Formation
LPS sensing by caspase-11 activates GSDMD-induced pyroptosis. It is well established that the NLRP3 inflammasome is also activated upon caspase-11 activation as the noncanonical NLRP3 inflammasome (3, 94). However, the precise mechanism of noncanonical NLRP3 activation downstream of caspase-11 activation is not known. The approximately 18-nm diameter of the inner core of the gasdermin-induced pore enables passive movement of various ions through the membrane barrier (Figure 3). The GSDMD pore facilitates K+ efflux in immune cells, which is an important activator of the NLRP3 inflammasome. Therefore, K+ efflux through GSDMD pores formed after caspase-11 activation likely engages noncanonical NLRP3 inflammasome activation to further potentiate pyroptosis and IL-1β and IL-18 secretion (3, 142) (Figure 3). Additionally, GSDMD pores on the cell membrane can act as a channel to facilitate mature IL-1β release into the extracellular space without cell membrane rupture (i.e., before pyroptosis execution) in living cells (91) (Figures 2 and 3). Two recent studies found that GSDMD activation is important for optimal activation of the DNA-sensing AIM2 inflammasome (143, 144). In addition, GSDMD pores formed after AIM2 inflammasome activation promote K+ efflux, which in turn inhibits DNA receptor and cyclic GMP-AMP synthase (cGAS)-induced type I interferon responses (143). Because of the relatively large inner diameter of gasdermin-induced pores in the cell membrane, they aid in the activation of several cellular functions, including pyroptosis by facilitating passage of various molecules. Once inflammatory caspases are activated, pyroptosis is induced within a few minutes of gasdermin processing. This point-of-no-return condition commits pyroptotic cells to an irreversible disruption process. However, a recent study purported that mammalian cells activate a cell-intrinsic membrane repair process via recruitment of the endosomal sorting complex required for transport III (ESCRT-III) machinery to the cell membrane to counteract pyroptosis execution (145). This process is similar to the membrane repair mechanism that rescues cells from MLKL-induced necroptosis to restore cell integrity (146).
Figure 3.
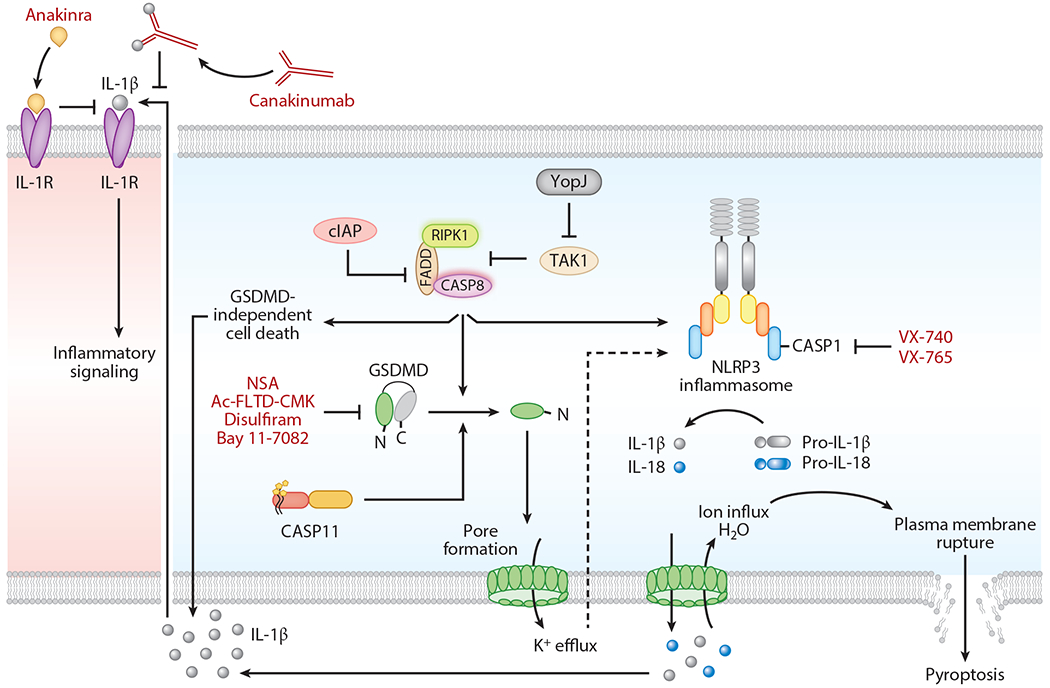
Intracellular regulation of caspase function to modulate inflammatory cell death. cIAPs inhibit caspase-8-dependent cell death functions. Degradation of cIAPs activates caspase-8 in the ripoptosome, which promotes maturation and release of IL-1β and activation of gasdermin D (GSDMD)-independent inflammatory cell death. TAK1 is necessary to inhibit the spontaneous induction of pyroptosis in immune cells. The absence of TAK1 or Yersinia YopJ-mediated inhibition of TAK1 leads to the assembly of the receptor-interacting serine/threonine protein kinase (RIPK1)–FADD–caspase-8 complex. This complex induces GSDMD activation either directly or through the activation of NLRP3 inflammasome assembly to trigger pyroptosis. Caspase-11-induced GSDMD pore formation facilitates K+ efflux, which activates the assembly of the noncanonical NLRP3 inflammasome complex. The GSDMD pores mediate release of matured IL-1β and IL-18. Necrosulfonamide (NSA), disulfiram, and Bay 11–7082 directly bind to GSDMD and inhibit inflammatory cell death. N-acetyl-Phe-Leu-Thr-Asp-chloromethylketone (Ac-FLTD-CMK) is a GSDMD-derived peptide inhibitor that inhibits cleavage of GSDMD by inflammatory caspases. Canakinumab is a human IL-1β-specific antibody that inhibits activation of IL-1R signaling. Anakinra is an antagonist of IL-1R that counteracts IL-1 cytokine signaling. VX-740 (Pralnacasan) and VX-765 (Belnacasan) are therapeutic inhibitors of caspase-1.
Role of Gasdermin-induced Pyroptosis in Disease
The discovery of GSDMD as an executioner of pyroptosis allowed the extension of research from in vitro induction of pyroptosis to in vivo studies focused on the specific role of pyroptosis in disease. The proinflammatory effects of gasdermin protein activation and pyroptosis can potentially influence the pathophysiology of autoinflammatory and infectious diseases, but will this play a protective or detrimental role in physiological disease settings? Mice lacking Gsdmd expression are resistant to lethality during LPS-induced septic shock (114). In vitro chemotherapeutic treatment induces GSDME (DFNA5) cleavage and pyroptosis, and lack of Gsdme (Dfna5) expression in mice confers resistance to chemotherapeutic-induced toxicity in multiple organs, suggesting a detrimental role for pyroptosis induced by chemotherapy (120). Both LPS-induced septic shock and chemotherapy treatment are artificially induced disease models that differ from spontaneous autoinflammatory or pathogen-induced diseases.
A new study shows that GSDMD plays a crucial role in a mouse model of familial Mediterranean fever (FMF) that recapitulates FMF in humans (147). FMF is an autoinflammatory disease caused by mutations in the Mefv gene, which confers constitutive activation of the pyrin inflammasome and release of proinflammatory cytokines (147). GSDMD-induced pyroptosis is crucial for the autoinflammatory phenotype in FMF mice. Loss of Gsdmd expression rescues FMF mice from the systemic inflammation, neutrophilia, and tissue damage that cause lethality. Neonatal-onset multisystem autoinflammatory disease (NOMID) is another autoinflammatory mouse model, and disease is induced by gain-of-function mutations in Nlrp3. Loss of Gsdmd expression also rescues NOMID mice from systemic inflammation (148).
GSDMD also plays roles in infection models. NLRP3 inflammasome-activated GSDMD cleavage and pyroptosis cause immunopathology after gastrointestinal murine norovirus (MNV) infection, and loss of GSDMD delays lethality in mice (149). In murine bacterial infections, type III secretion system rod proteins and LPS engage inflammasome-induced pyroptosis in macrophages to liberate tissue factor, which in turn initiates a coagulation cascade, massive thrombosis, and lethality; GSDMD deficiency prevents this coagulation and lethality (150). Collectively, these studies suggest that the activation of gasdermin-induced pyroptosis (and hence secretion of IL-1β and IL-18) causes exacerbated inflammatory responses, which confers lethality in mice. On the other hand, some studies have found that loss of Gsdmd expression is detrimental to the host during bacterial infections. Mice lacking Gsdmd expression are highly susceptible to Salmonella infection and have elevated bacterial load during infection, suggesting a critical function for GSDMD in host protection (151). Two additional studies found that mice lacking Gsdmd expression are highly susceptible to Francisella infection with high bacterial burden (143, 144). Therefore, induction of pyroptosis serves as an innate immune strategy to destroy a microbial niche of pathogens, but excessive gasdermin-induced pyroptosis is detrimental to the host.
Although the phenotypes observed reveal contrasting functions of GSDMD in disease settings, GSDMD-induced pyroptosis plays a critical role in driving immunological/pathological consequences. An intriguing question here is whether GSDMD-induced pyroptosis is self-sufficient to drive these phenotypes without the need to release DAMPs. In cancer cells, gasdermin proteins are known to exhibit pyroptosis-independent roles. A new study found decreased expression of Gsdmd in gastric cancer tissue, which is associated with gastric cancer cell proliferation (152, 153). To more fully understand the role of GSDMD in cancer, it is essential to determine the role of gasdermin pore-forming activity in tumor-associated macrophages that influence tumorigenesis and the tumor microenvironment (152).
EMERGING CASPASE MECHANISMS REGULATING INFLAMMATION AND GASDERMIN-INDUCED PYROPTOSIS
Tight regulation of inflammatory caspase function and pyroptosis is necessary to evade dysregulated inflammation and pathophysiologic consequences. Immune cells are equipped with additional upstream regulatory mechanisms to interrupt spontaneous activation of inflammatory cell death. Ripoptosomes, whose core components are RIPK1, FADD, and caspase-8, form in response to various stimuli that perturb cellular homeostasis (56, 57). The ripoptosome acts as a crucial cell fate decision checkpoint to enable activation of several cell survival or death-signaling complexes. TAK1 primarily regulates activation of extracellular signal-related kinase (ERK) and NF-κB in complex-I, which is essential for upregulating immune gene expression and conferring cellular homeostasis. TAK1 also restricts spontaneous activation of the NLRP3 inflammasome, pyroptosis, and other programmed cell death pathways (154) (Figure 3). The absence of TAK1 leads to ripoptosome formation and RIPK1-dependent spontaneous activation of the NLRP3 inflammasome and execution of programmed cell death pathways. This indicates that TAK1 plays a central role in protecting cells from spontaneous inflammatory cell death, mainly by engaging prosurvival signaling (154). FADD and caspase-8, the partners of RIPK1 in the ripoptosome, are also key regulators of NLRP3 inflammasome activation and pyroptosis (23). Ripoptosome-complex-dependent activation of pyroptosis during TAK1 inactivation implies that, under specific conditions, the ripoptosome associates with NLRP3 to activate the inflammasome, the release of IL-1β and IL-18, and pyroptosis. The NLRP3 inflammasome is also activated by diverse stress stimuli that ultimately execute pyroptosis. These stress stimuli can also activate formation of prosurvival stress granules. How immune cells coordinate both the NLRP3 inflammasome and stress granules, which engage contrasting cell fate decisions, is not clear. A new study suggests that stress granules inhibit NLRP3 inflammasome activation and pyroptosis (155). The protein DEAD-box helicase 3 X-linked (DDX3X) plays a crucial role in this stress granule-mediated inhibition of the NLRP3 inflammasome. DDX3X is required for the assembly of both the NLRP3 inflammasome and stress granules, and sequestration of DDX3X in stress granules inhibits NLRP3 inflammasome activation and pyroptosis (155).
An intriguing question based on these observations is whether ripoptosome-associated caspase-8 directly engages inflammatory cell death and promotes disease. Indeed, caspase-8 is redundant with caspase-1 and is important to drive IL-1β-mediated disease in Pstpip2cmo-mediated osteomyelitis in mice, which is comparable to chronic multifocal osteomyelitis (CRMO) in humans (156–158). While caspase-8 is an apoptotic caspase, and apoptosis is considered immunologically silent, caspase-8 promotes maturation of IL-1β and inflammation, suggesting that apoptosis can engage inflammatory cell death (5, 159–161). BAX/BAK-dependent intrinsic apoptosis triggers activation of proinflammatory IL-1β and the NLRP3 inflammasome (162, 163). During BAX/BAK-dependent apoptosis, degradation of cIAPs triggers activation of caspase-8 in the ripoptosome complex. Caspase-8 then directly participates in the maturation of IL-1β and its secretion (Figure 3). However, gasdermin proteins play no role in this caspase-8-activated IL-1β release. Caspase-8 activation and function in the ripoptosome complex are tightly controlled by antiapoptotic proteins, cIAPs. When caspase-1 is deleted in macrophages, inflammasome activation engages caspase-8-dependent apoptosis through recruitment of caspase-8 and FADD to the inflammasome complex (26). However, secretion of IL-1β and IL-18 is suppressed in this inflammasome-dependent caspase-8 activation. Additionally, inflammasome activation in the absence of caspase-1 engages GSDMD-independent inflammatory cell death and release of IL-1 cytokines (164). Nevertheless, these studies reveal that caspase-8 directly participates in maturation and secretion of IL-1β and inflammatory cell death in macrophages. In contrast to these findings, in mouse dendritic cells (DCs), lack of caspase-8 expression activates the RIPK1-RIPK3 complex and its downstream signaling events to promote activation of the NLRP3 inflammasome and IL-1β release (165). This study indicates that, in DCs, the association of caspase-8 and FADD counteracts inflammasome activation. These findings suggest cell type-specific regulation of caspase-8, NLRP3 activation, and inflammation. Collectively, these emerging studies establish that caspase-8 and FADD can be components of the inflammasome complex and facilitate apoptosis or inflammatory cell death in immune cells. However, activation of caspase-1 by the NLRP3 inflammasome during apoptosis is ambiguous. During intrinsic apoptosis, caspase-3 and -7 are proposed to induce K+ efflux and NLRP3 activation, which in turn activates caspase-1-dependent activation of IL-1β (162, 163). In contrast to this observation, a recent study argues that pannexin-1-mediated K+ efflux promotes NLRP3 inflammasome activation during extrinsic apoptosis (166). Interestingly, GSDMD- or GSDME-mediated lysis of the mitochondria also releases cytochrome c and promotes apoptosis, illustrating the existence of a feedback apoptotic program, downstream of inflammasome activation (167).
Does caspase-8 in the ripoptosome (RIPK1–FADD–caspase-8) also engage gasdermin-dependent pyroptosis? Recent studies showed that caspase-8, FADD, and RIPK1 kinase activity promote activation of the NLRP3 inflammasome and pyroptosis in response to gram-negative bacteria or under conditions of TAK1 inactivation (23, 154). YopJ, an effector protein of Yersinia, inhibits TAK1 function, inducing cell death in macrophages (168, 169). YopJ induces RIPK1–FADD–caspase-8–dependent cell death and innate immune defense mechanisms during Yersinia infection (170, 171). TAK1 inactivation by YopJ activates RIPK1- and caspase-8-dependent cleavage of GSDMD and GSDME to engage pyroptosis (172, 173) (Figure 3). Caspase-8, in addition to caspase-1 or caspase-4/-5/-11, directly regulates cleavage and activation of gasdermin proteins to induce pyroptosis (172, 173). Although caspase-1 and -8 cleave GSDMD at the same site, the processing capacity of GSDMD by caspase-8 is much less than that by caspase-1 (166). Another study found that caspase-8 is dispensable for caspase-11-mediated pyroptosis, but both caspase-8 and -11 are required to drive LPS-induced lethality in mice (174). These observations indicate that unlike cleavage by inflammatory caspase-1 and -11, apoptotic caspase-8-mediated cleavage of gasdermin proteins is observed under specific conditions, such as TAK1 inactivation or Yersinia infection. Although caspase-8 is classified as an apoptotic caspase, these new findings discussed above highlight that caspase-8 can also exhibit inflammatory caspase functions by proteolytically processing GSDMD, GSDME, and IL-1β. Furthermore, the role of TNF and ripoptosome components in NLRP3 and pyrin inflammasomes illustrates an important theme of functional connectivity between cell death pathways and inflammation (23, 154, 172, 173, 175, 176). Perhaps, caspase-8 should be considered a new member of the inflammatory caspase group with dual functions in both apoptosis and inflammation. Identification of the roles of caspase-8, FADD, and RIPK1 in NLRP3 inflammasome activation and GSDMD-induced pyroptosis suggests that the ripoptosome complex directly regulates inflammatory responses in association with the inflammasome.
ENDOGENOUS INHIBITORY MECHANISMS OF CASPASE ACTIVATION
Caspases are constitutively expressed in mammalian cells. Caspase activity needs to be regulated at homeostatic conditions to prevent cellular damage and induction of unrestrained inflammatory responses. There are several endogenous proteins and intracellular signaling processes that act as regulatory switches to control the activity of caspases. IAPs (XIAP, cIAP1, and cIAP2) are antiapoptotic proteins that are highly conserved from yeast to mammals. They are E3-ubiquitin ligases containing baculovirus IAP repeat (BIR) domains. IAPs are endogenous inhibitors of apoptotic caspases both in vitro and in vivo that directly bind to caspase-3, -7, -8, and -9 to inhibit their activities (177–179). The BIR domains of IAPs are important for apoptotic caspase inhibition. Overexpression of cIAPs in multiple cancer types confers resistance of cancer cells to apoptotic cell death activation (179). Therapeutic inhibition of cIAPs activates apoptosis in cancer cells (179). The role of cIAPs in controlling apoptotic caspases is a homeostatic process to maintain cell survival. While there is no direct evidence demonstrating that cIAPs control inflammatory caspases, a few studies indicate a complex regulation process. cIAP1 and cIAP2 mediate efficient activation of caspase-1 by K63-linked polyubiquitination, and deletion of cIAP1 or cIAP2 inhibits inflammasome-induced caspase-1 activation (180). This study suggests that cIAPs promote inflammatory caspase activation, in contrast to their inhibitory role with apoptotic caspases (180). Whether cIAPs target the enzymatic activity of inflammatory caspases is still an open question to be answered.
FLIP is a DD superfamily protein that contains DEDs and acts as an inhibitor of apoptosis by interfering with DISC assembly (1, 21). FLIPs were originally identified in gammaherpesviruses and molluscipoxviruses and termed vFLIPs. Viral genome-encoded vFLIPs directly interact with caspase-8 or FADD to counteract assembly of the DISC complex (21). Mammalian cells also encode cFLIPs that are similar to vFLIPs. The long splice variant of cFLIP (cFLIPL) is a homolog of caspase-8 that has an additional caspase-like domain at its N terminus along with DEDs. cFLIPL forms a caspase-8-cFLIPL heteromeric complex in the ripoptosome (complex-II) and inhibits the apoptotic function of caspase-8 (1, 21). Caspase-8 retains low catalytic activity in this heteromeric complex and cleaves cFLIPL into P43-cFLIP, leading to the recruitment of TRAF2 and RIPK1 to the complex to activate NF-κB signaling. The proteolytically active caspase-8-cFLIPL heteromeric complex also inhibits RIPK3-mediated necroptosis and protects cells from cell death activation (25). Overall, cFLIP directly regulates cell death and the inflammatory functions of caspase-8 to maintain cellular homeostasis.
The scaffolding function of RIPK1 acts as a prosurvival checkpoint in response to several immune stimuli. Ubiquitination of RIPK1 confers RIPK1 scaffolding function to assemble a cell survival signaling complex at the cell membrane associated with TNFR. Importantly TNF stimulation in the absence of RIPK1 triggers degradation of cIAPs, cFLIPL, and TRAF2, which are essential to restrict the cell death activity of caspases (181, 182). Interruption of the RIPK1 scaffolding function leads to the recruitment of FADD and caspase-8 to RIPK1 to form the ripoptosome complex. RIPK1 kinase activity in the ripoptosome complex facilitates caspase-8-dependent cellular functions. Thus, the ripoptosome complex, once formed, acts as a springboard to promote cellular homeostasis or to induce inflammation or cell death. An intriguing speculation is that RIPK1 scaffolding function also directly engages cell death, which is not well understood. RIPK1 is one of only four mammalian proteins that contains a RIP-homotypic interaction motif (RHIM); ZBP1, RIPK3, and Toll/IL-1 receptor domain–containing adaptor protein–inducing IFN-β (TRIF) are the others (183, 184). The RHIM domain facilitates homotypic interactions among RHIM-containing proteins to form macromolecular signaling complexes. ZBP1 activates multiple programmed cell death pathways via activation of caspase-8 and RIPK3 in response to viral infections (70–72, 185). ZBP1 directly participates in the activation of caspase-1 and -8 to induce cell death and inflammation during viral infections. During embryonic development, RIPK1 restricts the ZBP1-dependent activation of necroptosis to prevent embryonic lethality (186, 187). This suggests that RIPK1 critically regulates ZBP1-dependent activation of inflammatory cell death responses to retain cellular homeostasis. In general, RIPK1-mediated regulation of cell death is operated through the ripoptosome complex; however, ZBP1 regulation is an exception, since ZBP1 and RIPK1 do not directly interact in endogenous settings (186, 187). Perhaps RIPK1 employs a putative ripoptosome-independent molecular mechanism to control ZBP1 function, which needs in-depth investigation.
Most of the above-described mechanisms regulate primarily the enzymatic activity of caspases. It is interesting to learn whether endogenous processes target DED/CARD-mediated oligomerization of caspases to put the brakes on proximity-mediated activation of caspases. Serine protease inhibitors are grouped into a protein superfamily called serpins (188). Serpin family members share a similar tertiary structure and inhibit the activity of caspases through direct interaction. Cowpox virus encodes a serpin protein, cytokine response modifier A (CrmA), that inhibits caspase-1 activity and the maturation of IL-1β (189). CrmA also inhibits caspase-8 function and restricts apoptosis induction (190). The human-genome-encoded serpin PI-9 shares similar amino acid sequence with CrmA and inhibits caspase-1 activity and the processing of IL-1β and IL-18 in human vascular smooth muscle cells (191). A recent study suggests that, in addition to their inhibition of caspase catalytic activity, serpin proteins also restrict oligomerization of caspases to further restrict their function (192). SERPINB1 is expressed mainly in monocytes and neutrophils, binds to inflammatory caspases (caspase-1, -4, -5, -11), and inhibits CARD domain oligomerization along with enzymatic activity (192). Deletion of SERPINB1 leads to the spontaneous activation of these inflammatory caspases, release of IL-1β, and execution of pyroptosis. Flightless-I (Fli-I), an actin-remodeling protein, can also regulate inflammatory caspase activity through direct association with these caspases (193). Leucine-rich repeat Fli-I-interacting protein 2 (LRRFIP2) facilitates the interaction of Fli-I with caspase-1 during NLRP3 inflammasome activation to inhibit caspase-1 activity and the release of IL-1β (194). A new study suggests that cAMP production in macrophages inhibits LPS-induced activation of caspase-11 and pyroptosis (195). cAMP activates protein kinase, which in turn phosphorylates caspase-11 to attenuate caspase-11 function, further exemplifying the intracellular mechanisms that restrict inflammatory caspases (195). It appears that mammalian cells employ distinct endogenous mechanisms to regulate apoptotic and inflammatory caspases. Further studies are required to fully understand how inflammatory caspases (particularly caspase-1 and -11) are controlled endogenously to inhibit their spontaneous activation.
THERAPEUTIC TARGETING OF CASPASES, CELL DEATH, AND INFLAMMATION
Cancer cells attenuate activation of apoptosis to inhibit clearance of malignant cells and facilitate tumor development. This finding has led to the discovery of several therapeutic molecules inducing proapoptotic effects in tumor cells. BCL-2 protein family members are expressed in all cell types and are antiapoptotic effectors that promote cell survival by modulating mitochondrial outer membrane permeabilization (196). Several clinical trials have been done to target the cell-intrinsic caspase-inhibiting mechanisms, including both BCL-2 family members and cIAPs, to therapeutically activate caspase function in cancer cells (54, 196). Therapeutic activation of caspases in cancer is extensively reviewed elsewhere (54, 196); therefore, we focus here on targeting inflammatory caspases and their effector molecules.
Inflammatory caspases and proteins involved in pyroptosis became primary targets for therapeutic design because of their critical role in inflammatory and infectious diseases. The well-defined molecular pathways of inflammatory caspases provide a multitude of therapeutic opportunities to target inflammatory diseases and cancer. These include direct modulation of caspase activity (197); targeting of the upstream signaling complexes, such as inflammasomes (198–202); and neutralization of the caspase substrates, such as IL-1β and IL-18 (152, 203). While directly targeting caspases or upstream mechanisms has been challenging, blocking IL-1β/IL-1R has proved successful, and three such therapeutic agents are approved by the US Food and Drug Administration for the treatment of inflammatory diseases including arthritis, and gain-of-function mutations causing periodic fever syndromes and inflammasomopathies. The approved agents are anakinra, an endogenous, natural IL-1R antagonist; rilonacept, a chimeric decoy receptor designed from the fusion of the ligand-binding domains of IL-1R and IL-1RAcP to the FC portion of human IgG1; and canakinumab, a fully humanized, high-molecular-weight anti-IL-1β antibody with an extended half-life of 26 days, which compares favorably to the 4-h half-life of anakinra in the blood (203, 204). Although chronic low-grade inflammation is a known and well-recognized etiologic agent driving metabolic diseases and cancer, the therapeutic potential to target this process remained poorly understood until recently. A recent clinical study, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS study, clinicaltrials.gov number NCT01327846), focused on the application of canakinumab in a large cohort of over 10,000 patients with atherosclerosis and showed that IL-1β neutralization not only provided atherosclerotic benefits but also significantly protected these patients from lung cancer development (152, 203, 205).
As discussed above, activation of inflammatory caspases results in a lytic form of cell death, releasing several DAMPs and potent inflammatory mediators with the potential to drive immunopathology and inflammatory disease onset. Consistent with this notion, recent studies focused on blocking or eliminating the terminal executioners of pyroptosis have succeeded in providing therapeutic benefits. One study designed a peptide inhibitor, N-acetyl-Phe-Leu-Thr-Asp-chloromethylketone (Ac-FLTD-CMK), to mimic the caspase cleavage site of GSDMD (206). This inhibitor blocks cleavage of GSDMD by inflammatory caspases and inhibits pyroptosis and secretion of IL-1β (206). In addition, a chemical inhibitor of necroptosis, necrosulfonamide (NSA), also inhibits pyroptosis by directly interacting with the Cys191 residue of human GSDMD, further highlighting gasdermin blockade as a potential therapeutic strategy (207). Disulfiram, a drug used to treat alcohol addiction, and Bay 11–7082, a known NF-κB inhibitor, also inhibit inflammation and pyroptosis (208). These molecules, similar to NSA, target Cys191 of human GSDMD (Cys192 of mouse GSDMD), which is essential for its pore-forming activity. Of note, these compounds also directly inhibit inflammatory caspases activated by both canonical and noncanonical inflammasomes because of the critical function of the active site Cys in these caspases. The chemical inhibitors of caspases available to date primarily target their enzymatic activity. A potential caveat of these caspase inhibitors is their nonspecific targeting and tendency to inhibit the function of multiple caspases. Effective inflammatory caspase-specific therapeutic inhibitors are desired to target inflammatory and infectious diseases.
CONCLUDING REMARKS
Caspase activation is an evolutionarily conserved mechanism critical not only for organismal development but also for immune cell death and inflammation. The decisive potential of caspases in dismantling cells and driving potent inflammatory responses has necessitated the evolution of intricate caspase-regulatory mechanisms, which include the production of caspases as inactive zymogens and the requirement for the assembly of well-coordinated signaling platforms such as the apoptosome, ripoptosome, and inflammasome. Although early studies broadly accepted the paradigm of multiprotein signaling complexes being essential platforms for proximity-induced activation of initiator caspases, the last decade has witnessed tremendous progress in the field and revealed the existence of direct ligand binding and activation mechanisms, as seen in the case of inflammatory caspase-4 and -5 in humans and caspase-11 in mice. Recent studies have improved our understanding of the mechanisms of caspase-mediated inflammatory cell death pyroptosis, and the cross talk between apoptosis, pyroptosis, and necroptosis. Most importantly, these studies also revealed the critical role of caspase-mediated activation and inactivation of the gasdermin family of proteins and their essential role in lysis of cell membranes and subsequent inflammatory cell death. Another area of exponential progress has been the therapeutic targeting of inflammasomes and their substrates for better human health. The vast improvement in our knowledge of caspase-mediated cell death, immune mechanisms, and novel substrates fosters great hope for novel therapeutic opportunities in the future.
FUTURE DIRECTIONS.
Although proximity is known to induce activation of the initiator caspases, independent of their protease function, the molecular mechanism behind this nonproteolytic activation is poorly understood, and future studies should address the precise structural and biochemical requirements for this phenomenon.
Recent progress has revealed extensive cross talk between apoptotic and inflammatory caspases, yet it is not clear how cells avoid inflammatory cell death during the conditions of apoptosis and vice versa.
Future studies should delineate the nonenzymatic scaffolding functions of caspases to regulate cellular homeostasis and inflammation.
Apoptotic caspases are now increasingly recognized to engage pyroptosis; however, their physiological relevance in response to infection and/or metabolic diseases needs further investigation.
Do human/murine cells contain unknown pyroptosis executioners apart from gasdermin proteins? Which pyroptotic executioners are employed in epithelial cells/keratinocytes?
ACKNOWLEDGMENTS
Research investigations from our laboratory are supported by the US National Institutes of Health (AR056296, CA163507, AI124346, and AI101935 to T.-D.K.) and the American Lebanese Syrian Associated Charities (to T.-D.K.). Scientific editing and writing support were provided by Rebecca Tweedell, PhD. The authors of this review apologize to colleagues whose work was not cited owing to space limitations.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Pasparakis M, Vandenabeele P. 2015. Necroptosis and its role in inflammation. Nature 517:311–20 [DOI] [PubMed] [Google Scholar]
- 2.Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol 7:99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Man SM, Karki R, Kanneganti TD. 2017. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev 277:61–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, et al. 1996. Human ICE/CED-3 protease nomenclature. Cell 87:171. [DOI] [PubMed] [Google Scholar]
- 5.Man SM, Kanneganti TD. 2016. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol 16:7–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamkanfi M, Declercq W, Kalai M, Saelens X, Vandenabeele P. 2002. Alice in caspase land: a phylogenetic analysis of caspases from worm to man. Cell Death Differ. 9:358–61 [DOI] [PubMed] [Google Scholar]
- 7.Ramirez MLG, Salvesen GS. 2018. A primer on caspase mechanisms. Semin. Cell Dev. Biol 82:79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y 2004. Caspase activation: revisiting the induced proximity model. Cell 117:855–58 [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L, Lopez-Soto A, Kumar S, Kroemer G. 2016. Caspases connect cell-death signaling to organismal homeostasis. Immunity 44:221–31 [DOI] [PubMed] [Google Scholar]
- 10.Kesavardhana S, Kanneganti TD. 2017. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol 29:201–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fava LL, Schuler F, Sladky V, Haschka MD, Soratroi C, et al. 2017. The PIDDosome activates p53 in response to supernumerary centrosomes. Genes Dev. 31:34–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tinel A, Tschopp J. 2004. The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304:843–46 [DOI] [PubMed] [Google Scholar]
- 13.Wang XJ, Cao Q, Zhang Y, Su XD. 2015. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol 55:553–72 [DOI] [PubMed] [Google Scholar]
- 14.Guo H, Albrecht S, Bourdeau M, Petzke T, Bergeron C, LeBlanc AC. 2004. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease.Am. J. Pathol 165:523–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruchaud S, Korfali N, Villa P, Kottke TJ, Dingwall C, et al. 2002. Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 21:1967–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckhart L, Declercq W, Ban J, Rendl M, Lengauer B, et al. 2000Terminal differentiation of human keratinocytes and stratum corneum formation is associated with caspase-14 activation. J. Investig. Dermatol 115:1148–51 [DOI] [PubMed] [Google Scholar]
- 17.Lippens S, Kockx M, Knaapen M, Mortier L, Polakowska R, et al. 2000. Epidermal differentiation does not involve the pro-apoptotic executioner caspases, but is associated with caspase-14 induction and processing. Cell Death Differ. 7:1218–24 [DOI] [PubMed] [Google Scholar]
- 18.Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10:417–26 [DOI] [PubMed] [Google Scholar]
- 19.Park HH,Lo YC, Lin SC,Wang L,Yang JK,Wu H. 2007The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol 25:561–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry CM, Martin SJ. 2017. Caspase-8 acts in a non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol. Cell 65:715–29.e5 [DOI] [PubMed] [Google Scholar]
- 21.Budd RC, Yeh WC,Tschopp J. 2006. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol 6:196–204 [DOI] [PubMed] [Google Scholar]
- 22.Kang S, Fernandes-Alnemri T, Rogers C, Mayes L,Wang Y, et al. 2015. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun 6:7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, et al. 2014. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol 192:1835–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, et al. 2017. AIM2 engages active but unprocessed caspase-1 to induce noncanonical activation of the NLRP3 inflammasome. Cell Rep 20:794–805 [DOI] [PubMed] [Google Scholar]
- 25.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, et al. 2011. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471:363–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van Opdenbosch N, Van Gorp H, Verdonckt M, Saavedra PHV, de Vasconcelos NM, et al. 2017. Caspase-1 engagement and TLR-induced c-FLIP expression suppress ASC/caspase-8-dependent apoptosis by inflammasome sensors NLRP1b and NLRC4. Cell Rep. 21:3427–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boatright KM, Renatus M, Scott FL, Sperandio S, ShinH, et al. 2003. A unified model for apical caspase activation. Mol. Cell 11:529–41 [DOI] [PubMed] [Google Scholar]
- 28.Chang DW, Xing Z, Capacio VL, Peter ME, Yang X. 2003. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 22:4132–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muzio M, Stockwell BR, Stennicke HR, Salvesen GS, Dixit VM. 1998. An induced proximity model for caspase-8 activation. J. Biol. Chem 273:2926–30 [DOI] [PubMed] [Google Scholar]
- 30.Kerr JF, Wyllie AH, Currie AR. 1972. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26:239–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol 9:231–41 [DOI] [PubMed] [Google Scholar]
- 32.Arandjelovic S, Ravichandran KS. 2015. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol 16:907–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McIlwain DR, Berger T, Mak TW. 2013. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol 5:a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parrish AB, Freel CD, Kornbluth S. 2013. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol a008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol 11:621–32 [DOI] [PubMed] [Google Scholar]
- 36.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. 2002. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol. Cell 9:423–32 [DOI] [PubMed] [Google Scholar]
- 37.Cain K, Bratton SB, Cohen GM. 2002. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie 84:203–14 [DOI] [PubMed] [Google Scholar]
- 38.Shiozaki EN, Chai J, Shi Y. 2002. Oligomerization and activation of caspase-9, induced by Apaf-1 CARD. PNAS 99:4197–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiozaki EN, Shi Y. 2004. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem. Sci 29:486–94 [DOI] [PubMed] [Google Scholar]
- 40.Kondylis V, Kumari S, Vlantis K, Pasparakis M.2017The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev 277:113–27 [DOI] [PubMed] [Google Scholar]
- 41.Peltzer N,Walczak H. 2019. Cell death and inflammation—a vital but dangerous liaison.Trends Immunol 40:387–402 [DOI] [PubMed] [Google Scholar]
- 42.Ting AT, Bertrand MJM. 2016. More to Life than NF-γB in TNFR1 signaling.Trends Immunol 37:535–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horn S, Hughes MA,Schilling R, Sticht C,Tenev T, et al. 2017. Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep 19:785–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamy L, Ngo VN, Emre NC, Shaffer AL 3rd, Yang Y, et al. 2013. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell 23:435–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J, Zheng L, Lobito A, Chan FK, Dale J, et al. 1999. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell 98(1):47–58 [DOI] [PubMed] [Google Scholar]
- 46.Chun HJ, Zheng L, Ahmad M,Wang J, Speirs CK, et al. 2002. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419(6905):395–99 [DOI] [PubMed] [Google Scholar]
- 47.Juo P, Kuo CJ, Yuan J, Blenis J. 1998. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol 8:1001–8 [DOI] [PubMed] [Google Scholar]
- 48.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9:267–76 [DOI] [PubMed] [Google Scholar]
- 49.Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, et al. 2000. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12:633–42 [DOI] [PubMed] [Google Scholar]
- 50.Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, et al. 2009. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 460:1035–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olsson M, Vakifahmetoglu H, Abruzzo PM, Hogstrand K, Grandien A, Zhivotovsky B. 2009. DISC-mediated activation of caspase-2 in DNA damage-induced apoptosis. Oncogene 28:1949–59 [DOI] [PubMed] [Google Scholar]
- 52.Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, et al. 2008. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133:864–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Newton K, Manning G. 2016. Necroptosis and Inflammation. Annu. Rev. Biochem 85:743–63 [DOI] [PubMed] [Google Scholar]
- 54.Bertrand MJ, Milutinovic S, Dickson KM,Ho WC,Boudreault A, et al. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30:689–700 [DOI] [PubMed] [Google Scholar]
- 55.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, et al. 2008. Both cIAP1 and cIAP2 regulate TNFα-mediated NF-κB activation. PNAS 105:11778–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, et al. 2011. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43:449–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, et al. 2011. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43:432–48 [DOI] [PubMed] [Google Scholar]
- 58.Green DR. 2019. The coming decade of cell death research: five riddles. Cell 177:1094–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Opdenbosch N, Lamkanfi M. 2019. Caspases in cell death, inflammation, and disease. Immunity 50:1352–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalai M, Van Loo G, Vanden Berghe T, Meeus A, Burm W, et al. 2002. Tipping the balance between necrosis and apoptosis in human and murine cells treated with interferon and dsRNA. Cell Death Differ 9:981–94 [DOI] [PubMed] [Google Scholar]
- 61.Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, et al. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279:1954–58 [DOI] [PubMed] [Google Scholar]
- 62.Zhang J, Cado D, Chen A, Kabra NH,Winoto A. 1998. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392:296–300 [DOI] [PubMed] [Google Scholar]
- 63.Bossaller L, Chiang PI, Schmidt-Lauber C, Ganesan S, Kaiser WJ,et al. 2012. Cutting edge: FAS (CD95) mediates noncanonical IL-1β and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol 189:5508–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovalenko A, Kim JC, Kang TB, Rajput A, Bogdanov K, et al. 2009. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J. Exp. Med 206:2161–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kang TB, Ben-Moshe T,Varfolomeev EE, Pewzner-Jung Y, Yogev N, et al. 2004. Caspase-8 serves both apoptotic and nonapoptotic roles. J. Immunol 173:2976–84 [DOI] [PubMed] [Google Scholar]
- 66.Li C, Lasse S, Lee P, Nakasaki M, Chen SW, et al. 2010. Development of atopic dermatitis-like skin disease from the chronic loss of epidermal caspase-8. PNAS 107:22249–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, et al. 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471:368–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. 2011. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471:373–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, et al. 2011. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell Biol 13:1437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, et al. 2017. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med 214:2217–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, et al. 2016. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1:aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Upton JW, Kaiser WJ, Mocarski ES. 2012DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chu LH, Indramohan M, Ratsimandresy RA, Gangopadhyay A, Morris EP, et al. 2018. The oxidized phospholipid oxPAPC protects from septic shock by targeting the non-canonical inflammasome in macrophages. Nat. Commun 9:996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, et al. 2016. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352:1232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi J, Gao W, Shao F. 2017. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci 42:245–54 [DOI] [PubMed] [Google Scholar]
- 76.Boyden ED, Dietrich WF. 2006. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet 38:240–44 [DOI] [PubMed] [Google Scholar]
- 77.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, et al. 2006. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440:233–36 [DOI] [PubMed] [Google Scholar]
- 78.Mariathasan S,Weiss DS, Newton K, McBride J, O’Rourke K, et al. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–32 [DOI] [PubMed] [Google Scholar]
- 79.Martinon F, Agostini L, Meylan E, Tschopp J. 2004. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr. Biol 14:1929–34 [DOI] [PubMed] [Google Scholar]
- 80.Martinon F, Petrilli V, Mayor A,Tardivel A,Tschopp J. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–41 [DOI] [PubMed] [Google Scholar]
- 81.Franchi L,Amer A, Body-Malapel M,Kanneganti TD,Ozoren N, et al. 2006. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol 7:576–82 [DOI] [PubMed] [Google Scholar]
- 82.Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, et al. 2006. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol 7:569–75 [DOI] [PubMed] [Google Scholar]
- 83.Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, et al. 2010. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. PNAS 107:3076–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, et al. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600 [DOI] [PubMed] [Google Scholar]
- 85.Fernandes-Alnemri T, Yu JW, Datta P,Wu J, Alnemri ES. 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, et al. 2009. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, et al. 2009. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol 10:266–72 [DOI] [PubMed] [Google Scholar]
- 88.Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, et al. 2009. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–60 [DOI] [PubMed] [Google Scholar]
- 89.Chae JJ,Wood G, Masters SL, Richard K, Park G, et al. 2006. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1β production. PNAS 103:9982–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu H, Yang J, Gao W, Li L, Li P, et al. 2014. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513:237–41 [DOI] [PubMed] [Google Scholar]
- 91.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. 2018. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48:35–44.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Black RA, Kronheim SR, Sleath PR. 1989. Activation of interleukin-1β by a co-induced protease. FEBS Lett. 247:386–90 [DOI] [PubMed] [Google Scholar]
- 93.Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, et al. 1989. Identification of a monocyte specific pre-interleukin 1β convertase activity. PNAS 86:5227–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kayagaki N,Warming S, Lamkanfi M, Vande Walle L, Louie S, et al. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479:117–21 [DOI] [PubMed] [Google Scholar]
- 95.Shi J, Zhao Y,Wang Y, Gao W, Ding J, et al. 2014. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514:187–92 [DOI] [PubMed] [Google Scholar]
- 96.Lamkanfi M, Dixit VM. 2014. Mechanisms and functions of inflammasomes. Cell 157:1013–22 [DOI] [PubMed] [Google Scholar]
- 97.Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, et al. 2003. Regulation of the expression and processing of caspase-12. J. Cell Biol 162:457–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nakagawa T, Yuan J. 2000. Cross-talk between two cysteine protease families: activation of caspase-12 by calpain in apoptosis. J. Cell Biol 150:887–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Di Sano F, Ferraro E, Tufi R, Achsel T, Piacentini M, Cecconi F. 2006. Endoplasmic reticulum stress induces apoptosis by an apoptosome-dependent but caspase 12-independent mechanism. J. Biol. Chem 281:2693–700 [DOI] [PubMed] [Google Scholar]
- 100.Nakagawa T, Zhu H, Morishima N, Li E,Xu J, et al. 2000. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403:98–103 [DOI] [PubMed] [Google Scholar]
- 101.Saleh M, Mathison JC,Wolinski MK, Bensinger SJ, Fitzgerald P, et al. 2006. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440:1064–68 [DOI] [PubMed] [Google Scholar]
- 102.Salvamoser R, Brinkmann K, O’Reilly LA, Whitehead L, Strasser A,Herold MJ. 2019. Characterisation of mice lacking the inflammatory caspases-1/11/12 reveals no contribution of caspase-12 to cell death and sepsis. Cell Death Differ 26:1124–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vande Walle L, Jimenez Fernandez D, Demon D, Van Laethem N, Van Hauwermeiren F, et al. 2016. Does caspase-12 suppress inflammasome activation? Nature 534:E1–4 [DOI] [PubMed] [Google Scholar]
- 104.Fink SL, Cookson BT. 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 8:1812–25 [DOI] [PubMed] [Google Scholar]
- 105.Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, et al. 1998. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem 273:32895–900 [DOI] [PubMed] [Google Scholar]
- 106.Monack DM, Raupach B, Hromockyj AE, Falkow S. 1996. Salmonella typhimurium invasion induces apoptosis in infected macrophages. PNAS 93:9833–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.VandeWalle L, Lamkanfi M. 2016. Pyroptosis. Curr. Biol 26:R568–72 [DOI] [PubMed] [Google Scholar]
- 108.Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, et al. 2012. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490:288–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kang SJ,Wang S, Hara H, Peterson EP, Namura S, et al. 2000. Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J. Cell Biol 149:613–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, et al. 1996. Identification and characterization of Ich-3, a member of the interleukin-1β converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J. Biol. Chem 271:20580–87 [DOI] [PubMed] [Google Scholar]
- 111.Wang J, Sahoo M, Lantier L,Warawa J, Cordero H, et al. 2018. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLOS Pathog 14:e1007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, et al. 2014. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505:509–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Monroe KM, Yang Z, Johnson JR, Geng X, Doitsh G, et al. 2014. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 343:428–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, et al. 2015. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–71 [DOI] [PubMed] [Google Scholar]
- 115.Shi J, Zhao Y, Wang K, Shi X, Wang Y, et al. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–65 [DOI] [PubMed] [Google Scholar]
- 116.Agard NJ, Maltby D,Wells JA. 2010. Inflammatory stimuli regulate caspase substrate profiles. Mol. Cell Proteom 9:880–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O’Rourke K, et al. 2019. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal 12:eaax4917. [DOI] [PubMed] [Google Scholar]
- 118.Feng S, Fox D, Man SM. 2018. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J. Mol. Biol 430:3068–80 [DOI] [PubMed] [Google Scholar]
- 119.Kovacs SB, Miao EA. 2017. Gasdermins: effectors of pyroptosis. Trends Cell Biol 27:673–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang Y, Gao W, Shi X, Ding J, Liu W, et al. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547:99–103 [DOI] [PubMed] [Google Scholar]
- 121.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. 2017. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, et al. 2016. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. PNAS 113:7858–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ding J,Wang K, Liu W, She Y, Sun Q, et al. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535:111–16 [DOI] [PubMed] [Google Scholar]
- 124.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, et al. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, et al. 2016. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35:1766–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ. 2018. Mechanism of membrane pore formation by human gasdermin-D. EMBO J 37:e98321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, et al. 2019. Calpain drives pyroptotic vimentin cleavage, intermediate filament loss, and cell rupture that mediates immunostimulation. PNAS 116:5061–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. 2019. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 26:146–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nakanishi K, Maruyama M, Shibata T, Morishima N. 2001. Identification of a caspase-9 substrate and detection of its cleavage in programmed cell death during mouse development. J. Biol. Chem 276:41237–44 [DOI] [PubMed] [Google Scholar]
- 130.Byun Y, Chen F, Chang R,Trivedi M, Green KJ, Cryns VL. 2001. Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 8:443–50 [DOI] [PubMed] [Google Scholar]
- 131.Kambara H, Liu F, Zhang X, Liu P, Bajrami B, et al. 2018. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 22:2924–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, et al. 2018. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol 3:eaar6689. [DOI] [PubMed] [Google Scholar]
- 133.Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, et al. 2018. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol 3:eaar6676. [DOI] [PubMed] [Google Scholar]
- 134.Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, et al. 2016. Inflammasome dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 23:1219–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, et al. 2015. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J. Immunol 194:1763–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ,Tschopp J, et al. 2014. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 8:570–82 [DOI] [PubMed] [Google Scholar]
- 137.Ruan J, Xia S, Liu X, Lieberman J, Wu H. 2018. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu Z, Wang C, Yang J, Zhou B, Yang R, et al. 2019. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51:43–49.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu Z,Wang C, Rathkey JK, Yang J, Dubyak GR, et al. 2018. Structures of the gasdermin D C-terminal domains reveal mechanisms of autoinhibition. Structure 26:778–84.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuang S, Zheng J, Yang H, Li S, Duan S, et al. 2017. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. PNAS 114:10642–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu Z, Wang C, Yang J, Zhou B, Yang R, et al. 2019. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51(1):43–49.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ruhl S, Broz P. 2015. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K+ efflux. Eur. J. Immunol 45:2927–36 [DOI] [PubMed] [Google Scholar]
- 143.Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, et al. 2018. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 49:413–26.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhu Q, Zheng M,Balakrishnan A, Karki R, Kanneganti TD.2018. Gasdermin D promotes AIM2 inflammasome activation and is required for host protection against Francisella novicida. J. Immunol 201:3662–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. 2018ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362:956–60 [DOI] [PubMed] [Google Scholar]
- 146.Gong YN, Guy C, Olauson H, Becker JU, Yang M, et al. 2017. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169:286–300.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, et al. 2018. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. J. Exp. Med 215:1519–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xiao J,Wang C,Yao JC, Alippe Y,Xu C, et al. 2018. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLOS Biol. 16:e3000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dubois H, Sorgeloos F, Sarvestani ST, Martens L, Saeys Y, et al. 2019. Nlrp3 inflammasome activation and Gasdermin D-driven pyroptosis are immunopathogenic upon gastrointestinal norovirus infection. PLOS Pathog. 15:e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wu C, Lu W, Zhang Y, Zhang G, Shi X, et al. 2019. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 50:1401–11.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Karki R, Lee E, Place D, Samir P, Mavuluri J, et al. 2018. IRF8 regulates transcription of Naips for NLRC4 inflammasome activation. Cell 173:920–33.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karki R, Kanneganti TD. 2019. Diverging inflammasome signals in tumorigenesis and potential targeting. Nat. Rev. Cancer 19:197–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang WJ, Chen D, Jiang MZ, Xu B, Li XW, et al. 2018. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J. Dig. Dis 19:74–83 [DOI] [PubMed] [Google Scholar]
- 154.Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, et al. 2018. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med 215:1023–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, et al. 2019. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573:590–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, et al. 2014. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516:246–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cassel SL, Janczy JR, Bing X, Wilson SP, Olivier AK, et al. 2014. Inflammasome-independent IL-1β mediates autoinflammatory disease in Pstpip2-deficient mice. PNAS 111:1072–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lukens JR, Gross JM, Calabrese C, Iwakura Y, Lamkanfi M, et al. 2014. Critical role for inflammasome-independent IL-1β production in osteomyelitis. PNAS 111:1066–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Maelfait J, Vercammen E, Janssens S, Schotte P, Haegman M, et al. 2008. Stimulation of Toll-like receptor 3 and 4 induces interleukin-1β maturation by caspase-8. J. Exp. Med 205:1967–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gurung P,Lamkanfi M,Kanneganti TD.2015. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol 194:2064–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rickard JA, Anderton H, Etemadi N,Nachbur U, Darding M, et al. 2014. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 3:e03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chauhan D, Bartok E, Gaidt MM,Bock FJ,Herrmann J, et al. 2018. BAX/BAK-induced apoptosis results in caspase-8-dependent IL-1β maturation in macrophages. Cell Rep 25:2354–68.e5 [DOI] [PubMed] [Google Scholar]
- 163.Vince JE, De Nardo D, Gao W, Vince AJ, Hall C, et al. 2018. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1β activation. Cell Rep 25:2339–53.e4 [DOI] [PubMed] [Google Scholar]
- 164.Schneider KS, Gross CJ, Dreier RF, Saller BS, Mishra R, et al. 2017. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep 21:3846–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kang TB, Yang SH, Toth B, Kovalenko A,Wallach D. 2013. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38:27–40 [DOI] [PubMed] [Google Scholar]
- 166.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, et al. 2019. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38:e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. 2019. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun 10:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mukherjee S, Keitany G, Li Y,Wang Y, Ball HL, et al. 2006. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 312:1211–4 [DOI] [PubMed] [Google Scholar]
- 169.Paquette N, Conlon J, Sweet C, Rus F,Wilson L, et al. 2012. Serine/threonine acetylation of TGF beta-activated kinase (TAK1) by Yersinia pestis YopJ inhibits innate immune signaling. PNAS 109:12710–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Philip NH, Dillon CP, Snyder AG, Fitzgerald P,Wynosky-Dolfi MA, et al. 2014. Caspase-8 mediates caspase-1 processing and innate immune defense in response to bacterial blockade of NF-kappaB and MAPK signaling. PNAS 111:7385–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, et al. 2014. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. PNAS 111:7391–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Orning P, Weng D, Starheim K, Ratner D, Best Z, et al. 2018. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362:1064–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, et al. 2018. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. PNAS 115:E10888–E97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, et al. 2018. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity 49:42–55 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sharma D, Malik A, Guy C, Vogel P, Kanneganti TD. 2019. TNF/TNFR axis promotes pyrin inflammasome activation and distinctly modulates pyrin inflammasomopathy. J. Clin. Invest 129:150–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, et al. 2017. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J. Clin. Invest 127:4488–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yang YL, Li XM. 2000. The IAP family: endogenous caspase inhibitors with multiple biological activities. Cell Res 10:169–77 [DOI] [PubMed] [Google Scholar]
- 178.Zhang J,Webster JD, Dugger DL, Goncharov T, Roose-Girma M, et al. 2019. Ubiquitin ligases cIAP1 and cIAP2 limit cell death to prevent inflammation. Cell Rep 27:2679–89 e3 [DOI] [PubMed] [Google Scholar]
- 179.Fulda S, Vucic D. 2012. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov 11:109–24 [DOI] [PubMed] [Google Scholar]
- 180.Labbe K, McIntire CR, Doiron K, Leblanc PM, Saleh M. 2011. Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity 35:897–907 [DOI] [PubMed] [Google Scholar]
- 181.Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, et al. 2014. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513:90–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Takahashi N, Vereecke L, Bertrand MJ, Duprez L, Berger SB, et al. 2014. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513:95–9 [DOI] [PubMed] [Google Scholar]
- 183.Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O. 1999. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 240:157–63 [DOI] [PubMed] [Google Scholar]
- 184.Rebsamen M, Heinz LX, Meylan E, Michallet MC, Schroder K, et al. 2009. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 10:916–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Thapa RJ, Ingram JP, Ragan KB,Nogusa S, Boyd DF, et al. 2016DAI senses influenza A virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20:674–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, et al. 2016. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540:124–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Newton K,Wickliffe KE, Maltzman A, Dugger DL, Strasser A, et al. 2016. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540:129–33 [DOI] [PubMed] [Google Scholar]
- 188.Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, et al. 2006. An overview of the serpin superfamily. Genome Biol. 7:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ray CA, Black RA, Kronheim SR, Greenstreet TA, Sleath PR, et al. 1992. Viral inhibition of inflammation: Cowpox virus encodes an inhibitor of the interleukin-1 beta converting enzyme. Cell 69:597–604 [DOI] [PubMed] [Google Scholar]
- 190.Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS. 1997. Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J. Biol. Chem 272:7797–800 [DOI] [PubMed] [Google Scholar]
- 191.Young JL, Sukhova GK, Foster D, Kisiel W, Libby P, Schonbeck U. 2000. The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1beta-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med 191:1535–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Choi YJ, Kim S, Choi Y, Nielsen TB, Yan J, et al. 2019. SERPINB1-mediated checkpoint of inflammatory caspase activation. Nat. Immunol 20:276–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Li J, Yin HL, Yuan J. 2008. Flightless-I regulates proinflammatory caspases by selectively modulating intracellular localization and caspase activity. J. Cell Biol 181:321–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Jin J, Yu Q,Han C, Hu X, Xu S, et al. 2013. LRRFIP2 negatively regulates NLRP3 inflammasome activation in macrophages by promoting Flightless-I-mediated caspase-1 inhibition. Nat. Commun 4:2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen R, Zeng L, Zhu S, Liu J, Zeh HJ, et al. 2019cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv 5:eaav5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fuentes-Prior P, Salvesen GS. 2004. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem. J 384:201–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.MacKenzie SH, Schipper JL, Clark AC. 2010. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Devel 13:568–76 [PMC free article] [PubMed] [Google Scholar]
- 198.Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. 2018. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov 17:588–606 [DOI] [PubMed] [Google Scholar]
- 199.Perera AP, Fernando R, Shinde T, Gundamaraju R, Southam B, et al. 2018. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep 8:8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lamkanfi M, Dixit VM. 2017. A new lead to NLRP3 inhibition. J. Exp. Med 214:3147–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Ahn H, Kang SG, Yoon SI, Ko HJ, Kim PH, et al. 2017. Methylene blue inhibits NLRP3, NLRC4, AIM2, and non-canonical inflammasome activation. Sci. Rep 7:12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, et al. 2009. Glyburide inhibits the Cryopyrin/ Nalp3 inflammasome. J. Cell Biol 187:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Van Gorp H, Lamkanfi M. 2019. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 20:e47575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, et al. 2009. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med 360:2416–25 [DOI] [PubMed] [Google Scholar]
- 205.Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, et al. 2017. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390:1833–42 [DOI] [PubMed] [Google Scholar]
- 206.Yang J, Liu Z, Wang C, Yang R, Rathkey JK, et al. 2018. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. PNAS 115:6792–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, et al. 2018. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol 3:eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hu JJ, Liu X, Zhao J, Xia S, Ruan J, et al. 2019. Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D. bioRxiv 365908. 10.1101/36590 [DOI] [Google Scholar]